

Abstract
Purpose of review.
The purpose of this review is to provide a brief summary of recent advances in our understanding of liver metabolism. The critical role of the liver in controlling whole body energy homeostasis makes such understanding crucial to efficiently design new treatments for metabolic syndrome diseases, including Type 2 Diabetes (T2D).
Recent findings.
Significant advances have been made regarding our understanding of the direct and indirect effects of insulin on hepatic metabolism and the communication between the liver and other tissues. Moreover, the catabolic functions of glucagon, as well as the importance of hepatic redox status for the regulation of glucose production, are emerging as potential targets to reduce hyperglycemia.
Summary.
A resolution to the long-standing question “insulin suppression of hepatic glucose production, direct or indirect effect?” is starting to emerge. New advances in our understanding of important fasting-induced hepatic metabolic fluxes may help design better therapies for T2D.
Keywords: Liver, insulin, glucagon, glucose homeostasis, diabetes, metabolism
Introduction
Maintenance of blood glucose at a relatively constant concentration in healthy individuals requires an orchestrated response to fasting and fed signals. This response is perturbed in T2D, one of the most challenging medical conditions of the 21st century. T2D is expected to affect over 50 million people in the United States with an associated economic burden of >$600 billion by 2030 [1]. Fasting hyperglycemia characteristic of T2D is largely a result of the inability of insulin to control blood glucose, a phenomenon termed insulin resistance [2, 3].
The liver plays a crucial role in regulating metabolic homeostasis; perturbations in liver metabolism have major implications for systemic glucose and lipid homeostasis [4, 5]. During fasting, high glucagon levels stimulate hepatic glucose production (HGP) by promoting glycogen breakdown (glycogenolysis) and de-novo glucose synthesis from 3-carbon precursors (gluconeogenesis). Following a meal, elevated levels of insulin signal the liver to accumulate glycogen (glycogenesis), suppress HGP and synthesize triglycerides (TG) for storage. Mechanistically, the hormonal regulation of hepatic metabolism includes allosteric activation/inhibition, changes in metabolic fluxes and transcriptional regulation of key gluconeogenic/lipogenic components to support long-term activity of these pathways [6, 4, 7, 8]. In this review we will describe recent advances in our understanding of how insulin and glucagon regulate liver metabolism under normal physiology as well as under pathophysiological conditions such as T2D.
Insulin regulation of liver metabolism
Insulin regulates HGP both directly and indirectly.
Insulin, secreted by β cells in the pancreas, is considered the master anabolic hormone as it controls the mobilization and storage of carbohydrates, lipids and amino acids in basically all metabolic tissues. Insulin resistance is a hallmark of T2D and other components of the metabolic syndrome, which reflects inability of insulin to appropriately stimulate glucose uptake and to suppress HGP, contributing to hyperglycemia [9, 2, 10, 11]. The direct effects of insulin in hepatocytes includes stimulation of glycogen deposition, de-novo lipogenesis (DNL), and TG accumulation, and suppression of glycogenolysis, fatty acid oxidation (FAO) and gluconeogenic gene expression [11, 12]. Following binding of insulin to its cognate receptor, a downstream cascade of signaling is initiated; phosphorylation of insulin receptor substrates (IRSs) and recruitment of PI 3-kinase leads to the generation of phosphatidylinositol (3,4,5)-triphosphate (PIP3) and recruitment of Akt and PDK1 to the membrane. Subsequent phosphorylation-induced activation of Akt by PDK1 and mTORC2 mediates many of the direct metabolic effects of insulin in hepatocytes [7]. These include (1) stimulation of glycogen synthesis by promoting glycolytic flux and releasing the GSK-3β inhibition of glycogen synthase, (2) suppression of FoxO1-driven expression of gluconeogenic genes by Akt-mediated nuclear exclusion, and (3) stimulation of lipogenesis by increasing SREBP1c activity and glycolytic flux (Fig. 1).
Figure 1. The effects of insulin and glucagon on hepatic metabolism.
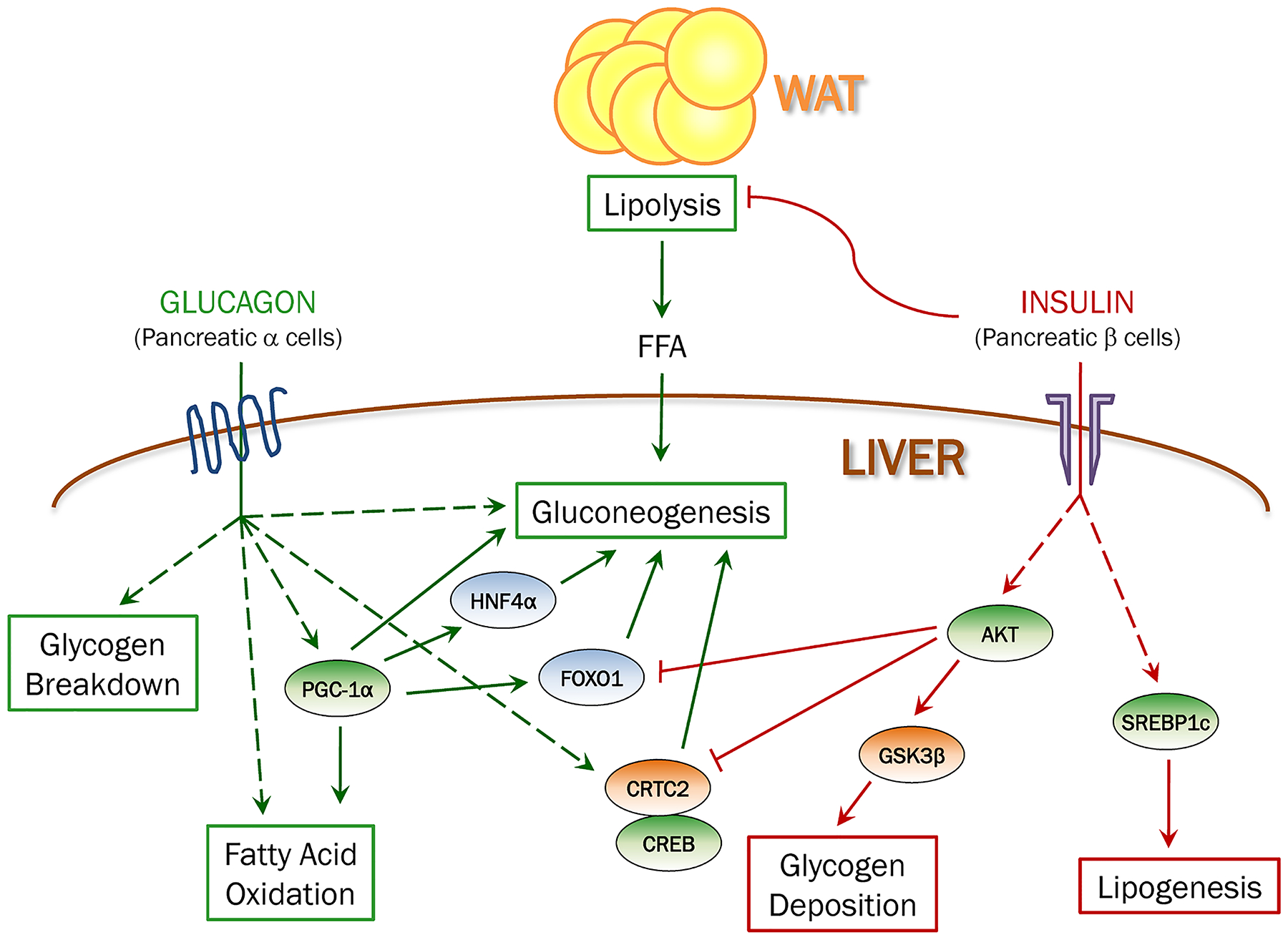
Insulin suppresses hepatic glucose production both directly by inhibiting glycogen breakdown and gluconeogenesis, and indirectly by inhibiting lipolysis in fat and limiting free fatty acids (FFA) supply to liver. In addition, insulin strongly stimulates glycogenesis and lipogenesis. Glucagon promotes hepatic glucose production by stimulating glycogenolysis and gluconeogenesis. Glucagon also has catabolic effects in liver by promoting fatty acid oxidation.
The importance of direct insulin signaling in the liver is underscored by the severe glucose intolerance and inability of insulin to suppress HGP in mice lacking insulin receptor in the liver (LIRKO mice) [13]. Consistent with the importance of Akt in mediating the hepatic response to insulin, liver specific deletion of Akt 1 and 2 phenocopies to a large extent the metabolic phenotype of LIRKO mice, including the inability of insulin to suppress the expression of gluconeogenic genes [14]. Surprisingly however, when FoxO1 is concomitantly deleted in the liver Akt-1/2 KO mice, insulin’s ability to suppress HGP is restored, raising the possibility that under certain conditions Akt is dispensable for insulin-mediated suppression of HGP. Several independent studies have confirmed this observation and showed that completely depleting hepatic insulin signaling, by either knocking out the insulin receptor or the insulin receptor substrates, renders HGP unresponsive to insulin but only when FoxO1 is present [15, 14, 16–19].
These studies show that in the absence of both hepatic insulin signaling and FoxO1, the direct effect of insulin is dispensable for the ability of insulin to block HGP, and support an important role for indirect insulin regulation of extrahepatic pathways to suppress HGP. It is important to note however that these studies were done under the extreme conditions of completely abrogating direct insulin action in the liver. It is plausible that only under these conditions the indirect effects of insulin on HGP become dominant over the direct effects. This notion is supported by studies in dogs that show that when hepatic insulin signaling is intact the direct effect of insulin on liver is dominant in controlling HGP [20].
The indirect regulation of HGP by insulin.
What are the extrahepatic pathways by which insulin controls HGP? It has been known for many years that free fatty acids (FFA) can augment HGP by multiple mechanisms [21, 22]. Given that insulin strongly suppresses lipolysis in fat, FFA mobilization from fat to liver may be a major contributor to the insulin-mediated control of HGP (Fig. 1). Perry et al. suggest that the intrahepatic increase in acetyl-CoA, induced by elevated hepatic FFA, drives gluconeogenesis and HGP by activating Pyruvate Carboxylase (PC). They further show that acetate infusion, by leading to an increase in hepatic acetyl-CoA levels, increases HGP and that inhibition of lipolysis in fat normalizes HGP in high fat diet-fed rats [23]. Titchenell et al. used a more physiologic substrate and show that infusion of FFA completely abrogates the ability of insulin to suppress HGP, but only when insulin receptor and FoxO1 are deleted in the liver [19]. Both studies agree that the ability of insulin to suppress lipolysis is a crucial component in regulation of HGP; however Titchenell et al. emphasize that this pathway becomes dominant only when direct hepatic insulin signaling is perturbed [19]. These studies are particularly important in the context of T2D due to the strong association between T2D and increased levels of serum FFA. Under these conditions, the inability of insulin to suppress lipolysis in fat, due to insulin resistance, probably becomes a dominant contributor to the uncontrolled HGP in the insulin resistant liver.
FoxO1 has been considered for many years the main transcription factor that controls HGP by regulating the expression of gluconeogenic enzymes [24]. Under fasting conditions, FoxO1 is localized to the nucleus where it drives the expression of PEPCK and G-6-Pase to promote HGP and at the same time represses the transcription of Glucokinase (Gck), leading to reduced glycolytic flux and reduced lipogenesis [25, 26]. FoxO1 transcriptional activity is strongly inhibited by insulin via Akt-dependent phosphorylation and nuclear exclusion. The observation that concomitant ablation of hepatic insulin signaling and FoxO1 is required to restore insulin’s ability to suppress HGP highlights the importance of FoxO1 in also mediating the communication between liver and fat tissues to regulate systemic glucose homeostasis.
One potential effector of hepatic FoxO1 activity is follistatin (Fst), which was identified by Tao et al. as a hepatokine that is controlled by liver FoxO1 activity and is elevated in serum of Irs1/2 liver specific KO mice [17]. Overexpression of Fst in liver promotes glucose intolerance; conversely, depletion of hepatic follistatin normalizes the ability of insulin to suppress HGP and also normalizes WAT insulin sensitivity in IRS1/2 DKO mice. Most interestingly, follistatin levels are elevated in T2D and reduced after bariatric surgery, emphasizing the potential relevance of follistatin in regulating HGP in insulin resistant humans as well [17].
The lipogenic effects of insulin.
The direct effects of insulin on liver also include a strong stimulation of de-novo lipogenesis (DNL), TG synthesis and inhibition of FAO. Indeed, LIRKO mice, while demonstrating severe hyperglycemia and glucose intolerance, are protected from hepatic steatosis [27, 28]. These effects are mediated to a large extent by activation of SREBP1c, which activates a transcription program that promotes lipogenic flux [29, 30]. Insulin activates SREBP1c by increasing its transcription levels and by facilitating its processing and nuclear translocation, which is mediated by mTORC1. However, activation of mTORC1 alone is not enough to promote lipogenesis since activation of Akt is also necessary, as recently demonstrated [19].
Another important mediator of insulin’s effect on hepatic DNL is FoxO1, which in addition to its strong positive effects on gluconeogenesis can also act as a repressor of Gck expression leading to reduced glycolytic flux and substrate availability for DNL. A recent study shows that SIN3A is the insulin-regulated FoxO1 corepressor that facilitates inhibition of Gck expression but does not affect the gene activation function of FoxO1 [31]. This type of selectivity is specifically interesting as small molecules that can specifically reduce gluconeogenic function of FoxO1 without affecting its ability to suppress Gck theoretically exist. Indeed, Langlet et al. identified a small molecule that is able to clear FoxO1 from G6pc promoter but not from Gck promoter. This type of small molecule can potentially be used to reduce glucose production without promoting lipogenesis; this outcome would be highly desirable as a potential adverse effect of insulin sensitizers is accumulation of fat in liver. Selectivity in suppressing HGP without resulting in hepatic lipid accumulation has also been recently shown by using a small molecule that selectively inhibits the gluconeogenic function of PGC-1α, an important transcription coactivator that promotes gluconeogenesis by activating the transcriptional activity of FoxO1 and HNF4α during fasting [32]. By enhancing the acetylation of PGC-1α, a chemical modification that was shown to strongly inhibit its activity [33–36], this small molecule attenuates the ability of PGC-1α to promote gluconeogenesis, leading to strong inhibition of HGP and improved insulin sensitivity [32]. Thus, these data suggest that targeting corepressors or coactivators, rather the transcription factors themselves, might be a key strategy in the design of future drugs that will only sensitize the liver to the effects of insulin on glucose metabolism.
Glucagon regulation of liver metabolism
Glucagon regulation of glucose homeostasis.
Hepatic glucagon actions include coordinated regulation of transcription factors and signal transduction networks that control carbohydrate, lipid and amino acid metabolism [6]. Glucagon is secreted by pancreatic α-cells and fasting hyperglucagonemia is often associated with T2D, where it contributes substantially to fasting hyperglycemia [37]. Binding of glucagon to the glucagon receptor, a 7 transmembrane G-protein coupled receptor (GPCR), leads to elevation in intracellular cAMP levels and activation of downstream signaling pathways. Like insulin, glucagon exerts its metabolic functions in the liver by controlling not only gene expression but also metabolic fluxes and enzyme activity. Glucagon positively regulates HGP by promoting both glycogen breakdown and gluconeogenesis (Fig. 1). Accordingly, mice with liver-specific deletion of glucagon receptor exhibit reduction in fasting blood glucose and improved insulin sensitivity and glucose tolerance [38].
Interfering with hepatic glucagon signaling, via either genetic [38] or chemical inhibition [39], leads to pancreatic α-cell hyperplasia, potentially via the action of a liver-derived factor promoting α-cell proliferation [38]. While these secondary effects may compromise to some extent the usage of glucagon signaling blockade as a clinical approach, identification of mechanisms contributing to liver-alpha-cell communication will be important. Recently, it has been shown that blocking glucagon signaling perturbs amino acid (AA) catabolism, leading to elevated levels of AA in liver and serum [39, 40]; increases in AA especially glutamine, in turn may potently stimulate proliferation of these cells in culture [39].
Glucagon is processed from the proglucagon peptide, which also gives rise to glucagon-like peptide 1 (GLP-1), an intestinal hormone that is secreted after a meal, promotes insulin secretion and contrary to glucagon, reduces glycemia [41]. A very exciting therapeutic approach is offered by dual glucagon/GLP-1 agonists, which provide enhanced efficacy as compared with GLP-1 agonists alone [42]. This approach takes advantage of the less appreciated effects of glucagon on satiety and increased energy expenditure, and the antidiabetic effects of GLP-1. This synthetic glucagon/GLP-1 peptide agonist results in a dramatic improvement in glucose homeostasis, reduced body weight, and increased energy expenditure [43, 44]. It will be interesting to see whether other antidiabetic drugs can be used in conjunction with glucagon to mitigate its hyperglycemic effect while taking advantage of its catabolic functions.
Glucagon regulation of lipid homeostasis.
Glucagon regulates lipid homeostasis, independently of its control over glucose homeostasis [45]. It promotes lipolysis in white adipocytes as well as energy expenditure both in rodents and human [45]. During fasting, hepatic FAO is elevated in WT mice but not in glucagon receptor KO mice (Gcgr −/− mice), pointing to the catabolic effect of glucagon in the liver [46]. TG secretion is elevated in the Gcgr −/− KO mice as well, further demonstrating the important contribution of direct hepatic glucagon signaling to lipid homeostasis. The effects of glucagon on TG synthesis and FAO are abolished in PPARα−/− hepatocytes, suggesting that these effects are PPARα-driven [46].
Glucagon regulation of metabolic fluxes and redox state.
Pyruvate/lactate, as well as alanine, glutamine and glycerol, are required as a carbon source for glucose production during gluconeogenesis. The importance of AA catabolism in the coordinated response to glucagon is underscored by a recent study showing that while quantitatively, lactate is a greater source for glucagon-stimulated glucose production, the contribution of carbons from glutamine is more enriched following glucagon stimulation in hepatocytes [47]. The authors propose that calcium entry to the mitochondria following glucagon treatment stimulates the activity of α-ketoglutarate dehydrogenase (AKGDH) leading to increased anaplerotic flux from glutamine. Consistent with the importance of glutamine flux to glucose production, deletion of glutaminase (Gls2), the enzyme converting glutamine to glutamate, leads to reduced glutamine utilization in response to glucagon and to reduced fasting blood glucose in mice. Importantly, a polymorphism at the locus containing the human GLS2, results in elevated glutaminase activity, increased glutamine flux and is associated with higher fasting blood glucose in humans [47].
Mitochondrial anaplerosis/cataplerosis function is critical in regulating gluconeogenesis in liver. The anaplerotic enzyme pyruvate carboxylase (PC) catalyzes the first step in gluconeogenesis by converting pyruvate into oxaloacetate (OAA); cytosolic PEPCK (PEPCK-C) subsequently converts OAA to phosphoenolpyruvate (PEP) in a cataplerotic reaction. The transcriptional regulation of PEPCK-C (encoded by the gene Pck1) by glucagon has been studied extensively under the prevailing hypothesis that inhibition of this of regulation, and reduction in PEPCK activity, can reduce gluconeogenesis and HGP. Indeed, numerous studies have correlated reduced Pck1 expression with reduced HGP and improved glucose tolerance [8]. This hypothesis was challenged by the finding that PEPCK depletion in liver has a small effect on gluconeogenic flux [48]. However, a more recent study shows that depletion of PEPCK-C can indeed reduce gluconeogenesis but only in the setting of high fat diet (HFD), when both cataplerotic and cataplerotic fluxes are elevated due to increased levels of circulating FFA [49]. Under these conditions, reduced cataplerotic flux by depletion of PEPCK-C improves hepatic insulin sensitivity and the ability to suppress HGP. The importance of anaplerosis/cataplerosis flux in regulation of hepatic function is also demonstrated by the importance of PC activity in liver. Loss of PC results in reduced gluconeogenesis, as can be predicted, but also perturbs hepatic AA catabolism [50]. Similar to PEPCK-C depletion, liver specific knockout of PC also results in reduced oxidative flux, improved insulin sensitivity and protects against HFD-induced hyperglycemia. Interestingly, while PEPCK-C depletion results in reduced redox state (lower NAD+/NADH ratio), PC depletion has an opposite effect with a more oxidized redox state (higher NAD+/NADH ratio). Moreover, while PEPCK-C depletion increases antioxidant capacity and improves inflammation, PC depletion does the opposite [49, 50]. These findings provide important information on the relationship between TCA cycle fluxes and antioxidant mechanisms. Inhibition of anaplerotic/catapleroic flux in liver might be beneficial in obese individuals with non-alcoholic fatty liver disease (NAFLD) since oxidative flux strongly correlates with the severity of NAFLD and inflammation in humans [50].
Metformin, the first line drug for treatment of T2DM, improves hyperglycemia primarily by reducing gluconeogenesis. The importance of hepatic redox state to the regulation of gluconeogenesis is also highlighted by a recent hypothesis suggesting that metformin exerts its anti-gluconeogenic function by controlling hepatic redox status [51, 52]. By non-competitively inhibiting the mitochondrial glycerol-3-phosphate dehydrogenase (mGPD), metformin increases mitochondrial NAD+/NADH ratio and reduces cytoplasmic NAD+/NADH ratio. This creates conditions that are unfavorable to conversion of lactate into pyruvate and prevents contribution of glycerol to gluconeogenic flux by inhibiting mGPD and thus formation of dihydroxyacetone phosphate (DHAP), a necessary entry point of glycerol into gluconeogenesis.
Conclusions
Regulation of hepatic gluconeogenesis is currently the most efficient way to control glucose homeostasis, as evident by the usage of metformin as the first line drug to treat hyperglycemia. Recent data improve our understanding of how insulin controls this very important process and suggest that under conditions of hepatic insulin resistance, as evident in obese and T2D patients, the contribution of circulating FFA originated from lipolysis in fat, and specifically the inability of insulin to suppress it, becomes dominant in the regulation of HGP. Liver-fat communication also seems to play an important role in controlling insulin resistance in fat. Thus, perturbing this harmful communication may provide a potential therapeutic target. The importance of several hepatic metabolic fluxes to the regulation of HGP was also underscored by several recent studies. Specifically, new data shed light on the importance of glutamine to the hepatic response to glucagon and on the implications of anaplerotic/cataplerotic flux regulation on oxidative stress and liver inflammation.
Acknowledgments
This work was supported by NIH grants (R01DK081418, R01DK089883 and R01DK117655) to P.P, The Chares King Postdoctoral Fellowship to K.S and the American Diabetes Association (ADA) Postdoctoral Fellowship to C.D.J.T.
Footnotes
Conflict of Interest
Kfir Sharabi, Clint D. J. Tavares, and Pere Puigserver declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Rowley WR, Bezold C, Arikan Y, Byrne E, Krohe S. Diabetes 2030: Insights from Yesterday, Today, and Future Trends. Popul Health Manag. 2017;20(1):6–12. doi: 10.1089/pop.2015.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiological Reviews. 2018;98(4):2133–223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nature Reviews Endocrinology. 2017;13(10):572–87. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14(1):9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr. 2012;3(3):286–94. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab. 2003;284(4):E671–8. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- 7.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 8.Sharabi K, Tavares CD, Rines AK, Puigserver P. Molecular pathophysiology of hepatic glucose production. Mol Aspects Med. 2015;46:21–33. doi: 10.1016/j.mam.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell. 2013;152(4):673–84. doi: 10.1016/j.cell.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 10.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–71. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends in endocrinology and metabolism: TEM. 2017;28(7):497–505. doi: 10.1016/j.tem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatting M, Tavares CDJ, Sharabi K, Rines AK, Puigserver P. Insulin regulation of gluconeogenesis. Ann N Y Acad Sci. 2018;1411(1):21–35. doi: 10.1111/nyas.13435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6(1):87–97. [PubMed] [Google Scholar]
- 14.Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nature medicine. 2012;18(3):388–95. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA et al. Inactivation of Hepatic Foxo1 by Insulin Signaling Is Required for Adaptive Nutrient Homeostasis and Endocrine Growth Regulation. Cell Metabolism. 2008;8(1):65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O-Sullivan I, Zhang W, Wasserman DH, Liew C, Liu J, Paik J et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nature Communications. 2015;6(1). doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.*.Tao R, Wang C, Stöhr O, Qiu W, Hu Y, Miao J et al. Inactivating hepatic follistatin alleviates hyperglycemia. Nature medicine. 2018;24(7):1058–69. doi: 10.1038/s41591-018-0048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; Findings from this study identify a potential link between FoxO1 activity in liver and insulin resistance in WAT. These findings help to clarify the relation between the direct and indirect effects of insulin on hepatic glucose production.
- 18.Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nature Communications. 2015;6(1):7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metabolism. 2016;23(6):1154–66. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgerton DS, Kraft G, Smith M, Farmer B, Williams PE, Coate KC et al. Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight. 2017;2(6):e91863. doi: 10.1172/jci.insight.91863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherrington AD. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48(5):1198–214. [DOI] [PubMed] [Google Scholar]
- 22.Cherrington AD, Moore MC, Sindelar DK, Edgerton DS. Insulin action on the liver in vivo. Biochem Soc Trans. 2007;35(Pt 5):1171–4. doi: 10.1042/BST0351171. [DOI] [PubMed] [Google Scholar]
- 23.Perry RJ, Camporez J-PGG, Kursawe R, Titchenell PM, Zhang D, Perry CJ et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160(4):745–58. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6(3):208–16. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Haeusler RA, Hartil K, Nature … VB. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Patil S, Chauhan B, Guo S, of Biological … P-DR. FoxO1 regulates multiple metabolic pathways in the liver effects on gluconeogenic, glycolytic, and lipogenic gene expression. Journal of Biological. 2006. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 27.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Aleman JO, Suzuki R et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7(2):125–34. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012;15(6):873–84. doi: 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moon YA, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012;15(2):240–6. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langlet F, Haeusler RA, Lindén D, Ericson E, Norris T, Johansson A et al. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell. 2017;171(4):824. doi: 10.1016/j.cell.2017.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharabi K, Lin H, Tavares CDJ, Dominy JE, Camporez JP, Perry RJ et al. Selective Chemical Inhibition of PGC-1alpha Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell. 2017;169(1):148–60 e15. doi: 10.1016/j.cell.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 34.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3(6):429–38. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510(7506):547–51. doi: 10.1038/nature13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dominy JE Jr., Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell. 2012;48(6):900–13. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122(1):4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Longuet C, Robledo AM, Dean ED, Dai C, Ali S, McGuinness I et al. Liver-Specific Disruption of the Murine Glucagon Receptor Produces α-Cell Hyperplasia. Evidence for a Circulating α-Cell Growth Factor. 2013;62(4):1196–205. doi: 10.2337/db11-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dean DE, Li M, Prasad N, Wisniewski SN, Deylen A, Spaeth J et al. Interrupted Glucagon Signaling Reveals Hepatic α Cell Axis and Role for L-Glutamine in α Cell Proliferation. Cell Metabolism. 2017;25(6):1362–137300000. doi: 10.1016/j.cmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Okamoto H, Huang Z, Anguiano G, Chen S, Liu Q et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic α Cell Hyperplasia in Mice. Cell Metabolism. 2017;25(6):1348. doi: 10.1016/j.cmet.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17(6):819–37. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Soni H. Peptide-based GLP-1/glucagon co-agonists: A double-edged sword to combat diabesity. Med Hypotheses. 2016;95:5–9. doi: 10.1016/j.mehy.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 43.Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5(10):749–57. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- 44.Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58(10):2258–66. doi: 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschop MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6(12):689–97. doi: 10.1038/nrendo.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Longuet C, Sinclair EM, Maida A, Baggio LL, Maziarz M, Charron MJ et al. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 2008;8(5):359–71. doi: 10.1016/j.cmet.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.*.Miller RA, Shi Y, Lu W, Pirman DA, Jatkar A, Blatnik M et al. Targeting hepatic glutaminase activity to ameliorate hyperglycemia. Nature medicine. 2018;24(4):518. doi: 10.1038/nm.4514. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study rigorously charechterizes changes in hepatic metabolic fluxes in response to glucagon stimulation, and identifies glutaminase as a potential target to control hyperglycemia.
- 48.Burgess SC, He T, Yan Z, Lindner J, Sherry AD, Malloy CR et al. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007;5(4):313–20. doi: 10.1016/j.cmet.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, Sunny NE et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest. 2015;125(12):4447–62. doi: 10.1172/JCI82204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cappel DA, Deja S, Duarte JAG, Kucejova B, Inigo M, Fletcher JA et al. Pyruvate-Carboxylase-Mediated Anaplerosis Promotes Antioxidant Capacity by Sustaining TCA Cycle and Redox Metabolism in Liver. Cell Metab. 2019;29(6):1291–305 e8. doi: 10.1016/j.cmet.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madiraju AK, Erion DM, Rahimi Y, Zhang X-M, Braddock DT, Albright RA et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506). doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang X-M, Zhang D et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nature medicine. 2018:1–11. doi: 10.1038/s41591-018-0125-4. [DOI] [PMC free article] [PubMed] [Google Scholar]