

Abstract
Trypsin is the major serine protease responsible for intestinal protein digestion. An inhibitor, camostat (CS), reduced weight gain, hyperglycemia, and dyslipidemia in obese rats; however, the mechanisms for these are largely unknown. We reasoned that CS creates an apparent dietary protein restriction, which is known to increase hepatic fibroblast growth factor 21 (FGF21). Therefore, metabolic responses to CS and a gut-restricted CS metabolite, FOY-251, were measured in mice. Food intake, body weight, blood glucose, branched-chain amino acids (LC/MS), hormone levels (ELISA), liver pathology (histology), and transcriptional changes (qRT-PCR) were measured in ob/ob, lean and diet-induced obese (DIO) C57BL/6 mice. In ob/ob mice, CS in chow (9–69 mg/kg) or FOY-251 (46 mg/kg) reduced food intake and body weight gain to a similar extent as pair-fed mice. CS decreased blood glucose, liver weight, and lipidosis and increased FGF21 gene transcription and plasma levels. In lean mice, CS increased liver FGF21 mRNA and plasma levels. Relative to pair feeding, FOY-251 also increased plasma FGF21 and induced liver FGF21 and integrated stress response (ISR) transcription. In DIO mice, FOY-251 (100 mg/kg po) did not alter peak glucose levels but reduced the AUC of the glucose excursion in response to an oral glucose challenge. FOY-251 increased plasma FGF21 levels. In addition to previously reported satiety-dependent (cholecystokinin-mediated) actions, intestinal trypsin inhibition engages non-satiety-related pathways in both leptin-deficient and DIO mice. This novel mechanism improves metabolism by a liver-integrated stress response and increased FGF21 expression levels in mice.
NEW & NOTEWORTHY Trypsin inhibitors, including plant-based consumer products, have long been associated with metabolic improvements. Studies in the 1980s and 1990s suggested this was due to satiety hormones and caloric wasting by loss of protein and fatty acids in feces. This work suggests an entirely new mechanism based on the lower amounts of digested protein available in the gut. This apparent protein reduction may cause beneficial metabolic adaptation by the intestinal-liver axis to perceived nutrient stress.
Keywords: camostat, gluconeogenesis, integrated stress response, liver, obesity, protein dilution, small intestine, triglycerides, type 2 diabetes
INTRODUCTION
Camostat (CS), a serine protease trypsin inhibitor used for chronic pancreatitis in Japan, has been shown to have beneficial metabolic effects. In an 8-year study, CS reduced the incidence of newly diagnosed type 2 diabetes (T2D) in pancreatitis patients from 32.7% to 24.3% (16). Camostat and plant-based trypsin inhibitors also reduce body weight gain by decreasing food intake in various obese rodent models (33, 35) and in other species (46). This satiety-mediated effect is due to increased secretion of the peptide cholecystokinin (CCK) (33). In rats, protein digestion in the small intestine is a potent stimulus for CCK release, which acts on CCK receptor 1 (CCK1R) to delay gastric emptying and inhibit food intake (27). CS has been used experimentally in rats as a nonnutrient stimulant of endogenous CCK release to reduce food intake, delay gastric emptying, and increase gall bladder emptying and pancreatic zymogen secretion (26, 37).
There are also studies demonstrating satiety-independent weight loss by trypsin inhibition in different animal models. For example, long-term CS administration reduced weight gain and hyperglycemia in Otsuka Long-Evans Tokushima fatty (OLETF) rats (18, 19). These rats are CCK1R deficient (34) and are thus insensitive to the feeding-inhibitory actions of CCK. They gain weight and do not compensate for eating larger meals by decreased meal frequency (5). In OLETF rats, CS reduced weight gain without reducing food intake (19). In mice, novel boropeptide inhibitors of enteropeptidase, which is the serine protease converting trypsinogen to its active form, reduced weight gain without reducing food intake (7). In both studies, caloric excretion in feces was increased (7, 19), but additional energy balance mechanisms had not, to our knowledge, been investigated.
In the present study we first characterized the metabolic changes and transcriptional responses to CS in a hyperphagic mouse model. In contrast to OLETF rats, CCK1R−/− mice do not overeat or gain weight when fed normal chow and respond to a high-fat diet (HFD) similarly to wild-type mice (4). Therefore, we selected ob/ob mice, which are leptin deficient, obese, and insulin resistant, as a hyperphagic model. In our initial findings, CS administered to ob/ob mice decreased food intake, reduced hyperglycemia, caused pancreatic hypertrophy, and increased protein excretion.
In humans, CS given intraduodenally abolished proteolysis in the duodenum and reduced pancreatic responses to albumin (50). These effects would be consistent with reduced availability of digested protein in the gut or amino acids entering the circulation. Therefore, we reasoned that intraduodenal CS creates an environment that is perceived by the body as protein restriction. Amino acid deprivation initiates a signal transduction cascade through integrated stress response (ISR) target genes (30, 31). The ISR is so named because different environmental stresses converge at the level of phosphorylated eukaryotic translation initiation factor 2α (eIF2α). Nutrient stress-specific kinases modulate a wide spectrum of genes involved in the adaptation to dietary stress, such as essential amino acid deprivation (32).
Fibroblast growth factor 21 (FGF21) is a hormone induced in the liver by amino acid deprivation or dietary protein restriction (30). In mice, liver FGF21 mRNA expression is increased within 24 h of reduced protein intake, resulting in higher circulating FGF21 levels (23). FGF21 induces hepatic fat oxidation, ketogenesis, and gluconeogenesis to provide for metabolic adaptation to starvation (9). Constitutively increased FGF21 expression in mice also protects them from HFD-induced obesity, hepatic steatosis, and insulin resistance (54). In obese primates and humans, FGF21 improves weight loss and reduces triglycerides (TGs) (21), and FGF21 may play a role in glycemic control in T2D patients (43). All together, this has fueled the interest in therapeutic agents targeting the FGF21 pathway. Therefore, to assess whether trypsin inhibition was acting on an endogenous ISR-FGF21 pathway, we measured ISR target genes in liver, circulating amino acids [branched-chain amino acids (BCAA) valine, isoleucine, and leucine], and FGF21 in plasma after administration of CS and the active metabolite of CS, 4-(4-guanidinobenzoyloxy phenyl acetic acid) (FOY-251). Relative to pair feeding, this increased FGF21 and ISR transcription in liver. FOY-251 increased circulating FGF21 in ob/ob, lean and diet-induced obese (DIO) mice. These data have been published in a preliminary form (17).
METHODS
Animal studies were performed in accordance with the Federal Animal Welfare Act, and protocols were approved by the Institutional Animal Care and Use Committee at Janssen Pharmaceutical R&D (Spring House, PA). Male ob/ob mice, aged 6–7 wk and with range of weight 43–56 g (Jax Laboratories, Bar Harbor, ME), were single housed in a temperature-controlled room with a 12:12-h light-dark cycle and allowed ad libitum access to water and food. DIO C57BL/6 mice were fed HFD diet (Research Diets 12492; 20% protein, 60% fat, 20% carbohydrate) and tested at 18–19 wk (weight range 46.4 ± 0.42 g, n = 32) after oral gavage of trypsin inhibitor.
The trypsin inhibitor CS also inhibits proteases, including plasmin, kallikrein, and thrombin (49). To confirm selectivity, IC50 for CS were generated against a panel of 63 proteases in singlet using five concentrations, where 10 µM CS was the lowest. Trypsin inhibition by CS was IC50 < 1.27E−07 in this assay, and only eight other proteases had similar potencies (thrombin, plasmin, elastase, factor XIa, plasma kallikrein, matriptase, tryptase, and urokinase).
Mice (ob/ob) were acclimated to powdered chow [Formula B Diet 5008 (16.5% protein; 17.0% fat; 56.5% carbohydrate), Purina LabDiet] for 5–7 days. Afterward, mice were fed powdered chow formulated with vehicle alone [20% HP-β-cyclodextran (HP-β-CD)] or CS (0.08, 0.25 or 0.8 mg/g food) or FOY-251 (0.8 mg/g food) for the indicated time periods.
During experiments, blood glucose was assessed by tail snip on a glucometer (AlphaTrak2 Glucometer, Abbott Laboratories). DIO mice were challenged with oral glucose (2 g/kg d-glucose; 20% glucose, Teknova), and blood glucose was measured (T0-T120 min) with samples collected (t = 0 and t = 15 min) for insulin measurement.
At the end of the experiment, mice were euthanized with CO2. Blood was collected by cardiac puncture and centrifuged at 10,000 rpm for 10 min at 4°C to obtain plasma, which was aliquoted into 96-well plates and stored at −80°C until further analysis. Small intestinal mucosa was obtained by isolating 5-cm sections (starting 2 cm below the pyloric sphincter for duodenum, and 5 cm above the cecum for ileum) and flushing the luminal contents with ice-cold 2× PBS, before longitudinally opening the intestinal segments and mechanically scraping the mucosa from the tissue using sterile microscope slides. Isolated mucosa was snap-frozen and stored at −80°C until further use. Pancreatic tissue (~60 mg) was collected, and whole livers were dissected and weighed, and then several 60-mg pieces of liver were isolated and stored for further use. Minced liver pieces and pancreatic tissue were placed into RNALater (Qiagen, Valencia, CA) at 4°C overnight and then −20°C until further use.
Fecal analysis.
Fecal pellets were collected and frozen at −20°C. For protein extraction, fecal pellets were diluted in PBS and homogenized (Tissue Lyser II; Qiagen to obtain a 1:10 dilution. Tubes were centrifuged for 5 min at 12,000 g to remove solid material, and then ~250 μl of each supernatant was transferred to a 96-well plate. PBS was added for a final dilution of 1:1000. For protein analysis, 150 μl of each diluted sample was analyzed in triplicate by Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Protein concentration (μg/ml) was determined by comparing samples to a standard curve of known values as read on a plate reader (SpectraMAX Plate Reader). Blank well values were subtracted from the sample results. This dye-based BCA assay is routinely used for protein concentration estimates but has caveats, as reviewed (39). In addition, dietary protein was unlabeled; thus, fecal protein estimates could not differentiate between ingested and endogenous protein sources. For fecal TG species (n = 18) and total TG levels, liquid chromatography-mass spectrometry (LC-MS) was used.
Insulin, FGF21, GLP1, TG, and amino acid measurement from blood.
Plasma and serum were collected at termination and for peptide analysis into EDTA tubes containing DPP IV inhibitor (Millipore, St. Louis, MO) and protease inhibitor cocktail (Roche). Insulin was assessed with a mouse/rat insulin ELISA kit (Meso Scale Discovery) according to manufacturer protocols. Plasma FGF21 was assessed with a rat/mouse FGF21 ELISA kit (Millipore) according to the manufacturer’s protocols. For both hormones, absolute concentration (pg/ml) was determined by comparing samples with a standard curve using manufacturer-provided standards. A total glucagon-like peptide-1 (GLP-1) Mesoscale Discovery (MSD v.2) assay kit was used according to its protocol using standards to generate standard curves. Samples were undiluted and tested as single data points, which could be back-calculated from the standard curve. Plasma amino acid levels were measured by LC-MS. For plasma TG, an L-Type Triglyceride M assay (Wako Life Sciences) was used following the manufacturer’s guidelines and using the stock calibrator standards from absorbance read at 600 nm using a four-parameter logistic curve fit to create a standard curve. Then, each sample TG concentration was determined using this standard curve. In all cases, unknown samples that were below the quantification threshold were reported as 1/2 the lower limit of quantification (LLOQ). For individual (18 species) TG measurements LC-MS was used.
Liver analysis and histology.
Whole livers were dissected and weighed. Liver mass was normalized to total body mass. Liver percent fat was determined by gently thawing flash-frozen liver sections of known weight and analyzing fat content by nuclear magnetic resonance. Formalin-fixed liver sections were prepared by histology for hematoxylin and eosin (H&E) staining and microscopic evaluation. Representative digital images were unedited other than for cropping and orientation. Lipidosis grading was determined by a board-certified pathologist who was blinded as to the treatment groups.
Tissue mRNA isolation and transcriptional analysis.
RNA was extracted from cells using the RNeasy mini kit and on-column DNA digestion (Qiagen) following the manufacturer’s protocols. Mouse tissue was removed from RNALater, and excess RNALater was gently dabbed off using sterile wipes. RNA was extracted by first homogenizing tissue with 5 mM sterile steel ball bearings in a Tissue Lyser II (Qiagen), following by RNeasy mini kit utilization the per the manufacturer’s protocols. For liver RNA extraction, the RNA isolation protocol was modified to use 50% EtOH in place of 70% EtOH for column binding (posthomogenization and lysis steps). RNA was converted to cDNA by using a reverse-transcriptase mastermix (Thermo Fisher, SuperScript VILO IV, no. 11756050) according to the manufacturer’s protocols. qRT-PCR was performed on the resulting cDNA using the viia-7 Real Time PCR machine and SYBR Green Master Mix (Applied Biosystems; Foster City, CA). Custom oligos obtained for qRT-PCR analysis (Life Technologies) were utilized to analyze the indicated genes. The run method for qRT-PCR analysis consisted of 1) Hold state: 50°C 2 min, 95°C 3 min; 2) PCR stage: 95°C 15 s (Melt stage), 60°C 30 s (annealing and amplification) × 40 cycles; and 3) Melt curve: for primer quality control. Results are represented as fold change (where fold change = 2−ΔΔCT). Data were normalized to vehicle-treated controls by utilizing cyclophilin A or β-actin as the normalizing housekeeping gene.
Statistics.
Compiled data were analyzed statistically (GraphPad Prism 7) as reported in results using unpaired Student’s t-test, by one-way ANOVA with Tukey’s multiple or Dunnett’s multiple comparison posttest or Kruskal-Wallis ANOVA with Dunn’s posttest comparison, as appropriate. P values are shown when P < 0.05 and considered to be statistically significant.
RESULTS
The metabolic changes of chronic CS treatment in ob/ob mice were measured in a 7-day study. Camostat was formulated in feed (0.08, 0.25, and 0.8 mg/g powdered Laboratory Diet 5008; 23% protein, 6.5% fat, 56.5% carbohydrate). This was predicted to be equivalent to doses of 10, 30, and 100 mg/kg, but the reduced consumption of food resulted in CS of average 9.2, 25.5, and 69.0 mg·kg−1·day−1 (Fig. 1A). CS food intake reduction was maintained over the course of 7 days (Fig. 1B), and, to compare for food intake changes alone, a pair-fed group was matched to the highest dose of CS. The starting body weights of each group of ob/ob mice were similar (veh = 46.8 ± 0.8 g; 9 mg/kg CS = 49.3 ± 1.4 g; 26 mg/kg CS = 49.3 ± 1.1 g, 69 mg/kg CS = 47.8 ± 0.8 g; pair-fed = 47.4 ± 0.9 g). Body weight decreased by 4% relative to baseline in CS (69 mg/kg) and pair-fed groups, whereas control mice gained ~5% body weight (Fig. 1C). Five-hour-fasted blood glucose levels in the 69 mg/kg CS-dosed group were lower than both vehicle control and pair-fed groups (Fig. 1D). Terminal plasma insulin (Fig. 1E) and total GLP1 (Fig. 1F) levels were similar across groups. Fecal protein levels were increased by CS relative to pair-fed (as well as vehicle-treated) mice (Fig. 1G). In vehicle- and CS-treated mice, weight loss on day 7 was positively correlated with fecal protein (P < 0.0001), fasted blood glucose (Fig. 1H), and plasma TG levels (Fig. 1I).
Fig. 1.

Metabolic changes in ob/ob mice given a range of doses of camostat (CS) in chow (0.08, 0.25, and 0.8 mg/g) for 7 days. A: compound intake resulted in average daily doses of 9.2, 25.5, and 69.0 mg/kg CS. B: food intake was reduced and maintained at a lower level through day 7 in CS-treated (69 and 26 mg·kg−1·day−1) mice; pair-fed mice (chow containing vehicle) were matched for the highest CS dose. C: %decreased body weight was similar in both CS and pair-fed mice compared with vehicle-treated mice. D: at termination, fasted blood glucose was reduced in 69 mg/kg group compared with pair-fed, but similar in vehicle, pair-fed and the lower CS-dosed groups. Terminal plasma insulin levels (E) and total glucagon-like peptide-1 (GLP1; F) were similar across the groups. G: fecal protein levels were increased by all CS doses relative to pair-fed mice. ○, Vehicle; ■, CS (9, 26, and 69 mg/kg); ◇ and dashed line, Pair-Fed; n = 8/group. Data are illustrated as box-and-whisker plots with individual plots in this and all subsequent figures. Bars indicate significance level by ANOVA with Tukey’s or Dunnett’s posttest. Percent weight loss correlated with fasted blood glucose (H) and plasma triglyceride (TG) levels (G).
In the same study, liver weight (as %body weight) was decreased by pair feeding alone relative to vehicle but was further reduced by consumption of the highest CS dose (Fig. 2A). The reduction in liver weight on day 7 was correlated with fasted blood glucose (Fig. 2B), body weight loss (P < 0.0001), fecal protein (P < 0.001), and plasma TG (P < 0.05). Liver fat (by NMR) tended to be reduced by 69 mg/kg CS compared with vehicle (P = 0.08; data not shown). Furthermore, lower liver lipidosis in CS-treated mice (Fig. 2C) was noted in H&E-stained liver sections scored semiquantitatively by lipidosis grade (1–4 of increasing severity). Representative digital images at low-power magnification (Fig. 2D) illustrate the decreased size of livers in CS-treated mice. This smaller size reflects the decreased liver weights shown in Fig. 2A. In a vehicle-treated mouse liver, widespread vacuolization was noted (grade 4 in Fig. 2D), which was not seen in CS-treated mice (representative images of grades 1–3 in Fig. 2D). Higher-power magnification illustrates the accumulation of lipid droplets in a vehicle-treated mouse, which was reduced in the CS-treated mice (Fig. 2D, insets). Pancreatic weight was increased by the highest CS dose (Veh = 0.5 ± 0.04 g, Pair-fed = 0.41 ± 0.04 g, CS 69 mg/kg = 1.1 ± 0.08 g, P < 0.0001 compared with vehicle- and pair-fed by ANOVA and Tukey’s posttest). Pancreatic weight changes correlated with microscopic findings of diffuse acinar cell hypertrophy with rare scattered mitoses. The hypertrophic pancreatic response to trypsin inhibitors, including CS, has been reported previously (12, 40). Microscopic findings were limited to the exocrine pancreas, with islets not obviously affected.
Fig. 2.

Liver changes in ob/ob mice after 7 days of camostat (CS) in chow. A: liver weight (as %body weight) was reduced by the highest dose of CS compared with vehicle and pair-fed groups, where pair-fed was also lower than vehicle weight. P value by ANOVA and Tukey’s posttest comparisons. B: liver weight was correlated with 5-h-fasted blood glucose levels in vehicle- and CS-treated groups. C: CS-treated mice had lower lipidosis grade compared with pair-fed in hematoxylin-eosin-stained livers. P value by Kruskal-Wallis ANOVA and Dunn’s posttest comparison. D: representative digital images illustrate decreased size of livers in CS-treated mice (69 mg/kg) compared with vehicle-treated mice. Widespread vacuolization and diffuse lipid droplet accumulation noted in vehicle-treated is reduced in CS-treated examples, also shown in insets at higher-power magnification. Scale bars, 2.5 mm at low-power magnification and 250 µm in insets.
In summary, this 7-day CS study in ob/ob mice resulted in dose-related reductions in food intake and body weight gain. Compared with pair-fed mice, the high CS dose reduced blood glucose by 50% without marked changes in insulin or GLP1, increased fecal protein content by twofold, decreased liver weight and improved liver lipidosis, and induced pancreatic hypertrophy.
Acute 24-h CS treatment (in chow) in ob/ob mice reduced food intake (Fig. 3A), body weight gain (Fig. 3B), and fed blood glucose (Fig. 3C) compared with vehicle. Insulin levels at termination were not reduced (vehicle = 29.5 ± 3.3 ng/ml vs. CS = 20.8 ± 4.6 ng/ml, P = 0.15 by unpaired t-test). Fecal protein concentration was increased modestly (Fig. 3D). To elucidate potential mechanisms for these CS-induced effects, metabolically relevant gene expression in liver, pancreas, and GI tract was measured by qRT-PCR analysis in a repeat acute study. This study resulted in similar reductions in food intake and body weight gain. In liver (Fig. 3E), CS treatment decreased glucose-6-phosphatase catalytic subunit (G6PC) gene expression (consistent with reduced gluconeogenesis) and decreased ATP citrate lyase (ACLY) expression without changing expression of fructose-bisphosphatase-1 (FBP1; consistent with reduced liver lipidosis and TG synthesis), and increased FGF21 transcription. In the pancreas (Fig. 3F), CS treatment transcriptionally increased FGF21, trypsin (PRSS1), mesotrypsin (PRSS3), and pancreatic trypsin inhibitor (SPINK). The transcriptional zymogen increases reflect the known mechanism of action of CS on the gut lumen to increase CCK secretion, resulting in feedback mechanisms to the acinar pancreas [reviewed by Liddle (26)]. Because of the increased transcription of FGF21 in liver (Fig. 3E) and pancreas (Fig. 3F), plasma was analyzed for FGF21 levels, which were significantly increased by CS treatment (Fig. 3G). In ileum mucosa, the same gene expression analysis showed increased transcription of multiple target genes (Fig. 3H). This included peptide hormones (CCK, secretin, and gastrin) and metabolically relevant transcription factors [farnesoid X receptor (FXR), peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α (PGC1α), and aryl hydrocarbon receptor (AHR)]. In contrast to the liver, the gluconeogenic enzyme G6PC, the lipogenic enzyme ACLY, and the gluconeogenic/glyceroneogenic enzyme FBP1 were increased. In duodenal mucosa, CS treatment increased transcription of only the peptide transporter PEPT1 (SLC15A1; Fig. 3I) and had no effect using the same panel of metabolically relevant target genes (data not shown).
Fig. 3.

Metabolic changes in ob/ob mice are recapitulated after 24-h treatment with camostat (CS; 69 mg/kg in feed) with tissue transcriptional responses. Reduced food intake (A), decreased body weight gain (B), decreased fed blood glucose (C), and increased fecal protein (D) compared with vehicle-treated. P value by Student’s t-test, n = 8/group. In a separate repeat experiment (n = 6/group), tissue was obtained for transcriptional level of metabolic genes. E: liver had relatively reduced gluconeogenic mRNA [glucose-6-phosphatase catalytic subunit (G6PC)] and increased fibroblast growth factor 21 (FGF21) mRNA. F: pancreas had increased expression of trypsinogens [protease, serine 1 and 3 (PRSS1, PRSS3)], serine peptidase inhibitor Kazal type (SPINK), and FGF21 mRNA. G: plasma FGF21 levels were increased by CS. H: ileum mucosa had increased transcription of multiple metabolic target genes. I: duodenal mucosa transcriptional responses were unaltered using the same panel as for ileum, except for a small increase in peptide transporter 1 (SLC15A1). P < 0.05 by unpaired Student’s t-test. ACLY, ATP citrate lyase; PC, phosphocreatine; PCK1, phosphoenolpyruvate carboxykinase-1; SCT, secretin; GIP, gastric inhibitory polypeptide; SST, somatostatin; GAST, gastrin; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1α; LCT, long-chain triglyceride; ACLY, ATP citrate lyase; ISG15, interferon-stimulated gene 15; AHR, aryl hydrocarbon receptor; FXR, farsenoid X receptor.
To determine whether these changes were specific to the leptin deficiency of ob/ob mice, the effect of 24-h CS treatment was assessed in lean C57BL/6 mice compared with vehicle (Fig. 4). In lean mice, acute CS treatment reduced food intake (Fig. 4A), body weight gain (Fig. 4B), and liver weight (vehicle = 5.2 ± 0.12 g, CS = 4.2 ± 0.14 g; n = 6/gp; P < 0.001 by Student’s t-test) but did not change fed blood glucose (vehicle = 105 ± 19; CS = 113 ± 4 mg/dl; n = 6). In liver, the gluconeogenesis gene G6PC was not altered, but lipogenesis gene ACLY was decreased (Fig. 4C). In this tissue, the induction of genes associated with the ISR response were assessed. The ISR includes environmental stress-specific kinases, e.g., general control nonderepressible 2 (GCN2), and phosphorylation of eIF2α. Phosphorylation of eIF2α increases activation transcription factor 4 (ATF4). In these lean mice, CS increased several liver ISR target genes: muscle, intestine, and stomach expression 1 (MIST1), asparagine synthetase (ASNS), and FGF21 (Fig. 4C). Plasma FGF21 levels were below the LLOQ but was increased and quantifiable in all CS-treated mice (Fig. 4D). There was no difference in plasma valine, isoleucine, and leucine (Fig. 4E).
Fig. 4.

In lean C57BL/6 mice, 24-h camostat (CS) treatment (69 mg/kg) compared with vehicle reduced food intake (A), decreased body weight (B), and increased liver fibroblast growth factor 21 (FGF21) mRNA fivefold (C) with increased integrated stress response (ISR) target gene transcription, such as asparagine synthetase (ASNS). ATP citrate lyase (ACLY) was the only gluconeogenic gene decreased. D: plasma FGF21 was detected in all CS-treated mice, whereas FGF21 was beneath quantification threshold of 98.8 pg/ml in vehicle-treated (shown as ½ lower limit of quantification). E: plasma branched-chain amino acids (BCAA) valine, isoleucine, and leucine were unchanged by CS in lean mice. Bar indicates P < 0.05 by unpaired Student’s t-test, n = 6/group. MIST1, muscle, intestine, and stomach expression 1; ATF4, activation transcription factor 4; GADD45, growth arrest and DNA damage inducible-45α; G6PC, glucose-6-phosphatase catalytic subunit; NUPR1, nuclear protein-1.
CS is rapidly absorbed (11) and converted to its active metabolite FOY-251 in plasma (3, 41). When given orally, FOY-251 had low bioavailability (F < 2% in mice). Thus, FOY-251 provided a tool to restrict serine protease inhibition primarily to the gut. In ob/ob mice, acute 24-h FOY-251 administered in feed (0.8 mg/g feed; 46 mg·kg−1·day−1) reduced food intake (Fig. 5A) and body weight (Fig. 5B). FOY-251 did not increase pica behavior shown by kaolin consumption and reflective of taste aversion/illness (Fig. 5A). Twenty-four-hour FOY-251 increased fecal protein concentration (Fig. 5C) and decreased fed blood glucose (Fig. 5D). Plasma FGF21 levels were increased twofold (Fig. 5E), and valine, isoleucine, and leucine levels were reduced compared with vehicle-fed ob/ob mice (Fig. 5F).
Fig. 5.

Metabolic changes in ob/ob mice administered gut-restricted camostat (CS) metabolite 4-(4-guanidinobenzoyloxy phenyl) acetic acid (FOY-251) in feed for 24 h compared with vehicle-treated mice. FOY-251 reduced food intake but did not increase kaolin intake (A), decreased body weight (B), increased fecal protein concentration (C), decreased fed blood glucose (D), and increased plasma fibroblast growth factor 21 (FGF21; E) (n = 7–8/group). F: in a separate group of mice given the same treatment, plasma branched-chain amino acids (BCAA) were reduced (n = 6/group). ○, vehicle; ■, FOY-251; Bar indicates P value by unpaired Student’s t-test.
On the basis of these acute data, ob/ob mice were given FOY-251 (0.8 mg/g in chow) for 7 days. FOY-251 reduced food intake (Fig. 6A) and body weight gain (Fig. 6B) compared with vehicle and to a similar extent as the pair-fed group. Fed blood glucose levels in FOY-251-treated mice were decreased compared with vehicle and pair-fed mice (Fig. 6C), with a trend toward lower insulin levels (Fig. 6D). FOY-251 treatment decreased liver weight (Fig. 6E) and increased plasma FGF21 levels (Fig. 6F) compared with both vehicle and pair-fed groups. However, both FOY-251 and pair-fed groups had lower plasma BCAA levels than vehicle-treated mice (Fig. 6G). The levels of BCAAs were similar to those detected in lean mice (Fig. 5E).
Fig. 6.

Metabolic effects in ob/ob mice after camostat (CS) metabolite (CSM) 4-(4-guanidinobenzoyloxy phenyl) acetic acid (FOY-251) in feed for 7 days compared with vehicle or pair-fed groups (n = 5–6/group). A: food intake was reduced in FOY-251-treated mice; pair-fed mice were matched to this. B: pair-fed and FOY-251 treatment reduced %body weight (BW) to the same extent. C: FOY-251 decreased terminal fed blood glucose (C) but terminal plasma insulin levels were not different between groups (D). E: FOY-251 treatment reduced liver weight compared with pair-fed mice. F: plasma fibroblast growth factor 21 (FGF21) levels were increased by FOY-251 compared with vehicle and pair-fed groups. G: plasma branched-chain amino acids (BCAA) were decreased by both FOY-251 and pair feeding relative to vehicle. ○, Vehicle; ■, FOY-251; □, Pair-Fed. Bar indicates P value; *P < 0.05 vs. pair-fed and FOY-251-treated groups, by one-way ANOVA and Tukey’s post hoc test, n = 6/group.
Alterations in target genes were assessed to identify mechanisms related to nutrient signaling in liver, pancreas, and ileum, white adipose tissue (WAT), and hindbrain dorsal vagal complex (DVC)/dorsomedial medulla (Fig. 7). In liver, FOY-251 only decreased ACLY transcription (Fig. 7A). In pancreas FOY-21 only tended to increase FGF21 mRNA, while INS1 and glucagon mRNA were transcriptionally suppressed (Fig. 7B). In liver, FOY-251 induced a 15-fold increase in FGF21, along with increases in all key ISR target genes [MIST1, activation transcription factor 4 (ATF4), nuclear protein-1 (NUPR1), growth arrest and DNA damage inducible 45α (GADD45a), and ASNS; Fig. 7C]. In ileal mucosa, transcriptional effects were variable (Fig. 7D). In WAT, uncoupling protein-1 (UCP1) was not altered (Fig. 8D). In the DVC, vagal signaling peptide receptor mRNA was not altered by FOY-251 treatment (Fig. 7E).
Fig. 7.

Transcriptional changes in various metabolic tissues in ob/ob mice from the same experiment as Fig. 6. A: liver ATP citrate lyase (ACLY) was decreased by camostat (CS) metabolite 4-(4-guanidinobenzoyloxy phenyl) acetic acid (FOY-251), whereas glucose-6-phosphatase catalytic subunit (G6PC) and phosphoenolpyruvate carboxykinase-1 (PCK1) were increased in the pair-fed group. B: in pancreas, fibroblast growth factor 21 (FGF21) mRNA was not significantly increased, whereas insulin (INS1) and glucagon (GCG) were strongly suppressed by FOY-251 treatment. C: liver FGF21 transcription was increased 15-fold, accompanied by induction of all integrated stress response target genes in FOY-251-treated mice. D: ileum mucosa of hormones (gastrin was removed because of widely variable fold increases due to very low control levels), gluconeogenic enzymes, and transcription factors and UCP1 in white adipose tissue (WAT) were not altered. F: in dorsal vagal complex (DVC), satiety hormone and vagal signaling peptide receptor mRNA was not altered by FOY-251 treatment, whereas G protein-coupled receptor 65 (GPR65) was decreased in the pair-fed group. Bar indicates P < 0.05 by unpaired Student’s t-test. PC, phosphocreatine; MIST1, muscle, intestine, and stomach expression 1; ATF4, activation transcription factor 4; NUPR1, nuclear protein-1; GADD45α, growth arrest and DNA damage inducible-α; ASNS, asparagine synthetase; SCT, ; SST, ; GIP, gastric inhibitory polypeptide; FXR, farsenoid X receptor; UCP1, uncoupling protein-1; NYP2R, neuropeptide Y2 receptor; CNR1, cannabinoid receptor 1; HCRTR1, hypocretin receptor 1; CCKRR, cholecystokinin A receptor; CCKRB, cholecystokinin receptor B; PAR2, protease-activated receptor 2; GLP1R, glucagon-like peptide-1 receptor.
Fig. 8.

Metabolic changes in diet-induced obese mice after camostat (CS) metabolite 4-(4-guanidinobenzoyloxy phenyl) acetic acid (FOY-251) given by oral gavage (100 mg/kg) for 4 days (A–F, n = 8/group) and in response to oral glucose challenge after a single dose (G–I, n = 10/group). In FOY-251-treated DIO mice, there was decreased food intake (A) and body weight gain (B) with increased fecal protein (C) compared with vehicle-treated mice. Plasma samples from bleeds taken 15 and 60 min after the last dose of FOY-251 had modestly reduced insulin at 60 min time point (D) and no change in total GLP1, glucagon-like peptide-1 (tGLP1; E) but markedly increased fibroblast growth factor 21 (FGF21) levels (F) at 15 and 60 min. Acute single dose of FOY-251 given before oral glucose challenge had (G) no effect on peak glucose levels (H) but modestly reduced the area under the curve (AUC) of the glucose excursion with increased plasma insulin levels (I) at 15 min compared with baseline.
We also assessed the effect of intestinal trypsin inhibition in a nutritional model of obesity and diabetes, which may be more relevant to overnutrition-related obesity and comorbidities in humans. In DIO mice, FOY-251 (100 mg/kg po twice a day for 4 days) decreased food intake and body weight and increased fecal protein compared with vehicle-treated mice (Fig. 8,A–C). After the last dose of FOY-251, two blood samples (taken at 15 and 60 min postdosing) had modestly reduced insulin (Fig. 8D; at 60-min time point) without a change in basal GLP1 (Fig. 8E), whereas FGF21 levels were increased (Fig. 8F). To assess glycemic control, a different group of DIO mice were given a single oral dose of FOY-251 before an oral glucose challenge. This resulted in no difference in peak blood glucose levels (Fig. 8G) but a modest reduction in the AUC of the glucose excursion (Fig. 8H) and an increase over baseline of plasma insulin levels at 15 min (Fig. 8I).
DISCUSSION
In ob/ob mice, 1 wk of CS treatment reduced food intake and body weight in a dose-related fashion. The highest dose also improved hyperglycemia and liver pathology similarly to that reported in OLETF rats fed CS (18, 19). This was despite the fact that the highest dose reduced food consumption resulting in approximately one-half the dose of CS compared with the OLETF rat studies (18, 19). Reduced food intake alone could not account for these improvements, because they were not observed in ob/ob mice matched for food intake to the CS-treated group, and food intake was not reduced in OLETF rats (19). The latter is presumably because CCK1R deficiency abrogated CS-evoked inhibition of food intake, where CS activates this satiety pathway in Sprague-Dawley rats (24, 47, 52). Furthermore, an enteropeptidase inhibitor (OBE-2008), while having no effect on food intake, reduced weight gain in HFD-fed mice (7). Thus, different obese models (OLETF rat and HFD-fed and ob/ob mice) using different trypsin inhibitors (despite differences in selectivity) provide evidence for non-satiety-mediated effects.
A novel finding of our study is that gut-restricted serine protease inhibition is associated with an ISR target gene induction and liver FGF21 transcription with increased FGF21 plasma levels. The key supporting data are that 1) increased liver FGF21 transcription and secretion noted acutely in both ob/ob and lean mice, suggesting that it is not dependent on leptin-deficient regulatory mechanisms; 2) in lean mice, CS increased FGF21 mRNA as well as ISR target genes; 3) FOY-251 replicated the metabolic and transcriptional effects of CS acutely, with low systemic exposure; and 4) increased FGF21 transcription (15-fold) and plasma (10-fold) were maintained with induction of liver ISR transcription target genes up to 1 wk of treatment. Chronic FOY-251 dosed to DIO mice also increased plasma FGF21 levels. The discussion will focus on these findings with respect to the known literature on the metabolic benefits and nutrient stress pathways involving FGF21.
The ISR is a signaling system in response to environmental stress-specific kinases associated with eIF2α phosphorylation to help restore cellular homeostasis. Ablating the regulatory control of the eIF2α phosphatase complex results in constitutive phosphorylation in the liver, with activation of ATF4 transcription (54). While genes induced by ATF4 include NUPR1 (20) and ASNS (32), a majority of studies illustrate that ATF4 induction leads to increased FGF21 production, which is associated with improved insulin sensitivity and protection from HFD-induced obesity and hepatic steatosis (54). FOY-251 administration markedly induced all liver ISR transcription target genes and increased liver FGF21 transcription and plasma FGF21 levels for at least 1 wk. In other insulin target tissues, such as skeletal muscle, mitochondrial stress also contributes to FGF21 induction through eIF2α-ATF4 (25). In the case of starvation, circulating FGF21 levels can be increased directly through PPARα in the liver (14). However, amino acid deprivation alone is sufficient to induce ATF4 and FGF21 in liver (9, 32), and conversely, high-protein diets correlate negatively with plasma FGF21 levels (10). Reduction of specific amino acids increases FGF21 transcription in vitro, and liver FGF21 mRNA and plasma FGF21 levels are increased in vivo (30, 44). Repletion of BCAAs to attempt to offset dietary protein restriction attenuated the improvements in metabolism (29) but did not reverse the increased levels of serum FGF21 protein and liver FGF21 mRNA (29). In our studies, CS- or FOY-251-treated ob/ob mice had reduced systemic plasma BCAAs, which was also noted in pair-fed mice, but never below the level relative to lean mice. Liver substrate utilization changes in response to altered amino acid uptake from the gut could result in circulating amino acid levels that may not be reflective of hepatic portal vein levels. However, BCAAs are primarily catabolized in muscle, not the liver. Additional studies are required to differentiate between BCAA reduction due to reduced food intake or available amino acids due to trypsin inhibition.
It is possible that a pathway other than liver ISR induction is responsible for plasma FGF21 increase, such as through glucose/insulin/glucagon signaling. For example, glucagon and insulin interact cooperatively to induce hepatocyte FGF21 gene expression in culture through direct binding of ATF4 to FGF21 promoter (2). It has been suggested that postprandial glucose is the signal to the liver that increases FGF21 in rodents (51); however, postprandial insulin (not glucose) levels directly increase FGF21 levels in humans (43). In our studies, both fasted and fed blood glucose levels were decreased by FOY-251 and CS compared with pair feeding. There was no change in insulin levels in treated mice. In the pancreas of FOY-521-treated mice, INS1 and glucagon transcription were decreased compared with pair-fed controls. Together these data suggest that increased systemic FGF21 levels may be insulin and glucagon independent.
We performed focused transcriptional analysis in various tissues to identify potential additional pathways induced or repressed in response to trypsin inhibition. CS increased pancreatic zymogen and SPINK transcriptional activity. This is consistent with the known actions of CS to increase pancreatic trypsinogen and chymotrypsinogen (26, 35). Pancreatic weight increased and acinar cell hypertrophy was noted in ob/ob mice, which is consistent with a trypsin inhibition-mediated CCK signaling that has been reported in rats (18) and mice (36). Pharmacological FGF21 is associated with increased energy expenditure, but FOY-251 did not enhance UCP1 transcription in WAT. It should be noted that there is controversy in rodents and primate species as to the role of mitochondrial stress-related FGF21 induction in metabolic homeostasis improvements related to energy expenditure. FOY-251 did not alter hindbrain DVC expression of receptors involved in gut-brain feedback such as CCK1R, GLP1R, and others. However, transcriptional changes in the intestine were suggestive of hormonal and gluconeogenic induction after CS treatment. In the ileum, but not in proximal duodenum, induction of genes associated with gluconeogenesis were noted, which has been proposed to improve metabolic health (45). Interestingly, there were opposite effects in the liver versus the ileum mucosa on expression of genes involved in gluconeogenesis (G6PC) and lipogenesis (ACLY). Whereas CS treatment decreased G6PC and ACLY gene expression in liver, they were both increased in the ileum, as was FBP1, all of which play roles in both glyceroneogenesis and gluconeogenesis. Thus, inhibition of gut serine proteases appears to be sufficient to shift glucose and lipid anabolism away from the liver and toward the ileum mucosa. A potential explanation for this is that protease inhibition in the gut leads to a shift of amino acid precursor availability that favors both gluconeogenesis and lipogenesis in the ileum mucosa over the liver. Tracer, diet, and histological studies would be needed to better understand the pathways and cell types involved.
Overall, a working model of the potential metabolic benefits of reducing protein digestion through intestinal protease inhibition is summarized (Fig. 9). In addition to the known effects of trypsin inhibition to increase CCK release and satiety-mediated pathways to reduce food intake, partially hydrolyzed protein entering the small intestine may signal through non-satiety-mediated pathways. This protein would not be digested further to peptones and constituent amino acids when trypsin and other serine proteases are inhibited. Thus, despite normal protein ingestion, the body could perceive this environment as a state of protein dilution. Lipogenesis (ACLY) and possibly gluconeogenesis (G6PC) are increased in distal small intestine; consequently, phosphoenolpyruvate carboxykinase-1 (PCK1) and G6PC are reduced in the liver, which contributes to improved glycemia. This speculation could be tested by determining protein expression and activity of liver PCK1 and G6PC. Furthermore, a pyruvate tolerance test would determine if there is a decrease in hepatic gluconeogenesis.
Fig. 9.
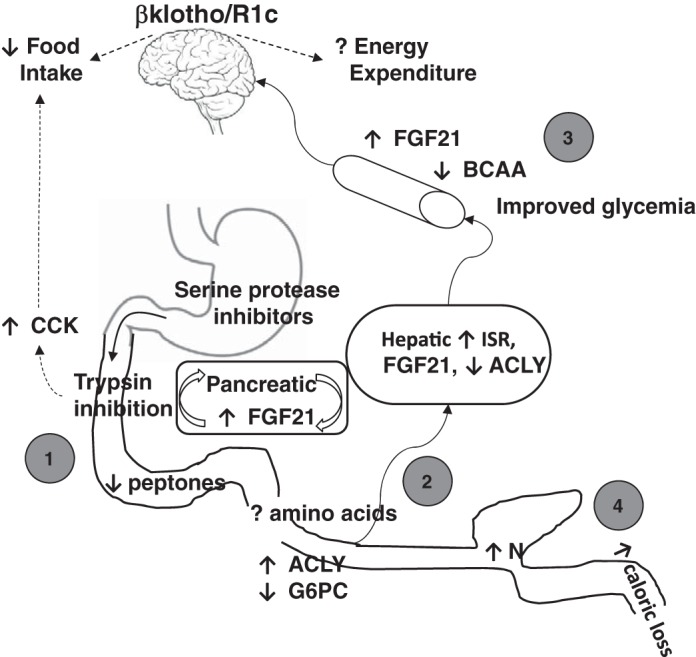
Working model of the potential metabolic benefits of reducing protein digestion through a trypsin-fibroblast growth factor 21 (FGF21) axis. First, partially hydrolyzed protein in the presence of trypsin inhibition reduces formation of peptones, mimicking a state of intestinal protein dilution. This adds to trypsin inhibitor-evoked cholecystokinin (CCK) secretion to reduce food intake. Second, apparent protein dilution is detected by the liver, decreasing lipogenesis [(ACLY)] and substrate utilization and inducing integrated stress response (ISR) target genes to increase FGF21. In the pancreas, FGF21 mRNA may be locally induced in an insulin- and glucagon-independent fashion with no change in circulating insulin levels. Overall, branched-chain amino acid (BCAA) levels in obese mice are reduced to the same extent as in pair-fed and lean mice. These mechanisms could all contribute to reduced liver weight, liver lipidosis, and hyperglycemia. Third, circulating FGF21 primarily acts on central nervous system βklothoR1c to reduce food intake and would also be expected to increase energy expenditure, warranting further evaluation in future studies. Fourth, in large intestine, increased protein may alter bacterial nitrogen utilization and nutrient signaling, especially bioactives related to putrefaction. ACLY, ATP citrate lyase; G6PC, glucose-6-phosphatase catalytic subunit.
The potentially reduced amino acids detected by the ileum and liver would be expected to result in reduced lipogenesis and induction of liver ISR target genes and FGF21 mRNA. In the pancreas, FGF21 mRNA may be locally induced in an insulin- and glucagon-independent fashion, with no change in circulating insulin levels. Overall, BCAAs in obese mice were reduced to the same extent as calorie restriction (pair feeding), which matched the BCAA levels detected in lean mice. These mechanisms could all contribute to reducing liver weight, liver lipidosis, and hyperglycemia. To demonstrate causality of a trypsin inhibition-evoked ISR response and increased FGF21 for these metabolic improvements, further studies are necessary. Proposed studies include demonstration of FGF21 dependence by using FGF21−/− mice similarly to those performed using a low-protein diet (13). Alternatively, trypsin inhibition in an amino acid-rich (Ensure) diet should not result in a liver ISR response. For example a CCK-mediated pancreatic response to amino acid intake was absent (compared with protein) during intraduodenal CS in humans (1). Finally, the site of action of circulating FGF21 is thought to primarily be on central nervous system (CNS) βklothoR1c (6, 42). Activation of CNS βklothoR1c results in metabolic effects that can include reduced food intake and increased energy expenditure via sympathetic nerve changes. Experiments utilizing CNS-specific βklotho knockout mice would be important to establish whether the trypsin-liver-brain pathway is sufficient for metabolic changes.
Protein levels increased modestly (<2-fold) in feces, and this caloric excretion is thought to be part of the long-term weight loss mechanism of serine protease inhibition (7, 19). The increased entry of partially digested protein into the large intestine may alter bacterial nitrogen utilization and nutrient signaling, especially bioactives related to putrefaction. Studies to assess microbially derived metabolites were beyond the scope of this work but would help elucidate cause and effect. For example, agmatine (a decarboxylation product of arginine) fed chronically to rats alters liver transcription and decreases caloric intake (38).
Perspective.
Obesity is a growing health and economic global burden with few pharmacological treatment options. Adding to the burden of obesity is that it can increase the risk of developing other serious comorbidities. Therefore, potential therapies need to be efficacious for weight loss and reduce the incidence of comorbidities as well as be associated with minimal adverse effects. The Food and Drug Administration-approved weight-loss medication orlistat acts by inhibiting the intestinal digestive enzyme lipase. It has low systemic exposure after oral administration. However, orlistat can be associated with a fecal continence problem or intermittent loss of oil in obese subjects. It also decreases high-density lipoprotein cholesterol (22), which may be an unfavorable cardiometabolic risk profile. An alternative approach would be to inhibit trypsin and related serine protease inhibitors such as enteropeptidase. However, efforts to optimize compounds to reduce weight by this mechanism are hampered by lack of understanding of mechanisms underlying efficacy. The present work is important because it identified a novel mechanism, ISR gene induction and FGF21 induction in response to serine protease inhibition. At present, it is not clear whether the ISR contributes to disease pathology or whether it is part of a defense mechanism against metabolic stress. However, FGF21 analogs have been pursued for their therapeutic metabolic benefits (48, 53). Trypsin inhibition could be a way to enhance endogenous FGF21, resulting in beneficial effects. However, caution is warranted, since, despite reports demonstrating that adaptive starvation (15) or protein restriction (13) induce FGF21 in mice, in the context of dietary interventions the regulation of FGF21 may vary between mouse and human (8, 28).
DISCLOSURES
All coauthors were employed by Janssen Pharmaceutical R&D during these studies.
AUTHOR CONTRIBUTIONS
K.A., M.J., J.M.L., and P.J.H. conceived and designed research; K.A., M.J., C.R.C., W.L., B.S., and P.J.H. performed experiments; K.A., M.J., C.R.C., W.L., B.S., and P.J.H. analyzed data; K.A., M.J., B.S., J.C.L., J.M.L., and P.J.H. interpreted results of experiments; K.A., M.J., C.R.C., and P.J.H. prepared figures; K.A., M.J., and P.J.H. drafted manuscript; K.A., J.M.L., and P.J.H. edited and revised manuscript; K.A. and P.J.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We express our appreciation to Corey Bender (Kelly Scientific) for assay analysis and to Sachin Lohani (Janssen Pharmaceutical R&D) for formulating CS and FOY-251 in feed and providing stability measurements. We also thank Shobha Seetharam and David Gutstein (CVM, Janssen Pharmaceutical R&D) for discussions on translation of these findings to humans, and to James N. Leonard (CVM, Janssen Pharmaceutical R&D) for review of the manuscript.
REFERENCES
- 1.Adler G, Müllenhoff A, Bozkurt T, Göke B, Koop I, Arnold R. Comparison of the effect of single and repeated administrations of a protease inhibitor (Camostate) on pancreatic secretion in man. Scand J Gastroenterol 23: 158–162, 1988. doi: 10.3109/00365528809103961. [DOI] [PubMed] [Google Scholar]
- 2.Alonge KM, Meares GP, Hillgartner FB. Glucagon and insulin cooperatively stimulate fibroblast growth factor 21 gene transcription by increasing the expression of activating transcription factor 4. J Biol Chem 292: 5239–5252, 2017. doi: 10.1074/jbc.M116.762922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beckh K, Göke B, Müller R, Arnold R. Elimination of the low-molecular weight proteinase inhibitor camostate (FOY 305) and its degradation products by the rat liver. Res Exp Med (Berl) 187: 401–406, 1987. doi: 10.1007/BF01852177. [DOI] [PubMed] [Google Scholar]
- 4.Bi S, Chen J, Behles RR, Hyun J, Kopin AS, Moran TH. Differential body weight and feeding responses to high-fat diets in rats and mice lacking cholecystokinin 1 receptors. Am J Physiol Regul Integr Comp Physiol 293: R55–R63, 2007. doi: 10.1152/ajpregu.00002.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bi S, Moran TH. Obesity in the Otsuka Long Evans Tokushima fatty rat: mechanisms and discoveries. Front Nutr 3: 21, 2016. doi: 10.3389/fnut.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, Ding X, Elmquist JK, Takahashi JS, Mangelsdorf DJ, Kliewer SA. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med 19: 1147–1152, 2013. doi: 10.1038/nm.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braud S, Ciufolini MA, Harosh I. Enteropeptidase: a gene associated with a starvation human phenotype and a novel target for obesity treatment. PLoS One 7: e49612, 2012. doi: 10.1371/journal.pone.0049612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christodoulides C, Dyson P, Sprecher D, Tsintzas K, Karpe F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J Clin Endocrinol Metab 94: 3594–3601, 2009. doi: 10.1210/jc.2009-0111. [DOI] [PubMed] [Google Scholar]
- 9.De Sousa-Coelho AL, Marrero PF, Haro D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem J 443: 165–171, 2012. doi: 10.1042/BJ20111748. [DOI] [PubMed] [Google Scholar]
- 10.Garcia Caraballo SC, Comhair TM, Dejong CHC, Lamers WH, Koehler SE. Dietary treatment of fatty liver: High dietary protein content has an antisteatotic and antiobesogenic effect in mice. Biochim Biophys Acta Mol Basis Dis 1863: 1789–1804, 2017. doi: 10.1016/j.bbadis.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 11.Göke B, Stöckmann F, Müller R, Lankisch PG, Creutzfeldt W. Effect of a specific serine protease inhibitor on the rat pancreas: systemic administration of camostate and exocrine pancreatic secretion. Digestion 30: 171–178, 1984. doi: 10.1159/000199102. [DOI] [PubMed] [Google Scholar]
- 12.Gumbmann MR, Dugan GM, Spangler WL, Baker EC, Rackis JJ. Pancreatic response in rats and mice to trypsin inhibitors from soy and potato after short- and long-term dietary exposure. J Nutr 119: 1598–1609, 1989. doi: 10.1093/jn/119.11.1598. [DOI] [PubMed] [Google Scholar]
- 13.Hill CM, Laeger T, Albarado DC, McDougal DH, Berthoud HR, Münzberg H, Morrison CD. Low protein-induced increases in FGF21 drive UCP1-dependent metabolic but not thermoregulatory endpoints. Sci Rep 7: 8209, 2017. doi: 10.1038/s41598-017-07498-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab 5: 415–425, 2007. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Inagaki T, Lin VY, Goetz R, Mohammadi M, Mangelsdorf DJ, Kliewer SA. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab 8: 77–83, 2008. doi: 10.1016/j.cmet.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito T, Otsuki M, Itoi T, Shimosegawa T, Funakoshi A, Shiratori K, Naruse S, Kuroda Y; Research Committee of Intractable Diseases of the Pancreas . Pancreatic diabetes in a follow-up survey of chronic pancreatitis in Japan. J Gastroenterol 42: 291–297, 2007. doi: 10.1007/s00535-006-1996-6. [DOI] [PubMed] [Google Scholar]
- 17.Jennis M, Albarazanji K, Seetharam S, Lanter JC, Cavanaugh CR, Lenhard J, Hornby PJ. Intestinal serine protease inhibition increases liver FGF21 secretion in diabetic obese mice. Gastroenterology 154: S218, 2018. doi: 10.1016/S0016-5085(18)31119-3. [DOI] [Google Scholar]
- 18.Jia D, Taguchi M, Otsuki M. Preventive and therapeutic effects of the protease inhibitor camostat on pancreatic fibrosis and atrophy in CCK-1 receptor-deficient rats. Pancreas 30: 54–61, 2005. [PubMed] [Google Scholar]
- 19.Jia D, Taguchi M, Otsuki M. Synthetic protease inhibitor camostat prevents and reverses dyslipidemia, insulin secretory defects, and histological abnormalities of the pancreas in genetically obese and diabetic rats. Metabolism 54: 619–627, 2005. doi: 10.1016/j.metabol.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Jin HO, Seo SK, Woo SH, Choe TB, Hong SI, Kim JI, Park IC. Nuclear protein 1 induced by ATF4 in response to various stressors acts as a positive regulator on the transcriptional activation of ATF4. IUBMB Life 61: 1153–1158, 2009. doi: 10.1002/iub.271. [DOI] [PubMed] [Google Scholar]
- 21.Kharitonenkov A, DiMarchi R. Fibroblast growth factor 21 night watch: advances and uncertainties in the field. J Intern Med 281: 233–246, 2017. doi: 10.1111/joim.12580. [DOI] [PubMed] [Google Scholar]
- 22.Khera R, Pandey A, Chandar AK, Murad MH, Prokop LJ, Neeland IJ, Berry JD, Camilleri M, Singh S. Effects of weight-loss medications on cardiometabolic risk profiles: a systematic review and network meta-analysis. Gastroenterology 154: 1309–1319.e7, 2018. doi: 10.1053/j.gastro.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, Münzberg H, Hutson SM, Gettys TW, Schwartz MW, Morrison CD. FGF21 is an endocrine signal of protein restriction. J Clin Invest 124: 3913–3922, 2014. doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lateef DM, Washington MC, Raboin SJ, Roberson AE, Mansour MM, Williams CS, Sayegh AI. Duodenal myotomy blocks reduction of meal size and prolongation of intermeal interval by cholecystokinin. Physiol Behav 105: 829–834, 2012. doi: 10.1016/j.physbeh.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Lee MS. Effect of mitochondrial stress on systemic metabolism. Ann NY Acad Sci 1350: 61–65, 2015. doi: 10.1111/nyas.12822. [DOI] [PubMed] [Google Scholar]
- 26.Liddle RA. Regulation of cholecystokinin secretion by intraluminal releasing factors. Am J Physiol Gastrointestinal Liver Physiol 269: G319–G327, 1995. doi: 10.1152/ajpgi.1995.269.3.G319. [DOI] [PubMed] [Google Scholar]
- 27.Liou AP, Chavez DI, Espero E, Hao S, Wank SA, Raybould HE. Protein hydrolysate-induced cholecystokinin secretion from enteroendocrine cells is indirectly mediated by the intestinal oligopeptide transporter PepT1. Am J Physiol Gastrointest Liver Physiol 300: G895–G902, 2011. doi: 10.1152/ajpgi.00521.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundsgaard AM, Fritzen AM, Sjøberg KA, Myrmel LS, Madsen L, Wojtaszewski JFP, Richter EA, Kiens B. Circulating FGF21 in humans is potently induced by short term overfeeding of carbohydrates. Mol Metab 6: 22–29, 2017. doi: 10.1016/j.molmet.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maida A, Chan JSK, Sjøberg KA, Zota A, Schmoll D, Kiens B, Herzig S, Rose AJ. Repletion of branched chain amino acids reverses mTORC1 signaling but not improved metabolism during dietary protein dilution. Mol Metab 6: 873–881, 2017. doi: 10.1016/j.molmet.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maida A, Zota A, Sjøberg KA, Schumacher J, Sijmonsma TP, Pfenninger A, Christensen MM, Gantert T, Fuhrmeister J, Rothermel U, Schmoll D, Heikenwälder M, Iovanna JL, Stemmer K, Kiens B, Herzig S, Rose AJ. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. J Clin Invest 126: 3263–3278, 2016. doi: 10.1172/JCI85946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maida A, Zota A, Vegiopoulos A, Appak-Baskoy S, Augustin HG, Heikenwalder M, Herzig S, Rose AJ. Dietary protein dilution limits dyslipidemia in obesity through FGF21-driven fatty acid clearance. J Nutr Biochem 57: 189–196, 2018. doi: 10.1016/j.jnutbio.2018.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Mazor KM, Stipanuk MH. GCN2- and eIF2α-phosphorylation-independent, but ATF4-dependent, induction of CARE-containing genes in methionine-deficient cells. Amino Acids 48: 2831–2842, 2016. doi: 10.1007/s00726-016-2318-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaughlin CL, Peikin SR, Baile CA. Trypsin inhibitor effects on food intake and weight gain in Zucker rats. Physiol Behav 31: 487–491, 1983. doi: 10.1016/0031-9384(83)90071-9. [DOI] [PubMed] [Google Scholar]
- 34.Miyasaka K, Masuda M, Funakoshi A. Regulation of cholecystokinin release and transcription in a rat without gene expression of cholecystokinin-A receptor. Digestion 58: 104–110, 1997. doi: 10.1159/000201431. [DOI] [PubMed] [Google Scholar]
- 35.Müller MK, Goebell H, Alfen R, Ehlers J, Jäger M, Plümpe H. Effects of camostat, a synthetic protease inhibitor, on endocrine and exocrine pancreas of the rat. J Nutr 118: 645–650, 1988. doi: 10.1093/jn/118.5.645. [DOI] [PubMed] [Google Scholar]
- 36.Niederau C, Liddle RA, Williams JA, Grendell JH. Pancreatic growth: interaction of exogenous cholecystokinin, a protease inhibitor, and a cholecystokinin receptor antagonist in mice. Gut 28, Suppl: 63–69, 1987. doi: 10.1136/gut.28.Suppl.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishi T, Hara H, Tomita F. Soybean beta-conglycinin peptone suppresses food intake and gastric emptying by increasing plasma cholecystokinin levels in rats. J Nutr 133: 352–357, 2003. doi: 10.1093/jn/133.2.352. [DOI] [PubMed] [Google Scholar]
- 38.Nissim I, Horyn O, Daikhin Y, Chen P, Li C, Wehrli SL, Nissim I, Yudkoff M. The molecular and metabolic influence of long term agmatine consumption. J Biol Chem 289: 9710–9729, 2014. doi: 10.1074/jbc.M113.544726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noble JE, Knight AE, Reason AJ, Di Matola A, Bailey MJ. A comparison of protein quantitation assays for biopharmaceutical applications. Mol Biotechnol 37: 99–111, 2007. doi: 10.1007/s12033-007-0038-9. [DOI] [PubMed] [Google Scholar]
- 40.Otsuki M, Fujii M, Nakamura T, Tani S, Okabayashi Y: Chronic oral administration of synthetic trypsin inhibitor camostate reduces amylase release from isolated rat pancreatic acini. Int J Pancreatol 18:135–143, 1995. doi: 10.1007/BF02785887. [DOI] [PubMed] [Google Scholar]
- 41.Otsuki M, Tani S, Okabayashi Y, Fuji M, Nakamura T, Fujisawa T, Itoh H. Beneficial effects of the synthetic trypsin inhibitor camostate in cerulein-induced acute pancreatitis in rats. Dig Dis Sci 35: 242–250, 1990. doi: 10.1007/BF01536770. [DOI] [PubMed] [Google Scholar]
- 42.Owen BM, Bookout AL, Ding X, Lin VY, Atkin SD, Gautron L, Kliewer SA, Mangelsdorf DJ. FGF21 contributes to neuroendocrine control of female reproduction. Nat Med 19: 1153–1156, 2013. doi: 10.1038/nm.3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samms RJ, Lewis JE, Norton L, Stephens FB, Gaffney CJ, Butterfield T, Smith DP, Cheng CC, Perfield JW II, Adams AC, Ebling FJP, Tsintzas K. FGF21 Is an insulin-dependent postprandial hormone in adult humans. J Clin Endocrinol Metab 102: 3806–3813, 2017. doi: 10.1210/jc.2017-01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimizu M, Li J, Maruyama R, Inoue J, Sato R. FGF19 (fibroblast growth factor 19) as a novel target gene for activating transcription factor 4 in response to endoplasmic reticulum stress. Biochem J 450: 221–229, 2013. doi: 10.1042/BJ20121393. [DOI] [PubMed] [Google Scholar]
- 45.Soty M, Penhoat A, Amigo-Correig M, Vinera J, Sardella A, Vullin-Bouilloux F, Zitoun C, Houberdon I, Mithieux G. A gut-brain neural circuit controlled by intestinal gluconeogenesis is crucial in metabolic health. Mol Metab 4: 106–117, 2015. doi: 10.1016/j.molmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Struthers BJ, MacDonald JR. Comparative inhibition of trypsins from several species by soybean trypsin inhibitors. J Nutr 113: 800–804, 1983. doi: 10.1093/jn/113.4.800. [DOI] [PubMed] [Google Scholar]
- 47.Sullivan CN, Raboin SJ, Gulley S, Sinzobahamvya NT, Green GM, Reeve JR Jr, Sayegh AI. Endogenous cholecystokinin reduces food intake and increases Fos-like immunoreactivity in the dorsal vagal complex but not in the myenteric plexus by CCK1 receptor in the adult rat. Am J Physiol Regul Integr Comp Physiol 292: R1071–R1080, 2007. doi: 10.1152/ajpregu.00490.2006. [DOI] [PubMed] [Google Scholar]
- 48.Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, Weng Y, Clark R, Lanba A, Owen BM, Brenner MB, Trimmer JK, Gropp KE, Chabot JR, Erion DM, Rolph TP, Goodwin B, Calle RA. A long-acting FGF21 molecule, PF-05231023, decreases body weight and improves lipid profile in non-human primates and type 2 diabetic subjects. Cell Metab 23: 427–440, 2016. doi: 10.1016/j.cmet.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Tamura Y, Hirado M, Okamura K, Minato Y, Fujii S. Synthetic inhibitors of trypsin, plasmin, kallikrein, thrombin, C1r-, and C1 esterase. Biochim Biophys Acta 484: 417–422, 1977. doi: 10.1016/0005-2744(77)90097-3. [DOI] [PubMed] [Google Scholar]
- 50.Thimister PW, Hopman WP, Sloots CE, Rosenbusch G, Willems HL, Trijbels FJ, Jansen JB. Role of intraduodenal proteases in plasma cholecystokinin and pancreaticobiliary responses to protein and amino acids. Gastroenterology 110: 567–575, 1996. doi: 10.1053/gast.1996.v110.pm8566605. [DOI] [PubMed] [Google Scholar]
- 51.von Holstein-Rathlou S, BonDurant LD, Peltekian L, Naber MC, Yin TC, Claflin KE, Urizar AI, Madsen AN, Ratner C, Holst B, Karstoft K, Vandenbeuch A, Anderson CB, Cassell MD, Thompson AP, Solomon TP, Rahmouni K, Kinnamon SC, Pieper AA, Gillum MP, Potthoff MJ. FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the liver. Cell Metab 23: 335–343, 2016. doi: 10.1016/j.cmet.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Washington MC, Williams K, Sayegh AI. The feeding responses evoked by endogenous cholecystokinin are regulated by different gastrointestinal sites. Horm Behav 78: 79–85, 2016. doi: 10.1016/j.yhbeh.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 53.Weng Y, Ishino T, Sievers A, Talukdar S, Chabot JR, Tam A, Duan W, Kerns K, Sousa E, He T, Logan A, Lee D, Li D, Zhou Y, Bernardo B, Joyce A, Kavosi M, O’Hara DM, Clark T, Guo J, Giragossian C, Stahl M, Calle RA, Kriz R, Somers W, Lin L. Glyco-engineered long acting FGF21 variant with optimal pharmaceutical and pharmacokinetic properties to enable weekly to twice monthly subcutaneous dosing. Sci Rep 8: 4241, 2018. doi: 10.1038/s41598-018-22456-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu X, Krumm C, So JS, Bare CJ, Holman C, Gromada J, Cohen DE, Lee AH. Preemptive activation of the integrated stress response protects mice from diet-induced obesity and insulin resistance via fibroblast growth factor 21 induction. Hepatology 68: 2167–2181, 2018. doi: 10.1002/hep.30060. [DOI] [PMC free article] [PubMed] [Google Scholar]