

Abstract
BACKGROUND
More than a million Americans harbor a cerebral cavernous angioma (CA), and those who suffer a prior symptomatic hemorrhage have an exceptionally high rebleeding risk. Preclinical studies show that atorvastatin blunts CA lesion development and hemorrhage through inhibiting RhoA kinase (ROCK), suggesting it may confer a therapeutic benefit.
OBJECTIVE
To evaluate whether atorvastatin produces a difference compared to placebo in lesional iron deposition as assessed by quantitative susceptibility mapping (QSM) on magnetic resonance imaging in CAs that have demonstrated a symptomatic hemorrhage in the prior year. Secondary aims shall assess effects on vascular permeability, ROCK activity in peripheral leukocytes, signal effects on clinical outcomes, adverse events, and prespecified subgroups.
METHODS
The phase I/IIa placebo-controlled, double-blinded, single-site clinical trial aims to enroll 80 subjects randomized 1-1 to atorvastatin (starting dose 80 mg PO daily) or placebo. Dosing shall continue for 24-mo or until reaching a safety endpoint.
EXPECTED OUTCOMES
The trial is powered to detect an absolute difference of 20% in the mean percent change in lesional QSM per year (2-tailed, power 0.9, alpha 0.05). A decrease in QSM change would be a signal of potential benefit, and an increase would signal a safety concern with the drug.
DISCUSSION
With firm mechanistic rationale, rigorous preclinical discoveries, and biomarker validations, the trial shall explore a proof of concept effect of a widely used repurposed drug in stabilizing CAs after a symptomatic hemorrhage. This will be the first clinical trial of a drug aimed at altering rebleeding in CA.
Keywords: Cavernous angioma with symptomatic hemorrhage (CASH), Cavernous angioma, Cerebral cavernous malformation (CCM), Symptomatic hemorrhage, Atorvastatin, Clinical trial
ABBREVIATIONS
- CA
cavernous angioma
- DCEQP
dynamic contrast enhanced quantitative perfusion
- MRI
magnetic resonance imaging
- pMLC
phosphorylated myosin light chain
- SH
symptomatic hemorrhage
- WMF
white matter far
GENERAL INFORMATION
Cavernous angiomas (CAs) are neurovascular lesions comprised of clustered dilated capillaries, causing chronic hemorrhage in the central nervous system.1 They occur either in a sporadic form harboring a solitary lesion, or in a familial form (20%-30% of cases) manifesting multifocal lesions.2,3 CAs are detected on magnetic resonance imaging (MRI), often following seizures, focal neurologic deficits, headache, or as an incidental finding.
CAs have a less than 0.5% annual risk of clinically significant hemorrhage, which has led to the consensus that symptomatic management is acceptable in most cases.4 However, once a patient has experienced a symptomatic hemorrhage (SH), there is a 42.4% (CI 26.8-58.0) rate of recurrent bleed or focal neurologic deficit within 5 yr.4–6 Furthermore, brainstem lesions are more likely than other CAs to bleed, rebleed, and cause severe disability.5–8 While brain surgery for excision of CA may benefit some patients, it can be associated with significant cost and morbidity.
The rates of new CA formation (in familial cases), and of a first SH, are too low to develop meaningful primary prevention strategies. However, CAs with recent SH (CASH) are at a higher likelihood for causing significant morbidity and mortality. Therefore, when surgical resection is not undertaken, mostly in deep and brainstem locations, these lesions are the most likely to be followed expectantly per current evidence-based guidelines, with clinical equipoise for testing novel therapies aimed at preventing rebleeding.4 A drug that stabilizes the CASH lesion would be highly desirable for preventing serious sequelae of this disease.
RATIONALE AND BACKGROUND INFORMATION
Preliminary Studies: Mechanistic Rationale, Preclinical Studies, and Biomarker Validations
CCM Gene Loss Results in Endothelial Permeability Through RhoA/ROCK Activation
The loss of any of the 3 CA genes (KRIT1/CCM1, CCM2, and PDCD10/CCM3) in endothelial cells activates the GTPase protein RhoA, resulting in increased actin stress fiber formation and increased permeability.9–12 Recent evidence indicates that CAs arise from endothelial gain of MEKK3-KLF2/4 signaling, resulting in downstream RhoA activation.13 The activity of Rho kinase (ROCK), a RhoA effector, is also increased in these systems, reflected by increased phosphorylated myosin light chain (pMLC), a ROCK target. Therefore, inhibition of ROCK was proposed as a potential therapeutic target countering the aforementioned effects.9–11 Another exciting discovery implicated somatic mutations in the same genes and associated ROCK activity, in excised human sporadic and familial CCM lesions, implicating that ROCK inhibition therapy could target both forms of the disease.14–19
Animal Models Show a Significant Response to Atorvastatin
Preclinical studies supporting the biologic premise were conducted using well validated animal models,18,20,21 recapitulating the human disease and deploying the highest standards of rigor per NINDS guidelines, including randomized concurrent treatment assignment and blinded assessment of lesion burden and bleeding using prespecified adjudicated criteria.22 These experiments investigated the effects of ROCK inhibitor fasudil,23,24 currently not approved for chronic use in humans, and statins,11,24 commonly used as cholesterol lowering drugs, with known pleiotropic effects including inhibition of RhoA prenylation, a critical early step in RhoA/ROCK activation.25,26 In subsequent research in two preclinical CCM mouse models (Ccm1+/−Msh2−/− mice and a more aggressive model Ccm3+/−Trp53−/−), oral atorvastatin at 80 mg/kg/day achieved significant inhibition of lesional development and hemorrhage (Figure 1) (manuscript in preparation). This effect recapitulated previously published benefit with specific ROCK inhibitor fasudil, and was more dramatic than with lower dose/potency simvastatin.24 There were significantly fewer pMLC immunopositive leukocytes within the lesions in atorvastatin-treated mice. These studies reassured us that atorvastatin effectively inhibits ROCK, decreasing iron leak in murine CA lesions, with no hint of increased hemorrhage or attrition.
FIGURE 1.

Representative microCT image of CCM mouse model brains with and without AT treatment. A, Lesion burden in murine models in control animal. B, Lesion burden in murine model mouse brain following atorvastatin treatment. C, High levels of Perls staining (blue) in placebo control animal lesion. D, Decreased Perls staining in lesion in animal treated with atorvastatin.
Equivalency of mouse versus human atorvastatin dose, effecting ROCK inhibition, is discussed in the Supplemental Digital Content.
The Case for Atorvastatin, at Doses Affecting ROCK Inhibition in Humans
A Phase I safety trial with simvastatin at doses up to 40 mg/day (trials.gov# NCT0176445) noted no deleterious effects in open label enrollment including no increase in rates of SH. Unlike simvastatin, atorvastatin is approved for higher doses by the US Food and Drug Administration, and is twice as potent as simvastatin in measurable effects. Atorvastatin doses at 40 to 80 mg/day, but not lower dose/potency simvastatin, achieves robust pleiotropic ROCK inhibition as assayed in human peripheral blood leukocytes.25–27
We also investigated CA subjects who were already taking statin medication (cardiovascular indicated) in an on-going CA biomarker study (n = 24). Among these patients followed for 247 lesion-years, there was only one documented SH (0.4 SH per 100 lesion-years) which is substantially lower than 173 bleeds during 1604 lesion-years of statin naïve cases in the same database (10.7 SH per 100 lesion-years). While this data does not control for treatment assignment or dose effect, it does not raise any concern about hemorrhagic tendency among CA patients receiving statins, nor in comparison to bleed rates previously reported.5
Quantitative Susceptibility Mapping on MRI as a Biomarker of CA Lesional Hemorrhage
Since iron deposition in lesions is modified by ROCK inhibitors and statins in mice, the Awad group recently applied quantitative susceptibility mapping (QSM), a novel technique to estimate iron deposition in CAs by quantifying their local magnetic susceptibility on MRI (Figure 2). We published validations of the technique including precise correlations of QSM with iron concentration in ferric, ferrous, and ferumoxytol phantom solutions, and the exact iron content in excised CA lesions assayed by mass spectroscopy.28 We showed strong interobserver agreement in QSM measurements in human CAs, stability of QSM in clinically stable lesions, reproducibility of QSM across MRI instrument platforms at two hospitals, and higher lesional QSM in older patients and in cases with prior SH.28,29
FIGURE 2.

A, T2-weighted image used to assess changes in size (diameter) and any new bleeding. B, QSM image of the same CA shown with gray-scale map of QSM iron content (ppm). C, DCEQP permeability map of the same lesion with color scale Ki units in ml/100 g/min. Reproduced with permission from Polster et al, Trial Readiness In CAs With Symptomatic Hemorrhage (CASH), Neurosurgery, 2018, [published online ahead of print]36 by the Congress of Neurological Surgeons.
In other pilot work, we showed a correlation of mean lesional QSM in mouse brains imaged ex vivo in the human 3T MRI magnet, with histologic iron densitometry of Perls staining of the same lesions upon subsequent sectioning (Figure S1 in the Supplemental Digital Content). This indicates that differences in QSM may be used to assess the same parameter of treatment effect (lesional iron density) impacted in preclinical studies.
We reported the first prospective assessment of lesional QSM change during 1-yr epochs of longitudinal follow-up of human subjects, in correlation with new clinical events.30 Among cases without recent SH within a year of the first measurement, CA lesions that remained stable during longitudinal follow-up had a tiny average decrease (−3.2%) in mean lesional QSM. CAs with demonstrated growth or SH exhibited a significant and large increase (+44%) in mean lesional QSM. Receiver operating characteristic curve in individual subjects demonstrated a sensitivity of 82.29% and a specificity of 88.89%, highly significant (P = .0004), with a mere +5.81% QSM change. These results link a robust QSM change directly to clinical events, and define a threshold QSM change as a sensitive and specific biomarker event (Figure 3).
FIGURE 3.

QSM changes during person-year epochs in longitudinal follow-up of previously stable CA lesions. A, Lesions manifesting growth or symptomatic bleeding during follow-up exhibited a significant increase in QSM (+ 44%, *P = .01). B, Lesions which remained stable had a slight significant decrease in QSM (-3.2%, **P = .003). In cases with multiple lesions, the unstable and stable lesions were analyzed separately. C, Receiver operating characteristic curve of sensitivity and specificity of QSM change in association with clinical events. Reproduced with permission from Girard R, Fam MD, Zeineddine HA, et al. Vascular permeability and iron deposition biomarkers in longitudinal follow-up of cerebral cavernous malformations. J Neurosurg. Jul 2017;127(1):102-110 (https://thejns.org/).30
QSM Change and Clinical Events in CAs after Recent SH (Trial Candidates)
We recently published further pilot studies assessing lesional QSM change during prospective follow-up of a pilot cohort of CA patients who had suffered a SH within the prior year (meeting the proposed trial criteria).31 During 22 patient-year epochs of follow-up in 16 subjects, the mean lesional QSM change in the index hemorrhagic lesion was +7.93% per patient-year in the whole cohort. Closer examination revealed 10/22 epochs with a documented threshold biomarker event (≥6% QSM increase). This was twice as frequent as clinical events. And no clinical event occurred without such threshold increase in QSM (Figure 4). A primary aim of the proposed trial shall explore whether the change in QSM is altered by statin treatment. A secondary aim shall explore the relationship of QSM change to clinical events in statin and placebo treated subjects.
FIGURE 4.
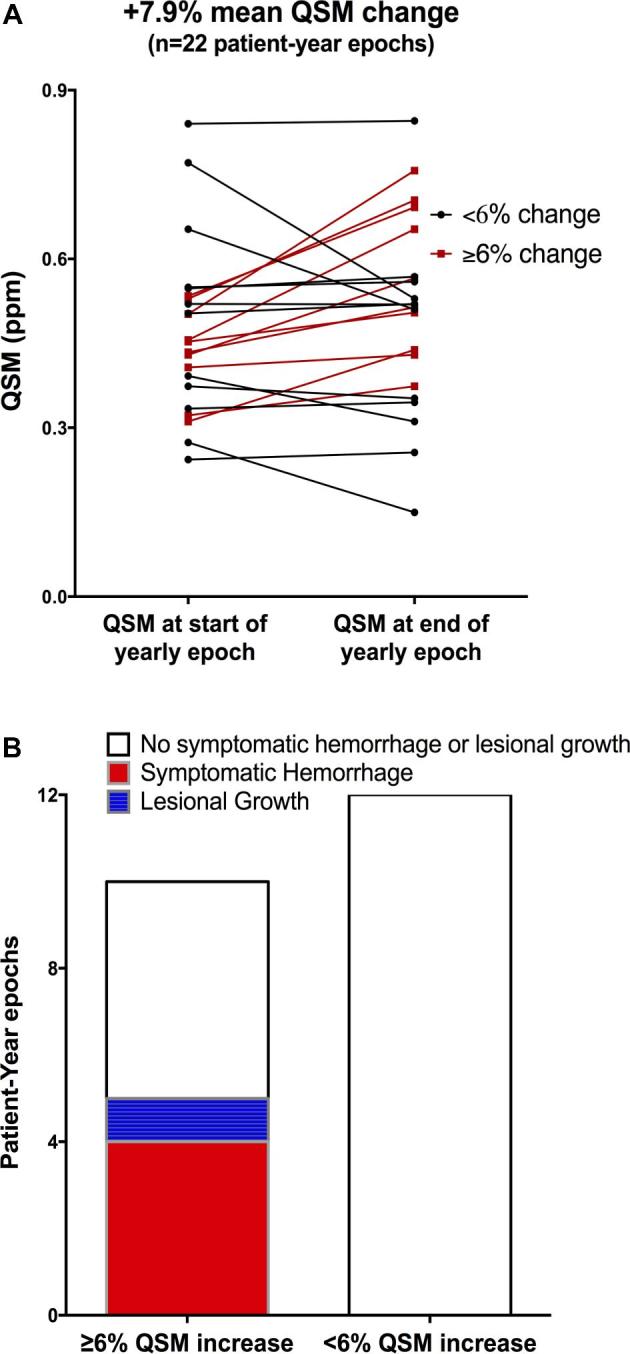
A, QSM measurements at the beginning and end of 22 person-year epochs of follow-up of cases with CA that had bled within 1 yr, and followed for 1 to 2 yr. Cases underwent annual QSM, allowing paired measurements at the beginning and end of each person-year epoch. In cases with multiple lesions, only QSM measurements in the lesion with initial hemorrhage (index lesion) were considered herein. Paired measurements in red identify epochs with a recorded threshold QSM increase by > 6% in the index lesion. B, Number of person-year epochs with and without threshold QSM change, and clinical events during these epochs. All clinical events occurred in the setting of > 6% increase in QSM. Reproduced with permission from Zeineddine et al.31
Quantitative Vascular Permeability in Lesion and Brain as a Potential Biomarker of Statin Therapy
Previous work by our group quantified vascular permeability in human CAs, using dynamic contrast enhanced quantitative perfusion (DCEQP) MRI technique with gadolinium.32,33 Our group further showed that more permeable CA lesions by DCEQP are associated with greater mean QSM.32 And we recently demonstrated, as with QSM, that CAs that bleed during longitudinal surveillance also manifest significant increases in lesional vascular permeability (mean change + 85.9%; P = .005).30 See Supplemental Digital Content for detailed information regarding imaging protocol.
Our group also reported greater permeability in normal brain white matter far (WMF) from lesions in familial cases (consistent with germline hemizygosity of CCM genes in those patients), than in sporadic cases (whose normal brain is unaffected by CCM genes).34 Nine CA subjects, already taking statins for routine cardiovascular indications unrelated to CA (5 familial, 4 sporadic), had lower mean WMF permeability than 60 cases not taking statins (34 familial, 26 sporadic).34 This effect was observed in both sporadic and familial cases (Figure 5), and we can postulate that it may reflect ROCK inhibition by statin in background brain. The impact of statin on lesional and brain vascular permeability, at doses affecting ROCK inhibition, shall be examined as a secondary aim in the proposed trial.
FIGURE 5.
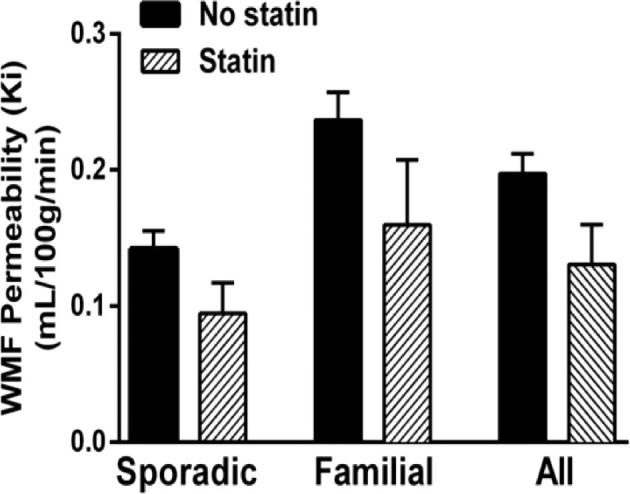
The effect of concomitant statin use on vascular permeability measured by DCEQP in WFM from lesions (WMF). There was a trend towards lower WMF permeability (Ki) in patients taking statins for cardiovascular disease than those not taking statins, in sporadic and familial CA subjects (P = .07).34 Reproduced with permission from Mikati AG, Khanna O, Zhang L, et al. Vascular permeability in cerebral cavernous malformations. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. Oct 2015;35(10):1632-1639.
STUDY GOALS AND OBJECTIVES
Primary Aim
To assess whether the proposed intervention produces evidence of QSM biomarker activity (indicative of reduced or increased lesional bleeding) during 2 yr of prospective follow-up after a recent SH. The primary outcome is mean percent change in QSM (change score) per year using an intention-to-treat analysis. QSM measurement in the initial SH lesion will be used for analysis should there be multiple lesions. Reduced CA hemorrhage would signal proof of concept of potential benefit, while increased hemorrhage a potential signal of risk. Secondary analysis shall examine treatment rendered considering attrition and compliance.
Secondary Aims
(1) Changes in DCEQP vascular permeability in the index lesion and WMF from initial lesion; (2) Rates of QSM and DCEQP biomarker events, representing increases in index lesional QSM or DCEQP above previously articulated biomarker thresholds; rates of clinically overt hemorrhages in the index lesion; and rates of lesional expansion (defined as an increase in maximum lesion diameter on T2-weighted sequences ≥ 2 mm); (3) adverse event rates and drug compliance; (4) Changes in functional outcome (modified Rankin Scale, Mini-Mental State Examination, Euro-Quality of Life [QoL] 5D, Euro-QoL, Visual Analogue Scale, Neuro-QoL); (5) ROCK activity assay on peripheral leukocytes; and (6) Impact of sex, genotype, and lesion location on primary and secondary outcomes (pre-specified subgroup analyses).
STUDY DESIGN
Atorvastatin treatment of CAs with SH exploratory proof of concept (AT-CASH-EPOC) trial is a phase I-IIa randomized, placebo-controlled, double-blinded, and single-site clinical trial. A single site is chosen where the outcomes biomarkers have been carefully validated. The study will enroll 80 adult patients 18 to 80 yr of age (inclusive) with untreated CAs of all genotypes who suffered an adjudicated symptomatic lesional hemorrhage within one year of enrollment.35 The CASH lesion would need to have been considered for surgery, and ultimately not resected. Dosing shall continue for a 24-mo follow-up period or until reaching a safety endpoint. Inclusion and exclusion criteria are listed in Table.
TABLE.
Inclusion and Exclusion Criteria
| Inclusion criteria | |
| Age 18 to 80 (inclusive). | |
| Asymptomatic, mild or moderate disability requiring some help but able to attend own bodily needs without assistance. Defined as modified Rankin score (mRS) 0 to 3. | |
| Diagnosis of CCM of any genotype supported by relevant imaging studies, where surgical resection is not performed after due consideration per current standard of care. | |
| Symptomatic CCM bleeding event within 1 yr prior to enrollment. | |
| Must be willing/able to travel to the study site for study visits (baseline, 12 and 24 mo) over the course of the study. | |
| Exclusion Criteria | |
| Premenopausal women who are breastfeeding, pregnant or likely to get pregnant during the study period. | |
| Prior surgical treatment of the hemorrhagic CCM lesion. | |
| Failure to pass MRI safety screening (claustrophobic, metal implant, etc.). | |
| Known allergy or intolerance to gadolinium. | |
| Severely impaired renal function (eGFR < 60 ml/min), active renal disease or status postkidney transplants. | |
| Statin therapy, for any indication, within 12 mo preceding enrollment. | |
| Indication to use statin medication for current approved indication, unrelated to CCM. | |
| Known allergy or intolerance to statins. | |
| Liver dysfunction or active liver disease (including chronic viral hepatitis) defined as baseline serum transaminases levels twice the upper range of normal. | |
| Previous diagnosis of skeletal muscle disorders of any cause (myopathy), or baseline creatine kinase levels five times the upper range of normal. | |
| Currently treated with or likely to need treatment with a prohibited medication (Cyclosporine, fibrates, niacin, Azol antifungals, HIV/HCV protease inhibitors, macrolides or colchicine). | |
| Active drug or alcohol use or dependence that, in the opinion of the site investigator, would interfere with adherence to study requirements. | |
| Serious illness (requiring systemic treatment and/or hospitalization) until subject either completes therapy or is clinically stable on therapy, in the opinion of the site investigator, for at least 30 d prior to study entry. | |
| Any other condition that the investigator believes would pose a significant hazard to the subject if the investigational therapy were initiated, including conditions resulting in or precipitating myopathy (eg, HIV, uncontrolled hypothyroidism). | |
| In the investigator's opinion, the patient is unstable, and would benefit from a specific intervention rather than treatment with atorvastatin. | |
| Inability or unwillingness of subject or legal guardian/representative to give written informed consent. | |
| No documentation of valid healthcare insurance. | |
| No medical record confirmation of a primary care physician or other provider of ongoing medical care. | |
| Previous cranial irradiation or radiosurgical treatment. |
METHODOLOGY
Screening and Enrollment
Pre-screening will be conducted for known or referred CA patients who may be eligible for the trial. All potential subjects will undergo a detailed screening visit at the University of Chicago involving collection of medical history, prior and concomitant medications, and a neurological examination to confirm eligibility. Surgical resection will need to have been considered in every case as an alternative to medical therapy, per current standard of care and evidence-based guidelines.4 As part of the screening, blood tests will be performed, which in the case of successful enrollment, will serve as baseline laboratory results. With verification of eligibility, informed consent will be finalized. A recruitment plan to secure the number of eligible subjects has been developed with regional academic centers in Chicago, and with the Angioma Alliance (see the Supplemental Digital Content).
Blinding/Randomization/Method of Dosing
Enrolled subjects will be blinded to treatment rendered, as will the trial staff. Atorvastatin (at 40 or 80 mg) and placebo capsules will be formulated to appear indistinguishable. Enrolled subjects will be randomized to atorvastatin or placebo at a 1:1 ratio. Randomization shall be stratified by sex, with separate permuted blocks for each sex. The dose may be de-escalated based on subject's tolerance, with subjects reporting moderate adverse events per pre-specified Category B criteria (Table S1 in the Supplemental Digital Content) allowed a lower dose (40 mg/day) of atorvastatin (or matching placebo pill, without breaking the blind).
DISCUSSION
There is no proven therapy to prevent rebleeding and the neurologic sequelae in CAs with prior SH, impacting up to 200 000 Americans.36 Taking advantage of firm mechanistic rationale, rigorous preclinical discoveries, and novel biomarker validations, we propose the first exploratory proof of concept trial aimed at stabilizing a CASH lesion using a repurposed, widely available, and well-tolerated drug. Go/No-go implications of the trial results and a therapeutic development roadmap are outlined in the Supplemental Digital Content.
TRIAL STATUS
Patients are actively being enrolled at the University of Chicago Medical Center, Chicago, Illinois. Clinicaltrials.gov Registration number NCT02603328.
SAFETY CONSIDERATIONS
See Supplemental Digital Content for detailed information regarding safety and quality.
FOLLOW-UP
Follow-up Schedule and Dosing
All enrolled subjects will go through the same schedule of investigations and follow-up visits throughout the study period (Figure 6). Dosing shall continue for the 24-mo follow-up period or until reaching a safety endpoint, whichever comes first. The study shall continue until all enrolled subjects, including those reaching safety endpoints, have undergone their 24-mo MRI and clinical assessment. Laboratory studies will also be performed at 3 mo after dose initiation. Dose de-escalation to 40 mg/day or identical placebo capsule shall be allowed at any time during the course of the study in the case of prearticulated side effects or laboratory abnormalities at the initial dose. Clinical evaluation, lesional QSM, brain/lesional DCEQP permeability, and peripheral blood leukocyte ROCK activity will be assessed to determine the response to atorvastatin or placebo at the 12 and 24-mo follow-ups.
FIGURE 6.

Trial enrollment course by visits.
Criteria for Intervention Discontinuation
Atorvastatin/placebo administration will be discontinued for any of the following reasons: symptomatic intracerebral hemorrhage, severe allergic or adverse reaction to study drug, or persistent moderate adverse reactions after dose reduction (See Table S1 in the Supplemental Digital Content). Furthermore, if the patient develops clinical indications for statin use for cholesterol or cardiovascular indications unrelated to the CA they will be considered to have reached a safety endpoint. A Trial Nurse (Kristina Piedad), with experience in CA disease, shall be available to the subjects, and Matthew Sorrentino, senior cardiologist and expert in statin therapy shall advise the clinical site team about management of statin side effects.
While the proposed trial is underpowered to detect a significant difference in symptomatic rebleeding rates between groups, differential and absolute rates of hemorrhage in the placebo and treatment cohorts will be critically analyzed throughout the trial. Continuous monitoring will be initiated with the enrollment of the tenth subject in the study, and a SH will be considered a safety endpoint for each subject. Based on the data previously reported in the literature, and our own pilot data, we will implement a running safety threshold after the 10th case receiving active drug is enrolled, to preemptively suspend trial enrollment for a full safety review if > 40% of subjects in the treatment group suffer a SH (for further detailed safety considerations see Supplemental Digital Content).
DATA MANAGEMENT AND STATISTICAL ANALYSIS
Sample Size and Treatment Effect Estimation
Sample size calculations were based on providing sufficient power to test the primary hypothesis during 2 yr of prospective follow-up after recent CASH. Evaluation of the intervention on this outcome will be performed as a time-averaged difference between the two arms using a repeated measures analysis implemented as a linear mixed model using the intention-to-treat principle.37
Based on pilot data linking QSM change to clinical events, we estimate a minimum, clinically meaningful and mechanistically plausible treatment effect to be a 20% absolute difference in the mean change score.31 It would be “a minimum expected effect”, < 1/2 the mean 44% QSM increase observed with CA bleed or growth during longitudinal 1-yr follow-up of previously stable lesions; and also “clinically meaningful”, > 3X the threshold of QSM change associated with maximum biomarker sensitivity-specificity.30 The absolute QSM effect size postulated herein is also “plausible mechanistically”, as it would be 0.0068 ppm mean change score [20% of 0.0340 ppm mean QSM change observed during follow-up of CA with SH], 6 to 22 fold lower than the observed range of atorvastatin treatment effect on Perls staining in murine models.
One-year % difference in mean QSM change score in 16 pilot study patients (22 patient-year epochs) was used to estimate the 1-yr/patient standard deviation and within-patient correlation of the primary outcome in trial candidates. A sample size calculation was performed using a repeated measures analysis of two annual percent QSM change scores, assuming a two-tailed test, power = 0.9 and alpha = 0.05.38 The actual within person correction from the pilot data was estimated at approximately -0.6 (Spearman). However, we used a more conservative within person correlation of 0 for our sample size estimates. In addition, we assumed a common standard deviation of 30 for the mean percent change score, which is 20% larger than the pilot data's standard deviation. To detect a 20% absolute difference between the mean change score in each arm, based on two annual change scores per patient (at years 1 and 2), 25 patients per arm would be required. To detect a smaller or larger effect size of 15% or 25%, 44 or 17 patients per arm would be required, respectively (Hintze 2014, PASS v13, NCSS, Kaysville, Utah; www.ncss.com).
Potential Missing Data, Attrition, and Premature Trial Endpoint
Using preliminary data, we conservatively estimated up to 10% missing biomarker data related to imaging (claustrophobia, etc) and an additional 10% due to imperfect follow-up. Up to 20% of subjects randomized to atorvastatin (10% of trial subjects) may not tolerate the 80-mg/day dose, while some will continue the drug at the lower dose. In fact, serious side effects or intolerance of the lower dose are expected to result in attrition in less than 5% of cases, per extensive meta-analysis.39 In addition, 20% to 30% of cases might reach a prespecified study endpoint of SH. However, all subjects who reach a pre-defined endpoint will still be followed for the full 2 yr for the primary intention-to-treat analysis. Taking all of the above factors into consideration at the highest combined estimates of attrition and missing data we expanded the planned sample size by 37.5%. Therefore, we shall aim to enroll 40 patients/arm with 80 total subjects.
Data Analyses
QSM acquisitions and data processing shall be overseen by bioengineer Nicholas Hobson and senior neuroradiologist Gregory Christoforidis, at the Chicago site. Data analysis is overseen by senior statistician Richard Thompson of Baltimore, assisted by the Chicago site statistician Ying Cao with experience in biomarkers data, both with dedicated time effort for the duration of this project. Evaluation of the intervention will be performed as a time-averaged difference between two arms using a repeated measures analysis implemented as a linear mixed model.37 The model accounts for within-patient correlation between the measurements.40,41 Patients with outcome measurements in both periods will be included in the primary intention-to-treat analysis. A secondary analysis of the percent QSM change per year shall be conducted per treatment rendered for all patients with at least one annual epoch of measurements. To address secondary aims, each of the other outcomes will be analyzed with a similar approach, first per intention-to-treat sample in patients receiving two annual epochs of follow-up, and second by treatment-rendered in all patients with at least one annual epoch of measurements. We articulate a detailed methodology of analyzing adverse events, and of querying potentially biased effects in 3 prespecified subgroups (male/female, sporadic/familial genotype, and brainstem/other lesion location) in secondary analyses.
Futility Analysis and Adaptive Adjustments to Sample Size
Due to concerns that the treatment may not be effective in reducing QSM measurements, futility analyses will occur once outcome data has been logged on 80 of the planned 160 person-year epochs. Within person correlations between year 1 and 2 will be controlled for in the futility assessment. The intervention will be deemed futile if the difference in QSM change scores is 10% or less between groups. The futility analysis will be performed using a linear mixed-effects model adjusted for gender, and a one-sided confidence interval will be created around the estimated regression coefficient from this model, which represents the mean group difference in the percentage change of QSM scores between treatment arms. Based on recommendations by Freidlin and Korn, we will consider a 99% one-side confidence on the difference in change scores between treatment groups.42 A recommendation to stop the trial would be made if the 99% confidence surrounding the true effect size does not include, and is less than 10%.
In addition, we will also assess the actual within person correlation in the year 1 and 2 outcome measures, and the variance in the change scores within each arm. Since the sample size is strongly related to these two statistics, estimating the true correlation and variances will allow for sample size adjustment at the trial midpoint. A larger than expected correlation and/or a smaller than expected variance of the change scores will allow for sample size reduction, while retaining the same estimated power. A higher sample size than initially proposed is unlikely due to the conservative assumptions. See Table S2 in theSupplemental Digital Content for sample size simulations with different within person correlations and variance in the primary outcome.
QUALITY ASSURANCE
See Supplemental Digital Content for detailed information regarding safety and quality.
EXPECTED OUTCOMES OF THE STUDY
See Supplemental Digital Content for Go/No-Go discussion regarding the implications of positive, negative or inconclusive trial results.
DURATION OF THE PROJECT
This trial will enroll 27 subjects per year over a 3-yr period. The follow-up duration of each individual patient will be 24 mo (see follow-up section).
PROJECT MANAGEMENT
The PI of the study (Issam Awad) will oversee the trial and enroll each patient through the University of Chicago's clinical and research infrastructure (www.uchospitals.edu/ccm), assisted by Trial Manager Agnieszka Stadnik and the Trial Nurse. Trial co-chair and the Data Coordinating Center are overseen by Daniel Hanley and the Brain Injury Outcomes Section (BIOS) at Johns Hopkins Medical Institution and senior manager Karen Lane. The data and safety monitoring team will comprise the local IRB, the trial's safety compliance officer (Nichol McBee of BIOS), and the Independent Medical Safety Officer (Kelly Flemming of Mayo Clinic). The latter is not associated with the study or data management teams, and shall be assisted by two external medical safety consultants with expertise in CA disease and in statin therapy respectively (Leslie Morrison of New Mexico, and Kevin Whitehead of Utah). Data management, and the monitoring and safety plans are detailed in the Supplemental Digital Content.
ETHICS
This study and the informed consent process have been reviewed and approved by the University of Chicago's Institutional Review Board for ethical compliance.
Disclosures
Protocol Title (ID#): Atorvastatin Treatment of CAs with SH Exploratory Proof of Concept (AT CASH EPOC) Trial (1 R01 NS107887). U.S. F.D.A. IND Exemption #126 840. Clinicaltrials.gov registration number NCT02603328. This protocol is sponsored/funded by NINDS/NIH R01 NS107887 (2018-2023). The authors have no personal, financial, or institutional interest in any of the drugs, materials, or devices described in this article.
Supplementary Material
Supplemental Digital Content. Rationale, Methods, 1 Figure, 2 Tables. The supplemental digital content expands upon the rationale for the dosing used in the trial, more detailed mechanistic support for the study, further safety considerations, and logistics of data collection. Figure S1, QSM and Perls iron deposition. Table S1, statin-related adverse events. Table S2, postulated effect and sample size.
REFERENCES
- 1. Robinson JR Jr, Awad IA, Masaryk TJ, Estes ML. Pathological heterogeneity of angiographically occult vascular malformations of the brain. Neurosurgery. 1993;33(4):547–554. [DOI] [PubMed] [Google Scholar]
- 2. Abdulrauf SI, Kaynar MY, Awad IA. A comparison of the clinical profile of cavernous malformations with and without associated venous malformations. Neurosurgery. 1999;44(1):41–46. [DOI] [PubMed] [Google Scholar]
- 3. Gault J, Sain S, Hu LJ, Awad IA. Spectrum of genotype and clinical manifestations in cerebral cavernous malformations. Neurosurgery. 2006;59(6):1278–1285. [DOI] [PubMed] [Google Scholar]
- 4. Akers A, Al-Shahi Salman R, Awad IA et al.. Synopsis of guidelines for the clinical management of cerebral cavernous malformations: consensus recommendations based on systematic literature review by the angioma alliance scientific advisory board clinical experts panel. Neurosurgery. 2017;80(5):665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al-Shahi Salman R, Hall JM, Horne MA et al.. Untreated clinical course of cerebral cavernous malformations: a prospective, population-based cohort study. The Lancet Neurology. 2012;11(3):217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horne MA, Flemming KD, Su IC et al.. Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. The Lancet Neurology 2016;15(2):166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robinson JR Jr, Awad IA, Magdinec M, Paranandi L. Factors predisposing to clinical disability in patients with cavernous malformations of the brain. Neurosurgery. 1993;32(5):730–736. [DOI] [PubMed] [Google Scholar]
- 8. Porter PJ, Willinsky RA, Harper W, Wallace MC. Cerebral cavernous malformations: natural history and prognosis after clinical deterioration with or without hemorrhage. J Neurosurg. 1997;87(2):190–197. [DOI] [PubMed] [Google Scholar]
- 9. Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 2010;207(4):881–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borikova AL, Dibble CF, Sciaky N et al.. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem. 2010;285(16):11760–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whitehead KJ, Chan AC, Navankasattusas S et al.. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15(2):177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crose LE, Hilder TL, Sciaky N, Johnson GL. Cerebral cavernous malformation 2 protein promotes smad ubiquitin regulatory factor 1-mediated RhoA degradation in endothelial cells. J Biol Chem. 2009;284(20):13301–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou Z, Tang AT, Wong WY et al.. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature. 2016;532(7597):122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McDonald DA, Shi C, Shenkar R et al.. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet. 2014;23(16):4357–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18(5):911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gault J, Shenkar R, Recksiek P, Awad IA. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke. 2005;36(4):872–874. [DOI] [PubMed] [Google Scholar]
- 17. Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet. 2009;18(5):919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McDonald DA, Shenkar R, Shi C et al.. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum Mol Genet. 2011;20(2):211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shenkar R, Shi C, Rebeiz T et al.. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet Med. 2015;17(3):188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Plummer NW, Gallione CJ, Srinivasan S, Zawistowski JS, Louis DN, Marchuk DA. Loss of p53 sensitizes mice with a mutation in Ccm1 (KRIT1) to development of cerebral vascular malformations. Am J Pathol. 2004;165(5):1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shenkar R, Venkatasubramanian PN, Wyrwicz AM et al.. Advanced magnetic resonance imaging of cerebral cavernous malformations: part II. Imaging of lesions in murine models. Neurosurgery. 2008;63(4):790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Landis SC, Amara SG, Asadullah K et al.. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490(7419):187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McDonald DA, Shi C, Shenkar R et al.. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke. 2012;43(2):571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shenkar R, Shi C, Austin C et al.. RhoA kinase inhibition with fasudil versus simvastatin in murine models of cerebral cavernous malformations. Stroke. 2017;48(1):187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rawlings R, Nohria A, Liu PY et al.. Comparison of effects of rosuvastatin (10 mg) versus atorvastatin (40 mg) on rho kinase activity in caucasian men with a previous atherosclerotic event. Am J Cardiol. 2009;103(4):437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nohria A, Prsic A, Liu PY et al.. Statins inhibit Rho kinase activity in patients with atherosclerosis. Atherosclerosis. 2009;205(2):517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weng TC, Yang YH, Lin SJ, Tai SH. A systematic review and meta-analysis on the therapeutic equivalence of statins. J Clin Pharm Ther. 2010;35(2):139–151. [DOI] [PubMed] [Google Scholar]
- 28. Tan H, Liu T, Wu Y et al.. Evaluation of iron content in human cerebral cavernous malformation using quantitative susceptibility mapping. Invest Radiol. 2014;49(7):498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan H, Zhang L, Mikati AG et al.. Quantitative susceptibility mapping in cerebral cavernous malformations: clinical correlations. Am J Neuroradiol. 2016;37(7):1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Girard R, Fam MD, Zeineddine HA et al.. Vascular permeability and iron deposition biomarkers in longitudinal follow-up of cerebral cavernous malformations. J Neurosurg. 2017;127(1):102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeineddine HA, Girard R, Cao Y et al.. Quantitative susceptibility mapping as a monitoring biomarker in cerebral cavernous malformations with recent hemorrhage. J Magn Reson Imaging. 2018;47(4):1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mikati AG, Tan H, Shenkar R et al.. Dynamic permeability and quantitative susceptibility: related imaging biomarkers in cerebral cavernous malformations. Stroke. 2014;45(2):598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hart BL, Taheri S, Rosenberg GA, Morrison LA. Dynamic contrast-enhanced MRI evaluation of cerebral cavernous malformations. Transl. Stroke Res. 2013;4(5):500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mikati AG, Khanna O, Zhang L et al.. Vascular permeability in cerebral cavernous malformations. J Cereb Blood Flow Metab. 2015;35(10):1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Al-Shahi Salman R, Berg MJ, Morrison L, Awad IA. Angioma Alliance Scientific Advisory Board Hemorrhage from cavernous malformations of the brain: definition and reporting standards. Angioma Alliance Scientific Advisory Board. Stroke. 2008;39(12):3222–3230. [DOI] [PubMed] [Google Scholar]
- 36. Polster SP, Cao Y, Carroll T et al.. Trial readiness in cavernous angiomas with symptomatic hemorrhage (CASH). Neurosurgery. 2019;84(4):954-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Varosanec M, Uher T, Horakova D et al.. Longitudinal mixed-effect model analysis of the association between global and tissue-specific brain atrophy and lesion accumulation in patients with clinically isolated syndrome. AJNR Am J Neuroradiol 2015;36(8):1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu H, Wu T. Sample size calculation and power analysis of time-averaged difference. J. Mod. App. Stat. Meth. 2005;4(2):434–445. [Google Scholar]
- 39. Amarenco P, Labreuche J. Lipid management in the prevention of stroke: review and updated meta-analysis of statins for stroke prevention. The Lancet Neurology 2009;8(5):453–463. [DOI] [PubMed] [Google Scholar]
- 40. Brown H, Prescott R. Applied mixed models in medicine. 2nd ed.Chichester, West Sussex, England: John Wiley & Sons; 2006, Chapter 6. [Google Scholar]
- 41. Diggle P, Liang K-Y, Zeger SL. Analysis of longitudinal data. New York, New York: Oxford University Press; 1994, Chapter 2. [Google Scholar]
- 42. Freidlin B, Korn EL. Monitoring for lack of benefit: a critical component of a randomized clinical trial. J Clin Oncol. 2009;27(4):629–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.