

Abstract
Inflammatory breast cancer (IBC) is the most pro‐metastatic form of breast cancer. Better understanding of its pathophysiology and identification of actionable genetic alterations (AGAs) are crucial to improve systemic treatment. We aimed to define the DNA profiles of IBC vs noninflammatory breast cancer (non‐IBC) clinical samples in terms of copy number alterations (CNAs), mutations, and AGAs. We applied targeted next‐generation sequencing (tNGS) and array‐comparative genomic hybridization (aCGH) to 57 IBC and 50 non‐IBC samples and pooled these data with four public datasets profiled using NGS and aCGH, leading to a total of 101 IBC and 2351 non‐IBC untreated primary tumors. The respective percentages of each molecular subtype [hormone receptor‐positive (HR+)/HER2−, HER2+, and triple‐negative] were 68%, 15%, and 17% in non‐IBC vs 25%, 35%, and 40% in IBC. The comparisons were adjusted for both the molecular subtypes and the American Joint Committee on Cancer (AJCC) stage. The 10 most frequently altered genes in IBCs were TP53 (63%), HER2/ERBB2 (30%), MYC (27%), PIK3CA (21%), BRCA2 (14%), CCND1 (13%), GATA3 (13%), NOTCH1 (12%), FGFR1 (11%), and ARID1A (10%). The tumor mutational burden was higher in IBC than in non‐IBC. We identified 96 genes with an alteration frequency (p < 5% and q < 20%) different between IBC and non‐IBC, independently from the molecular subtypes and AJCC stage; 95 were more frequently altered in IBC, including TP53, genes involved in the DNA repair (BRCA2) and NOTCH pathways, and one (PIK3CA) was more frequently altered in non‐IBC. Ninety‐seven percent of IBCs displayed at least one AGA. This percentage was higher than in non‐IBC (87%), notably for drugs targeting DNA repair, NOTCH signaling, and CDK4/6, whose pathways were more frequently altered (DNA repair) or activated (NOTCH and CDK4/6) in IBC than in non‐IBC. The genomic landscape of IBC is different from that of non‐IBC. Enriched AGAs in IBC may explain its aggressiveness and provide clinically relevant targets.
Keywords: copy number profiling, DNA repair, inflammatory breast cancer, NOTCH, sequencing, targeted therapy
We compared the DNA mutational and copy number profiles and mRNA expression profiles of a large series of IBC and noninflammatory breast cancer (non‐IBC) untreated primary tumors. Compared to non‐IBC samples, IBC samples displayed more frequent alterations in the NOTCH and DNA repair pathways independently form the unbalance in term of molecular subtypes and American Joint Committee on Cancer stages.

Abbreviations
- aCGH
array‐comparative genomic hybridization
- AGA
actionable genetic alteration
- AJCC
American Joint Committee on Cancer
- CIViC
Clinical‐Interpretation‐of‐Variants‐in‐Cancer
- CNA
copy number alteration
- GDKD
Gene‐Drug‐Knowledge‐database
- HR+
hormone receptor‐positive
- HRD
homologous recombination deficiency
- IBC
inflammatory breast cancer
- IHC
immunohistochemistry
- Mb
megabase
- Metabric
Molecular Taxonomy of Breast Cancer International Consortium
- NGS
next‐generation sequencing
- non‐IBC
noninflammatory breast cancer
- SNP
single nucleotide polymorphisms
- SNV
single nucleotide variant
- TARGET
Tumor‐Alterations‐Relevant‐for‐Genomics‐driven‐Therapy
- TCGA
The Cancer Genome Atlas
- TMB
tumor mutational burden
- TN
triple‐negative
- tNGS
targeted next‐generation sequencing
- WES
whole‐exome sequencing
- WGS
whole‐genome sequencing
1. Introduction
Inflammatory breast cancer (IBC) is the most aggressive clinical form of breast cancer (Dawood et al., 2011). Despite therapeutic progresses, ~ 50% of patients die from metastatic relapse. The distinct clinical presentation and aggressive behavior have not translated in design of differential treatment that remains similar to that of stage 3 noninflammatory breast cancer (non‐IBC). Identification of new therapeutic targets and better understanding of the pathophysiology are crucial (Charafe‐Jauffret et al., 2008). Because of the scarcity of disease, ‘omics’ studies remain rare in IBC (Bertucci et al., 2014a). The largest series reported to date is the one that we had collected within the International IBC Consortium (Bertucci et al., 2014b; Masuda et al., 2013; Van Laere et al., 2013), in which we notably showed the overrepresentation of aggressive molecular subtypes (basal, HER2‐enriched, luminal B) when compared with non‐IBC, justifying the need to stratify the IBC/non‐IBC comparison upon the molecular subtypes (Van Laere et al., 2013).
During the last decade, next‐generation sequencing (NGS) led to identification of driver alterations in non‐IBC (Banerji et al., 2012; Ellis et al., 2012; Ferrari et al., 2016; Nik‐Zainal et al., 2012a; Nik‐Zainal et al., 2016; Nik‐Zainal et al., 2012b; Shah et al., 2012; Stephens et al., 2012; The Cancer Genome Atlas, 2012). Precision medicine trials have shown the potential of DNA‐based genomics screening to identify clinically actionable genetic alterations (AGAs) for guiding treatment (Andre et al., 2014; Le Tourneau et al., 2015). Regarding IBC, five NGS‐based studies have been published since 2015 (Goh et al., 2016; Hamm et al., 2016; Liang et al., 2018; Matsuda et al., 2017; Ross et al., 2015). Except the most recent contribution (Liang et al., 2018), they concerned small series ranging from 19 to 53 IBCs, including both untreated primary tumors (between 16 and 25 cases only) and pretreated relapses. The number of tested genes varied between 50 and 255 for the studies using targeted NGS (Hamm et al., 2016; Liang et al., 2018; Matsuda et al., 2017; Ross et al., 2015) and whole‐exome sequencing (WES) (Goh et al., 2016). Few studies directly compared the genomic portraits of primary IBC and non‐IBC, and comparison was never stratified upon the molecular subtypes. However, three of the most recurrently mutated genes (TP53, PIK3CA, and HER2) have clear ties with molecular subtypes [i.e., triple‐negative (TN), luminal, and HER2‐enriched respectively]. The main finding of these studies was an increased tumor mutational burden (TMB) in IBC that translated in the presence of many AGAs with low frequency, but without identification of IBC‐specific driver genes.
Here, we present a large comparative study of untreated primary tumors of IBC and non‐IBC based on NGS data from Institut Paoli‐Calmettes (IPC; Marseille, France) and TCRU (Antwerp, Belgium), pooled with publicly available data (Hamm et al., 2016; Pereira et al., 2016; Ross et al., 2015; The Cancer Genome Atlas, 2012). After adjustment upon both the molecular subtypes and American Joint Committee on Cancer (AJCC) stage, we compared the genomic profiles of IBC and non‐IBC by in terms of DNA mutations and copy number alterations (CNA), TMB, and presence of AGAs.
2. Materials and methods
2.1. Patients and samples selection
All clinical samples were pretreatment diagnostic samples of primary breast cancers. IBC was clinically defined as T4d according to the international consensus criteria (Dawood et al., 2011), and the samples were diagnostic biopsies (AJCC stages 3–4). Non‐IBC samples were surgical specimens in case of early‐stage disease (stages 1–2) and diagnostic biopsies in case of advanced stage disease (locally advanced: stage 3, and metastatic: stage 4). The whole series included 101 IBCs and 2351 non‐IBCs, collected from six different sources (Table S1).
Forty‐four IBC and 50 non‐IBC samples were from patients consecutively treated at IPC, and 13 IBC samples were from patients consecutively treated at the General Hospital Sint‐Augustinus (TCRU). Extraction of tumor DNA, quality control, and concentration assessment were done as described (Bertucci et al., 2016). Each patient gave written informed consent, and the study was approved by the respective institutional review boards. The study methodology conformed to the standards set by the Declaration of Helsinki. The selection criteria included available frozen sample, tumor cellularity assessment to guide DNA extraction (> 50%), good‐quality extracted tumor DNA, and available clinicopathological data. These samples were pooled with four public series of similarly defined IBC and non‐IBC samples profiled by NGS [and array‐comparative genomic hybridization (aCGH) for two series]. The Ross’ (Ross et al., 2015) and Hamm's (Hamm et al., 2016) series included 25 and 17 IBC samples, respectively; the TCGA series (Cancer Genome Atlas, 2012) included two IBC and 988 non‐IBC samples; the Molecular Taxonomy of Breast Cancer International Consortium (Metabric) series included 1313 non‐IBC samples (Pereira et al., 2016). The molecular subtype of tumors based upon immunohistochemistry (IHC) was defined as HR+/HER2− when ER and/or PR were positive and HER2 negative, HER2+ when HER2 was positive, and TN when the three receptors were negative.
We also included NGS and aCGH data of metastatic samples from 468 non‐IBC patients pooled from our PERMED‐01 prospective clinical trial (NCT02342158) (N = 174) and from two public sets: Lefebvre et al. (2016) (N = 216) and the Metastatic Breast Cancer Project (2018) (N = 78). Moreover, we used the gene expression data from the International IBC Consortium (137 IBC and 252 non‐IBC samples) (Van Laere et al., 2013) to apply gene expression signatures of NOTCH (Villanueva et al., 2012) and E2F4 (Guerrero‐Zotano et al., 2018) activation.
2.2. DNA copy number profiling
In three series (IPC, TCGA, Metabric), the DNA copy number profiles were established by using whole‐genome aCGH: high‐resolution 4 × 180K CGH microarrays (SurePrint G3‐Human CGH‐Microarray; Agilent Technologies, Massy, France) for IPC (Bertucci et al., 2016), and Affymetrix single nucleotide polymorphisms (SNP) 6.0 arrays (Santa Clara, CA, USA) for TCGA and Metabric. All aCGH probes were mapped according to UCSC Build 37 (hg19). In the other series (TCRU, Ross, Hamm), the DNA copy number of tumors was derived from targeted NGS (tNGS) data generated by Foundation Medicine. The CNA results of those public sets were collected as processed data from the GDC Data Portal for the TCGA series, cBioPortal for Metabric, and the journal websites for Ross and Hamm series. Across all series, we used one threshold value (log2 ratio > |1|) to define amplifications and deletions. The homologous recombination deficiency score (HRD) (Marquard et al., 2015) was defined on segmented data processed with circular binary segmentation and considered positive above 10 (Olshen et al., 2004). We searched for chromothripsis in IBC by applying the CTLPScanner (Yang et al., 2016).
2.3. Mutational profiling
All series were sequenced using Illumina platforms. Except the TCGA series, which used WES, the other ones used tNGS. IPC samples were sequenced with a home‐made panel of 493 ‘cancer‐associated’ genes (CCP‐V8 panel, Table S2). The DNA libraries of all coding exons and intron–exon boundaries of all genes were prepared using the HaloPlex Target‐Enrichment‐System (Agilent, Santa Clara, CA, USA) as described (Bertucci et al., 2016), and sequencing was done using the 2 × 150‐bp paired‐end technology on the NextSeq500 Illumina platform (Illumina, San Diego, CA, USA). All sequence data were aligned to UCSC hg19 and analyzed as described (Bertucci et al., 2016). Pathogenicity scores for the single nucleotide variant (SNVs) were obtained with Annovar. Mutations were classified as ‘neutral’ or ‘damaging’ using the majority rule of predictor softwares (provided by dbnsfp: Sift, Polyphen2, LRT, MutationTaster, MutationAssesor, FATHMM, RadialSVM, LR). The TCRU, Ross's and Hamm's series were sequenced by Foundation Medicine (Cambridge, MA, USA) for, respectively, 324, 195/255, and 225 genes. The Metabric series (Pereira et al., 2016) was analyzed on a 173‐gene panel. Sequencing data of the public sets and TCRU were collected and processed as indicated above. The TMB was defined as the number of nonsilent mutations per megabase (Mb) of genome sequenced (Bertucci et al., 2016).
2.4. Definition of actionable gene alterations
We defined the AGAs by using the Perera‐Bel's algorithm (Perera‐Bel et al., 2018), which matches patient‐specific genomic alterations to treatment options. This model is based upon public knowledge of somatic variants with predictive evidence on drug response. It is based upon several public data including Gene‐Drug‐Knowledge‐database (GDKD), Clinical‐Interpretation‐of‐Variants‐in‐Cancer (CIViC), and Tumor‐Alterations‐Relevant‐for‐Genomics‐driven‐Therapy (TARGET). The molecular alterations of 312 actionable genes are classified into a six‐level system to rank the associations according to their evidence. The system uses two axes representing the cancer‐type (axis A/B) and the strength of clinical evidence (axis 1/2/3). Levels A and B mean evidence in the same cancer‐type (here breast cancer) and in any other cancer‐type, respectively. Level‐1 means supported by drug approval organizations/clinical guidelines, level‐2 contains clinical evidence, in which late clinical trials are ranked higher followed by early clinical trials and case reports, and level‐3 consists of preclinical evidence. The highest level is A1, followed by B1, then A2, B2, A3, and B3. Our analysis was limited to alterations noted as associated with ‘sensitivity’ to drugs or ‘response’.
2.5. Statistical analysis
Correlations between tumor classes and clinicopathological and molecular variables were analyzed using Student's t‐test or Fisher's exact test when appropriate. Uni‐ and multivariate analyses comparisons of the frequency of molecular alterations between the tumor groups adjusted for the molecular subtypes and the AJCC stage were done using logit link function. Genes with p‐value inferior to 0.05 and q‐value inferior to 0.2 in uni‐ and multivariate analyses were considered as significant. Ontology analysis (DAVID database: https://david.ncifcrf.gov/) of the gene list was limited to the Reactome pathways. Hypergeometric test assessed the significance of enrichment of genes common to the different gene lists. The significance of the P‐values threshold was set at 5% and analyzes used the R‐software (version 2.15.2: http://www.cran.r-project.org/).
3. Results
3.1. Population and genes analyzed
We analyzed 101 IBCs and 2351 non‐IBCs (Table 1). As expected, IBCs were associated with more unfavorable prognostic features than non‐IBCs: younger age, prevalent ductal type, higher AJCC stage (including stage 4), higher pathological grade, and more frequent HER2+ and TN subtypes. Forty percent of samples were TN, and 60% were non‐TN in IBC, vs 17% and 83%, respectively, in non‐IBC. By definition, all IBC were stage 3 or 4, but the precise stage (3 or 4) was available for 59/101 cases, including 33 stage 3 (59%) and 23 stage 4 (41%). Across all six data sets included, there were five different targeted gene panels and one whole‐exome. The CCP‐V8 panel gene list was compared with the four other lists retrieved from the Foundation Medicine website for TCRU, Ross and Hamm series, and the journal website for Metabric. Because there were only 41 genes common to all panels, we focused our analysis on 756 different genes defined as being present in at least one targeted panel (Table S2).
Table 1.
Clinicopathological characteristics of patients and samples.
| Characteristics | N | All cases | Type | P‐value | |
|---|---|---|---|---|---|
| non‐IBC | IBC | ||||
| Age | 2397 | 59.75 (24–96.29) | 60 (26–96.29) | 49.5 (24–80) | 2.43E‐07 |
| Pathological type | |||||
| Ductal | 1808 | 1808 (76%) | 1763 (75%) | 45 (98%) | 2.61E‐03 |
| Lobular | 277 | 277 (12%) | 276 (12%) | 1 (2%) | |
| Other | 309 | 309 (13%) | 309 (13%) | 0 (0%) | |
| Pathological grade | |||||
| 1 | 146 | 146 (11%) | 146 (11%) | 0 (0%) | 1.02E‐03 |
| 2 | 509 | 509 (38%) | 498 (39%) | 11 (25%) | |
| 3 | 682 | 682 (51%) | 649 (50%) | 33 (75%) | |
| AJCC stage | |||||
| 1–2 | 1615 | 1615 (82%) | 1615 (87%) | 0 (0%) | < 1.00E‐06 |
| 3–4 | 349 | 349 (18%) | 248 (3%) | 101 (100%) | |
| ER status | |||||
| Negative | 587 | 587 (25%) | 540 (23%) | 47 (64%) | 3.44E‐13 |
| Positive | 1791 | 1791 (75%) | 1765 (77%) | 26 (36%) | |
| PR status | |||||
| Negative | 1069 | 1069 (45%) | 1017 (44%) | 52 (72%) | 2.94E‐06 |
| Positive | 1305 | 1305 (55%) | 1285 (56%) | 20 (28%) | |
| ERBB2 status | |||||
| Negative | 1937 | 1937 (85%) | 1891 (85%) | 46 (65%) | 3.09E‐05 |
| Positive | 355 | 355 (15%) | 330 (15%) | 25 (35%) | |
| Molecular subtype | |||||
| HR+/HER2− | 1520 | 1520 (66%) | 1502 (68%) | 18 (25%) | < 1.00E‐06 |
| HER2+ | 355 | 355 (16%) | 330 (15%) | 25 (35%) | |
| TN | 415 | 415 (18%) | 387 (17%) | 28 (40%) | |
3.2. Gene alterations in IBC
We identified 1101 gene alterations through the 101 IBCs, including 228 amplifications (21% of all alterations), 15 deletions (1%), and 857 mutations (78%), comprising 730 SNVs (nonsynonymous, stop‐gains, splice‐site; 66%), and 127 indels (12%). They corresponded to 1013 different alterations involving 331 different genes (Table S3). The distribution of alterations of the top 50 genes altered in at least two IBCs is shown in Fig. 1. The 10 most frequently altered genes were TP53 (63%), HER2 (30%), MYC (27%), PIK3CA (21%), BRCA2 (14%), CCND1 (13%), GATA3 (13%), NOTCH1 (12%), FGFR1 (11%), and ARID1A (10%). For HER2, there was 93% concordance between the clinical status and the CNA. Ninety‐eight percent of IBC samples (99/101) harbored at least one alteration. The mean number of alterations per sample was 11 (CI95, 9–13). The mean TMB was six mutations per Mb (CI95, 4–8) (Fig. S1). Chromothripsis was present in 20 out of 44 tested IBC (45%). The most affected chromosomes were chromosome 17 (8% of samples), followed by chromosomes 11 (5%) and 8 (3%). The presence of chromothripsis tended to be associated with the molecular subtype: 69% of HER2+ samples displayed chromothripsis, vs 35% of HR+/HER2− and 30% of TN (P = 0.092; Fisher's exact test).
Figure 1.
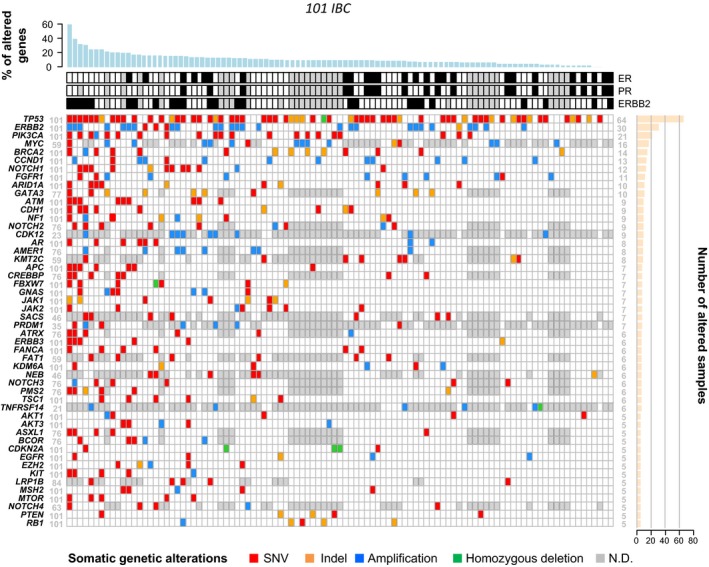
Distribution of alterations of the top 50 genes altered in IBC. Oncoprint of the top 50 genes altered in at least two IBC samples and analyzed in at least 20 samples. Top: immunohistochemical status for ER, PR, and ERBB2 (white: negative; black: positive; gray: unavailable). Bottom: somatic gene alterations (mutations and CNA) color‐coded according to the legend. The genes are ordered from top to bottom by decreasing number of altered tumors (right panel) and the tumors from left to right by decreasing percentage of altered genes (top panel). ND: not defined.
3.3. Comparison of gene alterations between IBC and non‐IBC
Similar analysis was done in the 2351 non‐IBCs. We identified 22 936 gene alterations, corresponding to 14 448 different alterations (Table S3). The distribution of the types of alterations was different from that of IBC (P = 1.24E‐17, Fisher's exact test) with a lesser percent of mutations (70% vs 78%, corresponding to 62% vs 66% for SNVs, and 8% vs 12% for indels). The gene alterations identified in non‐IBC confirmed the literature data (Banerji et al., 2012; Ellis et al., 2012; Ferrari et al., 2016; Nik‐Zainal et al., 2012a; Nik‐Zainal et al., 2016; Nik‐Zainal et al., 2012b; Shah et al., 2012; Stephens et al., 2012; Cancer Genome Atlas, 2012), that is, the most frequently altered genes including PIK3CA (39%), TP53 (34%), HER2 (13%), GATA3 (13%), KMT2C (11%), CDH1 (10%), and MAP3K1 (10%). The mean TMB for all variants was higher in IBC (six mutations/Mb; CI95, 4–8) than in non‐IBC (2; CI95, 2–2; Student's t‐test, P = 6.29E‐05; Fig. S1). Sixteen percent of IBC samples presented a TMB > 10 vs only 1% of non‐IBC samples (P = 3.36E‐12, Fisher's exact test). The same difference was observed when SNVs and indels were analyzed separately (Fig. S1), and all those differences persisted in multivariate analysis (MV) adjusted for the molecular subtypes, the type of NGS (targeted vs WES), and the AJCC stage.
We then applied similarly adjusted supervised analysis to search for genes with differential frequency of alterations between IBC and non‐IBC. Of note, when a sample was not informative for the gene tested, it was excluded from analysis. We identified 96 genes differentially altered (p < 0.05 and q < 0.20 in both univariate and multivariate analyses), including 95 more frequently altered in IBC and only one (PIK3CA) more frequently altered in non‐IBC (Table S4).
The most differentially altered gene was CYP2D6. Four genes (CYP2D6, FOXO3, TP53, and ZNF217) were altered in > 20% of IBCs and 57 genes such as BRCA2, ATM, ATRX, EMSY, NOTCH2, and NOTCH4 were altered in 5–20% of cases. Ontology analysis of the 96 differential genes revealed several pathways associated with IBC genes, such as NOTCH‐related pathways, interleukins and interferon signal, and KIT signaling (Fig. 2B). Genes involved in chromatin remodeling were also more frequently altered in IBC, such as EZH2 and SMARCA4, altered in 5% of IBC, providing a rationale for the evaluation of epigenetic modifiers for the treatment of IBC. Of note, the use of the PAM50‐based genomic definition of molecular subtypes and the use of the IHC definition applied to the 1773 samples (41 IBC and 1732 non‐IBC) informative for both definitions and for the MV showed similar results with the two definitions: 54 and 51 genes were identified as differential with the IHC definition and the PAM50 definition, respectively, with 49 (91% and 96%, respectively) common genes (Fig. S2).
Figure 2.
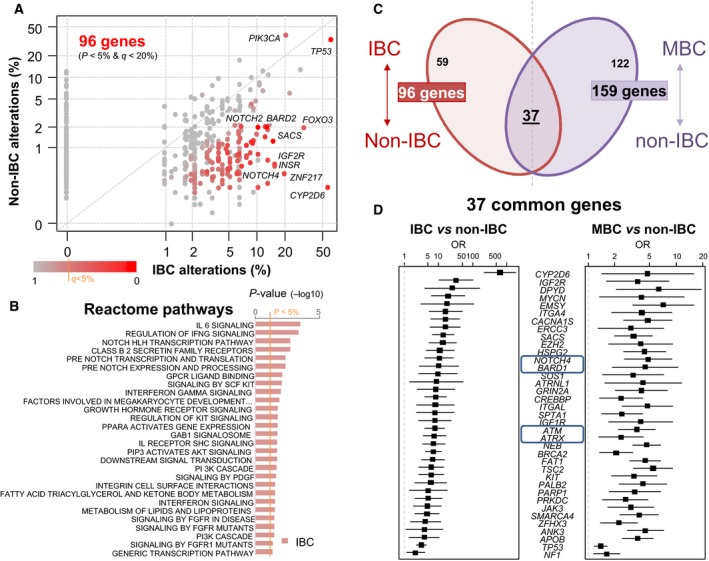
Identification of genes with differential frequency of alterations between samples. (A) Scatter plot depicting the alteration frequency (% of patients) between IBC and non‐IBC. Each dot represents one gene, and dots are color‐coded according to the P‐values (−log10 P‐values) according to the legend below. Significantly mutated genes in either IBC or non‐IBC are included. A few genes differentially mutated are labeled. (B) Ontology analysis revealed several Reactome pathways significantly associated with the 95 IBC genes. (C) Crossings of the lists of genes differentially altered in IBC vs non‐IBC (96 genes) and of genes differentially altered in metastatic (MBC) vs primary non‐IBC (159 genes). (D) List of 37 genes common to the two gene lists. OR: odds ratio of frequencies of alterations in the tumor subgroups.
Supposing that these 96 differentially altered genes might be related to IBC aggressiveness, we tested whether they were also differentially altered in metastatic vs primary non‐IBC. We compared the frequency of alterations between 468 metastatic samples of non‐IBC patients and the 2351 non‐IBC primary samples. By using the same significance threshold as above, we found 159 differentially altered genes, most of them being more frequently altered in metastatic samples (Table S5). The comparison with the above‐quoted 96‐gene list identified 37 genes more frequently altered in both IBC vs non‐IBC samples and in metastatic vs primary non‐IBC samples (Fig. 2C,D). Such overrepresentation was significant (P = 5.58E‐06, hypergeometric test) and indirectly validated the association of our 96‐gene list with IBC, known for its stronger metastatic potential than non‐IBC. These 37 common genes included genes involved in DNA repair (ATM, ATRX, BARD1, BRCA2, EMSY, PALB2) and in NOTCH pathway (NOTCH4). By contrast, the same analysis between the 468 metastatic samples of non‐IBC patients and the 101 IBC samples identified only one gene differentially altered (HER2), indicating that IBC and metastatic non‐IBC samples are not so different at a genomic level when compared head‐on.
3.4. Actionable genetic alterations in IBC versus non‐IBC
We assessed the distribution of AGAs in IBC, comparatively to non‐IBC, using the Perera‐Bel's algorithm (Perera‐Bel et al., 2018). The percentage of IBC patients with AGAs was high (97%) with 26% of A1 alterations, which corresponded to HER2 amplification, 24% of B1, 18% of A2, and 29% of B2 (Table S3). Examples of B1 alterations included BRCA2, JAK2, and EGFR alterations observed in 13 (13%), five (5%), and three (3%) patients, respectively. Examples of A2 alterations included PIK3CA, FGFR1, and PTEN alterations observed in 21 (21%), eight (8%), and four (4%) patients, respectively. Examples of B2 alterations included CCND1 and ATM (nine cases each: 9%), NF1 (seven cases: 7%), MTOR and TSC2 (five cases each: 5%), AKT1, RB1, and TSC1 (four cases each: 4%), and ERBB3 (three cases: 3%). Figure 3 shows the distribution of 44 genes with AGA in at least four IBC samples. The most frequent actionable targets with evidence‐level between A1 and B2 were TP53, HER2, PIK3CA, BRCA2, CCND1, FGFR1, ATM, and NF1. Many samples had several AGAs simultaneously. This percentage of patients with AGAs was higher than the one observed in non‐IBC (87%; P = 4.65E‐20, logit link; Fig. S3A), and the difference remained significant in MV (P = 5.65E‐14, logit link). There were significantly more A1 and B1 alterations in IBC and more A2 and A3 alterations in non‐IBC (Fig. S3B).
Figure 3.
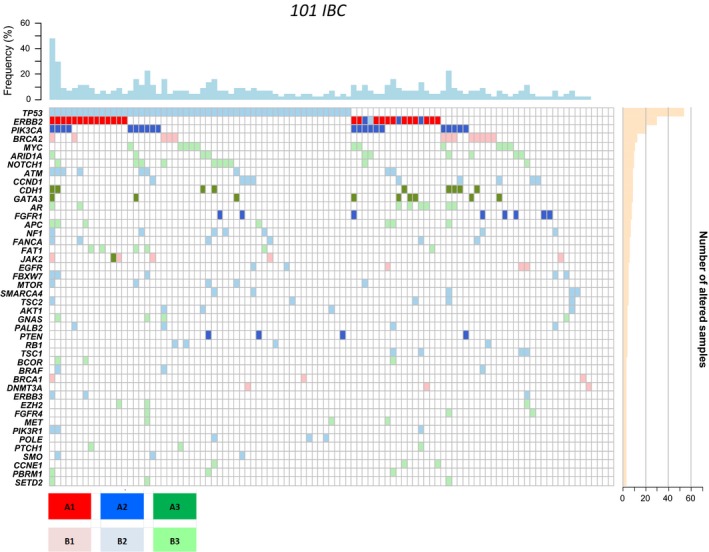
Distribution of genes with actionable alterations in IBC. The 44 genes with actionable alterations in at least four IBC are shown. The genes are ordered from top to bottom by decreasing frequency of mutations. The degree of evidence of actionable alterations according to the Perera‐Bel's algorithm (2018) is color‐coded as indicated in the color scale.
3.5. Enrichment of actionable genetic alterations for different therapeutic classes
We analyzed whether there was enrichment in patients with AGAs in IBC vs non‐IBC in specific drug classes and functional pathways (Fig. S4). Regarding the class of PI3K/AKT/mTOR inhibitors, the percentage of patients with AGAs was higher in non‐IBC patients (52% vs 40%; P = 1.97E‐02), but this difference disappeared in MV (P = 0.185). The percentage of ‘actionable patients’ in the class of HER/EGFR inhibitors was higher in IBC (36% vs 23% in non‐IBC, P = 2.68E‐03) and tended to be significant in MV (P = 0.091). This percentage regarding the class of other tyrosine kinase receptors inhibitors, higher in IBC (27% vs 18% in non‐IBC) in univariate analysis (P = 2.01E‐02), but did not remain significant in MV (P = 0.565). The same was observed regarding the class of CDK inhibitors with higher percentage of patients with AGAs in IBC (29% vs 15%, P = 4.48E‐04), not significant in MV (P = 0.180). We applied an E2F4 activation 24‐gene signature associated with sensitivity to the palbociclib CDK4/6 inhibitor and resistance to aromatase inhibitor (Guerrero‐Zotano et al., 2018) to the 389 samples of the International IBC Consortium expression dataset (Fig. S5). The corresponding metagene score was higher in IBC than in non‐IBC samples (P = 3.68E‐04, Student's t‐test; P = 5.93E‐03, Fisher's exact test), and this difference remained independent from the molecular subtypes and the AJCC stage (P = 0.055, glm; Fig. S5A). This enrichment concerned the HR+/HER2− subtype, which is currently the subtype candidate for CDK4/6 inhibitors (Fig. S5B).
3.6. DNA repair more frequently altered in IBC
Several genes more frequently altered in IBC such as ATM, ATRX, BARD1, BRCA2, and EMSY are involved in DNA repair. Pathway analysis confirmed such enrichment: The percentage of patients with alterations of DNA repair genes was 33% in IBC vs 17% in non‐IBC (P = 7.03E‐06, logit link), even after adjustment in MV (P = 2.20E‐02, logit link; Fig. 4A). BRCA2 was the most frequently altered DNA repair gene in IBC with 13 mutations, including eight truncating mutations (Fig. 4B), suggesting possible enrichment in HRD in IBC. This was confirmed with a higher HRD score in IBCs than in non‐IBCs (P = 2.36E‐02, Student's t‐test). The OR for high HRD score (≥ 10) was 2.27 (95% CI: 1.19–4.26) in IBC compared with non‐IBC (P = 9.45E‐03, Fisher's exact test; Fig. 4C).
Figure 4.
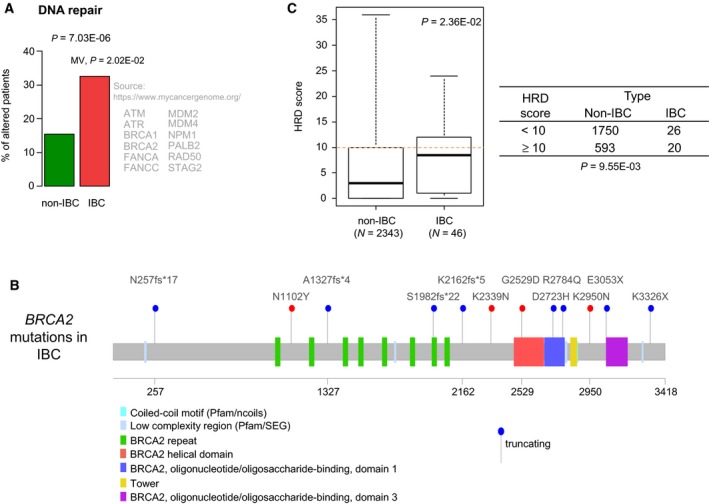
DNA repair genes are more frequently altered in IBC than in non‐IBC. (A) Plot showing the percentage of patients with AGAs in genes involved in DNA repair in IBC vs non‐IBC patients. The P‐values are for the logit link in univariate analysis and in MV. Beside the plot, are indicated the 12 genes common to the pathway in the indicated bibliographic source and to our list of 756 genes tested. (B) Lolliplot of BRCA2 gene showing the 12 mutations identified in IBC (blue: truncating mutation; red: nontruncating mutation). (C) Left: Box‐plot of HRD score in non‐IBC and IBC samples. Right: Contingency table between HRD score and IBC/non‐IBC status.
3.7. NOTCH pathway more frequently altered in IBC
NOTCH pathway alterations were also enriched in patients with IBC (30% vs 17% in non‐IBC patients) in univariate (P = 1.76E‐03, logit link) and multivariate analyses (P = 4.49E‐04, logit link; Fig. 5A). Whereas NOTCH1 was among the most frequently altered genes in IBC (12%), it was not differentially altered compared with non‐IBC. By contrast, NOTCH2 and NOTCH4 were significantly more frequently mutated in IBC with a total of 12 mutations, including nine predicted as damaging in silico. There was a mutual exclusivity in IBC samples between alterations in the three genes (NOTCH2, NOTCH4, and CREBBP) found differentially altered in IBC vs non‐IBC, present in the KEGG NOTCH pathway, and tested in at least 30% of samples (Fig. S6). Other genes involved in the NOTCH pathway and frequently altered in IBC included MAML1 (11%), MED12 (9%), and FBXW7 (8%).
Figure 5.
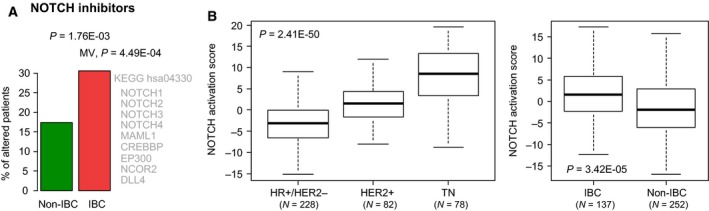
NOTCH pathways genes are more frequently altered in IBC than in non‐IBC. (A) Plot showing the percentage of patients with AGAs involved in the NOTCH pathway in IBC vs non‐IBC patients. The P‐values are for the logit link in univariate analysis and in MV. Beside the plot, are indicated the nine genes common to the pathway in the indicated bibliographic source and to our list of 756 genes tested. (B) Left: Box‐plot of NOTCH activation score in the breast cancer samples of the International IBC Consortium dataset according to the molecular subtypes. The P‐value is for the one‐way ANOVA test. Right: Box‐plot of NOTCH activation score in the breast cancer samples of the International IBC Consortium dataset according to the non‐IBC and IBC statutes. The P‐value is for the Student's t‐test.
Such enrichment led to search for signs of NOTCH pathway activation in IBC. We applied a 384‐gene signature of NOTCH activation (Villanueva et al., 2012) to expression data of the International IBC Consortium set. As expected (Shen et al., 2017), the activation score was higher in the TN samples than in the HR+/HER2− samples (P = 2.41E‐50, one‐way ANOVA test Fig. 5B), validating its robustness. Interestingly, this score was higher in IBCs than in non‐IBCs (P = 3.42E‐05, Student's t‐test; Fig. 5B), and this difference remained significant in MV (Table 2).
Table 2.
Uni‐ and multivariate analyses for IBC vs non‐IBC. OR, odds ratio.
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| N | OR (95% CI) | P‐value | N | OR (95% CI) | P‐value | |
| Villanueva's NOTCH activation score | 389 | 1.06 (1.04–1.09) | 9.27E‐05 | 384 | 1.07 (1.03–1.12) | 5.82E‐03 |
| Molecular subtype, HER2+ vs HR+/HER2− | 388 | 2.81 (1.81–4.36) | 1.04E‐04 | 384 | 1.40 (0.79–2.48) | 0.336 |
| TN vs HR+/HER2‐TN | 388 | 1.96 (1.25–3.08) | 1.36E‐02 | 384 | 0.77 (0.39–1.53) | 0.532 |
| AJCC stage, 3–4 vs 1–2 | 389 | 1.06 (1.04–1.09) | 9.27E‐05 | 384 | 4.8E8 (0.00–Inf) | 0.981 |
3.8. Comparison with literature
Finally, we compared our results to those of two tNGS studies for which data, publicly unavailable, were not included in our analysis. The Liang et al. (2018) study analyzed 91 genes in a series of non‐pretreated primary tumors including 156 IBC and 197 stage 3–4 TCGA non‐IBC: Seventeen genes were more frequently mutated in IBC than in non‐IBC, including TP53, NOTCH2, MYH9, BRCA2, ERBB4, POLE, FGFR3, ROS1, NOTCH4, LAMA2, EGFR, BRCA1, TP53BP1, ESR1, THBS1, CASP8, and NOTCH1, and one gene, CDH1, more frequently mutated in non‐IBC. The Matsuda et al. series analyzed 50 genes in non‐pretreated primaries and pretreated relapses of 24 IBC and 376 non‐IBC (Matsuda et al., 2017): Two genes (TP53, HER2) were more frequently mutated in IBC. In both studies, the comparison was not stratified upon the molecular subtypes.
We compared these lists of differential genes with ours (Fig. S7). Between the Liang et al. list and ours, 84 genes were common to both panels tested, with only five differential genes in common: BRCA2, NOTCH2, NOTCH4, POLE, and TP53. Between the Matsuda et al. list and ours, 50 genes were common to both panels tested, with only one differential gene (TP53) in common. Thirty genes were common to the Matsuda et al. and Liang et al. panels, with only one differential gene in common: TP53. These results revealed low concordance between all three gene lists.
4. Discussion
We compared the DNA copy number and mutational profiles of untreated primary tumors of 101 IBCs and 2351 non‐IBCs. Ninety‐seven percent of IBCs displayed at least one AGA. This percentage, higher than in non‐IBC, suggests that personalized therapy is a relevant approach for this aggressive disease, in particular with drugs targeting the DNA repair and NOTCH pathways.
We focused our study on untreated primary tumors to avoid biases induced by previous systemic treatments that induce changes in mutations and subclonal structure between primary tumor and relapses (Bertucci et al., 2019; McGranahan et al., 2015). The scarcity of IBC and diagnostic samples, and the need to adjust analyses upon the molecular subtypes and AJCC stage because of the unbalance between IBC and non‐IBC led us to pool our own bicentric data with available public data. As expected for breast cancers, the genomic profiles were heterogeneous in IBC. We found higher TMB in IBC compared to non‐IBC, possibly related to the higher genomic instability and complexity of the disease (Bekhouche et al., 2011), suggesting that immune checkpoint inhibitors warrant further investigation in IBC (Bertucci et al., 2015; Bertucci and Goncalves, 2017; Van Berckelaer et al., 2019; Van Laere et al., 2013). Such difference was independent from the molecular subtypes and the disease stage. Chromothripsis was identified in 45% of 44 tested IBC, a percentage close to that previously reported in a series of 28 non‐IBC (Przybytkowski et al., 2014).
The 10 most frequently altered genes in IBC are TP53 (63%), HER2 (30%), MYC (27%), PIK3CA (21%), BRCA2 (14%), CCND1 (13%), GATA3 (13%), NOTCH1 (12%), FGFR1 (11%), and ARID1A (10%), which are also altered in non‐IBC. But the comparison with non‐IBC identified 95 genes more frequently altered in IBC in a molecular subtype and stage‐independent way, including 37 that were also more frequently altered in metastases vs primary tumors of non‐IBC patients. This suggests a possible link of these genes with disease aggressiveness and proclivity to metastasize, although functional studies are warranted. Interestingly, the pairwise comparisons of IBC, non‐IBC primaries, and non‐IBC metastatic samples showed that IBC and metastatic non‐IBC are much less different at a genomic level when compared head‐on than are IBC vs non‐IBC primaries and primary vs metastatic non‐IBC. CYP2D6 was the most differentially altered gene (58% of IBC vs 0.2% in non‐IBC). This gene codes for the cytochrome P‐450 2D6, which oxidizes tamoxifen to its most active metabolite. Many CYP2D6 polymorphisms, such as the one found in our series (435T>S), have been identified, leading to the decrease of CYP2D6 enzymatic activity. Several data suggest that poor metabolizers of CYP2D6 do not benefit as much from tamoxifen therapy as other patients do; however, conflicting results were published (Hoskins et al., 2009). Absence of analysis of constitutional DNA impedes us to conclude on the SNP nature of our variant. However, the large difference in frequency with non‐IBC potentially reveals an important role for this variant in the predisposition to IBC and/or the known resistance of the disease to standard hormone therapy. Analysis of a larger series is needed, including both tumor and matched normal DNA.
Several therapeutically actionable targets were frequently altered in IBC, including a few ones more frequently than in non‐IBC and independently from the molecular subtypes and AJCC stage. For example, we found frequent alterations in genes involved in DNA repair. ATM, ATRX, BARD1, BRCA2, ERCC3, MSH2, MSH6, PMS2, and POLE were more frequently altered in IBC, in which we found also frequent alterations of TP53, FANCA, and FANCB. This observation confirms recent findings (Liang et al., 2018) of frequent alterations in BRCA1/BRCA2/POLE genes in IBC. It is likely that deficient DNA repair contributes to disease progression, as well as to the high TMB observed in IBC and indirectly to its peculiar immune microenvironment (Bertucci et al., 2015; Van Berckelaer et al., 2019; Van Laere et al., 2013). In addition, IBC showed more frequently a HRD and alterations in genes involved in mismatch repair, supporting the ongoing development of PARP inhibitors in IBC as radiosensitizers in phase I–II trials with veliparib (Jagsi et al., 2018) and olaparib (NCT03598257) (Michmerhuizen et al., 2019). This observation warrants a deeper NGS study of IBC, using whole‐genome sequencing (WGS) to investigate structural variations. A recent study assessed the prevalence of germline variants in cancer predisposition genes in 368 patients with IBC (Rana et al., 2019). Germline mutations were identified in 53 cases (14.4%). BRCA1 or BRCA2 mutations were found in 7.3% of the subjects, 6.3% had a mutation in other breast cancer genes (PALB2, CHEK2, ATM, and BARD1), and 1.6% had mutations in genes not associated with breast cancer.
Alterations in the NOTCH pathway were almost twice as enriched in IBC (30% vs 17% in non‐IBC), independently from the molecular subtypes and AJCC stage. NOTCH receptors are transmembrane receptors that play an essential role in cell fate decisions, such as proliferation, differentiation, and apoptosis, and in the maintenance of breast cancer stem‐like cells (Mollen et al., 2018). NOTCH1 was the most frequently altered NOTCH gene in IBC (12%), whereas NOTCH2 and NOTCH4 were more frequently altered in IBC compared with non‐IBC, with a total of 12 mutations including 9 predicted as damaging. Functional studies measuring NOTCH pathway activation, transformation potential, and sensitivity to pathway inhibition are required to better define the relevance of these alterations. Interestingly, we found a mutual exclusivity in IBCs for NOTCH2, NOTCH4, and CREBBP alterations, present in a total of 19 out of 76 (25%) informative samples. Of note, NOTCH2 and NOTCH4 were also reported as more frequently mutated in IBC in the recent Liang's study (Liang et al., 2018). We also found a NOTCH pathway activation score (Villanueva et al., 2012) higher in IBC than in non‐IBC independently from the molecular subtypes and AJCC stage, further supporting a role for the NOTCH pathway in IBC. Several data in literature, based on preclinical models, have also related IBC and NOTCH alterations. In the MARY‐X model, the lymphovascular emboli of IBC exhibit a NOTCH3 addiction (Xiao et al., 2011). The FC‐IBC02 cell line shows NOTCH3 amplification (Fernandez et al., 2013). Pregnant mice expressing higher levels of an activated intracellular form of NOTCH3 develop luminal mammary tumors resembling IBC that frequently metastasize (Ling et al., 2013). Thus, the NOTCH targeting might be an option for IBC treatment. Accordingly, a preclinical study in IBC showed that a gamma‐secretase inhibitor, RO4929097, was able to block the NOTCH signaling and to attenuate the stem‐like phenotype of IBC cells and regulate the inflammatory environment (Debeb et al., 2012). All this taken together, the NOTCH pathway may constitute the most prominent difference between IBC and non‐IBC.
We also analyzed four other classes of targeted therapies approved or tested in breast cancer. Regarding the PI3K/AKT/mTOR inhibitors, even if PIK3CA was the only gene more frequently altered in non‐IBC compared to IBC, its frequency of alteration was relatively high in IBC (21%), with many hotspot mutations. Other actionable genes of the PI3K/AKT/mTOR pathway were frequently altered in IBC with likely loss‐of‐function mutations in PTEN, TSC1, and TSC2 and gain‐of‐function mutations in AKT1, AKT3, MTOR, RPTOR, and RICTOR. Thus, like non‐IBC patients, IBC patients may benefit from inhibition of the pathway with the PI3K/AKT/mTOR inhibitors approved and in development (Kenna et al., 2018).
Regarding the CDK4/6 inhibitors class, we found twice as higher percentage of patients with AGAs in IBC than in non‐IBC (29% vs 15%). The most frequent AGA in this group was CCND1 amplification (10% of IBCs), followed by CDKN2A deletion/mutation (5%). Mutations in the FAT1 and RB1 tumor suppressors, potentially associated with resistance to CDK4/6 inhibitors (Li et al., 2018), were also observed in 10% and 4% of IBC, respectively. Interestingly, an E2F4 activation expression signature associated with sensitivity to palbociclib and resistance to aromatase inhibitors (Guerrero‐Zotano et al., 2018) was higher in IBC than in non‐IBC samples, notably in HR+/HER2− patients. Clearly, CDK4/6 inhibitors deserve to be tested in IBC.
The percentage of ‘actionable patients’ in the class of HER/EGFR inhibitors was higher in IBC (36% vs 23%), and the difference tended toward significance in MV including the molecular subtypes and AJCC stage. As expected, ERBB2 amplification was observed in 26% of IBC, and activating ERBB2 mutations (Petrelli et al., 2017) were much less frequent. Five IBC patients displayed such mutations, including two with (HER2+ patient) and three without (HER2− patient) simultaneous amplification. Mutations were located in the extracellular domain and the kinase domain and have been associated with sensitivity to ERBB2 tyrosine kinase inhibitors such as neratinib (Bose et al., 2013) with which clinical trials are ongoing. Three IBC patients (3%) displayed an ERBB3 mutation identified as AGA vs 42 non‐IBC patients (1.8%). In their small series of cases, Hamm et al. (2016) previously reported frequent co‐occurrence of ERBB3 mutations and ERBB2 amplification in IBC and suggested possible contribution to resistance to anti‐HER2 therapy. In our larger series, such co‐occurrence was found in 2% of IBC and only 0.2% of non‐IBC, supporting investigation of ERBB3‐inhibitors in combination with ERBB2‐inhibitors in IBC.
5. Conclusions
Our study confirms the hypothesis that IBC is distinct from non‐IBC at the genomic level, independently from the molecular subtypes and disease stage. We found higher TMB in IBC than in non‐IBC and 95 genes more frequently altered in IBC in a molecular subtype‐ and stage‐independent way. Ninety‐seven percent of IBC samples displayed at least one AGA. This percentage, higher than in non‐IBC (87%), suggests that precision medicine is a bona fide option in this aggressive disease, notably with drugs targeting DNA repair, NOTCH signaling, and CDK4/6. The strengths of our study are follows: (a) the largest comparison of IBC vs non‐IBC samples (total of 2452 samples), and the largest number of genes tested (756 genes) in such comparison, (b) a consensual uniform case definition for IBC, (c) a homogeneous series of non‐pretreated primary tumors, (d) an adjustment upon both the molecular subtypes and AJCC stage, and (e) a statistical correction (FDR with q‐values) for multiple tests in the gene‐by‐gene supervised analysis. To our knowledge, these two last points have never been combined in studies comparing the molecular alterations of IBC and non‐IBC. Other strengths include the use of an algorithm to define more objectively the AGAs, the validation of differential activation of potentially targetable pathways (NOTCH, HRD) using transcriptomic and genomic data, and the demonstration that our supervised analysis gave very similar results whatever was the definition of molecular subtypes included in the MV, either IHC‐ or PAM‐based. However, like most of other studies published in the field, it also displays a few limitations: (a) its retrospective nature and associated biases, (b) absence of matched normal DNA sequenced for the tNGS‐based series and absence of information regarding eventual germline mutations and family and personal histories of cancer, (c) presence of missing data for several genes because of the variation in genes targeted across the cohorts examined, leading to a loss of sensitivity regarding identification of genes differentially altered, (d) the use of different sequencing platforms including tNGS and WES, even if that did not impact our comparative analysis as suggested by the MV, and (e) no further analysis of structural variations, driver mutations, intra‐tumor heterogeneity, and mutational signatures. Of course, analysis of a larger and homogeneous series of untreated primary tumors analyzed with WES, WGS, and RNA‐Seq is warranted. Such analysis could also reveal etiology of IBC by identifying DNA sequences not matching to the human genome, such as viral or bacterial infection, as suggested (El‐Shinawi et al., 2016). But yet, our results suggest targeted therapies that have the potential to bring benefit to IBC patients and encourage prospective clinical trials.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Concept, design, and supervision: FB and SVL. Data analysis and interpretation: all authors. Writing and review of manuscript: FB, CR, PF, BD, XW, NU, MCr, DB, SVL. Reading and approval of the final manuscript: all authors.
Supporting information
Fig. S1 . Tumor mutational burden (TMB) in IBC and non‐IBC.
Fig. S2 . Absence of impact of the definition of molecular subtypes (IHC vs PAM50) on the differentially altered character of our 96 genes.
Fig. S3 . Percentage of patients with AGAs along IBC vs non‐IBC patients.
Fig. S4 . Percentage of patients with actionable alterations in four specific drug classes.
Fig. S5 . E2F4 activation signature enriched in IBC vs non‐IBC.
Fig. S6 . Mutual exclusivity of NOTCH pathway alterations in IBC.
Fig. S7 . Comparison of the lists of genes differentially altered in IBC vs non‐IBC across three studies.
Table S1 . IBC and non‐IBC data sets included in the present study.
Table S2 . List of 756 genes analyzed in the present study.
Table S3 . List of gene alterations identified in the 101 IBC and the 2351 non‐IBC.
Table S4 . List of 96 genes differentially altered in IBC vs non‐IBC in MV.
Table S5 . List of 159 genes differentially altered in MBC vs non‐IBC.
Table S6 . Detailed clinicopathological data and genomic data analyzed in the present study.
Acknowledgements
We thank the computing facilities DISC (Datacenter IT and Scientific Computing, CRCM) and the IPC biobank (authorization number AC‐2018‐1905) for their support.
This work was supported by SIRIC (PV), label Ligue (FB/DB), Ruban Rose (DB), and Fondation Groupe EDF (DB).
Charlotte Rypens, Pascal Finetti and Arnaud Guille are equal second co‐authors
Data accessibility
All clinicopathological data and genomic data analyzed in the present study are available in this article in the Table S6.
References
- Andre F, Bachelot T, Commo F, Campone M, Arnedos M, Dieras V, Lacroix‐Triki M, Lacroix L, Cohen P, Gentien D et al (2014) Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol 15, 267–274. [DOI] [PubMed] [Google Scholar]
- Banerji S, Cibulskis K, Rangel‐Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L et al (2012) Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekhouche I, Finetti P, Adelaide J, Ferrari A, Tarpin C, Charafe‐Jauffret E, Charpin C, Houvenaeghel G, Jacquemier J, Bidaut G et al (2011) High‐resolution comparative genomic hybridization of inflammatory breast cancer and identification of candidate genes. PLoS ONE 6, e16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F, Finetti P, Colpaert C, Mamessier E, Parizel M, Dirix L, Viens P, Birnbaum D and van Laere S (2015) PDL1 expression in inflammatory breast cancer is frequent and predicts for the pathological response to chemotherapy. Oncotarget 6, 13506–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F, Finetti P, Guille A, Adelaide J, Garnier S, Carbuccia N, Monneur A, Charafe‐Jauffret E, Goncalves A, Viens P et al (2016) Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget 7, 27208–27219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F, Finetti P, Vermeulen P, Van Dam P, Dirix L, Birnbaum D, Viens P and Van Laere S (2014a) Genomic profiling of inflammatory breast cancer: a review. Breast 23, 538–545. [DOI] [PubMed] [Google Scholar]
- Bertucci F and Goncalves A (2017) Immunotherapy in breast cancer: the emerging role of PD‐1 and PD‐L1. Curr Oncol Rep 19, 64. [DOI] [PubMed] [Google Scholar]
- Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, Soria JC, Dien AT, Adnani Y, Kamal M et al (2019) Genomic characterization of metastatic breast cancers. Nature 569, 560–564. [DOI] [PubMed] [Google Scholar]
- Bertucci F, Ueno NT, Finetti P, Vermeulen P, Lucci A, Robertson FM, Marsan M, Iwamoto T, Krishnamurthy S, Masuda H et al (2014b) Gene expression profiles of inflammatory breast cancer: correlation with response to neoadjuvant chemotherapy and metastasis‐free survival. Ann Oncol 25, 358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S et al (2013) Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov 3, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charafe‐Jauffret E, Tarpin C, Viens P and Bertucci F (2008) Defining the molecular biology of inflammatory breast cancer. Semin Oncol 35, 41–50. [DOI] [PubMed] [Google Scholar]
- Dawood S, Merajver SD, Viens P, Vermeulen PB, Swain SM, Buchholz TA, Dirix LY, Levine PH, Lucci A, Krishnamurthy S et al (2011) International expert panel on inflammatory breast cancer: consensus statement for standardized diagnosis and treatment. Ann Oncol 22, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debeb BG, Cohen EN, Boley K, Freiter EM, Li L, Robertson FM, Reuben JM, Cristofanilli M, Buchholz TA and Woodward WA (2012) Pre‐clinical studies of Notch signaling inhibitor RO4929097 in inflammatory breast cancer cells. Breast Cancer Res Treat 134, 495–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC et al (2012) Whole‐genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Shinawi M, Mohamed HT, Abdel‐Fattah HH, Ibrahim SA, El‐Halawany MS, Nouh MA, Schneider RJ and Mohamed MM (2016) Inflammatory and non‐inflammatory breast cancer: a potential role for detection of multiple viral DNAs in disease progression. Ann Surg Oncol 23, 494–502. [DOI] [PubMed] [Google Scholar]
- Fernandez SV, Robertson FM, Pei J, Aburto‐Chumpitaz L, Mu Z, Chu K, Alpaugh RK, Huang Y, Cao Y, Ye Z et al (2013) Inflammatory breast cancer (IBC): clues for targeted therapies. Breast Cancer Res Treat 140, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari A, Vincent‐Salomon A, Pivot X, Sertier AS, Thomas E, Tonon L, Boyault S, Mulugeta E, Treilleux I, MacGrogan G et al (2016) A whole‐genome sequence and transcriptome perspective on HER2‐positive breast cancers. Nature Commun 7, 12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh G, Schmid R, Guiver K, Arpornwirat W, Chitapanarux I, Ganju V, Im SA, Kim SB, Dechaphunkul A, Maneechavakajorn J et al (2016) Clonal evolutionary analysis during HER2 blockade in HER2‐positive inflammatory breast cancer: a phase II open‐label clinical trial of afatinib +/‐ vinorelbine. PLoS Medicine 13, e1002136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero‐Zotano AL, Stricker TP, Formisano L, Hutchinson KE, Stover DG, Lee KM, Schwarz LJ, Giltnane JM, Estrada MV, Jansen VM et al (2018) ER(+) breast cancers resistant to prolonged neoadjuvant letrozole exhibit an E2F4 transcriptional program sensitive to CDK4/6 inhibitors. Clin Cancer Res 24, 2517–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm CA, Moran D, Rao K, Trusk PB, Pry K, Sausen M, Jones S, Velculescu VE, Cristofanilli M and Bacus S (2016) Genomic and immunological tumor profiling identifies targetable pathways and extensive CD8+/PDL1+ immune infiltration in inflammatory breast cancer tumors. Mol Cancer Ther 15, 1746–1756. [DOI] [PubMed] [Google Scholar]
- Hoskins JM, Carey LA and McLeod HL (2009) CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer 9, 576–586. [DOI] [PubMed] [Google Scholar]
- Jagsi R, Griffith KA, Bellon JR, Woodward WA, Horton JK, Ho A, Feng FY, Speers C, Overmoyer B, Sabel M et al (2018) Concurrent veliparib with chest wall and nodal radiotherapy in patients with inflammatory or locoregionally recurrent breast cancer: the TBCRC 024 phase I multicenter study. J Clin Oncol 36, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenna MM, McGarrigle S and Pidgeon GP (2018) The next generation of PI3K‐Akt‐mTOR pathway inhibitors in breast cancer cohorts. Biochim Biophys Acta Rev Cancer 1870, 185–197. [DOI] [PubMed] [Google Scholar]
- Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, Campone M, Tredan O, Massiani MA, Mauborgne C et al (2015) Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol 16, 1324–1334. [DOI] [PubMed] [Google Scholar]
- Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, Massard C, Levy C, Arnedos M, Lacroix‐Triki M et al (2016) Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Medicine 13, e1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, Hsieh W, Sanchez‐Vega F, Brown DN, Da Cruz Paula AF et al (2018) Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell 34, 893–905. e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Vacher S, Boulai A, Bernard V, Baulande S, Bohec M, Bieche I, Lerebours F and Callens C (2018) Targeted next‐generation sequencing identifies clinically relevant somatic mutations in a large cohort of inflammatory breast cancer. Breast Cancer Res 20, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Sylvestre JR and Jolicoeur P (2013) Cyclin D1‐dependent induction of luminal inflammatory breast tumors by activated notch3. Cancer Res 73, 5963–5973. [DOI] [PubMed] [Google Scholar]
- Marquard AM, Eklund AC, Joshi T, Krzystanek M, Favero F, Wang ZC, Richardson AL, Silver DP, Szallasi Z and Birkbak NJ (2015) Pan‐cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H, Baggerly KA, Wang Y, Iwamoto T, Brewer T, Pusztai L, Kai K, Kogawa T, Finetti P, Birnbaum D et al (2013) Comparison of molecular subtype distribution in triple‐negative inflammatory and non‐inflammatory breast cancers. Breast Cancer Res 15, R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Lim B, Wang Y, Krishnamurthy S, Woodward W, Alvarez RH, Lucci A, Valero V, Reuben JM, Meric‐Bernstam F et al (2017) Identification of frequent somatic mutations in inflammatory breast cancer. Breast Cancer Res Treat 163, 263–272. [DOI] [PubMed] [Google Scholar]
- McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z and Swanton C (2015) Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 7, 283ra254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metastatic Breast Cancer Project (2018) The metastatic breast cancer project. http://www.cbioportal.org/study?xml:id=brca_mbcproject_wagle_2017. Accessed 20 April 2018.
- Michmerhuizen AR, Pesch AM, Moubadder L, Chandler BC, Wilder‐Romans K, Cameron M, Olsen E, Thomas DG, Zhang A, Hirsh N et al (2019) PARP1 inhibition radiosensitizes models of inflammatory breast cancer to ionizing radiation. Mol Cancer Ther 18, 2063–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollen EWJ, Ient J, Tjan‐Heijnen VCG, Boersma LJ, Miele L, Smidt ML and Vooijs M (2018) Moving breast cancer therapy up a notch. Front Oncol 8, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik‐Zainal S, Alexandrov LB, Wedge DC, Van Loo P , Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA (2012a) Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik‐Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC et al (2016) Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature 534, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik‐Zainal S, Van Loo P , Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M (2012b) The life history of 21 breast cancers. Cell 149, 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R and Wigler M (2004) Circular binary segmentation for the analysis of array‐based DNA copy number data. Biostatistics 5, 557–572. [DOI] [PubMed] [Google Scholar]
- Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ et al (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature Commun 7, 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera‐Bel J, Hutter B, Heining C, Bleckmann A, Frohlich M, Frohling S, Glimm H, Brors B and Beissbarth T (2018) From somatic variants towards precision oncology: evidence‐driven reporting of treatment options in molecular tumor boards. Genome Med 10, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli F, Tomasello G, Barni S, Lonati V, Passalacqua R and Ghidini M (2017) Clinical and pathological characterization of HER2 mutations in human breast cancer: a systematic review of the literature. Breast Cancer Res Treat 166, 339–349. [DOI] [PubMed] [Google Scholar]
- Przybytkowski E, Lenkiewicz E, Barrett MT, Klein K, Nabavi S, Greenwood CM and Basik M (2014) Chromosome‐breakage genomic instability and chromothripsis in breast cancer. BMC Genom 15, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana HQ, Sacca R, Drogan C, Gutierrez S, Schlosnagle E, Regan MM, Speare V, LaDuca H, Dolinsky J, Garber JE et al (2019) Prevalence of germline variants in inflammatory breast cancer. Cancer. 10.1002/cncr.32062 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Ross JS, Ali SM, Wang K, Khaira D, Palma NA, Chmielecki J, Palmer GA, Morosini D, Elvin JA, Fernandez SV et al (2015) Comprehensive genomic profiling of inflammatory breast cancer cases reveals a high frequency of clinically relevant genomic alterations. Breast Cancer Res Treat 154, 155–162. [DOI] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G et al (2012) The clonal and mutational evolution spectrum of primary triple‐negative breast cancers. Nature 486, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Cohen B, Zheng W, Rahbar R, Martin B, Murakami K, Lamorte S, Thompson P, Berman H, Zuniga‐Pflucker JC et al (2017) Notch shapes the innate immunophenotype in breast cancer. Cancer Discov 7, 1320–1335. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik‐Zainal S, Martin S, Varela I, Bignell GR et al (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Berckelaer C, Rypens C, van Dam P, Pouillon L, Parizel M, Schats KA, Kockx M, Tjalma WAA, Vermeulen P, van Laere S et al (2019) Infiltrating stromal immune cells in inflammatory breast cancer are associated with an improved outcome and increased PD‐L1 expression. Breast Cancer Res 21, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laere S, Ueno NT, Finetti P, Vermeulen PB, Lucci A, Robertson F, Marsan M, Iwamoto T, Krishnamurthy S, Masuda H et al (2013) Uncovering the molecular secrets of Inflammatory Breast Cancer biology: an integrated analysis of three distinct Affymetrix gene expression data sets. Clin Cancer Res 19, 4685–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S, Rodriguez‐Carunchio L, Sole M, Thung S, Stanger BZ et al (2012) Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 143, 1660–1669 e1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Ye Y, Zou X, Jones S, Yearsley K, Shetuni B, Tellez J and Barsky SH (2011) The lymphovascular embolus of inflammatory breast cancer exhibits a Notch 3 addiction. Oncogene 30, 287–300. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu J, Ouyang L, Chen Y, Liu B and Cai H (2016) CTLPScanner: a web server for chromothripsis‐like pattern detection. Nucleic Acids Res 44, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 . Tumor mutational burden (TMB) in IBC and non‐IBC.
Fig. S2 . Absence of impact of the definition of molecular subtypes (IHC vs PAM50) on the differentially altered character of our 96 genes.
Fig. S3 . Percentage of patients with AGAs along IBC vs non‐IBC patients.
Fig. S4 . Percentage of patients with actionable alterations in four specific drug classes.
Fig. S5 . E2F4 activation signature enriched in IBC vs non‐IBC.
Fig. S6 . Mutual exclusivity of NOTCH pathway alterations in IBC.
Fig. S7 . Comparison of the lists of genes differentially altered in IBC vs non‐IBC across three studies.
Table S1 . IBC and non‐IBC data sets included in the present study.
Table S2 . List of 756 genes analyzed in the present study.
Table S3 . List of gene alterations identified in the 101 IBC and the 2351 non‐IBC.
Table S4 . List of 96 genes differentially altered in IBC vs non‐IBC in MV.
Table S5 . List of 159 genes differentially altered in MBC vs non‐IBC.
Table S6 . Detailed clinicopathological data and genomic data analyzed in the present study.
Data Availability Statement
All clinicopathological data and genomic data analyzed in the present study are available in this article in the Table S6.