

Here, Rahman et al. looked for a specific role of mutant SRSF2 in nonsense-mediated mRNA decay (NMD). They show that SRSF2 Pro95 hot spot mutations elicit enhanced mRNA decay, which is dependent on sequence-specific RNA binding and splicing, and thus provide new insight into the critical effects of SRSF2 mutants in hematologic malignancies.
Keywords: leukemia, mutation, myelodysplastic syndromes, nonsense-mediated mRNA decay, SRSF2, splicing, splicing factor
Abstract
Oncogenic mutations in the RNA splicing factors SRSF2, SF3B1, and U2AF1 are the most frequent class of mutations in myelodysplastic syndromes and are also common in clonal hematopoiesis, acute myeloid leukemia, chronic lymphocytic leukemia, and a variety of solid tumors. They cause genome-wide splicing alterations that affect important regulators of hematopoiesis. Several mRNA isoforms promoted by the various splicing factor mutants comprise a premature termination codon (PTC) and are therefore potential targets of nonsense-mediated mRNA decay (NMD). In light of the mechanistic relationship between splicing and NMD, we sought evidence for a specific role of mutant SRSF2 in NMD. We show that SRSF2 Pro95 hot spot mutations elicit enhanced mRNA decay, which is dependent on sequence-specific RNA binding and splicing. SRSF2 mutants enhance the deposition of exon junction complexes (EJCs) downstream from the PTC through RNA-mediated molecular interactions. This architecture then favors the association of key NMD factors to elicit mRNA decay. Gene-specific blocking of EJC deposition by antisense oligonucleotides circumvents aberrant NMD promoted by mutant SRSF2, restoring the expression of PTC-containing transcript. Our study uncovered critical effects of SRSF2 mutants in hematologic malignancies, reflecting the regulation at multiple levels of RNA metabolism, from splicing to decay.
Recurrent heterozygous mutations in RNA splicing factors are the most frequent class of mutations in patients with myelodysplastic syndromes (MDS) and also commonly occur in clonal hematopoiesis, acute myeloid leukemia (AML), chronic lymphocytic leukemia, and a variety of solid tumors (Yoshida et al. 2011; Papaemmanuil et al. 2013; Darman et al. 2015). SF3B1, SRSF2, U2AF1, or ZRSR2 splicing factors are mutated in ∼60% of MDS patients (Kennedy and Ebert 2017). These mutations occur in highly restricted residues (except in ZRSR2) and in a mutually exclusive manner (Yoshida et al. 2011). Several studies have shown that mutations in SRSF2 and U2AF1 alter their normal RNA-binding and splicing properties (Ilagan et al. 2015; Kim et al. 2015; Komeno et al. 2015; Zhang et al. 2015), whereas mutations in SF3B1 promote the selection of cryptic 3′ splice sites (Darman et al. 2015; Obeng et al. 2016). In contrast, mutations in ZRSR2 impair splicing of U12-type introns by the minor-spliceosome pathway (Madan et al. 2015). Some of this aberrant regulation of alternative splicing (AS) is predicted to drive pathogenic phenotypes by affecting multiple physiological functions and pathways, including hematopoiesis. For example, a recent study reported a cascade of physiological events triggered by multiple splicing factor mutations, especially high-risk alleles in SRSF2 and U2AF1, which include elevated R-loop formation, replication stress, and activation of the ataxia telangiectasia and Rad3-related protein (ATR)–Chk1 pathway (Chen et al. 2018).
One of the earliest observations about spliceosomal mutant MDS is that several mRNA isoforms promoted by the various splicing factor mutants harbor a premature termination codon (PTC) and are therefore potential targets of nonsense-mediated mRNA decay (NMD). This AS-coupled NMD is also known as “AS-NMD.” Although down-regulation of several AS targets by NMD has been predicted to be associated with the pathogenesis of MDS (Darman et al. 2015; Kim et al. 2015; Obeng et al. 2016), a specific role of mutant splicing factors in the NMD pathway has not been addressed. Previously, we showed that overexpression of certain SR proteins (including SRSF2) enhances NMD (Zhang and Krainer 2004; Aznarez et al. 2018). In contrast, to our knowledge, there is no evidence of a direct role of SF3B1, U2AF1, or ZRSR2 in the NMD pathway. We therefore systematically investigated the activity of the Pro95 hot spot MDS mutants of SRSF2 (SRSF2Mut) in NMD.
NMD is an important posttranscriptional RNA-processing pathway, initially identified as an mRNA surveillance, quality control pathway that selectively degrades transcripts with a PTC (Hwang and Maquat 2011). Later, it became evident that NMD also promotes posttranscriptional down-regulation of various endogenous mRNA isoforms (Huang et al. 2011). Therefore, NMD regulates gene expression by (1) buffering the activity of abnormal truncated proteins with compromised activity, deleterious functions, or dominant-negative effects, and (2) by controlling the cellular abundance of many mRNA isoforms. Pre-mRNA splicing is important for NMD (Lykke-Andersen et al. 2001; Maquat and Li 2001). Concomitantly with, or immediately after, splicing, a multiprotein complex termed the exon junction complex (EJC) is deposited ∼20–24 nt upstream of ∼80% of exon–exon junctions (Le Hir et al. 2000; Saulière et al. 2012; Singh et al. 2012). The core of the EJC consists of four proteins: the anchor eIF4A3, the Y14/MAGOH or Y14/MAGOH B heterodimer, and BARENTSZ (BTZ, CASC3, or MLN51) (Tange et al. 2005).
The EJC serves as an anchor point for NMD factors. UPF3B is the first NMD factor assembled on the spliced mRNA in the nucleus, through interactions with Y14 at EJC sites (Kim et al. 2001), and it travels with mRNA-protein complexes (mRNPs) to the cytoplasm. Then, UPF2 is recruited through interactions with UPF3B (Lykke-Andersen et al. 2000). In the cytoplasm, the mRNP bound by either the cap-binding complex (CBP80–CBP20) or eIF4E undergoes translation, which is a prerequisite step for NMD (Maquat et al. 2010; Hentze and Izaurralde 2013). When a translating ribosome encounters a stop codon, the transient SURF complex—consisting of SMG1, UPF1, and the release factors eRF1 and eRF3—is recruited to the mRNP (Kashima et al. 2006). The presence of one or more downstream EJCs discriminates a PTC from the authentic termination codon and initiates the SMG1-mediated phosphorylation of the RNA helicase UPF1 (Kashima et al. 2006). This in turn promotes the degradation of the PTC-containing mRNA by endonucleases and exonucleases (Schoenberg and Maquat 2012).
Splicing is further involved in NMD. A weak splice site downstream from (but not upstream of) a PTC often decreases NMD efficiency (Gudikote et al. 2005). Overexpression of certain SR protein splicing factors enhances NMD efficiency (Zhang and Krainer 2004). SRSF1 can switch the cellular site of the pioneer round of translation to nuclear-associated NMD, thereby enhancing NMD (Sato et al. 2008). SRSF1 promotes UPF1 binding to nuclear-associated mRNAs, bypassing the requirement for UPF2 (Aznarez et al. 2018). Recent studies showed that noncanonical EJC deposition overlaps with SR protein binding motifs (Saulière et al. 2012; Singh et al. 2012). However, the precise regulatory mechanisms through which exon definition, SR proteins, and cis-acting sequences influence the efficiency of NMD remained unclear.
Here, we investigated the effects and precise mechanisms of the cancer-associated mutations in Pro95 of SRSF2 that alter the fate of PTC-harboring mRNA, from splicing to NMD, in order to understand the relevant pathological consequences in MDS/leukemia. We uncovered key NMD-regulatory steps promoted by the SR protein-EJC nexus. In addition, we report that, in the context of MDS, SRSF2Mut has stronger NMD-inducing activity than SRSF2 wild type (SRSF2WT), reflecting the stabilization of EJC components downstream from the PTC, and the subsequent enhanced association of key NMD factors, to elicit mRNA decay. Finally, we present a method for targeted blocking of EJC deposition to inhibit the pathologic NMD promoted by mutant SRSF2.
Results
Aberrant splicing and NMD in SRSF2Mut AML
Recent studies collectively suggest that mutations in Pro95 of SRSF2 (P95H, P95L, or P95R) change its RNA-binding specificity from its normal preferred binding motif, GGWG, to another motif, C/GCWG (W = A/U) (Kim et al. 2015; Zhang et al. 2015), which in turn causes genome-wide splicing alterations. To extend this analysis to clinical samples, we analyzed the AML data set of The Cancer Genome Atlas (LAML-TCGA) (109 SRSF2WT vs. six SRSF2Mut). We identified 843 differential splicing events, including cassette exon splicing, ES (50%), alternative splice site selection, ASS (26%), and intron retention, IR (24%) (Fig. 1A; Supplemental Table S1). Among these events, 135 are predicted to target the resulting mRNA isoform for NMD (16%) (Fig. 1B). We next analyzed cassette exons to quantify the enrichment of each possible 5-mer motif in SRSF2Mut-regulated exons, using the MEME (multiple expectation maximization for motif elicitation) algorithm (Bailey et al. 2009). The most enriched motif in promoted exons was TGCAG (30%), and in repressed exons it was TGGAG (43%) (Fig. 1C; Supplemental Fig. S1A). The consensus motif for promoted exons was (T/G)(G/C)C(A/T)G, and for repressed exons it was (T/A)GG(A/T)G. This result is consistent with previous studies (Kim et al. 2015; Zhang et al. 2015). The altered SRSF2-binding specificity presumably enables recognition of different exonic splicing enhancers (ESEs), which drives differential splicing and probably differential activity in NMD.
Figure 1.

Splicing and NMD profile in AML patients with oncogenic mutations at Pro95 of SRSF2. (A) Stacked bar graph representing different types of splicing alterations in SRSF2Mut patients in LAML-TCGA RNA-seq data. (ASS) Alternative splice site, (ES) cassette exon, (IR) intron retention. (B) Pie chart showing the proportion of NMD targets among altered splicing events from A. (C) Logos of consensus binding motif enriched in promoted or repressed exons in SRSF2Mut patients from A. (D) Representative Sashimi plots showing EZH2 poison exon inclusion in AML samples from the genotypes indicated. (E) Radioactive RT-PCR of EZH2 poison exon inclusion and skipping in an independent set of SRSF2 WT and mutant AML samples and in three different human leukemia cells: K562, MOLM-13, and K052. Actin (ACTB) was amplified as an internal control. (F, top) Western blotting (WB) of SRSF2 P95H mutant K052 cells treated with the indicated shRNAs. (Bottom) Radioactive RT-PCR of SRSF3 (positive control), 18S rRNA, and EZH2 poison exon inclusion (percent spliced in [PSI]) (mean ± SD, n = 3). (*) P < 0.05, t-test. (G) Diagram of cDNA constructs and WB of total cell lysate, nuclear, or cytoplasmic fractions from the K562 cells transfected with the indicated cDNAs. (H, top) Diagram (not to scale) of WT (CCWG) and mutant (GGWG) EZH2 reporters harboring different combinations of motifs (W = A/U) within the poison exon. (Bottom) Radioactive RT-PCR of EZH2 reporters in K562 cells cotransfected with the indicated cDNAs. Quantification of poison exon inclusion (PSI) is shown by a bar graph (mean ± SD, n = 3). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, t-test.
We compared the splicing profiles of TCGA-LAML RNA-seq data with two previous RNA-seq studies of isogenic K562 cells with or without SRSF2 mutation: study 1 (Kim et al. 2015) and study 2 (Supplemental Fig. S1B; Zhang et al. 2015). One robust AS-NMD target in the SRSF2Mut LAML-TCGA data set is inclusion of a poison exon in EZH2 (Fig. 1D; Supplemental Fig. S1B), which was also identified in study 1. EZH2 encodes Enhancer of zeste homolog 2 protein, which catalyzes histone methylation and functions in chromatin remodeling. SRSF2Mut promotes inclusion of the EZH2 poison exon by binding to a C-rich exonic splicing enhancer (Kim et al. 2015). The resulting transcript is degraded by NMD, and the overall reduction in EZH2 impairs hematopoietic differentiation. It was further shown that restoring EZH2 expression partially rescues hematopoiesis in Srsf2 mutant cells. Besides EZH2, the number of total differential splicing events and splicing targets was also variable across each study, potentially related to the different cell contexts, methods of introducing the mutant allele (viral transduction vs. CRISPR/Cas9), methods used to infer changes in splicing from RNA-seq data, etc.
We performed radioactive RT-PCR in an independent panel of primary AML patient samples with or without SRSF2 mutations (three of each). We consistently observed inclusion of the EZH2 poison exon in all three SRSF2Mut samples but not in the SRSF2WT samples (Fig. 1E). We next analyzed three different human leukemia cells: K562, MOLM-13, and K052 (Kim et al. 2015). Sequencing of genomic DNA from these cell lines showed that K562 and MOLM-13 are SRSF2WT, whereas K052 is SRSF2Mut(P95H) (Kim et al. 2015). In K052 cells, we confirmed inclusion of the EZH2 poison exon but not in K562 or MOLM-13 cells (Fig. 1E). We next sought to confirm that the EZH2 poison exon indeed triggers NMD. We transfected K052 cells with a control or two different UPF1 (an essential factor for NMD) shRNAs. As an internal control, we tested a previously reported NMD isoform of SRSF3 (Ni et al. 2007). Knocking down UPF1 (Fig. 1F) increased the SRSF3 NMD mRNA isoform, compared with the control (1.72 ± 0.74 fold for shUPF1-1 and 2.99 ± 0.45 fold for shUPF1-2, mean ± SD, n = 3, normalized to 18S rRNA) (Fig. 1F). We then measured the EZH2 mRNA level. Knocking down UPF1 indeed significantly increased the level of the poison-exon-included mRNA isoform for both shRNAs (Fig. 1F). This result confirmed that the poison-exon-included mRNA isoform undergoes NMD.
Another robust AS-NMD target of SRSF2Mut across each data set studied was retention of two consecutive introns (introns 4 and 5) in INTS3, generating a PTC in intron 4 (Supplemental Figs. S1B, S2A). INTS3 (integrator complex subunit 3) is a member of the Integrator complex, which plays important roles in both transcription initiation and the release of paused RNA polymerase II (Gardini et al. 2014). We amplified the INTS3 mRNA region spanning exon 4 to exon 5 by radioactive RT-PCR, using primary AML patient RNA. We observed increased intron 4 retention in all three SRSF2Mut AML cohorts but not in SRSF2WT (Supplemental Fig. S2B). We also consistently observed intron 4 retention in K052 cells but not in K562 or MOLM-13 cells (Supplemental Fig. S2B). Knocking down UPF1 (with shUPF1-1 or shUPF1-2) also significantly increased the intron-4-retained mRNA (Supplemental Fig. S2C). These results confirmed that the intron-retained INTS3 mRNA isoform is indeed degraded by NMD.
Sequence-specific splicing regulation by SRSF2Mut
We used a cell-based cotransfection assay to investigate the activities of SRSF2Mut. We generated T7-tagged cDNA constructs, encoding SRSF2WT or SRSF2Mut (P95H, P95L, or P95R), and confirmed their expression at similar levels in K562 cells (Fig. 1G). Although several SR proteins shuttle between the nucleus and the cytoplasm, SRSF2 localizes exclusively in the nucleus in HeLa cells (Fu and Maniatis 1990; Spector et al. 1991; Cáceres et al. 1998). Nuclear and cytoplasmic fractionation revealed that mutations in Pro95 of SRSF2 do not alter its exclusive nuclear localization (Fig. 1G). We then cotransfected an EZH2 minigene into K562 cells together with T7-tagged cDNAs. The EZH2 minigene spans sequences from exons 8 to 9, including the alternative poison exon within intron 8. The poison exon in the EZH2 WT minigene (CCWG) has three preferred binding motifs for SRSF2Mut but no preferred motif for SRSF2WT. We included another mutant version of the minigene, EZH2 mutant (GGWG), in which we mutated all three motifs to match the preferred binding motifs of SRSF2WT (Fig. 1H). Radioactive RT-PCR revealed that transient expression of SRSF2Mut significantly promoted exon inclusion with the CCWG minigene but not the GGWG minigene (Fig. 1H). In contrast, inclusion of the poison exon was significantly promoted by SRSF2WT with the GGWG minigene but not the CCWG minigene. We repeated the experiment in HeLa cells and obtained consistent results (Supplemental Fig. S2D,E). These results not only confirmed the sequence-specific exon-inclusion activity of SRSF2Mut in EZH2 but also validated the suitability of our cell-based cotransfection assay.
SRSF2WT and SRSF2Mut elicit mRNA decay in a sequence-specific manner
Our previous work showed that overexpression of SRSF1 or SRSF2 enhances NMD of HBB and GPX1 reporters harboring a PTC at the 39th and 46th codons, respectively, which cause recessive forms of β-thalassemia and hemolytic anemia, respectively (Zhang and Krainer 2004; Aznarez et al. 2018). To investigate whether mutation at Pro95 of SRSF2 alters its NMD-inducing activity, we tested the well-characterized NMD substrate, HBB with a nonsense mutation in codon 39 (HBB T39). We cotransfected K562 cells with the HBB reporter (WT or T39) and T7-tagged cDNAs. Radioactive RT-PCR showed that the SRSF2 mutants stimulate NMD to a similar extent as the WT (Fig. 2A–C). SRSF1 served as a control and showed a similar extent of NMD-inducing activity, consistent with our previous observations (Zhang and Krainer 2004; Aznarez et al. 2018).
Figure 2.

Effect of SRSF2 WT and Pro95 mutants on NMD. (A) Diagram (not to scale) of wild-type (WT) and T39 nonsense mutant (T39) HBB reporters and two additional versions of each reporter, HBB CCWG and HBB GGWG, harboring different combinations of motifs (W = A/U) in exon 2. (TC) Normal termination codon, (PTC) premature termination codon. Arrows indicate the RT-PCR primers. (B) Radioactive RT-PCR of the HBB reporters cotransfected with the indicated T7-tagged cDNAs into K562 cells. An intron 1 retained transcript variant is marked by a hash (#). GFP products were loaded on the same gel a short time after loading HBB products. (C) mRNA bands in panel B were quantified, normalized to GFP, and plotted as T39/WT (%) (mean ± SD, n = 3). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (NS) not significant, t-test. (D) mRNA-decay assay of the HBB reporters cotransfected with the indicated T7-tagged cDNAs into HeLa cells. Cells were collected at the indicated times after actinomycin D treatment. mRNAs were quantified by RT-qPCR, normalized to transfected GFP RNA levels, and expressed as a percentage of the levels at time 0 h (mean ± SD, n = 3).
To test for sequence-specific NMD-stimulating activity, we constructed NMD reporters comprising the preferred binding motifs of SRSF2WT or SRSF2Mut. Each of the three exons of HBB has several consensus motifs for both SRSF2WT and SRSF2Mut. We first simultaneously mutated the three motifs corresponding to SRSF2WT in exon 2 to match the SRSF2Mut motif (HBB CCWG reporter) (Fig. 2A). Note that mutating these motifs did not generate any additional PTCs. With this reporter, we observed a significant loss of the NMD-stimulatory activity of SRSF2WT, compared with SRSF2Mut (Fig. 2B,C). We noticed that the activity of SRSF1 was also compromised with HBB CCWG. Detailed motif analysis revealed that the binding motifs of SRSF1 and SRSF2WT overlap in HBB exon 2, such that mutating the binding motifs of SRSF2WT also disrupted the preferred motifs of SRSF1 (Tacke and Manley 1995; Anczuków et al. 2015). We next simultaneously mutated the 10 motifs corresponding to SRSF2Mut in exon 2 to match the SRSF2WT motifs (HBB GGWG reporter). As expected, we observed a significant reduction of the NMD-stimulatory activity of SRSF2Mut, compared with SRSF2WT (Fig. 2B,C). We also performed similar assays in nonhematopoietic HeLa cells (Supplemental Fig. S3A,B) and observed consistent results with those in K562 cells. We further performed an actinomycin D chase assay to measure the mRNA decay rate (Fig. 2D; Nomakuchi et al. 2016). As expected, both SRSF2WT- and SRSF2Mut(P95H) transfected cells expressed HBB T39 mRNA with lower half-lives (0.90 h and 0.88 h, respectively) than empty vector transfected cells (1.43 h). For HBB CCWG T39 mRNA, the half-life in SRSF2WT transfected cells was higher (4.58 h) than in SRSF2Mut(P95H) transfected cells (0.63 h). In contrast, for HBB GGWG T39 mRNA, the half-life in SRSF2WT transfected cells was lower (0.67 h) than in SRSF2Mut(P95H) transfected cells (2.04 h). These results, taken together, suggest that SRSF2WT and SRSF2Mut elicit mRNA decay in a sequence-dependent manner.
SRSF2Mut elicits stronger NMD than SRSF2WT when tethered downstream from a PTC
To understand the mode of action of SRSF2Mut, we designed a set of reporter constructs to tether the protein to the mRNA at different positions with respect to the PTC. We used HBB reporters (WT and T39) with MS2 hairpins inserted upstream of or downstream from the PTC in such a way that a complete and functional hairpin is formed only after splicing at the exon–exon junction (E1/E2 or E2/E3) (Fig. 3B; Lacadie et al. 2006; Aznarez et al. 2018). We also generated MS2-fused T7-tagged SRSF2 (MS2-SRSF2) (Fig. 3A). We confirmed the expression of MS2-SRSF2WT and MS2-SRSF2Mut at similar levels in HeLa cells (Fig. 3A). To eliminate MS2-independent binding of MS2-SRSF2 (WT or mutant) in exon 2, we inactivated both GGWG and C/GCWG motifs to AAWG (which is not a preferred binding motif for either SRSF2WT or SRSF2Mut) (Fig. 3B; Daubner et al. 2012; Kim et al. 2015; Zhang et al. 2015). Note that mutating these motifs did not generate any additional PTCs. We cotransfected HeLa cells with the reporter and effector constructs encoding T7-tagged SR protein (T7-SR) or MS2-fused T7-tagged SR protein (MS2-SR), representing MS2-untagged and MS2-tagged versions of SR proteins, respectively. Radioactive RT-PCR of RNA extracted from the T7-tagged antibody immunoprecipitated samples confirmed the enrichment of HBB-MS2 reporter RNA from MS2-tagged cDNA transfected cells, compared with MS2-untagged cDNA transfected cells (Supplemental Fig. S4A).Tethering of MS2-SR showed stronger NMD efficiency for the downstream recruitment (E2/E3) but not the upstream recruitment (E1/E2) (Fig. 3C,D). In addition, the SRSF2 mutants showed significantly stronger NMD efficiency than SRSF2WT. In contrast, T7-SR did not markedly change NMD efficiency, with either E1/E2 or E2/E3 reporters (Supplemental Fig. S4B,C).
Figure 3.

SRSF2 promotes NMD when tethered downstream from a PTC, an effect that is dependent on splicing. (A) Diagram of cDNA constructs and Western blotting (right) of HeLa cells transfected with the indicated cDNAs using anti-T7-tagged and anti-β-tubulin (tubulin) antibodies. Note that MS2 cDNA does not have a T7 tag. (B) Diagram (not to scale) of HBB reporters with MS2-binding sites formed after splicing at the first or second exon–exon junctions, respectively (E1/E2-MS2 and E2/E3-MS2, respectively). Note that the binding motifs for both SRSF2 WT (GGWG) or SRSF2 mutant (CCWG) in exon 2 were inactivated to AAWG motifs (W = A/U). (C) Radioactive RT-PCR of the HBB reporters shown in B in HeLa cells cotransfected with the indicated cDNAs. (D) mRNA bands in C were quantified, normalized to GFP, and plotted as T39/WT (%) (mean ± SD, n = 3). (**) P < 0.01; (NS) not significant, t-test. (E) Diagram (not to scale) of HBB E2/E3-MS2 reporters shown in B, with sequential deletions of introns. (F) Radioactive RT-PCR of the HBB reporters shown in E in HeLa cells cotransfected with the indicated cDNAs. (G) mRNA bands in F were quantified, normalized to GFP, and plotted as T39/WT (%) (mean ± SD, n = 3). (*) P < 0.05; (NS) not significant, t-test. An intron 1 retained transcript variant is marked by a hash (#) in C. (TC) Normal termination codon; (PTC) premature termination codon.
Note that in these experiments (Fig. 3C,D), all the effectors were recruited to the same site through MS2, because all of the WT and mutant SRSF2 motifs in exon 2 were mutated in these reporters; therefore, the relative NMD-inducing activity of WT versus mutant SRSF2 can be directly compared. Another important implication of these results is that the MS2-SR proteins should exclusively bind to spliced mRNA, due to the lack of a functional hairpin-binding site in the pre-mRNA. Therefore, all of the effects of MS2-SR proteins should be mRNA-specific. These results clearly argue that mutations in SRSF2 exert more effective NMD induction by additional mechanisms besides differential substrate recognition.
One intriguing question is whether high-affinity binding of MS2-SRSF2 to the mRNA through the MS2 tag results in transport of MS2-SRSF2 to the cytoplasm, as part of the mRNPs, which could result in altered properties of MS2-SRSF2, compared with natural SRSF2. To evaluate this possibility, we cotransfected HeLa cells with HBB E2/E3-MS2 (WT and T39) reporters and plasmids expressing MS2-SR, fractionated the cells into nuclear and cytoplasmic fractions, and measured the levels of MS2-SR proteins by Western blotting. MS2-SRSF1 was present in both nuclear and cytoplasmic fractions, whereas MS2-SRSF2 (either WT or mutant) remained exclusively in the nucleus (Supplemental Fig. S4D). Therefore, the bulk of MS2-SRSF2 (either WT or mutant) has the same nuclear-retention properties as SRSF2, excluding possible artifacts due to altered subcellular distribution. These results, taken together, indicate that the SRSF2 mutants have stronger NMD-inducing activity than the WT and elicit NMD when tethered downstream from a PTC.
SRSF2-mediated NMD-inducing activity is dependent on RNA splicing
We next tried to understand whether RNA splicing is necessary for SRSF2-promoted NMD. We generated two additional versions of the HBB E2/E3-MS2 reporter, deleting intron 1 alone (HBB-Δi1 E2/E3-MS2) or both introns 1 and 2 (HBB-Δi1Δi2 E2/E3-MS2) (Fig. 3E). Cotransfection into HeLa cells, as above, revealed that intron 1 is not required for the NMD-inducing activity of MS2-SRSF2 (WT or mutant), although the overall NMD efficiency was slightly reduced, compared with the reporter with both introns (HBB E2/E3-MS2) (Fig. 3F,G). However, deletion of both introns completely abrogated the NMD-inducing activity of MS2-SRSF2, indicating that splicing is necessary for SRSF2 to promote NMD (Fig. 3F,G). Radioactive RT-PCR of RNA from nuclear and cytoplasmic fractions confirmed that the mRNA of the reporter lacking both introns is efficiently exported to the cytoplasm (Supplemental Fig. S4E). We conclude that splicing of the intron upstream of the PTC is dispensable, but splicing of the intron downstream from the PTC is essential for SRSF2-mediated NMD.
SRSF2-mediated NMD-inducing activity is dependent on the EJC
We next sought to identify the critical step(s) in the NMD pathway that are modulated by SRSF2 (WT vs. mutants). Considering the exclusive nuclear localization of SRSF2, we assumed that the primary regulation should occur in the nucleus. We investigated the proteins that interact with SRSF2 (WT and mutants) in the NMD pathway. We performed coimmunoprecipitations (co-IP) followed by mass spectrometry (MS) with isobaric tags for relative and absolute quantification (iTRAQ) (Perkins et al. 1999; Ross et al. 2004; Gilar et al. 2005). We used isogenic K562 cells expressing SRSF2WT or SRSF2Mut (P95H/P95L/P95R) with a C-terminal Flag tag (Supplemental Fig. S5A; Kim et al. 2015). We performed IPs via the Flag tag in the presence of both Benzonase Nuclease and RNase A + T1 cocktail (Supplemental Fig. S5B). MS revealed a large number of proteins that copurified with either WT or mutants of SRSF2 (Supplemental Fig. S6A lists those proteins relevant to splicing or NMD). The iTRAQ ratios did not reveal significantly different interactions between SRSF2WT and SRSF2Mut. Many splicing factors, including SR proteins, showed enrichment, both with WT and mutant SRSF2. Among the EJC components, we observed enrichment of two core factors, eIF4A3 and MAGOH B, and several peripheral factors, including RNPS1, Aly/REF, and PININ. Only UPF2 was enriched among the NMD factors. We performed IP-Westerns to validate several interactions (Supplemental Fig. S6B).
Although the observed protein–protein interactions were not significantly different between SRSF2WT and SRSF2Mut, their NMD-inducing activity could be differentially modulated in the context of their target mRNPs, based on their binding affinities. We next evaluated whether SRSF2-EJC interactions are functionally correlated with the NMD-inducing activity. We down-regulated the expression of several core EJC factors (eIF4A3, MAGOH, Y14) and one essential NMD factor (UPF1), using siRNA in HeLa cells, and measured the NMD of the HBB T39 reporter in the presence or absence of SRSF2 (WT or mutant). Down-regulation of UPF1 inhibited NMD and resulted in loss of NMD-inducing activity of all tested SR proteins, compared with control knockdown using siRNA against luciferase (Fig. 4A–D). We also observed NMD inhibition upon knocking down each of the three core EJC factors eIF4A3, Y14, and MAGOH. In agreement with our previous work (Aznarez et al. 2018), overexpression of SRSF1 still induced NMD to some extent, despite the reduction in each of the EJC core proteins, suggesting that these individual components are dispensable for SRSF1 to elicit NMD. However, SRSF2-enhanced NMD was exclusively dependent on the EJC.
Figure 4.

The NMD-inducing activity of SRSF2 is dependent on the EJC. (A) Western blotting of HeLa cells treated with the indicated siRNAs. Tubulin was used as a loading control. (B) Quantification of knockdown efficiency in A, relative to control siRNA against luciferase (%) (mean ± SD, n = 3). (C) Radioactive RT-PCR of the HBB reporters (WT or T39) of the indicated siRNA-treated HeLa cells cotransfected with the indicated cDNAs. (#) An intron 1 retained transcript variant. (D) mRNA bands in C were quantified, normalized to GFP, and plotted as T39/WT (%) (mean ± SD, n = 3). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, compared with corresponding empty vector-treated cells, t-test.
Activity of SRSF2WT and SRSF2Mut in deposition of EJC and NMD factors
To assess the individual ability of WT or mutant SRSF2 to modulate the deposition of EJC and NMD factors, we coexpressed an NMD reporter in HeLa cells expressing WT or mutant SRSF2 and purified the NMD substrate-specific mRNPs. We performed MS2-mediated mRNP purification (MMP) (see the Materials and Methods) adapted with modifications from previously reported methods (Zhou et al. 2002; Lejeune and Maquat 2004; Singh et al. 2012). We generated a modified version of the HBB T39 reporter (HBB-T39-2MS2) with two split MS2 hairpins, formed in the mRNA across the E1/E2 and E2/E3 junctions, respectively (Fig. 5A). We used two MS2 hairpins to improve the pull-down efficiency. We prepared recombinant MBP-MS2 fusion protein, comprising maltose-binding protein (MBP) and MS2 coat protein (MS2) (Supplemental Fig. S7; Jurica et al. 2002; Zhou et al. 2002). Using this fusion protein along with amylose resin beads, we captured mRNPs formed on spliced HBB-T39-2MS2 mRNA, and detected individual bound proteins by Western blotting (Fig. 5B). Note that the corresponding unspliced pre-mRNP complexes should not be captured, due to the lack of MS2 hairpins.
Figure 5.

SRSF2 Pro95 mutants enhance the deposition of EJC and NMD factors on mRNA. (A) Diagram (not to scale) of T39 nonsense mutant HBB reporter (HBB-T39-2MS2), with MS2 binding sites formed after splicing at both exon–exon junctions. An additional version of this reporter (HBB-T39-AAWG-2MS2) was generated in which the binding motifs of both SRSF2 WT (GGWG) and SRSF2 mutant (CCWG) in exon 2 were changed to AAWG motifs (W = A/U). (TC) Normal termination codon; (PTC) premature termination codon. (B) Diagram of MS2-mediated mRNP purification (MMP). (C) Representative Western blotting and RT-PCR of input or purified mRNA-protein complexes from HeLa cells cotransfected with the indicated reporters and cDNAs. (D) Quantification of the enrichment of individual EJC or NMD factors relative to the corresponding empty vector lane (normalized to the corresponding amount of enriched RNA) in C (mean ± SD, n = 3). (*) P < 0.05; (**) P < 0.01, compared with corresponding empty vector-treated cells, t-test. Enrichment of individual EJC or NMD factors was further compared (horizontal lines) between WT and each SRSF2 mutant (mean ± SD, n = 3). (*) P < 0.05, t-test.
To assess nonspecific background, we probed for tubulin in the purified samples but could not detect it in any of the samples. We observed significantly enhanced deposition of several EJC and NMD factors in cells expressing T7-SR proteins, compared with control cells expressing an empty vector (Fig. 5C,D). Moreover, the efficiency of deposition of three core EJC factors, eIF4A3, Y14, and MAGOH, was significantly enhanced in SRSF2 mutant cells, compared with WT cells. However, there was no enhanced deposition of the fourth EJC core factor, MLN51, or the peripheral EJC factor, RNPS1. In the case of NMD factors, we found significantly enhanced deposition of UPF1, UPF2, and UPF3B (except for SRSF2 P95H, P = 0.13) in SRSF2 mutant cells but no increment in the deposition of eRF1 and eRF3 release factors. To determine whether SRSF2Mut-mediated enhanced deposition of EJC and NMD factors is dependent on binding to RNA, we generated an additional version of the reporter (HBB-AAWG-T39-2MS2), in which we inactivated all the preferred binding motifs of either WT SRSF2 or mutant SRSF2 in exon 2 by changing them to the AAWG motif. As expected, SRSF2Mut-mediated enhanced deposition of EJC (eIF4A3, MAGOH, and Y14) and NMD (UPF1, UPF2, and UPF3B) factors was not seen with this reporter (Fig. 5C,D). We also compared the expression profile of EJC and NMD factors in the RNA-seq data of AML patients and K562 cells with or without SRSF2 mutation and found that they are expressed at similar levels (Supplemental Table S2). We conclude that the sequence-specific binding of SRSF2Mut to RNA enhances the deposition of EJC factors via RNA-mediated interactions. This in turn facilitates the association of the key NMD factors to enhance mRNA decay.
Antisense-mediated targeted blocking of EJC deposition downstream from the PTC inhibits SRSF2Mut-induced NMD
We next asked whether targeted blocking of EJC deposition downstream from a PTC circumvents SRSF2Mut-mediated NMD. We recently reported a gene-specific method of NMD inhibition using antisense oligonucleotides (ASOs) (Nomakuchi et al. 2016). We selected one of the 2′-O-(2-methoxyethyl) (MOE) ribose-modified 15-mer ASOs that inhibits NMD of the HBB T39 mRNA and a control ASO targeting MECP2 (Nomakuchi et al. 2016) for cotransfection in HeLa cells along with the HBB reporter (WT or T39) and T7-SR cDNAs. As expected, ASO targeting HBB efficiently abrogated SRSF2-mediated NMD of HBB T39, compared with the negative control ASO or nontreated cells (Fig. 6A–C). This result again confirms the EJC-dependence of the NMD stimulatory activity of SRSF2Mut.
Figure 6.
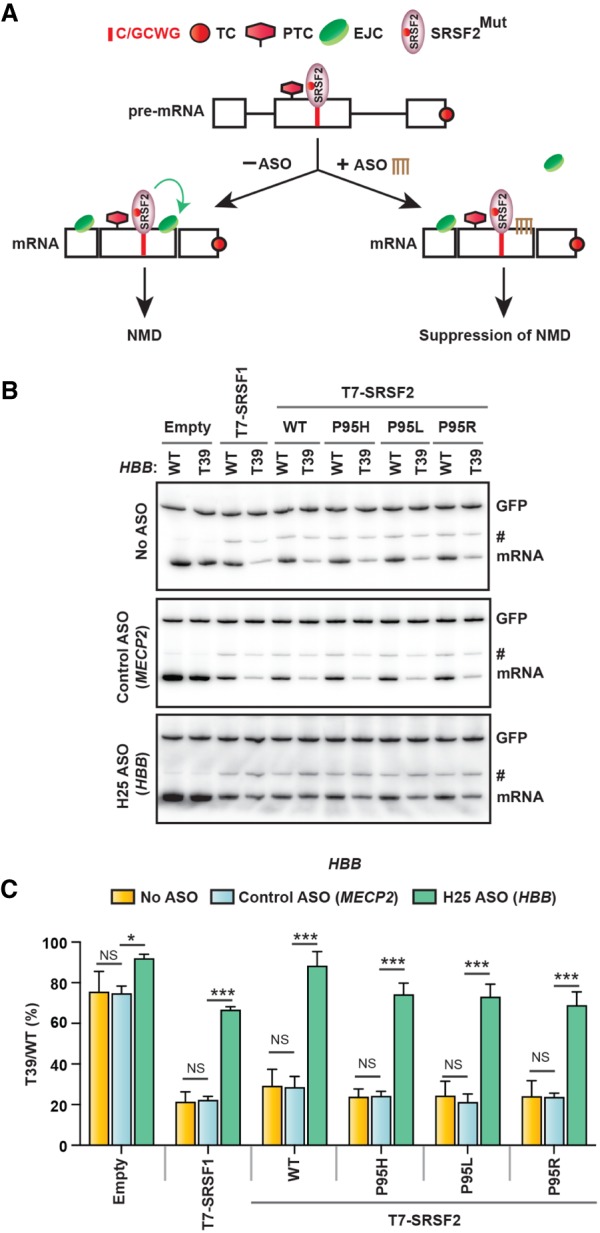
EJC targeting ASO inhibits NMD induction by mutant SRSF2. (A) Diagram of the antisense approach to inhibit mutant SRSF2-induced NMD. An ASO (brown) complementary to the predicted EJC-deposition site downstream from the PTC interferes with EJC assembly, inhibiting SRSF2Mut-promoted NMD. (TC) Normal termination codon, (PTC) premature termination codon. (B) Radioactive RT-PCR of the HBB reporters (WT or T39) of the indicated ASO-treated HeLa cells cotransfected with the indicated cDNAs. (#) An intron 1 retained transcript variant. (C) HBB mRNA bands in B were quantified, normalized to GFP, and plotted as T39/WT (%) (mean ± SD, n = 3). (*) P < 0.05; (***) P < 0.001; (NS) not significant, one-way ANOVA.
Discussion
We describe mechanistic features of SRSF2 in multiple aspects of RNA biogenesis, including splicing, remodeling mRNP structure, and NMD, which differ between WT and cancer-associated mutations in SRSF2, with broad implications both in normal physiology and in the context of hematologic malignancies. Our results confirmed the gain of binding of SRSF2 mutants to C-rich motifs, compared with G-rich motifs for WT SRSF2 in patients and the subsequent splicing misregulation throughout the genome. In addition, we demonstrated that mutation of SRSF2 results in stronger NMD-inducing activity, which also requires sequence-specific RNA binding (Fig. 7). This observation suggests an mRNA-specific gain of function, downstream from splicing. Binding of mutant SRSF2 to RNA enhances the deposition of EJCs to a greater extent than WT SRSF2, resulting in enhanced association of several key NMD factors to augment mRNA decay (Fig. 7). In Drosophila, cis-acting sequences distal to the EJC site are essential for the recruitment of EJCs and to promote NMD (Saulière et al. 2010), and our model supports and extends this observation.
Figure 7.

Model of oncogenic splicing factor mutation-induced altered splicing and aberrant NMD. Mutation at Pro95 of SRSF2 changes its RNA-binding preferences from a G-rich motif (GGWG) to a C-rich motif (C/GCWG) (W = A/U), causing transcriptome-wide splicing alterations. Several mRNA isoforms promoted by SRSF2 mutants harbor a premature termination codon and are therefore potential targets of NMD. In addition to altering RNA splicing, SRSF2 mutants further enhance the deposition of EJCs bound to mRNA downstream from a PTC. Specifically, mutant SRSF2 promotes the deposition of three core EJC factors (eIF4A3, MAGOH, and Y14) via RNA-mediated molecular interactions. This subsequently enhances the association of several NMD factors (UPF3B, UPF2, and UPF1), thereby enhancing mRNA decay.
We also observed wild-type SRSF1-mediated enhancement of EJC deposition, similar in extent to the activity of SRSF2Mut. In contrast, we consistently observed a comparatively lower extent of EJC deposition by SRSF2WT. One possible explanation is that physical association of SRSF2WT with the EJC might be weaker than that of SRSF2Mut or SRSF1. Analysis of the EJC interactome using RIPiT (RNA:protein immunoprecipitation in tandem) and MS revealed a robust enrichment of SR proteins (Singh et al. 2012). In that study, several SR proteins (SRSF1, SRSF3, SRSF7) were enriched in stoichiometric excess to the EJC core factors, whereas other SR proteins (SRSF9, SRSF10, SRSF12) were approximately stoichiometric with the EJC. SRSF2 was in the group of SR proteins with lower stoichiometry (with SRSF4, SRSF6, SRSF8). These data suggest a weaker association of SRSF2WT than SRSF1 with the EJC, in agreement with our results (Fig. 5). However, we did not observe a significant difference between WT and mutant SRSF2 in general protein–protein interactions with EJC core factors (eIF4A3 and MAGOH) in the presence of nuclease (Supplemental Fig. S6). These results are consistent with an RNA-dependent molecular interaction of the SRSF2Mut with the EJC, promoted by the presence of preferred binding sites on the RNA (Fig. 5).
How do SRSF2 mutants promote enhanced deposition of the EJC, compared with SRSF2WT? Structural analysis in solution by NMR indicated that Pro95 of the SRSF2WT RRM (RNA recognition motif) contacts the second nucleotide (marked by underline) of the GGWG motif or the G/CCWG motif, as shown by intermolecular nuclear Overhauser effects (NOEs) (Daubner et al. 2012). However, isothermal titration calorimetry (ITC) (Daubner et al. 2012; Kim et al. 2015) revealed that the SRSF2WT RRM has stronger binding affinity for GGWG (Kd = 0.22 µM for 5′-UGGAGU-3′) than for C/GCWG (Kd = 0.27 µM for 5′-UCCAGU-3′; Kd = 0.41 µM for 5′-UGCAGU-3′). In contrast, mutations in Pro95 of SRSF2 cause a conformational change in both termini of the RRM, which promote stronger contact with the second nucleotide (underlined) of the G/CCWG motif, compared with a weaker contact with the GGWG motif, as shown by the chemical-shift perturbations of proton resonances (Kim et al. 2015). ITC further showed that mutations at Pro95 enhance the binding affinity for the C/GCWG motif ∼3.9-fold to 4.5-fold relative to SRSF2WT. Considering our results, it is plausible that the stronger association of SRSF2Mut with RNA harboring a G/CCWG motif enhances the recruitment or stabilization of the EJC.
Our model is also consistent with previous findings about the distribution of EJC sites throughout the transcriptome. Two types of EJCs have been described (Saulière et al. 2012; Singh et al. 2012) on the basis of the RNA sites they occupy: canonical EJCs (cEJCs) centered at the −24 position, and noncanonical EJCs (ncEJCs) at other exonic locations, comprising ∼40% of the total EJCs. It is well established that cEJCs are platforms for recruitment of NMD factors, but the functional role(s) of ncEJC is not well understood. Sequence analysis of cEJC peaks did not reveal a consensus motif; in contrast, ncEJC peaks yielded identifiable consensus motifs. Two highly enriched ncEJC motif classes in internal exons are AG-rich motifs and C-rich motifs (Singh et al. 2012). In general, AG-rich motifs are preferred binding sites for SRSF1 and SRSF3, whereas C-rich motifs are low-affinity binding sites for wild-type SRSF2 and high-affinity binding sites for mutant SRSF2. These observations support the notion that SR proteins bind to ncEJC sites and that there is functional coordination between RNA-bound SR proteins and EJCs. Based on our findings, we propose that SR proteins are good candidates to function in EJC stabilization, being bound to the mRNA in close proximity. Interestingly, both cEJC and ncEJC signals are most enriched in alternatively spliced mRNAs linked to NMD (Singh et al. 2012), suggesting that the SR protein-EJC nexus is a key feature of AS-NMD.
Although multiple SR proteins are involved in NMD regulation, the mechanisms appear to be different for individual family members. For example, knocking down individual EJC core factors abolishes SRSF2 (WT or mutant)-stimulated NMD activity; in contrast, SRSF1 still promoted NMD to some extent (Fig. 4), suggesting that it does so via an alternative EJC-independent pathway. We recently reported that SRSF1 directly interacts with UPF1 and promotes binding of UPF1 to mRNAs, bypassing the requirement for the EJC and UPF2 to enhance NMD (Aznarez et al. 2018). In contrast, we did not detect an interaction of SRSF2 (WT or mutant) with UPF1 (Supplemental Fig. S6B), explaining its strict dependence on the EJC. The SRSF2 mutants had no effect on deposition of MLN51, the fourth member of the EJC core. Compared with the other three core EJC factors (eIF4A3, Y14, and MAGOH), MLN51 assembles onto the EJC after the completion of splicing. It was suggested that MLN51 may join the exported mRNP in the cytoplasm (Gehring et al. 2009), considering its predominantly cytoplasmic localization. Our results confirmed that the SRSF2 mutants, like SRSF2 WT, remain exclusively nuclear in both K562 and HeLa cells. Therefore, if MLN51 assembles onto the exported mRNP in the cytoplasm, the SRSF2 mutants would not have a direct effect on its deposition. Our findings are also consistent with Mabin et al. (2018), who showed that EJCs first assemble onto SR protein-rich megadalton-sized RNPs that lack MLN51 and then undergo a compositional switch into SR protein-devoid monomeric MLN51-containing EJCs. In addition, the authors showed that MLN51 is dispensable for NMD of certain transcripts.
We consistently observe an accumulation of pre-mRNA when overexpressing various SR proteins (Figs. 2–4, 6, marked by a hash), as also seen in our previous work (Zhang and Krainer 2004; Aznarez et al. 2018). A similar accumulation of pre-mRNA was previously observed by overexpression of the SR-like protein SRm160 (McCracken et al. 2002). One possible explanation is that overexpression could result in the bypass of processes that normally degrade unprocessed RNA in the nucleus (McCracken et al. 2002). However, this accumulation of pre-mRNA does not account for the NMD enhancement we observed, as we confirmed mRNA decay after transcriptional shut-off by actinomycin D treatment (Fig. 2D). Moreover, several SR proteins or deletion mutants thereof that promoted pre-mRNA accumulation failed to enhance NMD (Aznarez et al. 2018).
Tumor cells often exploit NMD for survival benefit through different mechanisms: down-regulating the expression of tumor suppressors via NMD-inducing mutations (such as BRCA1, BRCA2, TP53, RB1, WT1), altering the function of NMD factors (such as UPF1) via somatic mutation, or adjusting NMD activity to adapt their microenvironment via signaling pathways (e.g., phosphorylation of eIF2α during hypoxia) (Popp and Maquat 2018). Although degrading PTC-containing transcripts by NMD is a protective mechanism that reduces dominant-negative effects of truncated proteins, it can be detrimental when the truncated protein is partially functional (such as hypomorphic nonsense CFTR mutations in cystic fibrosis) (Wilschanski et al. 2003). Here, we present mechanistic insights into hematologic malignancies, in which tumor cells down-regulate the expression of proteins that affect hematopoiesis by leveraging AS-NMD (such as poison exon inclusion in EZH2 and intron retention in INTS3) via somatic mutations in an RNA-binding protein that functions in both alternative splicing and NMD.
Having characterized the detailed mechanisms, we showed that gene-specific blocking of EJC deposition by ASOs inhibits SRSF2Mut-mediated NMD and restores the expression of a PTC-containing transcript. Therefore, this approach could be used in genetic model cells or mice to determine whether a particular AS-NMD isoform retains functional activity or has different activity and whether stabilizing such an isoform could alter the pathogenic phenotypes in hematopoietic malignancies.
Materials and methods
Patient samples
All human studies were approved by the Institutional Review Boards of Cold Spring Harbor Laboratory and Memorial Sloan Kettering Cancer Center and conducted in accordance with the Declaration of Helsinki protocol.
Minigene reporters
Construction of HBB WT and HBB T39 reporters was reported previously (Cáceres et al. 1994; Aznarez et al. 2018). We modified these plasmids by introducing artificial mutations to change binding motifs. MS2 binding sites were inserted using a QuikChange site-directed mutagenesis kit (Agilent). The absence of artifacts was confirmed by sequencing the entire inserts. Construction of EZH2 minigenes was described previously (Kim et al. 2015).
cDNA expression vectors
pCGT7-SRSF1 and pCGT7-SRSF2 (WT) were reported previously (Cáceres et al. 1997). To construct pCIneo-MS2-T7-SRSF1 and pCIneo-MS2-T7-SRSF2 (WT), the ORFs of T7-SRSF1 and T7-SRSF2 were cloned into BamHI/EcoRI sites downstream from MS2. P95H, P95L, and P95R mutations were introduced into pCGT7-SRSF2 and pCIneo-MS2-T7-SRSF2 using a QuikChange site-directed mutagenesis kit (Agilent). The absence of artifacts was confirmed by sequencing the entire inserts.
Cell culture, transfection, RT-PCR, and Western blotting
K562, K052, and MOLM-13 cells were maintained in RPMI-1640 and HeLa cells in DMEM, both supplemented with 10% fetal bovine serum, at 37°C in 5% CO2. Plasmids were transfected into K562 cells using Lipofectamine LTX reagent with PLUS reagent (Thermo Fisher Scientific) and into HeLa cells using X-tremeGENE DNA transfection reagents (Roche), according to the manufacturer's instructions. Transfections included 1 μg of plasmid mixture (0.1 μg of GFP, 0.3 μg of SR protein expression plasmid, 0.6 μg of NMD reporter construct) per well of a six-well plate. Total RNA was isolated after 48 h (unless specified otherwise) using TRIzol reagent, followed by DNase I treatment. Reverse transcription was performed using oligo-dT primer or vector-specific reverse primer and ImProm-II reverse transcriptase (Promega). Radioactive PCR was conducted using 32P-α-dCTP, 1.25 U of AmpliTaq (Invitrogen), and 26 cycles. Products were separated by 5% PAGE, and the bands were detected using a Typhoon FLA 7000 (GE Healthcare). Primers for amplification of endogenous EZH2 or EZH2 minigene were as described (Kim et al. 2015). See other primer sequences in Supplemental Table S3. Cell fractionation and Western blotting were done as described before (Aznarez et al. 2018). See information about antibodies in Supplemental Table S4 (Mayeda et al. 1999).
RNA interference
siRNAs (100 nM) were transfected into HeLa cells using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions for 72 h. See information about siRNAs in Supplemental Table S5 (Gehring et al. 2003; Lee et al. 2009; Muromoto et al. 2009). shRNAs were transduced using lentivirus into K052 cells, as described (Kim et al. 2015).
Coimmunoprecipitations and iTRAQ mass spectrometry
Isogenic K562 cells expressing an empty vector or SRSF2 (WT/P95H/P95L/P95R) with a C-terminal Flag tag were used for IP and MS. For IP, two 10-cm flasks of K562 cells were used per condition. Cells were lysed using IP lysis buffer (50 mM HEPES, 200 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail). Nuclease treatment was performed with 2 U/mL extract of benzonase nuclease and 2 U/mL extract of RNase A + T1 cocktail for 30 min on ice. Anti-Flag M2 magnetic beads (Sigma-Aldrich) were prepared according to the manufacturer's instructions and incubated with cell lysate supernatant, with gentle agitation for 2 h at 4°C. The IP samples were washed three times with IP lysis buffer. Proteins for iTRAQ mass spectrometry were eluted using an on-bead digestion protocol (see detail in the Supplemental Material). Proteins for Western blotting were eluted with Flag peptide (200 ng/μL).
MS2-mediated mRNP purification (MMP)
HeLa cells transfected with the indicated reporter and cDNA construct were grown in three 15-cm plates for 24 h. Two hours prior to cell harvesting, cycloheximide (CHX) was added at 100 mg/mL. The cells were washed in phosphate-buffered saline (PBS) containing 100 mg/mL CHX. The cells were then resuspended in 3 mL of lysis buffer (20 mM Tris-HCl at pH 7.5, 15 mM NaCl, 10 mM EDTA, 0.5% NP-40, 0.1% Triton X-100, RNasin, protease inhibitor cocktail, 100 mg/mL CHX) and incubated on ice for 10 min. After lysing the cells by sonication on ice, the NaCl concentration was adjusted to 200 mM. The lysate was then cleared by centrifugation at 15,000g for 10 min at 4°C. The cleared lysate was diluted to 10 mL in lysis buffer with a final NaCl concentration of 200 mM. The diluted lysate was incubated for 4 h at 4°C with prewashed 150 µL of amylose resin beads (50% slurry) mixed with 150 µg of recombinant MBP-MS2 (MS2 coat protein-tagged maltose-binding protein) (Zhou et al. 2002). The RNA–protein (RNP) complexes captured on beads were washed three times with wash buffer (20 mMTris-HCl at pH 7.5, 200 mM NaCl, 0.1% NP-40). Then, the bound mRNP complexes were eluted with elution buffer (20 mM Tris-HCl at pH 7.5, 200 mM NaCl, 10 mM 2-mercaptoethanol, 10 mM maltose, 1 mM PMSF). The purified proteins were resolved by SDS-PAGE and detected by Western blotting.
ASO transfections
ASOs were purchased from Integrated DNA Technologies (IDT) and were uniformly modified with MOE sugars, phosphorothioate backbone, and 5-methyl cytosine, as described (Nomakuchi et al. 2016). ASO transfections in HeLa cells were carried out using Lipofectamine 2000 (Life Technologies) as described (Nomakuchi et al. 2016).
RNA-seq analysis
Alignment files of the TCGA-LAML data set were downloaded from National Cancer Institute's Genomic Data Commons (dbGaP accession no. phs000178.v10.p8.c1). The junction reads were extracted from the alignment files by using the tool kit of STAR aligner (Dobin et al. 2013). Splicing analysis was done using “PSI-Sigma” (https://github.com/wososa/PSI-Sigma; Lin and Krainer 2019).
Quantification and statistical analysis
Statistical significance (RNA-seq analysis) was determined by a two-sample t-test. For the comparison of proportions, the “N-1” χ2 test was used. Quantification of the isoforms in radioactive RT-PCR or Western blots was done using ImageJ (National Institutes of Health). Statistical significance was determined by (1) t-test or (2) one-way ANOVA followed by Tukey's multiple comparison test. Data were plotted using GraphPad Prism 7 software as mean values, with error bars representing the standard deviation (SD).
Supplementary Material
Acknowledgments
We thank Akila Mayeda for kindly sharing the RNPS1 antibody, J. Lykke-Andersen for the UPF3B antibody, and Robin Reed for the MS2-MBP construct. We also thank the Krainer laboratory members, especially Isabel Aznarez, Olga Anczuków, Tomoki Nomakuchi, and Young Jin Kim, for sharing reagents and helpful advice. A.R.K. and O.A.-W. acknowledge support from the National Cancer Institute (P01-CA13106), National Instiutes of Health/National Heart, Lung, and Blood Institute (R01-HL128239), Department of Defense Bone Marrow Failure Research Program (W81XWH-16-1-0059), Starr Foundation (I8-A8-075), Henry and Marilyn Taub Foundation, Leukemia and Lymphoma Society, and Pershing Square Sohn Cancer Research Alliance. We also acknowledge assistance from the CSHL Shared Resources, funded in part by NCI Cancer Center Support Grant 5P30CA045508.
Author contributions: A.R.K. and O.A.-W. conceived the project. M.A.R. and A.R.K designed the study. M.A.R. performed all experiments under the supervision of A.R.K. K.-T.L. performed computational analysis. O.A.-W. provided AML patient RNA samples, human leukemia cell lines, and shRNAs against UPF1. R.K.B. provided EZH2 minigenes and SRSF2 wild-type and mutant K562 cell lines. M.A.R. and A.R.K prepared the manuscript, with input from all coauthors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.332270.119.
References
- Anczuków O, Akerman M, Cléry A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al. 2015. SRSF1-regulated alternative splicing in breast cancer. Mol Cell 60: 105–117. 10.1016/j.molcel.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznarez I, Nomakuchi TT, Tetenbaum-Novatt J, Rahman MA, Fregoso O, Rees H, Krainer AR. 2018. Mechanism of nonsense-mediated mRNA decay stimulation by splicing factor SRSF1. Cell Rep 23: 2186–2198. 10.1016/j.celrep.2018.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME suite: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres JF, Stamm S, Helfman D, Krainer AR. 1994. Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science 265: 1706–1709. 10.1126/science.8085156 [DOI] [PubMed] [Google Scholar]
- Cáceres JF, Misteli T, Screaton GR, Spector DL, Krainer AR. 1997. Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J Cell Biol 138: 225–238. 10.1083/jcb.138.2.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres JF, Screaton GR, Krainer AR. 1998. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev 12: 55–66. 10.1101/gad.12.1.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen JY, Huang YJ, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H, et al. 2018. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol Cell 69: 412–425.e6. 10.1016/j.molcel.2017.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. 10.1016/j.celrep.2015.09.053 [DOI] [PubMed] [Google Scholar]
- Daubner GM, Cléry A, Jayne S, Stevenin J, Allain FHT. 2012. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J 31: 162–174. 10.1038/emboj.2011.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Maniatis T. 1990. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature 343: 437–441. 10.1038/343437a0 [DOI] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, Shiekhattar R. 2014. Integrator regulates transcriptional initiation and pause release following activation. Mol Cell 56: 128–139. 10.1016/j.molcel.2014.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Neu-Yilik G, Schell T, Hentze MW, Kulozik AE. 2003. Y14 and hUpf3b form an NMD-activating complex. Mol Cell 11: 939–949. 10.1016/S1097-2765(03)00142-4 [DOI] [PubMed] [Google Scholar]
- Gehring NH, Lamprinaki S, Hentze MW, Kulozik AE. 2009. The hierarchy of exon-junction complex assembly by the spliceosome explains key features of mammalian nonsense-mediated mRNA decay. PLoS Biol 7: e1000120 10.1371/journal.pbio.1000120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilar M, Olivova P, Daly AE, Gebler JC. 2005. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J Sep Sci 28: 1694–1703. 10.1002/jssc.200500116 [DOI] [PubMed] [Google Scholar]
- Gudikote JP, Imam JS, Garcia RF, Wilkinson MF. 2005. RNA splicing promotes translation and RNA surveillance. Nat Struct Mol Biol 12: 801–809. 10.1038/nsmb980 [DOI] [PubMed] [Google Scholar]
- Hentze MW, Izaurralde E. 2013. Making sense of nonsense. Nat Struct Mol Biol 20: 651–653. 10.1038/nsmb.2601 [DOI] [PubMed] [Google Scholar]
- Huang L, Lou CH, Chan W, Shum EY, Shao A, Stone E, Karam R, Song HW, Wilkinson MF. 2011. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell 43: 950–961. 10.1016/j.molcel.2011.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Maquat LE. 2011. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr Opin Genet Dev 21: 422–430. 10.1016/j.gde.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. 2015. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25: 14–26. 10.1101/gr.181016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ. 2002. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8: 426–439. 10.1017/S1355838202021088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S. 2006. Binding of a novel SMG-1–Upf1–eRF1–eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 20: 355–367. 10.1101/gad.1389006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JA, Ebert BL. 2017. Clinical implications of genetic mutations in myelodysplastic syndrome. J Clin Oncol 35: 968–974. 10.1200/JCO.2016.71.0806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Kataoka N, Dreyfuss G. 2001. Role of the nonsense-mediated decay factor hUpf3 in the splicing-dependent exon-exon junction complex. Science 293: 1832–1836. 10.1126/science.1062829 [DOI] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SCW, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. 2015. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27: 617–630. 10.1016/j.ccell.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, Chen L, Monterroza DD, Li H, DeKelver RC, et al. 2015. SRSF2 is essential for hematopoiesis, and its myelodysplastic syndrome-related mutations dysregulate alternative pre-mRNA splicing. Mol Cell Biol 35: 3071–3082. 10.1128/MCB.00202-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacadie SA, Tardiff DF, Kadener S, Rosbash M. 2006. In vivo commitment to yeast cotranscriptional splicing is sensitive to transcription elongation mutants. Genes Dev 20: 2055–2066. 10.1101/gad.1434706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Choe J, Chi SG, Kim YK. 2009. Exon junction complex enhances translation of spliced mRNAs at multiple steps. Biochem Biophys Res Commun 384: 334–340. 10.1016/j.bbrc.2009.04.123 [DOI] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ. 2000. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon–exon junctions. EMBO J 19: 6860–6869. 10.1093/emboj/19.24.6860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Maquat LE. 2004. Immunopurification and analysis of protein and RNA components of mRNP in mammalian cells. Methods Mol Biol 257: 115–124. [DOI] [PubMed] [Google Scholar]
- Lin KT, Krainer AR. 2019. PSI-Sigma: a comprehensive splicing-detection method for short-read and long-read RNA-seq analysis. Bioinformatics 35: 5048–5054. 10.1093/bioinformatics/btz438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, Shu M, Steitz JA. 2000. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103: 1121–1131. 10.1016/S0092-8674(00)00214-2 [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen J, Shu MD, Steitz JA. 2001. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science 293: 1836–1839. 10.1126/science.1062786 [DOI] [PubMed] [Google Scholar]
- Mabin JW, Woodward LA, Patton RD, Yi Z, Jia M, Wysocki VH, Bundschuh R, Singh G. 2018. The exon junction complex undergoes a compositional switch that alters mRNP structure and nonsense-mediated mRNA decay activity. Cell Rep 25: 2431–2446.e7. 10.1016/j.celrep.2018.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. 2015. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6: 6042 10.1038/ncomms7042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE, Li X. 2001. Mammalian heat shock p70 and histone H4 transcripts, which derive from naturally intronless genes, are immune to nonsense-mediated decay. RNA 7: 445–456. 10.1017/S1355838201002229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE, Tarn WY, Isken O. 2010. The pioneer round of translation: features and functions. Cell 142: 368–374. 10.1016/j.cell.2010.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda A, Badolato J, Kobayashi R, Zhang MQ, Gardiner EM, Krainer AR. 1999. Purification and characterization of human RNPS1: a general activator of pre-mRNA splicing. EMBO J 18: 4560–4570. 10.1093/emboj/18.16.4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S, Lambermon M, Blencowe BJ. 2002. SRm160 splicing coactivator promotes transcript 3′-end cleavage. Mol Cell Biol 22: 148–160. 10.1128/MCB.22.1.148-160.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muromoto R, Taira N, Ikeda O, Shiga K, Kamitani S, Togi S, Kawakami S, Sekine Y, Nanbo A, Oritani K, et al. 2009. The exon-junction complex proteins, Y14 and MAGOH regulate STAT3 activation. Biochem Biophys Res Commun 382: 63–68. 10.1016/j.bbrc.2009.02.127 [DOI] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. 10.1101/gad.1525507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomakuchi TT, Rigo F, Aznarez I, Krainer AR. 2016. Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat Biotechnol 34: 164–166. 10.1038/nbt.3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. 2016. Physiologic expression of Sf3b1K700E causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 30: 404–417. 10.1016/j.ccell.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. 2013. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122: 3616–3627. 10.1182/blood-2013-08-518886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- Popp MW, Maquat LE. 2018. Nonsense-mediated mRNA decay and cancer. Curr. Opin. Genet. Dev 48: 44–50. 10.1016/j.gde.2017.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et al. 2004. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3: 1154–1169. 10.1074/mcp.M400129-MCP200 [DOI] [PubMed] [Google Scholar]
- Sato H, Hosoda N, Maquat LE. 2008. Efficiency of the pioneer round of translation affects the cellular site of nonsense-mediated mRNA decay. Mol Cell 29: 255–262. 10.1016/j.molcel.2007.12.009 [DOI] [PubMed] [Google Scholar]
- Saulière J, Haque N, Harms S, Barbosa I, Blanchette M, Le Hir H. 2010. The exon junction complex differentially marks spliced junctions. Nat Struct Mol Biol 17: 1269–1271. 10.1038/nsmb.1890 [DOI] [PubMed] [Google Scholar]
- Saulière J, Murigneux V, Wang Z, Marquenet E, Barbosa I, Le Tonquèze O, Audic Y, Paillard L, Roest Crollius H, Le Hir H. 2012. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nat Struct Mol Biol 19: 1124–1131. 10.1038/nsmb.2420 [DOI] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE. 2012. Regulation of cytoplasmic mRNA decay. Nat Rev Genet 13: 246–259. 10.1038/nrg3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ. 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151: 750–764. 10.1016/j.cell.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL, Fu XD, Maniatis T. 1991. Associations between distinct pre-mRNA splicing components and the cell nucleus. EMBO J 10: 3467–3481. 10.1002/j.1460-2075.1991.tb04911.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke R, Manley JL. 1995. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J 14: 3540–3551. 10.1002/j.1460-2075.1995.tb07360.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange TØ, Shibuya T, Jurica MS, Moore MJ. 2005. Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA 11: 1869–1883. 10.1261/rna.2155905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, et al. 2003. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med 349: 1433–1441. 10.1056/NEJMoa022170 [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. 10.1038/nature10496 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Krainer AR. 2004. Involvement of SR proteins in mRNA surveillance. Mol Cell 16: 597–607. 10.1016/j.molcel.2004.10.031 [DOI] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. 2015. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci 112: E4726–E4734. 10.1073/pnas.1514105112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Licklider LJ, Gygi SP, Reed R. 2002. Comprehensive proteomic analysis of the human spliceosome. Nature 419: 182–185. 10.1038/nature01031 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.