

Abstract
A main group-catalyzed method for the modular synthesis of diverse N-aryl and N-alkyl azaheterocycles (i.e. indoles, oxindoles, benzimidazoles, and quinoxalinediones) is reported. The method employs a small ring organophosphorus-based catalyst (1,2,2,3,4,4-hexamethylphosphetane P-oxide) and a hydrosilane reductant to drive the conversion of ortho-functionalized nitroarenes to azaheterocycles via sequential intermolecular reductive C–N cross coupling with boronic acids, followed by intramolecular cyclization. This method provides for the rapid construction of azaheterocycles from readily available building blocks, including a regiospecific approach to N-substituted benzimidazoles and quinoxalinediones.
Keywords: Organocatalysis, Cross-coupling, Phosphorus, Nitrogen heterocycles, Cyclization
Graphical Abstract
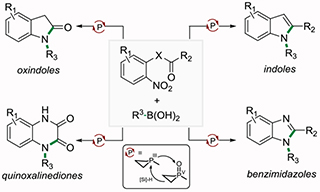
A main group-catalyzed method for the modular synthesis of diverse N-aryl and N-alkyl azaheterocycles (i.e. indoles, oxindoles, benzimidazoles, and quinoxalinediones) from readily available building blocks, including a regiospecific approach to N-substituted benzimidazoles and quinoxalinediones.
Introduction
N-Functionalized azaheterocycles are important constituents of the modern pharmacopeia (see Figure 1A),1 and synthetic methods that provide rapid and modular entry to this varied group of compounds contribute to the discovery of new drugs. Nitroarenes are attractive substrates for such heterocycle synthesis since—beyond the many nitroaromatic building blocks that are commercially available—the nitro functional group is easily and reliably introduced to arenes, where it exerts a powerful inductive effect that enables proximate transformations.2 The nitro moiety has many applications in well-known heterocyclization methods, 3,4 but less commonly as a strategic site for consecutive C–N bond formation at nitrogen.5 We report here a unified cascade approach to multiple classes of useful N-functionalized azaheterocycles by the conversion of readily available ortho-derivatized nitroarenes in a single main group-catalyzed operation.
Figure 1.

(A) Selected examples of investigational and FDA-approved N-functionalized azaheterocyclic drugs. (B) Present work: A unified PIII/PV=O-catalyzed cyclative coupling approach for the modular synthesis of oxindoles, indoles, quinoxalinediones, and benzimidazoles directly from o-functionalized nitroarene precursors.
The recognition that nitroarenes may serve as masked precursors of reactive nitrogen intermediates for direct azafunctionalization6,7 informed the view that a one-pot reaction sequence involving: (1) organophosphorus-catalyzed intermolecular reductive C–N cross coupling, 8 and (2) in situ intramolecular acyl substitution or carbonyl condensation would constitute an integrated approach to multiple heterocyclic classes from o-functionalized nitroarenes (Figure 1B). As compared to conceptually-related transition metal-catalyzed cyclative coupling of o-functionalized haloarenes, 9–12 the envisioned organophosphorus-catalyzed strategy would offer a cohesive synthesis of several distinct azaheterocycles that: (1) proceeds directly from readily-accessible substituted nitroarene precursors with diversifiable ortho-functionality, (2) leverages the increasingly vast store of bench stable aryl- and alkylboronic acids now available, and (3) exhibits unique chemoselectivities and functional group tolerance inherent to the all-main-group conditions of the PIII/PV=O catalyzed coupling method.13–14 The versatility of this main group-catalyzed strategy is exemplifed by the regiospecific preparation of multiple classes of useful heterocycles in a single operation with modular and precise control over positional substitution about the heterocyclic periphery.
Results and Discussion
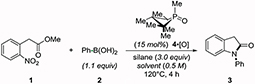
As an initial validation of the target tandem C–N coupling/cyclization reaction sequence, N-arylative cyclization of methyl 2-nitrophenylacetate (1) with phenylboronic acid (2) yielded oxindole product 3 in 85% NMR yield (80% isolated yield) on a 0.5 mmol scale within 4 h using 1,2,2,3,4,4-hexamethylphosphetane oxide 15 (4•[O]) as the catalyst and diphenylsilane as the terminal reductant (Table 1, entry 1). Control experiments (entries 2-4) are consistent with a reaction system that is operating via a PIII/PV=O redox cycling process and is under catalyst control; specifically, the use of PIII compound 4 instead of PV compound 4•[O] as catalyst is comparably efficient (entry 2, 88%), and omission of either catalyst 4•[O] (entry 3) or hydrosilane (entry 4) give no product 3. Among a brief survey of alternate organophosphorus compounds (See Table S2), commercially-available phosphine oxide 4•[O]16 was found to be most active. A variety of common hydrosilane reducing reagents (phenylsilane, entry 5; poly(methylhydro)siloxane, entry 6) can all similarly be employed.17 Practically, the method is not bounded by stringent operational constraints; the catalytic reaction is robust to a variety of solvents (entries 6-8) as well as the presence of both aerobic (entry 9) and aqueous (entry 10) contaminants.
Table 1.
Discovery and Optimization of Tandem Organophosphorus-Catalyzed Oxindole Synthesis.[a]
| ||||
|---|---|---|---|---|
| Entry | Solvent | Silane | R3P=O | Yield (%)[a] |
| 1 | m-xylene | Ph2SiH2 | 4•[O] | 85 |
| 2 | m-xylene | Ph2SiH2 | 4 | 88 |
| 3 | m-xylene | Ph2SiH2 | none | 0 |
| 4 | m-xylene | none | 4•[O] | 0 |
| 5 | m-xylene | PhSiH3 | 4•[O] | 83 |
| 6b | m-xylene | PMHS | 4•[O] | 87 |
| 7 | CPME | Ph2SiH2 | 4•[O] | 84 |
| 8b | PhCN | Ph2SiH2 | 4•[O] | 70 |
| 9c | m-xylene | Ph2SiH2 | 4•[O] | 79 |
| 10d | m-xylene | Ph2SiH2 | 4•[O] | 85 |
1H NMR yields compared to internal standard.
12 h reaction time.
Reaction run under air.
2 equiv. of H2O added.
CPME = cyclopentyl methyl ether. See Supporting Information for full synthetic details.
Although no long-lived intermediates en route from 1 to 3 are observed under optimized conditions, the use of tert-butyl 2-nitrophenylacetate (1a) as substrate (eq. 1) leads primarily to the C–N coupling intermediate 1b after 4 h (64%), which proceeds further to oxindole 3 only with prolonged heating (68% after 60 h). The initial C–N coupling does not proceed via the free aniline 1c, for which the optimized conditions did not result in the formation of oxindole 3; instead, only the parent N-H oxindole 3a was formed (eq. 2). Moreover, N-H oxindole 3a itself is not converted to 3 by the main-group catalyzed reaction conditions (eq. 3) but is recovered without N-arylation. Collectively, these probe experiments confirm a two-stage cascade sequence for the N-arylative cyclization of 1 involving initial reductive C–N coupling to form 1e followed by intramolecular cyclization to give 3 (Scheme 1, bottom).
Scheme 1.

Probe experiments and proposal for the reaction sequence leading to formation of oxindole 3.
Synthetic examples illustrating the scope of the organophosphorus-catalyzed heterocycle synthesis are collected in Table 2. With respect to oxindole synthesis (Table 2A),18 complete chemoselectivity for the desired tandem C–N bond constructions in preference to functionalization of aryl halides is observed; halogenation on either the nitroaryl substrate (5, 6) or the arylboronic acid partner (6, 8) result in halogenated oxindole products in good yield. Electronically diverse reaction partners are all incorporated in good yield within the developed scheme; electron-deficient nitroarenes can be paired with electron-rich boronic acids partners as in oxindole 7, or alternatively electron-rich nitroarenes can likewise be merged with electron-deficient boronic acids as in oxindole product 8. It was also found that alkylboronic acids can serve as a good partner in the tandem reaction sequence, for instance providing N-cyclopropyl substituted oxindole product (9) in 61% yield.
Table 2.
Synthetic scope of PIII/PV=O-catalyzed cyclative coupling method.
 |
Representative examples of (A) oxindole, (B) indole, (C) quinoxalinedione, and (D) benzimidazole synthesis via PIII/PV=O catalyzed tandem C–N coupling/cyclization. See SI for full experimental details and conditions. Yields are reported for pure isolated material following chromatography or recrystallization.
N-Arylative cyclization starting from α-(2-nitroaryl)ketones as substrates provides entry to substituted indole 19,20 products through an intramolecular carbonyl condensation of a first-formed reductive C–N coupled intermediate onto the pendant ketone moiety (Table 2B). Notably, many diverse 1,2-disubsituted indoles can be obtained via the developed method. It was found that 1-alkyl-2-aryl (10), 1-aryl-2-alkyl (11, 12), and 1-aryl-2-aryl (13-14) substituted indole products can all be synthesized by selection of the appropriate nitroarene and boronic acid reaction partners. The complementarity of the developed PIII/PV=O-catalyzed N-arylative cyclization with respect to transition metal approaches is amply demonstrated within this indole series of examples. Sulfur-containing containing products (i.e. N-thianthrenyl indole 13), potential poisons for late metal catalysis, are unproblematic under these main group conditions. Moreover, C–F, C–Cl, C–Br (13) and C–I (14) substituents are all tolerated without issue and carried through the tandem coupling and cyclization events. The retention of the reactive aryl halides thus permits their use as synthetic handles for the further diversification of the indole core via downstream coupling chemistry.
A family of 6-membered ring containing quinoxalinedione21 products are similarly accessible starting from oxalate amides of o-nitroaniline substrates (Table 2C). A useful feature for this class of molecules is that the products are insoluble in m-xylene and can be isolated by filtration following the reaction. Various N-functionality can be introduced from alkyl (i.e. cyclopropyl, 15), aryl (16, 18-19), and heteroaryl (17) boronic acids through the tandem sequence, in which intermolecular C–N bond formation leads to intramolecular addition to the pendant ester and regiospecific formation of the quinoxalinedione core. Similarly, not only are different nitroarene functionalities tolerated without issue (17-19), but also the method can be applied in the synthesis of the fused heterocyclic pyrido[2,3-b]pyrazinedione (16).N-acylated 2-nitroanilines of diverse substitution undergo PIII/PV=O catalyzed N-arylative cyclization process to provide N-functionalized benzimidazole22 products (Table 2D). In terms of C2-substitution originating from the amide fragment, (fluoro)alkyl (20-24) and heteroaryl substituents (25) could be successfully incorporated. Additionally, aryl (20-23), heteroaryl (24) and alkyl (i.e. cyclobutyl, 25) boronic acids could all be used as coupling partners in order to introduce a variety of N-substitution. Withrespect to functional group tolerance, it was found that esters (21), ethers (22), halogens (20-25), and thienyl units (24-25) could all be carried through the tandem reaction sequence without issue.
The PIII/PV=O catalyzed N-arylative cyclization method provides a regiospecific synthesis of N-aryl benzimidazoles in cases direct C–N coupling of the pseudosymmetric N-H precursor would be unselective.23 The full suite of regioisomeric fluorinated N-aryl benzimidazole products 26a-26d (Figure 2A) are accessible with programmed regiochemistry as dictated by the initial position of fluorination on the trifluoroacetyl-2-nitroanilide starting material. As a further practical point of utility, a modular and concise one-pot synthesis of benzimidazole product 28 via in situ acylation and N-arylative cyclization directly from 4-methyl-2-nitroaniline (27) is illustrated (Figure 2B). These results present the opportunity for this modular method to be applied in the synthesis of diverse benzimidazole compound libraries through a unified tandem reaction sequence directly from functionalized nitroanilines, boronic acids, and a suitable acylating reagent.
Figure 2.

Examples of catalytic reductive N-arylative cyclization. (A) Regiospecific benzimidazole synthesis. (B) Modular one pot amidation/reductive N-arylative cyclization. (C) Synthesis of linopiridine. See SI for full experimental details and conditions.
As a further demonstration of the synthetic versatility of the transformation and to demonstrate the potential application of this methodology in the context of medicinal chemistry, the KCNQ K+ ion channel blocker linopiridine (30) 24, 25 was synthesizing from methyl 2-nitrophenylacetate (1) using the developed sequential C–N coupling and cyclization approach, followed by in situ alkylation with 4-(bromomethyl)pyridine with 76% yield in one pot (Figure 2C).
Conclusion
The foregoing results constitute a practical, scalable, and operationally robust organophosphorus-catalyzed protocol for the modular (regiospecific) synthesis of azaheterocycles via a tandem intermolecular C–N coupling of nitroarenes and boronic acid partners, followed by intramolecular addition of the intermediary species by either carbonyl condensation (for benzimidazoles and indoles) or acyl substitution (for quinoxalinediones and oxindoles) moieties. The chemoselectivities and functional group tolerance enabled by the all-main-group conditions of the PIII/PV=O catalyzed coupling method establish this approach as a useful complement to existing methods to N-aryl heterocycles including those based on transition metal C–N coupling.26 In view of the prevalence of these types of nitrogen heterocycles in pharmaceuticals,1 bioactive natural metabolites,27 and organic materials28 among other applications, many possible implementations of this method can be envisioned.
Supplementary Material
Acknowledgements
Financial support was provided by NIH (GM114547), MIT, and Kallyope Inc.
Footnotes
Dedicated to Dr. Guy R. Humphrey (Merck & Co.) on the occasion of his 60th birthday.
References
- 1.a) Catarzi D, Colotta V, Varano F, Med. Res. Rev 2006, 2, 239; [DOI] [PubMed] [Google Scholar]; b) Kaushik NK, Kaushik N, Attri P, Kumar N, Kim CH, Verma AK, Choi EH, Molecules. 2013, 18, 6620; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Keri RS, Hiremathad A, Budagumpi S, Nagaraja BM, Chem. Biol. Drug Des 2015, 86, 19; [DOI] [PubMed] [Google Scholar]; d) Taylor AP, Robinson RP, Fobian YM, Blakemore DC, Jones LH, Fadeyi O, Org. Biomol. Chem 2016, 14, 6611; [DOI] [PubMed] [Google Scholar]; e) Nautiyal OH, OMCIJ 2018, 5, 555671; [Google Scholar]; f) Pallesen J, Møllerud S, Frydenvang K, Pickering DS, Bornholdt J, Nielsen B, Pasini D, Han L, Marconi L, Kastrup JS, Johansen TN. ACS Chem. Neurosci 2019, 10, 1841. [DOI] [PubMed] [Google Scholar]
- 2.Ono N, The Nitro Group in Organic Synthesis. Wiley, New York, 2001. [Google Scholar]
- 3.a) Cadogan JIG, Cameron-Wood M, Mackie RK, Searle RJG, J. Chem. Soc 1965, 4831; [Google Scholar]; b) Sundberg RJ, J. Org. Chem 1965, 30, 3604; [Google Scholar]; c) Cadogan JIG, Q. Rev., Chem. Soc 1968, 22, 222; [Google Scholar]; d) Cadogan JIG, Synthesis 1969, 11; [Google Scholar]; e) Cadogan JIG, Todd MJ, J. Chem. Soc. C 1969, 2808. [Google Scholar]
- 4.a) Bartoli G, Palmieri G, Bosco M, Dalpozzo R, Tetrahedron Lett. 1989, 30, 2129; [Google Scholar]; b) Bartoli G, Bosco M, Dalpozzo R, Palmieri G, Marcantoni E, J. Chem. Soc., Perkin Trans. 1. 1991, 11, 2757; [Google Scholar]; c) Dobbs AP, Voyle M, Whitall N, Synlett 1999, 10, 1594; [Google Scholar]; d) Bartoli G, Dalpozzo R, Nardi M, Chem. Soc. Rev 2014, 43, 4728. [DOI] [PubMed] [Google Scholar]
- 5.a) Výprachtický D, Kmínek I, Pokorná V, Cimrová V, Tetrahedron 2012, 68, 5075; [Google Scholar]; b) Kadam HK, Tilve SG, Eur. J. Org. Chem 2013, 4280. [Google Scholar]; c) Ames DE, Hansen KJ, Griffiths ND. J. Chem. Soc, Perkin Trans 1, 1973, 2818; [Google Scholar]; d) Tanaka A, Yakushijin K, Yoshina S. J. Heterocyclic Chem, 1979, 16, 785; [Google Scholar]; e) Jesudoss K, Srinivasan PC, Synth. Commun 1994, 24, 1701. [Google Scholar]
- 6.a) Sapountzis I, Knochel P, J. Am. Chem. Soc 2002, 124, 9390; [DOI] [PubMed] [Google Scholar]; b) Doyle W, Staubitz A, Knochel P, Chem. Eur. J 2003, 9, 5323; [DOI] [PubMed] [Google Scholar]; c) Kopp F, Sapountzis I, Knochel P, Synlett 2003, 885; [Google Scholar]; (d) Sapountzis I, Knochel P, Synlett 2004, 955; [Google Scholar]; e) Srivastava RS, Nicholas KM, Organometallics 2005, 24, 1563; [Google Scholar]; f) Fang X, Jackstell R, Beller M, Angew. Chem. Int. Ed 2013, 52, 14089; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 14339; [Google Scholar]; g) Gao H, Xu Q-L, Ess DH, Kürti L, Angew. Chem. Int. Ed 2014, 53, 2701; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 2739; [Google Scholar]; h) Gui J, Pan C-M, Jin Y, Qin T, Lo JC, Lee BJ, Spergel SH, Mertzman ME, Pitts WJ, La Cruz TE, Schmidt MA, Darvatkar N, Natarajan SR, Baran PS, Science 2015, 348, 886; [DOI] [PubMed] [Google Scholar]; i) Dhayalan V, Saemann C, Knochel P, Chem. Commun 2015, 51, 3239; [DOI] [PubMed] [Google Scholar]; j) Cheung CW, Hu X, Nat. Commun 2016, 7, 12494; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Cheung CW, Hu X, ACS Catal. 2017, 7, 7092; [Google Scholar]; l) Cheung CW, Ploeger ML, Hu X, Nat. Commun 2017, 8, 14878; [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Zhou F, Wang D-S, Guan X, Driver TG, Angew. Chem. Int. Ed 2017, 56, 4530; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 4601; [Google Scholar]; n) Rauser M, Ascheberg C, Niggemann M, Angew. Chem. Int. Ed. 2017, 56, 11570; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 11728; [Google Scholar]; o) Rauser M, Ascheberg C, Niggemann M, Chem. - A Eur. J 2018, 24, 3970; [DOI] [PubMed] [Google Scholar]; p) Cheung CW, Ploeger ML, Hu X, Chem. Sci 2018, 9, 655; [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Xiao J, He Y, Ye F, Zhu S, Chem. 2018, 4, 1645. [Google Scholar]; r) Suarez-Pantiga S, Hernandez-Ruiz R, Virumbrales C, Pedrosa MR, Sanz R, Angew. Chem. Int. Ed 2019, 58, 2129; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 2151; [Google Scholar]; s) Roscales S, Csáky AG, Adv. Synth. Catal 2019, DOI 10.1002/adsc.201901009. [DOI] [Google Scholar]
- 7.For C-N bond formation between nitrosoarenes and boronic acids.; a) Yu Y, Srogl J, Liebeskind LS, Org. Lett 2004, 6, 2631; [DOI] [PubMed] [Google Scholar]; b) Roscales S, Csáky AG, Org. Lett 2018, 20, 1667; [DOI] [PubMed] [Google Scholar]; c) Roscales S, Csákÿ AG, ACS Omega 2019, 4, 13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nykaza TV, Cooper JC, Li G, Mahieu N, Ramirez A, Luzung MR, Radosevich AT, J. Am. Chem. Soc 2018, 140, 15200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Zheng N, Anderson KW, Huang X, Nguyen HN, Buchwald SL, Angew. Chem. Int. Ed 2007, 46, 7509; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2007, 119, 7653; [Google Scholar]; b) Zheng N, Buchwald SL, Org. Lett 2007, 9, 4749. [DOI] [PubMed] [Google Scholar]; c) Jui NT, Buchwald SL, Angew. Chem. Int. Ed 2013, 52, 11624; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 11838. [Google Scholar]
- 10.Zou B, Yuan Q, Ma D, Angew. Chem. Int. Ed 2007, 46, 2598; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2007, 119, 2652. [Google Scholar]
- 11.a) Brain CT, Brunton SA, Tetrahedron Lett. 2002, 43, 1893; [Google Scholar]; b) Brain CT, Steer JT, J. Org. Chem 2003, 68, 6814. [DOI] [PubMed] [Google Scholar]
- 12.a) Saha P, Ramana T, Purkait N, Ali MA, Paul R, Punniyamurthy T, J. Org. Chem 2009, 74, 8719; [DOI] [PubMed] [Google Scholar]; b) Deng X, Mani NS, Eur. J. Org. Chem 2010, 680; [Google Scholar]; c) Saha P, Ali MA, Ghosh P, Punniyamurthy T, Org. Biomol. Chem 2010, 8, 5692; [DOI] [PubMed] [Google Scholar]; d) Alonso J, Halland N, Nazaré M, R’kyek O, Urmann M, Lindenschmidt A, Eur. J. Org. Chem 2011, 234; [Google Scholar]; e) Peng J, Ye M, Zong C, Hu F, Feng L, Wang X, Wang Y, Chen C, J. Org. Chem 2011, 76, 716. [DOI] [PubMed] [Google Scholar]
- 13.For a review of PIII/PV=O redox cycling, see:; a) Marsden SP, Catalytic Variants of Phosphine Oxide-Mediated Organic Transformations in Sustainable Catalysis; Dunn PJ, Hii KK, Krische MJ, Williams MT, Eds.; John Wiley & Sons, Inc.: New York, 2013; pp 339–361; [Google Scholar]; b) Guo H, Fan YC, Sun Z, Wu Y, Kwon O, Chem. Rev 2018, 118, 10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) O’Brien CJ, Tellez JL, Nixon ZS, Kang LJ, Carter AL, Kunkel SR, Przeworski KC, Chass GA, Angew. Chem. Int. Ed 2009, 48, 6836; [DOI] [PubMed] [Google Scholar]; Angew Chem, 2009, 121: 6968; [Google Scholar]; b) van Kalkeren HA, Leenders SHAM, Hommersom CRA, Rutjes FPJT, van Delft FL, Chem. Eur. J 2011, 17, 11290; [DOI] [PubMed] [Google Scholar]; c) van Kalkeren HA, Bruins JJ, Rutjes FPJT, van Delft FL, Adv. Synth. Catal 2012, 354, 1417; [Google Scholar]; d) O’Brien CJ, Lavigne F, Coyle EE, Holohan AJ, Doonan BJ, Chem. Eur. J 2013, 19, 5854. [DOI] [PubMed] [Google Scholar]; e) O’Brien CJ, Nixon ZS, Holohan AJ, Kunkel SR, Tellez JL, Doonan BJ, Coyle EE, Lavigne F, Kang LJ, Przeworski KC, Chem. Eur. J 2013, 19, 15281; [DOI] [PubMed] [Google Scholar]; f) Coyle EE, Doonan BJ, Holohan AJ, Walsh KA, Lavigne F, Krenske EH, O’Brien CJ, Angew. Chem. Int. Ed 2014, 53, 12907; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 13121; [Google Scholar]; g) Reichl KD, Dunn NL, Fastuca NJ, Radosevich AT, J. Am. Chem. Soc 2015, 137, 5292; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Zhao W, Yan PK, Radosevich AT, J. Am. Chem. Soc 2015, 137, 616; [DOI] [PubMed] [Google Scholar]; i) Lee C, Chang T, Yu J, Reddy GM, Hsiao M, Lin W, Org. Lett. 2016, 18, 3758; [DOI] [PubMed] [Google Scholar]; j) Lao Z, Toy PH, Beilstein J. Org. Chem 2016, 12, 2577; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Saleh N, Voituriez A, J. Org. Chem 2016, 81, 4371; [DOI] [PubMed] [Google Scholar]; l) Saleh N, Blanchard F, Voituriez A, Adv. Synth. Catal 2017, 359, 2304; [Google Scholar]; m) Lin Y-C, Hatzakis E, McCarthy SM, Reichl KD, Lai T-Y, Yennawar HP, Radosevich AT, J. Am. Chem. Soc 2017, 139, 6008; [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Nykaza TV, Harrison TS, Ghosh A, Putnik RA, Radosevich AT, J. Am. Chem. Soc 2017, 139, 6839; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Nykaza TV, Ramirez A, Harrison TS, Luzung MR, Radosevich AT, J. Am. Chem. Soc 2018, 140, 3103; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Zhang K, Cai L, Yang Z, Houk KN, Kwon O, Chem. Sci 2018, 9, 1867; [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Ghosh A, Lecomte M, Kim-Lee S-H, Radosevich AT, Angew. Chem. Int. Ed 2019, 58, 2864; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 2890; [Google Scholar]; r) Lecomte M, Lipshultz JM, Kim-Lee S-H, Li G, Radosevich AT, J. Am. Chem. Soc 2019. 141, 12507; [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Cai L, Zhang K, Chen S, Lepage RJ, Houk KN, Krenske EH, Kwon O, J. Am. Chem. Soc 2019, 141, 9537; [DOI] [PMC free article] [PubMed] [Google Scholar]; t) Lorton C, Castanheiro T, Voituriez A, J. Am. Chem. Soc 2019, 141, 10142; [DOI] [PubMed] [Google Scholar]; u) Longwitz L, Spannenberg A, Werner T, ACS Catal. 2019, 9, 9237. [Google Scholar]
- 15.For a preparation of 4 [O], see:; Nykaza TV, Cooper JC, Radosevich AT, Org. Synth 2019, 96, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Available from Strem Chemicals, item no. 15-8150.
- 17.When using phenylsilane, product 3 was found to convert to 1-phenyl-1H-indole (87%) at prolonged reaction times (24 h).
- 18.a) Goehring RR, Sachdeva YP, Pisipati JS, Sleevi MC, Wolfe JF, J. Am. Chem. Soc 1985, 107, 435; [Google Scholar]; b) Bowman WR, Heaney H, Jordan BM, Tetrahedron Lett. 1988, 29, 6657; [Google Scholar]; c) Hennessy EJ, Buchwald SL, J. Am. Chem. Soc 2003, 125, 12084; [DOI] [PubMed] [Google Scholar]; d) Poondra RR, Turner NJ, Org. Lett 2005, 7, 863; [DOI] [PubMed] [Google Scholar]; e) Dalpozzo R, Bartoli G, Bencivenni G, Chem. Soc. Rev 2012, 41, 7247; [DOI] [PubMed] [Google Scholar]; f) Kiser EJ, Magano J, Shine RJ, Chen MH, Org. Process Res. Dev 2012, 16, 255. [Google Scholar]
- 19.a) Humphrey GR, Kuethe JT, Chem. Rev 2006, 106, 2875. [DOI] [PubMed] [Google Scholar]; b) Taber DF, Tirunahari PK, Tetrahedron 2011, 67, 7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For reviews see:; a) Wexler RR, Greenlee WJ, Irvin JD, Goldberg MR, Prendergast K, Smith RD, Timmermans PBMWM, J. Med. Chem 1996, 39, 625; [DOI] [PubMed] [Google Scholar]; b) Morphy R, Rankovic Z, J. Med. Chem 2005, 48, 6523; [DOI] [PubMed] [Google Scholar]; c) Schnürch M, Flasik R, Khan AF, Spina M, Mihovilovic MD, Stanetty P, Eur. J. Org. Chem 2006, 3283. [Google Scholar]
- 21.a) Nikam SS, Cordon JJ, Ortwine DF, Heimbach TH, Blackburn AC, Vartanian MG, Nelson CB, Schwarz RD, Boxer PA, Rafferty MF, J. Med. Chem 1999, 42, 2266; [DOI] [PubMed] [Google Scholar]; b) Fray MJ, Bull DJ, Carr CL, Gautier ECL, Mowbray CE, Stobie A, J. Med. Chem 2001, 44, 1951. [DOI] [PubMed] [Google Scholar]
- 22.a) Panda SS, Malik RM, Jain SC, Curr. Org. Chem 2012, 16, 1905; [Google Scholar]; b) Kurhade S, Rossetti A, Dömling A, Synthesis 2016, 48, 3713. [Google Scholar]
- 23.Carvalho LCR, Fernandes E, Marques MMB, Chem. Eur. J 2011, 17, 12544. [DOI] [PubMed] [Google Scholar]
- 24.a) Schnee ME, Brown BS, J. Pharmacol. Exp. Ther 1998, 286, 709. [PubMed] [Google Scholar]; b) Robbins J, Pharmacol. Ther 2001, 90, 1. [DOI] [PubMed] [Google Scholar]
- 25.A three step synthesis from diphenylamine (57% overall yield) is reported.; Earl RA, Meyers MJ, Nickolson VJ (DuPont Merck Pharmaceutical Co; ), US 5173489(A), 1992.
- 26.For literature concerning N-H functionalization see:; a) Lam PYS, Clark CG, Saubern S, Adams J, Winters MP, Chan DMT, Combs A, Tetrahedron Lett. 1998, 39, 2941; [Google Scholar]; b) Lam PYS, Deudon S, Averill KM, Li R, He MY, DeShong P, Clark CG, J. Am. Chem. Soc 2000, 122, 7600; [Google Scholar]; c) Old DW, Harris MC, Buchwald SL, Org. Lett 2000, 2, 1403; [DOI] [PubMed] [Google Scholar]; d) Klapars A, Antilla JC, Huang X, Buchwald SL, J. Am. Chem. Soc 2001, 123, 7727; [DOI] [PubMed] [Google Scholar]; e) Antilla JC, Klapars A, Buchwald SL, J. Am. Chem. Soc 2002, 124, 11684; [DOI] [PubMed] [Google Scholar]; f) Choudary BM, Sridhar C, Kantam ML, Venkanna GT, Sreedhar B, J. Am. Chem. Soc 2005, 127, 9948; [DOI] [PubMed] [Google Scholar]; g) Altman RA, Buchwald SL, Org. Lett 2006, 8, 2779; [DOI] [PubMed] [Google Scholar]; h) Altman RA, Koval ED, Buchwald SL, J. Org. Chem 2007, 72, 6190; [DOI] [PubMed] [Google Scholar]; i) Correa A, Bolm C, Angew. Chem. Int. Ed 2007, 46, 8862; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2007, 119, 9018–9021; [Google Scholar]; j) Zhu L, Guo P, Li G, Lan J, Xie R, You J, J. Org. Chem 2007, 72, 8535; [DOI] [PubMed] [Google Scholar]; k) Altman RA, Hyde AM, Huang X, Buchwald SL, J. Am. Chem. Soc 2008, 130, 9613; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Zhu L, Li G, Luo L, Guo P, Lan J, You J, J. Org. Chem 2009, 74, 2200; [DOI] [PubMed] [Google Scholar]; m) Ganesh Babu S, Karvembu R, Ind. Eng. Chem. Res 2011, 50, 9594; [Google Scholar]; n) Karchava AV, Melkonyan FS, Yurovskaya MA, Chem. Heterocycl. Comp 2012, 48, 391; [Google Scholar]; o) Ueda S, Su M, Buchwald SL, J. Am. Chem. Soc 2012, 134, 700; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Davis OA, Hughes M, Bull JA, J. Org. Chem 2013, 78, 3470; [DOI] [PubMed] [Google Scholar]; q) Dar’in D, Krasavin M, J. Org. Chem 2016, 81, 12514; [DOI] [PubMed] [Google Scholar]; r) Jung S-H, Sung D-B, Park C-H, Kim W-S, J. Org. Chem 2016, 81, 7717; [DOI] [PubMed] [Google Scholar]; s) Miao B, Li S, Li G, Ma S, Org. Lett 2016, 18, 2556; [DOI] [PubMed] [Google Scholar]; t) Ye Y, Kim S-T, Jeong J, Baik M-H, Buchwald SL, J. Am. Chem. Soc 2019, 141, 3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Gul W, Hamann MT, Life Sci. 2005, 78, 442; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Millemaggi A, Taylor RJK, Eur. J. Org. Chem 2010, 4527; [Google Scholar]; c) Corsello MA, Kim J, Garg NK, Chem. Sci 2017, 8, 5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a) Asensio JA, Sánchez EM, Gómez-Romero P, Chem. Soc. Rev 2010, 39, 3210; [DOI] [PubMed] [Google Scholar]; b) Molina P, Tárraga A, Otón F, Org. Biomol. Chem 2012, 10, 1711; [DOI] [PubMed] [Google Scholar]; c) Rabbani MG, El-Kaderi HM, Chem. Mater 2012, 24, 1511; [Google Scholar]; d) Wu J, Chen J, Huang H, Li S, Wu H, Hu C, Tang J, Zhang Q, Macromolecules 2016, 49, 2145; [Google Scholar]; e) Demmer CS, Rombach D, Liu N, Nielsen B, Pickering DS, Bunch L, ACS Chem. Neurosci 2017, 8, 2477; [DOI] [PubMed] [Google Scholar]; f) Wang P, Arza CR, Zhang B, Polym. Chem 2018, 9, 4706; [Google Scholar]; (g) Roke D, Sen M, Danowski W, Wezenberg SJ, Feringa BL, J. Am. Chem. Soc 2019, 141, 7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.