

In this review, Cohen discusses NAD+ metabolism, how different subcellular pools of NAD+ are established and regulated, and how free NAD+ levels can control signaling by PARPs and redox metabolism.
Keywords: ADP-ribosylation, biosensor, NAD, NAD consumer, PARP
Abstract
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor for redox enzymes, but also moonlights as a substrate for signaling enzymes. When used as a substrate by signaling enzymes, it is consumed, necessitating the recycling of NAD+ consumption products (i.e., nicotinamide) via a salvage pathway in order to maintain NAD+ homeostasis. A major family of NAD+ consumers in mammalian cells are poly-ADP-ribose-polymerases (PARPs). PARPs comprise a family of 17 enzymes in humans, 16 of which catalyze the transfer of ADP-ribose from NAD+ to macromolecular targets (namely, proteins, but also DNA and RNA). Because PARPs and the NAD+ biosynthetic enzymes are subcellularly localized, an emerging concept is that the activity of PARPs and other NAD+ consumers are regulated in a compartmentalized manner. In this review, I discuss NAD+ metabolism, how different subcellular pools of NAD+ are established and regulated, and how free NAD+ levels can control signaling by PARPs and redox metabolism.
Nicotinamide adenine dinucleotide (NAD+): beyond redox reactions
Nicotinamide adenine dinucleotide (NAD+) is found in all living organisms and is essential for life. NAD+ was discovered by Sir Arthur Harden in 1906 and for the first half of the 20th century the only known role for NAD+ was as a coenzyme for redox reactions in metabolic processes (e.g., glycolysis). In this capacity, NAD+ binds to oxidoreductase enzymes where it undergoes a two-electron reduction to generate NADH and an oxidized substrate (Fig. 1). This reaction is catalytic and reversible (NADH is reoxidized to NAD+); therefore, NAD+ is not consumed by redox reactions in cells. However, in the mid-1960s NAD+ was shown to be a substrate for a nuclear enzyme (now known as poly-ADP-ribose-polymerase 1, PARP1) that cleaves the nicotinamide-glycosidic bond of NAD+ to generate a polymer of ADP-ribose (ADPr), a process known as poly-ADP-ribosylation or PARylation (more on this below) (Fig. 1; Chambon et al. 1963, 1966; Fujimura et al. 1967a,b). Unlike NAD+-mediated redox reactions, this glycosidic cleavage reaction is irreversible and leads to the consumption of NAD+. Consistent with this notion, in the 1970s it was shown that NAD+ exhibits a high turnover in human cells (Rechsteiner et al. 1976). We now know that there are many “NAD+ consumers” (e.g., other PARP family member, sirtuins, etc.) beyond PARP1, which are found in nearly all subcellular compartments, including the nucleus, cytoplasm, and mitochondria (Fig. 1; Verdin 2015). Because of these NAD+ consumers, continuous synthesis of NAD+ is required for maintaining NAD+ levels.
Figure 1.
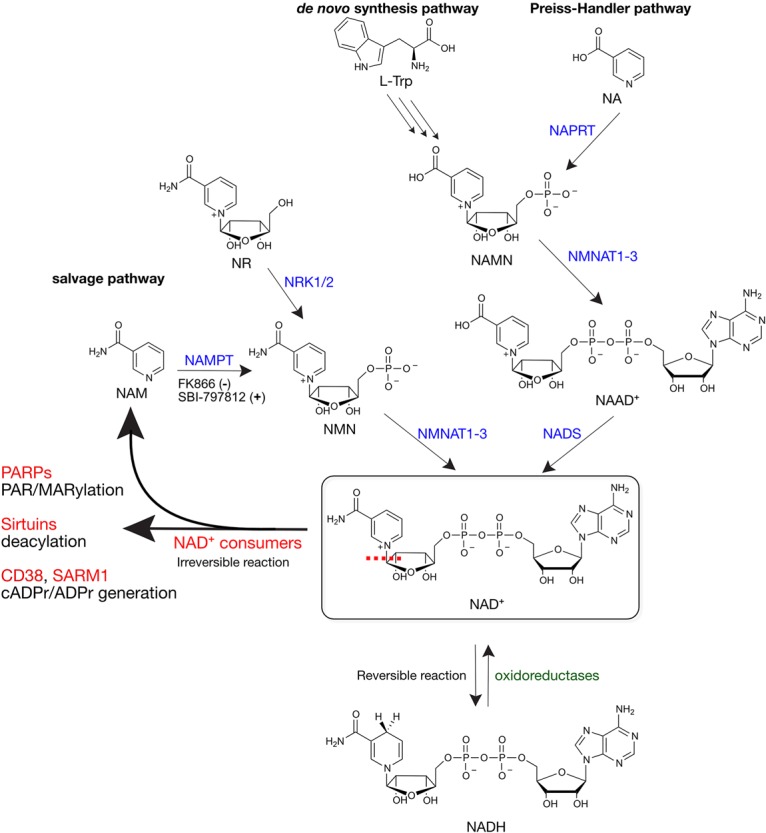
Pathways of NAD+ synthesis, consumption, and redox chemistry. The dashed red line indicates cleavage of the glycosidic bond of NAD+ by NAD+ consumers. NAD+ biosynthetic enzymes are shown in blue, NAD+ consumers are shown in red, and NAD+ redox enzymes are shown in green. (−) Inhibition; (+) activation.
Curiously, enzymes involved in NAD+ synthesis are localized to distinct subcellular compartments—like the NAD+ consumers themselves—suggesting that NAD+-dependent signaling is regulated in a compartmentalized manner. In this review I discuss the interplay between NAD+ synthesis and consumption by NAD+ consumers, with a particular focus on PARPs because they are the largest family of NAD+ consumers in cells. I first discuss NAD+ metabolism (i.e., its synthesis and consumption). I then discuss how NAD+ metabolism is compartmentalized. Last, I discuss how PARPs regulate—and are regulated by—changes in free NAD+ levels within subcellular compartments.
How is NAD+ synthesized in cells?
NAD+ can be synthesized in mammalian cells from precursors via three major pathways: (1) synthesis from tryptophan (the de novo pathway), (2) synthesis from nicotinic acid (NA; Preiss-Handler pathway), and (3) synthesis from nicotinamide (NAM; salvage pathway) (Fig. 1). In humans, the major source of NAD+ is from NA and NAM, collectively referred to as niacin (vitamin B3). Niacin is essential for maintaining NAD+ levels in vivo. Indeed, deficiencies in niacin causes Pellagra, a metabolic disease that results in diarrhea, dermatitis, dementia, and if untreated, death (Kirkland and Meyer-Ficca 2018). Hence, niacin is essential for the human diet. High levels of niacin are found in many foods, including meat, brown rice, and peanuts; however, because these niacin-rich foods were not commonplace in the American diet in the early part of the 20th century, especially in the South, grains were fortified with niacin beginning in the 1930s (Kirkland and Meyer-Ficca 2018). Recently, there has been much interest in using nicotinamide riboside (NR) to boost NAD+ levels in in vivo (Trammell et al. 2016). NR is a naturally occurring NAD+ precursor that feeds into the salvage pathway through the action of enzymes known as nicotinamide riboside kinase 1 and 2 (NRK1/2) (Fig. 1; Bieganowski and Brenner 2004). Notwithstanding the importance of dietary niacin and related NAD+ precursors, a recently published study found that in human patients the loss-of-function mutations in enzymes in the de novo tryptophan-to-NAD+ pathway results in congenital malformations (Shi et al. 2017). In these patients, serum NAD+ levels were significantly lower, demonstrating that dietary tryptophan is also an important source of NAD+ in vivo. Perhaps this result is not surprising considering the essentiality of NAD+ in human health.
In many cells, the salvage pathway via the precursor NAM plays an essential role in maintaining physiological NAD+ levels in cells. NAD+ synthesis from NAM in mammalian cells requires two enzymes: nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide mononucleotide adenylyltransferases (NMNAT) (Berger et al. 2005). NAMPT synthesizes nicotinamide mononucleotide (NMN) from NAM and α-D-5-phosphoribosyl-1-pyrophosphate (PRPP) (Fig. 1; Revollo et al. 2007). NMNAT, which exists as three distinct genes (NMNAT1–3) that have nonoverlapping functions (more on this below), synthesizes NAD+ directly from NMN and ATP (Fig. 1; Berger et al. 2005). Knockdown of NAMPT, or inhibition of NAMPT activity using a small molecule inhibitor (e.g., FK866, Fig. 1), substantially reduces NAD+ levels in most cells (Liu et al. 2018). Conversely, treatment of cells with a small molecule activator (e.g., SBI-797812) (Fig. 1) of NAMPT increases NAD+ levels in cells (Gardell et al. 2019). Therefore, NAMPT is a critical regulator of NAD+ levels in many cultured cells.
While NAMPT appears to be critical for maintaining NAD+ levels in many cells, NAD+ can be synthesized in an NAMPT-independent manner via the Preiss-Handler pathway. A recent study demonstrated that some cancer cells (e.g., OV4 ovarian cancer cells) are refractory to changes in NAD+ levels upon knockdown of NAMPT (Chowdhry et al. 2019). In OV4 cells, enzymes in the Preiss-Handler pathway (synthesis of NAD+ from NA) such as nicotinate phosphoribosyltransferase (NAPRT) are amplified. NAPRT synthesizes nicotinic acid mononucleotide (NAMN) from NA, and NAMN is subsequently converted into nicotinic acid dinucleotide (NAAD+) by NMNATs. Finally, NAAD+ is converted into NAD+ by NAD+ synthetase (NADS) (Fig. 1). Knockdown of NAPRT in OV4 cells, which were implanted subcutaneously in nude mice, decreased tumor NAD+ levels and tumor volume. In contrast, knockdown of NAPRT in subcutaneously implanted H460 lung cancer cells, which do not exhibit amplification of enzymes in the Preiss-Handler pathway, did not alter NAD+ levels; rather, knockdown of NAMPT (or treatment with FK866) decreased tumor NAD+ levels and tumor volume. Hence, cancer cells that have high levels of NAPRT depend on the Preiss-Handler pathway for survival, whereas cancer cells that have low levels of NAPRT depend on the salvage pathway. Cell type-dependent differences in NAD+ synthesis pathways have important implications in cancer therapeutic strategies aimed at lowering NAD+ levels in cells. Indeed, a recent study showed that, in a subclass of pediatric gliomas, mutations in the oncogene PPM1D (a protein phosphatase) resulted in epigenetic silencing of NAPRT, sensitizing these gliomas to NAMPT inhibitors (Fons et al. 2019).
What are the major NAD+ consumption pathways in cells?
The demonstration of enzymatic NAD+ consumption via nicotinamide-glycosidic bond cleavage first emerged in the 1960s. In one example, it was shown that NAD+ was consumed by diphtheria toxin, which was required for protein synthesis inhibition (Collier and Pappenheimer 1964). Later studies demonstrated that diphtheria toxin transferred the ADPr unit from NAD+ to elongation factor 2 (EF-2), which shuts down protein synthesis in host cells (Chung and Collier 1977). In another example, it was shown that a nuclear enzyme could transfer the ADPr moiety from NAD+ into poly-ADP-ribose (PAR) with concomitant release of NAM (Chambon et al. 1966; Fujimura et al. 1967a). This PAR synthase, now known as PARP1, was the first reported example of a mammalian NAD+ consumer. The presence of this NAD+ consumer in cells led to the hypothesis NAD+ undergoes rapid consumption, and as consequence, resynthesis (Gholson 1966). Indeed, in 1971 a study using radiolabeled NAD+ precursors in cultured cells demonstrated the rapid turnover (t1/2 = 5.8 h) of NAD+ in D98/AH2 cells (derived from HeLa cells) (Rechsteiner et al. 1976); however, the major NAD+ consumer under these conditions was not elucidated.
Over the next several decades other enzymes that consume NAD+ via nicotinamide-glycosidic bond cleavage were discovered (Fig. 1). These include PARP1-related enzymes that share a conserved PARP catalytic domain (PARP1–17) (Hottiger et al. 2010; Cohen and Chang 2018), sirtuins (SIRT1–7) (Gray and Ekström 2001), cluster of differentiation 38 (CD38) (Howard et al. 1993), and, most recently, sterile α and TIR motif-containing 1 (SARM1) (Gerdts et al. 2015; Essuman et al. 2017). The PARP family of enzymes catalyze the transfer of ADPr from NAD+ to a protein (Cohen and Chang 2018) or, as recently demonstrated, an RNA (Munnur et al. 2019) and DNA (Munnur and Ahel 2017; Belousova et al. 2018) substrate. Similar to PARP1, PARP2, PARP5a, and PARP5b catalyze poly-ADP-ribosylation (PARylation). Twelve PARP family members (PARP3, PARP6–12, and PARP14–16) catalyze the transfer of a single unit of ADPr to their targets, a process referred to as mono-ADP-ribosylation (MARylation) (Vyas et al. 2014; Yang et al. 2017). SIRTs are NAD+-dependent deacylases (Kosciuk et al. 2019), though some SIRT family members (e.g., SIRT4,6) have been shown to also catalyze ADP-ribosylation (Liszt et al. 2005; Haigis et al. 2006). The mechanism of sirtuin-mediated deacylation occurs via an ADPr-lysine-imidate intermediate generated from the attack of an acyl oxygen of lysine at the anomeric position of the nicotinamide ribose (Sauve et al. 2001). CD38 is a transmembrane enzyme that cleaves NAD+ to generate cADPr (and in some cases, ADPr), which acts as a second messenger to regulate calcium signaling in cells (Galione et al. 1991). Knockout of CD38 in mice increased NAD+ levels in several tissues, most prominently in the brain and heart, suggesting that CD38 is a major NAD+ consumer in vivo (Aksoy et al. 2006). Similar to CD38, SARM1 generates both cADPr and ADPr, and is thought to contribute to the loss of NAD+ during axonal injury in neurons (Essuman et al. 2017).
It is clear there are many possible routes to NAD+ consumption in cells, but what are the dominant NAD+ consumers in cultured cells? This question was recently addressed using NAD+ flux quantitation with isotopically labeled NAM in the presence of inhibitors of NAD+ consumers (Liu et al. 2018). Treatment of T47D breast cells under basal conditions with olaparib (10 µM), a clinically used inhibitor of PARP1/2, decreased NAD+ consumption by ∼33%. It should be noted that at 10 µM, olaparib appreciably inhibits several other PARPs, including PARP5a, PARP5b, PARP7, and PARP15 (Thorsell et al. 2017; Lu et al. 2019). In a previous study, expression of PARP7 (also known as TiPARP) in primary hepatocytes decreased total NAD+ levels (MacPherson et al. 2013). Hence, it is possible that PARP7 and other PARP targets of olaparib contribute to NAD+ consumption in T47D cells. In addition to PARPs, it appears that SIRTs are significant NAD+ consumers in T47D cells. Similar to results with olaparib, treatment of T47D cells with either 25 µM sirtinol (SIRT1/2 inhibitor) or 10 µM EX527 (also known as selisistat, SIRT1-selective inhibitor, currently in phase 2 clinical trials for improving endometrial receptivity before embryo transfer, NCT04184323) decreased NAD+ consumption by ∼33% (Liu et al. 2018). Given that SIRT1-selective inhibitor EX527 had the same effect as sirtinol, these results suggest that SIRT1 is the major SIRT NAD+ consumer. Treatment of T47D cells with both sirtinol (25 µM) and olaparib (10 µM) was additive, resulting in ∼66% decrease in NAD+ consumption. Similar results were obtained in other cell lines derived from different tissues supporting the notion that under basal conditions, PARPs (in particular PARP1/2, but perhaps other PARPs too) and SIRT1 are the major NAD+ consumers in cells. Interestingly, treatment of T47D cells with the CD38 flavonoid-based inhibitors quercetin (50 µM) and apigenin (25 µM) did not alter NAD+ flux despite previous studies showing that these compounds increase NAD+ levels in mouse embryonic fibroblasts (MEFs) in a CD38-dependent manner (Escande et al. 2013). This suggest that CD38 is not a major consumer of NAD+ in T47D cells; however, whether CD38 is expressed and/or active in these cells was not addressed. It is likely the case that the dominant NAD+ consumer(s) is cell type- and context-dependent (e.g., upon DNA damage, as I discuss below).
The aforementioned discussion focused on the role of NAD+ consumers under basal physiological conditions, but what happens under pathophysiological circumstances? Induction of DNA damage is a well-validated pathophysiological condition that profoundly stimulates PARP1-mediated PARylation. In T47D cells, the DNA double-strand break-inducer zeocin increased NAD+ consumption by approximately two times, resulting in a 60% decrease in total NAD+ levels (Liu et al. 2018). The increase in NAD+ consumption was completely blocked by olaparib demonstrating that, under conditions of DNA damage, PARP1 is the major NAD+ consumer in cells. Whether other PARP family members become major NAD+ consumers under a given pathophysiological condition is unknown but certainly an interesting area of further exploration.
As noted above, activation of PARP1 during DNA damage leads to a substantial decrease in NAD+ levels. But does this decrease in NAD+ affect the activity of other PARP family members, other NAD+ consumers, or NAD+-dependent redox pathways? To address this question we first need to consider the concentrations of free NAD+ in cells—that is, the NAD+ pool that is freely available for use as a substrate for other NAD+ consumers and redox enzymes—and this is where things become interesting.
NAD+ synthesis is compartmentalized: implications for NAD+ consumption
Intriguingly, the enzymes (NMNAT1–3) that catalyze the final step of NAD+ synthesis are subcellularly localized in cells: NMNAT1 localizes to the nucleus, NMNAT2 localizes to the Golgi complex and cytoplasm, and NMNAT3 localizes to mitochondria (Fig. 2; Zhang et al. 2003; Berger et al. 2005). In some commonly used cell lines, such as HEK 293T cells, all three isoforms are expressed at similar levels, whereas in others such as HeLa cells, NMNAT1 is the major paralog, and very little NMNAT2 and NMNAT3 are present (Berger et al. 2005; Cambronne et al. 2016). In mice, NMNAT1 is ubiquitously expressed, NMNAT2 is expressed predominately in the brain (Hicks et al. 2012), and NMNAT3 is expressed predominately in whole blood (Hikosaka et al. 2014). It is important to note that the role of NMNAT3 in regulating mitochondrial NAD+ levels has recently been questioned. In erythrocytes, which do not contain mitochondria, NMNAT3 regulates cytoplasmic NAD+ levels as shown by NMNAT3 knockout studies in mice (Hikosaka et al. 2014). NMNAT3 is also expressed, to a lesser extent, in skeletal muscle cells where it is localized predominately in the cytoplasm, and only slightly in mitochondria (Yamamoto et al. 2016). Moreover, mitochondrial NAD+ levels in skeletal muscle are only partially affected by the loss of NMNAT3. Notwithstanding these results, the differences in the subcellular expression of the three NMNAT paralogs lead to the hypothesis that NMNATs have distinct functions in regulating compartmentalized NAD+ synthesis in cells (Berger et al. 2005). Consistent with this notion, knockout of either NMNAT1 or NMNAT2 results in embryonic lethality, demonstrating that they cannot compensate for each other, further bolstering the idea that, at least in some cell types, NMNAT paralogs regulate distinct pools of NAD+ in cells (Conforti et al. 2011; Hicks et al. 2012).
Figure 2.
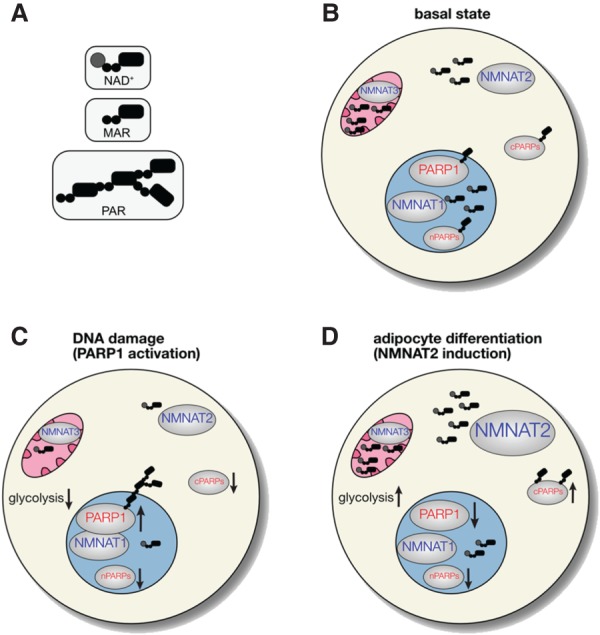
NAD+ synthesis is compartmentalized in mammalian cells and regulates—and is regulated by—the NAD+ consumer PARP1. NMNAT1–3, which synthesize NAD+, are distinctly localized in major subcellular compartments: NMNAT1 in the nucleus, NMNAT2 in the cytoplasm, and NMNAT3 in mitochondria. (cPARPs) Cytoplasmic PARPs; (nPARPs) nuclear PARPs. (A) Cartoon representation of NAD+, mono-ADP-ribose (MAR), and poly-ADP-ribose (PAR). (B) Under basal conditions, the concentration of NAD+ is highest in mitochondria (∼250 μM), followed by the cytoplasm and nucleus (both ∼100 μM). The number of NAD+ molecules indicates relative subcellular concentrations of NAD+. (C) During DNA damage, PARP1 associates with NMNAT1 and is activated (up arrow), leading to auto-poly-ADP-ribosylation (PARylation). This results in a temporal reduction of NAD+ levels in the nucleus, and likely the cytoplasm and mitochondria. The decrease in NAD+ levels in all compartments is expected to decrease the activity of nPARPs and cPARPs, as well as glycolytic flux and oxidative phosphorylation (down arrows). (D) Upon adipocyte differentiation, NMNAT2 is induced, which increases NAD+ levels in the cytoplasm and leads to an increase in glycolytic flux and perhaps the activity of cPARPs (up arrows). The increase in NMNAT2 levels depletes NMN from the nucleus (not shown), leading to lower NAD+ levels in the nucleus and a decrease in the activity of PARP1 and perhaps other nPARPs (down arrows).
Although the idea of compartmentalized regulation of NAD+ synthesis and consumption has existed in the literature for some time, experimental evidence for this type of regulation has only been obtained recently with the advent of biosensors that can measure free NAD+ levels in different subcellular compartments. Currently, there are two types of biosensors capable of detecting free NAD+ levels in cells. One biosensor is completely genetically encoded and is composed of a circularly permuted Venus fluorescent protein (cpVenus) and the bipartite NAD+-binding domain from bacterial DNA ligase (Cambronne et al. 2016). The other biosensor is a FRET-based, semisynthetic biosensor based on the “Snifit” concept (Sallin et al. 2018). Because these biosensors are genetically encoded, localization sequences can be used to target them to either the nucleus, cytoplasm, or mitochondria. Using organelle-targeted cpVenus-based NAD+ biosensor, the concentration of free NAD+ in various subcellular compartments was determined: 92–122 µM (95% CI [confidence interval]) in the cytoplasm, 87–136 µM (95% CI) in the nucleus, and 191–275 µM (95% CI) in the mitochondria (Cambronne et al. 2016). Remarkably, despite the very different designs of the two biosensors, there was a remarkable congruence in the measured concentration of free NAD+ (40–70 µM NAD+) in the cytoplasm in various cell lines (e.g., HeLa, HEK 293T, U2OS) (Cambronne et al. 2016; Sallin et al. 2018).
In addition to knowing the concentrations of free NAD+, the other two factors that need to be considered are the localization of PARPs and their Km values for NAD+ (KmNAD+). The 16 active PARP family members are expressed in various subcellular compartments, including the nucleus and cytoplasm, and distinct organelles/structures within these compartments (Table 1; Di Paola et al. 2012; Jwa and Chang 2012; Vyas et al. 2013; Bindesbøll et al. 2016; Carter-O'Connell et al. 2016, 2018; Catara et al. 2017). For example, PARP7 (also known as TiPARP) localizes to distinct foci in the nucleus (Vyas et al. 2013; Bindesbøll et al. 2016). PARP11 localizes to the nuclear envelope and colocalizes with nuclear pore complexes (NPCs), which are multiprotein channels that regulate nucleocytoplasmic transport (Meyer-Ficca et al. 2015; Carter-O'Connell et al. 2016; Kirby et al. 2018). PARP12 localizes to the Golgi, and under pathophysiological conditions that activate PARP1, it localizes to stress granules (Catara et al. 2017). PARP14 localizes to distinct foci in the cytoplasm, some of which colocalize with one of its MARylation targets DDX6, a protein that is associated with processing bodies (P-bodies, sites of mRNA turnover) (Carter-O'Connell et al. 2018). PARP16 is a tailed-anchored protein that contains a hydrophobic transmembrane domain and a C-terminal domain that are required for PARP16 recruitment to the endoplasmic reticulum (Jwa and Chang 2012). While no PARP family members contain obvious mitochondrial localization sequences, there are a few studies that report the presence of PARP1 or PARylation in mitochondria (Mosgoeller et al. 1996; Du et al. 2003; Lai et al. 2008; Rossi et al. 2009), but this remains controversial.
Table 1.
Enzymatically active human PARP family members

The KmNAD+ values for several PARPs (Table 1) are similar to or higher than the concentration of free NAD+ in the nucleus and cytoplasm (Amé et al. 1999; Thorsell et al. 2017; Yang et al. 2017). This suggests that the activity of PARPs, especially those with KmNAD+ values higher than the concentration of free NAD+ in a given subcellular compartment, will be influenced by changes in the levels of free NAD+. For example, activation of PARP1 during DNA damage, which lowers NAD+ levels substantially (∼60%) (Liu et al. 2018), is expected to decrease the activity of other nuclear PARPs (Fig. 2). Of course, the activity of other nuclear NAD+ consumers such as SIRT1, could decrease too, since it has a KmNAD+ value of 94 µM (Pacholec et al. 2010). If PARP1 activation during prolonged DNA damage lowers free NAD+ levels in the cytoplasm, then the activity of cytoplasmic PARPs and NAD+-dependent redox enzymes (e.g., GAPDH, KmNAD+ ∼20–140 µM) (BRENDA database, https://wwwbrenda-enzymes.org) could decrease (Fig. 2). Indeed, activation of PARP1 by the DNA alkylating agent MNNG in mouse astrocytes led to a block in glycolysis, which was attributed to a decrease in the availability of cytoplasmic NAD+ for GAPDH (Ying et al. 2003). A recent study, however, showed that the effects of PARP1 activation on glycolytic blockade are not due to decreases in NAD+, but rather a PARylation-dependent inhibition of hexokinase 1 (Fouquerel et al. 2014).
The above discussion focused on PARP1 as the dominant NAD+ consumer under DNA damage conditions and how the resulting decrease in the concentration of NAD+ could impact other NAD+-dependent signaling and redox pathways. It is currently not known whether other PARP family members become dominant NAD+ consumers under a given physiological or pathophysiological condition. The rate of NAD+ consumption by a PARP will depend on not only its concentration, but its activity. Most PARP family members are expressed at very low levels compared with PARP1 (e.g., the concentration of PARP1 is at least 100-fold higher than other PARP family members in HeLa cells (Wiśniewski et al. 2014), but can be induced. For example, treatment of bone marrow derived macrophages with interferon β (IFNβ) induces the expression of several PARP family members (PARP7, PARP10–12, and PARP14) that catalyze MARylation (Grunewald et al. 2019). Under these conditions one or several of these mono-PARPs could become dominant NAD+ consumers. One thing that remains unknown is whether any of these mono-PARPs also become “activated” under these conditions, which would be expected to further increase NAD+ consumption.
One might expect an increased demand for NAD+ synthesis under conditions that activate PARP1 in order to maintain homeostatic levels of NAD+. Consistent with this idea, a recent study demonstrated that PARP-mediated NAD+ consumption in inflammatory macrophages results in an enhanced dependence of the NAM salvage pathway for maintaining NAD+ levels in cells (Cameron et al. 2019). Treatment of mouse macrophages with lipopolysaccharide (LPS) and interferon-γ (IFN-γ) induces a proinflammatory state that leads to DNA damage and subsequent induction of PARP-mediated PARylation with a concomitant depletion in total NAD+ levels. The depletion of NAD+ stimulated the induction of NAMPT and NMNAT1, which was critical for maintaining NAD+ levels for redox signaling. This study nicely illustrates the interplay between PARP activity and the transcriptional regulation of NAD+ biosynthetic enzymes.
PARP1 regulation by NMNAT1 and compartmentalized NAD+ synthesis
Up to this point, I have discussed how a PARP can consume NAD+ and limit NAD+ availability in a compartmentalized manner. What about vice versa? Can NAD+ biosynthetic enzymes and compartmentalized NAD+ synthesis regulate the activity of PARPs? Studies on the interaction between PARP1 and NMNAT1 provide evidence for this type of regulation. In one study, NMNAT1 was shown to bind to auto-PARylated PARP1 (through direct PAR binding) and activate PARP1 catalytic activity in vitro (Berger et al. 2007). In another study, NMNAT1 was shown in vitro to not only synthesize NAD+ for PARP1-mediated PARylation, but also stimulate PARP1 activity independently of NAD+ synthesis (Zhang et al. 2012). In MCF-7 breast cancer cells, PARP1 and NMNAT1 interact at promoters of commonly regulated target genes (Zhang et al. 2012). Furthermore, NMNAT1 activity is required for PARP1-mediated PARylation at these promoters and the expression of PARP1/NMNAT1-target genes (Zhang et al. 2012). This last result supports the notion that PARP1 activity can be regulated by compartmentalized NAD+ synthesis.
Recently, the first experimental evidence for the regulation of PARP1 activity by compartmentalized NAD+ synthesis was demonstrated. Using an in vitro model of adipogenesis in 3T3-L1 cells, PARP1 activity decreased during the first several hours of adipocyte differentiation (Luo et al. 2017). It was hypothesized that the decrease in PARP1-mediated PARylation was due to a decrease in the availability of free NAD+ generated by the nuclear NMNAT1 (Ryu et al. 2018). Indeed, a significant drop in nuclear NAD+ levels was observed 4 h after adipocyte differentiation using the cpVenus-based NAD+ biosensor (Ryu et al. 2018). Moreover, knockdown of NMNAT1 caused a decrease in PARP1-mediated PARylation. Together, these results support the idea that changes in nuclear NAD+ biosynthesis regulates the activity of PARP1.
Although the levels of nuclear NAD+ decreased during adipocyte differentiation, the total levels of NAD+ were, surprisingly, unchanged (Ryu et al. 2018). It was hypothesized that NAD+ levels increased in another cellular compartment resulting in no net change in total levels of NAD+. Consistent with this notion, the levels of NMNAT2 and free NAD+ in the cytoplasm increased significantly 4 h after adipocyte differentiation. The increased levels of NMNAT2 were associated with enhanced glucose metabolism during adipocyte differentiation, perhaps because more cytoplasmic NAD+ was available for use by oxidoreductases in the redox-signaling pathway. These results are intriguing because they not only suggest that changes in the levels of free NAD+ in one cellular compartment can affect the levels of free NAD+ in another cellular compartment, but that compartmentalized pools of NAD+ exist. While it may seem like NAD+ should be able to freely diffuse from the cytoplasm to the nucleus, it is important to keep in mind that the diffusion of small molecule substrates in the crowded milieu of the cell can be attenuated compared with diffusion in a test tube (Zotter et al. 2017).
What is the mechanism for the crosstalk between compartmentalized pools of NAD+? Treating cells with NMN, the substrate for NMNATs, prevented the loss of free NAD+ in the nucleus, and enhanced PARP1 activity 8 h after adipocyte differentiation (Ryu et al. 2018). These results lead to a substrate competition model for establishing compartmentalized pools of NAD+: As adipocyte differentiation ensues, the increased levels of NMNAT2 cause a siphoning of NMN into the cytoplasm, leading to an increase in cytoplasmic NAD+ and a concomitant decrease in nuclear NAD+ (Ryu et al. 2018). An increase in the levels of free NAD+ in the cytoplasm results in an enhancement in cytoplasmic-mediated NAD+ redox signaling, whereas the decrease in the levels of free NAD+ in the nucleus results in a decrease in PARP1 activity (Fig. 2). A biosensor capable of detecting free NMN in cells would substantiate this substrate competition model.
Closing thoughts
Stimulating developments in the NAD+ field in the last few years have revealed a complex interplay between NAD+ synthesis and the activity of NAD+ consumers; namely, PARPs. In particular, the generation of biosensors capable of measuring free NAD+ in cells has enabled studies examining compartmentalized NAD+ metabolism in cells. Thus far, only the regulation of PARP1 activity by compartmentalized pools of NAD+ has been examined. However, as discussed, there are 16 other enzymatically active PARP family members. In future studies, it will be important to determine whether the activity of PARPs that catalyze MARylation are regulated by changes in the levels of free NAD+ in the subcellular compartment in which they are expressed; equally important will be to understand how NAD+ consumption by a PARP in one compartment influences the activity of PARPs (or other NAD+ consumers and redox enzymes) in another compartment.
On a final note, because of the widespread use of various PARP inhibitors (currently there are four FDA-approved PARP inhibitors) in the clinic for the treatment of ovarian and breast cancer, we need to understand how these inhibitors impact NAD+ consumption in vivo. Given that PARP1 is a major consumer of NAD+ in cells, it is conceivable that treatment with PARP inhibitors increases NAD+ levels in vivo. While this may be beneficial, it is possible that supraphysiologic levels of NAD+ are actually detrimental; for example, by altering redox pathways in cells. Clearly more studies are required to provide a deeper understanding of the impact of long-term PARP inhibition on NAD+ levels in vivo.
Acknowledgments
I thank members of the Cohen laboratory for many helpful discussions and careful reading of the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.335109.119.
References
- Aksoy P, White TA, Thompson M, Chini EN. 2006. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun 345: 1386–1392. 10.1016/j.bbrc.2006.05.042 [DOI] [PubMed] [Google Scholar]
- Amé JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Höger T, Ménissier-de Murcia J, de Murcia G. 1999. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274: 17860–17868. 10.1074/jbc.274.25.17860 [DOI] [PubMed] [Google Scholar]
- Belousova EA, Ishchenko АA, Lavrik OI. 2018. Dna is a new target of Parp3. Sci Rep 8: 1–12. 10.1038/s41598-018-22673-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. 2005. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280: 36334–36341. 10.1074/jbc.M508660200 [DOI] [PubMed] [Google Scholar]
- Berger F, Lau C, Ziegler M. 2007. Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci 104: 3765–3770. 10.1073/pnas.0609211104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C. 2004. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117: 495–502. 10.1016/S0092-8674(04)00416-7 [DOI] [PubMed] [Google Scholar]
- Bindesbøll C, Tan S, Bott D, Cho T, Tamblyn L, MacPherson L, Grønning-Wang L, Nebb HI, Matthews J. 2016. TCDD-inducible poly-ADP-ribose polymerase (TIPARP/PARP7) mono-ADP-ribosylates and co-activates liver X receptors. Biochem J 473: 899–910. 10.1042/BJ20151077 [DOI] [PubMed] [Google Scholar]
- Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, Goodman RH. 2016. Biosensor reveals multiple sources for mitochondrial NAD+. Science 352: 1474–1477. 10.1126/science.aad5168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AM, Castoldi A, Sanin DE, Flachsmann LJ, Field CS, Puleston DJ, Kyle RL, Patterson AE, Hässler F, Buescher JM, et al. 2019. Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species–mediated DNA damage. Nat Immunol 20: 420–432. 10.1038/s41590-019-0336-y [DOI] [PubMed] [Google Scholar]
- Carter-O'Connell I, Jin H, Morgan RK, Zaja R, David LL, Ahel I, Cohen MS. 2016. Identifying family-member-specific targets of mono-ARTDs by using a chemical genetics approach. Cell Rep 14: 621–631. 10.1016/j.celrep.2015.12.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-O'Connell I, Vermehren-Schmaedick A, Jin H, Morgan RK, David LL, Cohen MS. 2018. Combining chemical genetics with proximity-dependent labeling reveals cellular targets of poly(ADP-ribose) polymerase 14 (PARP14). ACS Chem Biol 13: 2841–2848. 10.1021/acschembio.8b00567 [DOI] [PubMed] [Google Scholar]
- Catara G, Grimaldi G, Schembri L, Spano D, Turacchio G, Lo Monte M, Beccari AR, Valente C, Corda D. 2017a. PARP1 -produced poly-ADP-ribose causes the PARP12 translocation to stress granules and impairment of Golgi complex functions. Sci Rep 7: 1–17. 10.1038/s41598-017-14156-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catara G, Grimaldi G, Schembri L, Spano D, Turacchio G, Lo Monte M, Beccari AR, Valente C, Corda D. 2017b. PARP1-produced poly-ADP-ribose causes the PARP12 translocation to stress granules and impairment of Golgi complex functions. Sci Rep 7: 1–17. 10.1038/s41598-017-14156-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon P, Weill JD, Mandel P. 1963. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun 11: 39–43. 10.1016/0006-291X(63)90024-X [DOI] [PubMed] [Google Scholar]
- Chambon P, Weill JD, Doly J, Strosser MT, Mandel P. 1966. On the formation of a novel adenylic compound by enzymatic extracts of liver nuclei. Biochem Biophys Res Commun 25: 638–643. 10.1016/0006-291X(66)90502-X [DOI] [Google Scholar]
- Chowdhry S, Zanca C, Rajkumar U, Koga T, Diao Y, Raviram R, Liu F, Turner K, Yang H, Brunk E, et al. 2019. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 569: 570–575. 10.1038/s41586-019-1150-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DW, Collier RJ. 1977. The mechanism of ADP-ribosylation of elongation factor 2 catalyzed by fragment A from diphtheria toxin. Biochim Biophys Acta 483: 248–257. 10.1016/0005-2744(77)90053-5 [DOI] [PubMed] [Google Scholar]
- Cohen MS, Chang P. 2018. Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat Chem Biol 14: 236–243. 10.1038/nchembio.2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier RJ, Pappenheimer AM. 1964. Studies on the mode of action of diphtheria toxin: II. Effect of toxin on amino acid incorporation in cell-free systems. J Exp Med 120: 1019–1039. 10.1084/jem.120.6.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Janeckova L, Wagner D, Mazzola F, Cialabrini L, Di Stefano M, Orsomando G, Magni G, Bendotti C, Smyth N, et al. 2011. Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J 278: 2666–2679. 10.1111/j.1742-4658.2011.08193.x [DOI] [PubMed] [Google Scholar]
- Di Paola S, Micaroni M, Di Tullio G, Buccione R, Di Girolamo M. 2012. PARP16/ARTD15 is a novel endoplasmic-reticulum-associated mono-ADP-ribosyltransferase that interacts with, and modifies Karyopherin-ß1. PLoS One 7: e37352 10.1371/journal.pone.0037352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabó C, Clark RSB. 2003. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem 278: 18426–18433. 10.1074/jbc.M301295200 [DOI] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O'Neil L, White TA, Sinclair DA, Chini EN. 2013. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 62: 1084–1093. 10.2337/db12-1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. 2017. The SARM1 Toll/Interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron 93: 1334–1343.e5. 10.1016/j.neuron.2017.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fons NR, Sundaram RK, Breuer GA, Peng S, McLean RL, Kalathil AN, Schmidt MS, Carvalho DM, Mackay A, Jones C, et al. 2019. PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nat Commun 10: 3790–3710. 10.1038/s41467-019-11732-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouquerel E, Goellner EM, Yu Z, Gagné J-P, de Moura MB, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, et al. 2014. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep 8: 1819–1831. 10.1016/j.celrep.2014.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura S, Hasegawa S, Shimizu Y, Sugimura T. 1967a. Polymerization of the adenosine 5′-diphosphate-ribose moiety of nicotinamide-adenine dinucleotide by nuclear enzyme. I. Enzymatic reactions. Biochim Biophys Acta 145: 247–259. 10.1016/0005-2787(67)90043-3 [DOI] [PubMed] [Google Scholar]
- Fujimura S, Hasegawa S, Sugimura T. 1967b. Nicotinamide mononucleotide-dependent incorporation of ATP into acidinsoluble material in rat liver nuclei preparation. Biochim Biophys Acta 134: 496–499. 10.1016/0005-2787(67)90036-6 [DOI] [Google Scholar]
- Galione A, Lee HC, Busa WB. 1991. Ca2+-induced Ca2+ release in sea urchin egg homogenates: modulation by cyclic ADP-ribose. Science 253: 1143–1146. 10.1126/science.1909457 [DOI] [PubMed] [Google Scholar]
- Gardell SJ, Hopf M, Khan A, Dispagna M, Hampton Sessions EH, Falter R, Kapoor N, Brooks J, Culver J, Petucci C, et al. 2019. Boosting NAD+ with a small molecule that activates NAMPT. Nat Commun 10: 1–12. 10.1038/s41467-019-11078-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. 2015. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348: 453–457. 10.1126/science.1258366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholson RK. 1966. The pyridine nucleotide cycle. Nature 212: 933–935. 10.1038/212933a0 [DOI] [PubMed] [Google Scholar]
- Gray SG, Ekström TJ. 2001. The human histone deacetylase family. Exp Cell Res 262: 75–83. 10.1006/excr.2000.5080 [DOI] [PubMed] [Google Scholar]
- Grunewald ME, Chen Y, Kuny C, Maejima T, Lease R, Ferraris D, Aikawa M, Sullivan CS, Perlman S, Fehr AR. 2019. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog 15: e1007756 10.1371/journal.ppat.1007756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. 2006. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell 126: 941–954. 10.1016/j.cell.2006.06.057 [DOI] [PubMed] [Google Scholar]
- Hicks AN, Lorenzetti D, Gilley J, Lu B, Andersson K-E, Miligan C, Overbeek PA, Oppenheim R, Bishop CE. 2012. Nicotinamide mononucleotide adenylyltransferase 2 (Nmnat2) regulates axon integrity in the mouse embryo. PLoS One 7: e47869 10.1371/journal.pone.0047869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka K, Ikutani M, Shito M, Kazuma K, Gulshan M, Nagai Y, Takatsu K, Konno K, Tobe K, Kanno H, et al. 2014. Deficiency of nicotinamide mononucleotide adenylyltransferase 3 (Nmnat3) causes hemolytic anemia by altering the glycolytic flow in mature erythrocytes. J Biol Chem 289: 14796–14811. 10.1074/jbc.M114.554378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger MO, Hassa PO, Lüscher B, Schüler H, Koch-Nolte F. 2010. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 35: 208–219. 10.1016/j.tibs.2009.12.003 [DOI] [PubMed] [Google Scholar]
- Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC. 1993. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 262: 1056–1059. 10.1126/science.8235624 [DOI] [PubMed] [Google Scholar]
- Jwa M, Chang P. 2012. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1α-mediated unfolded protein response. Nat Cell Biol 14: 1223–1230. 10.1038/ncb2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby IT, Kojic A, Arnold MR, Thorsell A-G, Karlberg T, Vermehren-Schmaedick A, Sreenivasan R, Schultz C, Schüler H, Cohen MS. 2018. A potent and selective PARP11 inhibitor suggests coupling between cellular localization and catalytic activity. Cell Chem Biol 25: 1547–1553.e12. 10.1016/j.chembiol.2018.09.011 [DOI] [PubMed] [Google Scholar]
- Kirkland JB, Meyer-Ficca ML. 2018. Niacin. Adv Food Nutr Res 83: 83–149. 10.1016/bs.afnr.2017.11.003 [DOI] [PubMed] [Google Scholar]
- Kosciuk T, Wang M, Hong JY, Lin H. 2019. Updates on the epigenetic roles of sirtuins. Curr Opin Chem Biol 51: 18–29. 10.1016/j.cbpa.2019.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, Jenkins LW, Szabó C, Clark RSB. 2008. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem 104: 1700–1711. 10.1111/j.1471-4159.2007.05114.x [DOI] [PubMed] [Google Scholar]
- Liszt G, Ford E, Kurtev M, Guarente L. 2005. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem 280: 21313–21320. 10.1074/jbc.M413296200 [DOI] [PubMed] [Google Scholar]
- Liu L, Su X, Quinn WJ, Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, et al. 2018. Quantitative analysis of NAD synthesis–breakdown fluxes. Cell Metab 27: 1067–1080.e5. 10.1016/j.cmet.2018.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AZ, Abo R, Ren Y, Gui B, Mo J-R, Blackwell D, Wigle T, Keilhack H, Niepel M. 2019. Enabling drug discovery for the PARP protein family through the detection of mono-ADP-ribosylation. Biochem Pharmacol 167: 97–106. 10.1016/j.bcp.2019.05.007 [DOI] [PubMed] [Google Scholar]
- Luo X, Ryu KW, Kim D-S, Nandu T, Medina CJ, Gupte R, Gibson BA, Soccio RE, Yu Y, Gupta RK, et al. 2017. PARP-1 controls the adipogenic transcriptional program by PARylating C/EBPβ and modulating its transcriptional activity. Mol Cell 65: 260–271. 10.1016/j.molcel.2016.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson L, Tamblyn L, Rajendra S, Bralha F, McPherson JP, Matthews J. 2013. 2,3,7,8-Tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acids Res 41: 1604–1621. 10.1093/nar/gks1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Ficca ML, Ihara M, Bader JJ, Leu NA, Beneke S, Meyer RG. 2015. Spermatid head elongation with normal nuclear shaping requires ADP-ribosyltransferase PARP11 (ARTD11) in mice. Biol Reprod 92: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosgoeller W, Steiner M, Hozák P, Penner E, Wesierska-Gadek J. 1996. Nuclear architecture and ultrastructural distribution of poly(ADP-ribosyl)transferase, a multifunctional enzyme. J Cell Sci 109: 409–418. [DOI] [PubMed] [Google Scholar]
- Munnur D, Ahel I. 2017. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J 284: 4002–4016. 10.1111/febs.14297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munnur D, Bartlett E, Mikolčević P, Kirby IT, Matthias Rack JG, Mikoč A, Cohen MS, Ahel I. 2019. Reversible ADP-ribosylation of RNA. Nucleic Acids Res 47: 5658–5669. 10.1093/nar/gkz305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, et al. 2010. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem 285: 8340–8351. 10.1074/jbc.M109.088682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Hillyard D, Olivera BM. 1976. Magnitude and significance of NAD turnover in human cell line D98/AH2. Nature 259: 695–696. 10.1038/259695a0 [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S-I. 2007. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol 23: 164–170. 10.1097/MOG.0b013e32801b3c8f [DOI] [PubMed] [Google Scholar]
- Rossi MN, Carbone M, Mostocotto C, Mancone C, Tripodi M, Maione R, Amati P. 2009. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J Biol Chem 284: 31616–31624. 10.1074/jbc.M109.025882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, Kraus WL. 2018. Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science 360: eaan5780 10.1126/science.aan5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallin O, Reymond L, Gondrand C, Raith F, Koch B, Johnsson K. 2018. Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. Elife 7: e32639 10.7554/eLife.32638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Celic I, Avalos J, Deng H, Boeke A JD, Schramm VL. 2001. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 40: 15456–15463. 10.1021/bi011858j [DOI] [PubMed] [Google Scholar]
- Shi H, Enriquez A, Rapadas M, Martin EMMA, Wang R, Moreau J, Lim CK, Szot JO, Ip E, Hughes JN, et al. 2017. NAD deficiency, congenital malformations, and niacin supplementation. Nat Commun 377: 544–552. 10.1056/NEJMoa1616361 [DOI] [PubMed] [Google Scholar]
- Thorsell A-G, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L, Moche M, Cohen MS, Schüler H. 2017. Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J Med Chem 60: 1262–1271. 10.1021/acs.jmedchem.6b00990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SAJ, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C. 2016. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun 7: 12948–12914. 10.1038/ncomms12948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. 2015. NAD+ in aging, metabolism, and neurodegeneration. Science 350: 1208–1213. 10.1126/science.aac4854 [DOI] [PubMed] [Google Scholar]
- Vyas S, Chesarone-Cataldo M, Todorova T, Huang Y-H, Chang P. 2013. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun 4: 2240 10.1038/ncomms3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P. 2014. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun 5: 4426 10.1038/ncomms5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski JR, Hein MY, Cox J, Mann M. 2014. A ‘proteomic ruler’ for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics 13: 3497–3506. 10.1074/mcp.M113.037309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Hikosaka K, Mahmood A, Tobe K, Shojaku H, Inohara H, Nakagawa T. 2016. Nmnat3 is dispensable in mitochondrial NAD level maintenance in vivo. PLoS One 11: e0147037 10.1371/journal.pone.0147037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C-S, Jividen K, Spencer A, Dworak N, Ni L, Oostdyk LT, Chatterjee M, Kuśmider B, Reon B, Parlak M, et al. 2017. Ubiquitin modification by the E3 Ligase/ADP-ribosyltransferase Dtx3L/Parp9. Mol Cell 66: 503–516.e5. 10.1016/j.molcel.2017.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Garnier P, Swanson RA. 2003. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem Biophys Res Commun 308: 809–813. 10.1016/S0006-291X(03)01483-9 [DOI] [PubMed] [Google Scholar]
- Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. 2003. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem 278: 13503–13511. 10.1074/jbc.M300073200 [DOI] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, Ryu KW, Gamble MJ, Kraus WL. 2012. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. J Biol Chem 287: 12405–12416. 10.1074/jbc.M111.304469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotter A, Bäuerle F, Dey D, Kiss V, Schreiber G. 2017. Quantifying enzyme activity in living cells. J Biol Chem 292: 15838–15848. 10.1074/jbc.M117.792119 [DOI] [PMC free article] [PubMed] [Google Scholar]