

Abstract
The ability to prevent blood loss in response to injury is a conserved function of all vertebrates. Complete deficiency of the central clotting enzyme prothrombin has never been observed in humans and is incompatible with postnatal life in mice, thus limiting the ability to study its role in vivo. Zebrafish are able to tolerate severe hemostatic deficiencies that are lethal in mammals. We have generated a targeted genetic deletion in the kringle 1 domain of zebrafish prothrombin. Homozygous mutant embryos develop normally into the mid-juvenile stage but demonstrate complete mortality by 2 months of age primarily due to internal hemorrhage. Mutants are unable to form occlusive venous and arterial thrombi in response to endothelial injury, a defect that was phenocopied using direct oral anticoagulants. Human prothrombin engineered with the equivalent mutation exhibits a severe reduction in secretion, thrombin generation, and fibrinogen cleavage. Together, these data demonstrate the conserved function of thrombin in zebrafish and provide insight into the role of kringle 1 in prothrombin maturation and activity. Understanding how zebrafish are able to develop normally and survive into early adulthood without thrombin activity will provide important insight into its pleiotropic functions as well as the management of patients with bleeding disorders.
Subject terms: Haematological diseases, Mechanisms of disease
Introduction
Maintaining blood flow in a closed circulatory system requires a delicate balance between pro- and anticoagulant factors. Disequilibrium of these factors in either direction can lead to pathology. In response to vascular injury, the balance shifts towards coagulation in an effort to stabilize blood clots and prevent exsanguination. Critical to this clot stabilization is the activation of prothrombin, a vitamin K-dependent clotting factor, to form the central clotting enzyme, thrombin1. Thrombin cleaves soluble fibrinogen into fibrin monomers, which then polymerize to form the insoluble fibrin clot2. Additionally, thrombin interacts with protease activated receptors (PARs) on the surface of various cells, including platelets, which makes it the most potent endogenous agonist of primary hemostasis3. Human prothrombin (F2) variants have been linked to both thrombophilia and bleeding diatheses. The most common variant (~2% in European populations)4 is a guanine to adenine transition in the 3’ untranslated region at position 20210 that leads to increased plasma prothrombin levels and a 2–3 fold elevated risk of deep vein thrombosis5,6. Congenital prothrombin deficiencies result in a bleeding diathesis, but are rare. Acquired deficiencies due to liver failure or vitamin K deficiency are more common7,8.
Structurally, prothrombin consists of six domains: signal peptide, propeptide, Gla, kringle 1, kringle 2, and serine protease; a domain composed of a light and heavy chain1. Following translation, the propeptide targets the protein for post-translational gamma-carboxylation of the glutamic acid residues within the Gla domain. This vitamin K-dependent process is necessary for proper localization of the mature zymogen to the activated membrane surface9. While relatively understudied, the kringle domains are thought to interact with activated factor V (FVa) during assembly of the prothrombinase complex (FVa and activated factor X (FXa))10–12. Cleavage of prothrombin by the prothrombinase complex results in fragment 1.2 (the Gla and 2 kringle domains) and thrombin (light and heavy chains).
Targeted mutagenesis of F2 in mice demonstrates 50% mortality by embryonic day 10.5 with hemorrhage associated with suspected defects in yolk sac vasculature. Mutants surviving to birth succumb to hemorrhage by postnatal day 113,14. Loss of other common pathway factors, including FV and FX, show a similar pattern of bimodal lethality15,16. These data are suggestive of a secondary role beyond the coagulation system during development. More recent studies reveal that conditional knockout of prothrombin in adult mice leads to mortality due to hemorrhagic events within 5–7 days, although there remains a residual level of prothrombin17. Combined with the long half-life of prothrombin (60–72 hours)18, it is likely that the adult mice do not achieve complete deficiency prior to lethality.
Due to early lethality of complete genetic knockouts of prothrombin in mice, the nuances of thrombin’s role in in vivo hemostasis and development are difficult to study. Zebrafish (Danio rerio) has advantages for the investigation of early development because of its high fecundity, external fertilization, and transparent development. Embryonic and adult studies have demonstrated the benefits of zebrafish for the study of hemostatic and other human diseases19,20. Conservation of coagulation factors in zebrafish is well characterized with a variety of techniques and genetic models21–27. FX knockout (f10−/−) zebrafish survive several months into adulthood before succumbing to lethal hemorrhage, suggesting that the model is more resistant to extreme disturbances in hemostasis than mammals28. Notably, evaluation of f10−/− fish vasculature shows no abnormalities in the embryonic period.
Complete prothrombin deficiency has never been seen in humans and is incompatible with life in mice, limiting the ability to understand the entirety of prothrombin’s in vivo functions. Therefore, we have now used genome editing TALENs (transcription activator-like effector nucleases) to produce a genetic knockout of the prothrombin gene (f2) in zebrafish. We show here that loss of prothrombin in zebrafish does not result in severe developmental defects but does prevent the formation of clots in response to endothelial injury and leads to early mortality at 2–3 months of age. Furthermore, the mutation generated lends insight into the role of the kringle 1 domain in prothrombin biosynthesis and activation.
Results
Zebrafish prothrombin demonstrates a high degree of sequence conservation
Mature zebrafish prothrombin is a 579 amino acid protein that shares 55% identity and 72% homology with human prothrombin, including 68% identity and 86% homology when comparing the regions corresponding to thrombin. Zebrafish prothrombin is detected within the first day of development and leads to measurable fibrin forming activity by 48 hours29. Overall, the prothrombin sequence is highly conserved across vertebrates, with complete conservation of cysteine residues, suggesting the conservation of major structural features such as the kringle domains (Fig. 1).
Figure 1.

Peptide sequence alignment shows strong conservation of prothrombin across a broad range of species. Sequences shown include the conserved domains. Numbering begins at the first residue after the propetide according to the prothrombin numbering scheme. Sequences are shaded to indicate degree of conservation. Green colored residues indicate conserved cysteines. Red colored residues represent amino acids altered by mutagenesis (Δ15; C138A) with the affected paired cysteines highlighted in yellow. Numbers above alignment indicate paired cysteines in human prothrombin.
Zebrafish thrombin activity is inhibited by direct oral anticoagulants
To test the relative conservation between zebrafish and human thrombin, larvae were treated with three direct oral anticoagulants: direct thrombin inhibitor (dabigatran) as well as direct FXa inhibitors (rivaroxaban and apixaban). Following 24 hours of treatment, 6 dpf larvae exposed to dabigatran etexilate, rivaroxaban, or apixaban were unable to form occlusive thrombi in response to injury (Fig. 2). These data show that the carboxylesterase function required to convert dabigatran etexilate to dabigatran is conserved in zebrafish, as is the ability of dabigatran to inhibit zebrafish thrombin. Apixaban and rivaroxaban also demonstrate conserved inhibition of FXa, which inhibits prothrombin activation. In humans, mean plasma concentrations of all 3 inhibitors are in the range of 100–600 nM30,31. Similar concentrations of dabigatran and 10–100 fold higher concentrations of rivaroxaban have been shown to impair clot formation following tail bleeds in mice32,33. Although difficult to predict, the low polarity of the 3 molecules leads us to estimate that the molecules will be poorly absorbed from the media and result in similar tissue levels to those seen in humans34.
Figure 2.
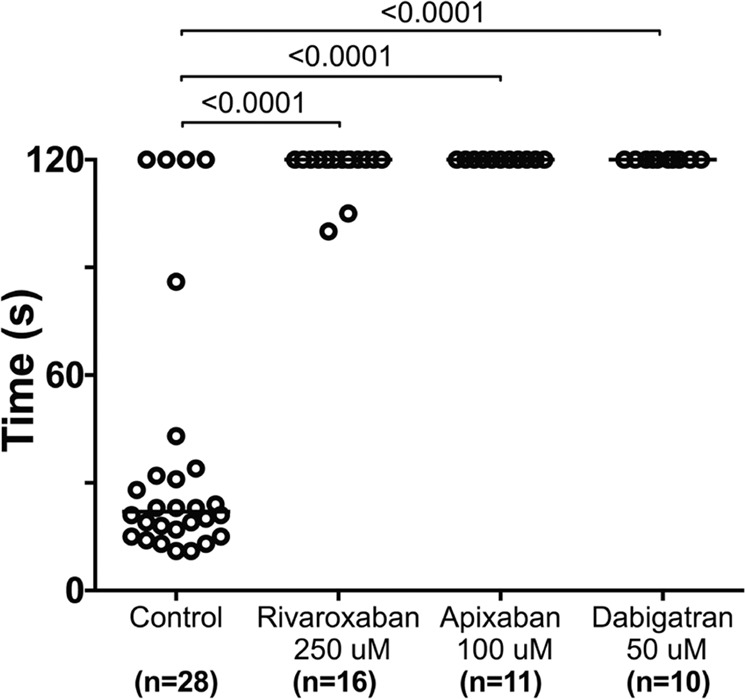
Direct oral anticoagulants prevent occlusive thrombus formation in zebrafish. 5 dpf larvae were exposed to chemical inhibitors of thrombin (dabigatran) and FXa (apixaban, rivaroxaban) for 24 hours. This resulted in the inability to form occlusive venous thrombi at 6 dpf in wild-type larvae following laser-mediated endothelial injury.
Targeted genome editing induces a deletion resulting in decreased f2 expression
Having established the functional conservation of prothrombin, we sought to analyze the long-term effects of thrombin deficiency using a genetic model. Utilizing TALEN-mediated genome editing, exon 6 of f2 was targeted with the aim of creating a frameshift and subsequent nonsense mutation prior to the protease domain. Sequencing data showed a 14 bp deletion within the genomic region homologous to the human prothrombin kringle 1 domain (Fig. 3A). In situ hybridization demonstrated decreased, but not absent f2 mRNA in homozygous mutants at 3 and 5 dpf compared to wild-type siblings (Fig. 3B). This is further supported by quantitative RT-PCR data demonstrating a 45% reduction in mRNA transcript in homozygous mutants (Fig. 3C). To characterize the residual mutant transcript, semi-quantitative RT-PCR was performed using primers flanking the mutation site. Only wild-type and mutant bands were seen in pools of wild-type and homozygous mutant embryos, respectively. In heterozygous embryos, the computed molar amount of the mutant band was only 26% of the total, with the remainder being wild-type (Fig. 3D). Notably, the homozygous mutant transcript was roughly 30 base pairs smaller than expected.
Figure 3.

Genome editing creates a 14 bp genomic deletion with a resulting decrease in mRNA expression. (A) Alignment of Sanger sequencing with the chromosome 7 genomic region showed an overall 17 bp genomic deletion replaced with a 3 bp insertion; outlined in red, resulting in a net 14 bp deletion. (B) in situ hybridization demonstrated reduction of transcript at 72 and 120 hours post fertilization in homozygous mutants compared to control siblings. Spatial regulation remained intact with expression restricted primarily to the liver. (C) qPCR data of f2 expression reveals significant decrease of 45% in the homozygous mutant embryos. (D) Semi-quantitative RT-PCR of embryos shows a mutant band ~30 bp smaller than expected (later shown to be a 45 bp deletion, Fig. 4). Quantitation of the bands reveal that the mutant band is only 26% of the total in heterozygotes.
Genomic deletion reveals a cryptic splice site that creates an alternative splice variant
To identify potential splice variants, full length f2 cDNA was sequenced from f2+/+ and f2Δ14/Δ14 larvae (Fig. 4, top). This revealed that there is a cryptic splice acceptor 3′ to the 14 bp genomic deletion in exon 6. In contrast to the canonical splicing which forms a transcript containing the 14 bp deletion, alternative splicing from the exon 5 splice donor site to the cryptic splice site generates a 45 bp deletion in kringle 1 of prothrombin (Fig. 4). Notably, this maintains the reading frame and replaces DEFNVTQLKLQENFCR with a single glutamic acid for a net loss of 15 amino acids. This deletion includes a highly conserved cysteine (C119 in the zebrafish peptide) predicted by homology to form a disulfide bond with zebrafish C143 (Fig. 1). The protein product of this alternatively spliced transcript is referred to as Δ15. To understand the distribution of splicing, we performed deep SMRT sequencing of f2 cDNA from f2+/Δ14 mutant larvae at 3 dpf. Overall, 3842 consensus sequences were analyzed. Using downstream allelic polymorphisms, the reads were sorted into wild-type and f2Δ14 haplotypes. Of the 2384 reads derived from the wild-type locus, 100% underwent canonical splicing (Fig. 4, middle). In contrast, only 17/1458 (1.2%) mutant locus reads displayed canonical splicing (Fig. 4, bottom) and the remainder made use of the cryptic splice site to form the Δ15 transcript (1441/1458, 98.8%).
Figure 4.

Single molecule real time sequencing of f2+/Δ14 mRNA demonstrates altered splicing in f2 following a deletion in exon 6. (top) Sanger sequencing of f2+/+ and f2 Δ14/Δ14 cDNA reveals a canonical splice donor but alternative splice acceptor (red dashed lines indicate splice junctions). Allelic SNPs in exon 7 allowed the sorting of transcripts by their genomic allele in mRNA extracted from heterozygotes. Wild-type transcripts (middle) solely demonstrated canonical splicing. Mutant transcripts (bottom) primarily exhibited alternative splicing to a cryptic splice site with a resulting 45 bp inframe deletion, as well as low frequency canonical splicing with a resulting 14 bp out of frame deletion. gDNA, genomic DNA.
Disruption of the prothrombin kringle 1 domain results in adult lethality due to internal hemorrhage
To examine the functional consequence of kringle 1 disruption, we evaluated homozygous mutant fish at various stages of development. Beginning around 1 month of age, f2Δ14/Δ14 fish showed signs of overt internal hemorrhage and the majority (23/24) died by adulthood, defined as 90 days of age (Fig. 5A,B). Gross intracranial and fin hemorrhage was visible, and histological analysis demonstrated the occurrence of bleeds within the head, jaw, muscle, fins, and around the heart and abdomen (Fig. 5C).
Figure 5.

Loss of prothrombin results in early lethality due to hemorrhage. (A) Survival curve demonstrating significant mortality by 2 months of age in f2−/− siblings, log-rank (Mantel-Cox) analysis. (B) Examples of grossly visible intracranial, intramuscular, and fin bleeds (arrows). (C) Histological sections of wild-type and f2−/− siblings demonstrated microscopic bleeds in the brain, jaw, heart, muscle, and fins. Arrows point to pools of erythrocytes and unaffected comparable tissue in the control.
Homozygous mutant larvae fail to form thrombi in response to endothelial injury
Given that 80% of f2Δ14/Δ14 fish were able to survive development to the juvenile stage (30–90 days of age), we assessed the possibility that there could be residual detectable thrombin activity in early embryos and larvae. Thrombin has roles in both primary and secondary hemostasis, platelet activation and fibrin formation, respectively. These roles were evaluated using laser-mediated endothelial injury models of arterial and venous thrombus formation (Fig. 6A). Homozygous mutant larvae were unable to form occlusive thrombi in the venous system in response to injury. This was refractory to treatment with the fibrinolytic inhibitor ε-aminocaproic acid, further confirming a lack of fibrin generation (Fig. 6B). To confirm that defects in hemostasis were not due to off target effects of genome editing, embryos were injected at the one-cell stage with plasmid expressing human F2 cDNA driven by the constitutively active cytomegalovirus promoter35,36. Endothelial injury at 3 dpf induced clot formation within 2 minutes in 50% of homozygous embryos in contrast to uninjected controls (Fig. 6C). To assess the role of thrombin in the zebrafish arterial system, the f2Δ14 mutation was bred into the Tg(cd41-egfp) background in which circulating thrombocytes express GFP. At 5 and 6 dpf, f2Δ14/Δ14 larvae had a decreased ability to form occlusive arterial thrombi (Fig. 6D). The time to thrombocyte attachment was not statistically different between groups (Fig. 6E) while the number of attached thrombocytes appeared to be slightly increased in the f2Δ14/Δ14 larvae, but only at 6 dpf (Fig. 6F). Overall these data reveal intact primary hemostasis but a loss of secondary hemostasis. Despite this, erythrocyte staining with o-dianisidine revealed no overt signs of hemorrhage in 1-week old f2+/+ or f2Δ14/Δ14 larvae (data not shown).
Figure 6.

Loss of thrombin activity results in defects in secondary hemostasis. (A) Larvae were immobilized in agarose, subjected to laser-mediated endothelial injury (green arrow) of the venous (PCV, blue) or arterial (dorsal aorta, red) circulation, and followed for 2 minutes by a blinded observer. (B) Genetic ablation of f2 resulted in the inability to form induced PCV thrombi at 3 dpf and was not influenced by inhibiting fibrinolysis (ɛ-aminocaproic acid treatment, blue). (C) Overexpression of human F2 cDNA (blue) rescued the ability to form thrombi in the PCV at 3 dpf. (D) Homozygous mutant larvae demonstrated a significant impairment in arterial thrombus formation at 5 and 6 dpf without any changes in the time to initial thrombocyte attachment (E). (F) The number of thrombocytes attached to the site of injury in 2 minutes was significantly increased at 6 dpf in f2 homozygous mutants. Statistical significance assessed by Mann-Whitney U testing.
An intact kringle 1 domain is important for normal prothrombin levels
To interrogate how partial loss of the kringle 1 domain alters prothrombin function, a Δ15 version of human prothrombin was generated, corresponding to the zebrafish deletion. As this deletion removes C114 and results in a free-thiol at C138, two additional prothrombin variants were generated (C138A and C138A/Δ15). Transient expression in HEK293T cells led to wild-type secretion >100 ng/mL while all mutants were <3 ng/mL and not significantly different from the non-transfected control (Fig. 7A). Furthermore, measurement of prothrombin in the cell lysate revealed >10 ng/mL for wild-type expression while two of the three mutants (C138A and C138A/Δ15) were <5 ng/mL and not significantly different from control. The remaining mutant, Δ15, was significantly different than control but on average produced less protein than the wild-type product (Fig. 7B). Overall, this suggests that the loss of these residues within kringle 1 impaired biosynthesis or stability and led to a significant reduction in overall secretion.
Figure 7.

Expression and activation of prothrombin variants and resulting activity of thrombin upon activation. (A) Measurement of prothrombin in the cell media by ELISA demonstrated that expression of prothrombin variants in HEK293T cells resulted in reduced secretion levels that were indistinguishable from control and (B) corresponding measurement of prothrombin in the cell lysate demonstrated decreased biosynthesis. (C) Rates of prothrombin activation by prothrombinase measured using DAPA. Once prothrombin variants were fully activated by prothrombinase, their ability to cleave synthetic substrate S-2238 (D) or fibrinogen (E) were monitored, with significant differences in the latter, but not the former. For both S-2238 and fibrinogen, the rate was calculated during the initial reaction phase when the substrate is non-limiting and the conditions are presumed to be steady-state. Rate was then normalized to enzyme concentration. (F) Clotting profile between wild-type and C138A/Δ15 demonstrated delayed clot initiation and altered turbidity. (G) The rates of clot formation were determined from the clotting profile and reduced in the mutant.
The kringle 1 mutant has impaired endogenous activity despite an intact protease domain
The C138A/Δ15 prothrombin was expressed from a stably transfected HEK293 cell line, purified, and its activation by prothrombinase was quantified. The quantum yield of fluorescence signal change observed upon thrombin-DAPA complex formation was comparable between wild-type (3262 ± 347 RFU/µM) and C138A/Δ15 (3354 ± 180 RFU/µM), consistent with the protease domain remaining intact. However, the initial rate of activation, defined as the linear phase of steady state kinetics during the first 15–20% of the reaction, was 3.3-fold faster for wild-type (19.5 ± 1.4 s−1) compared with C138A/Δ15 (5.8 ± 0.5 s−1) (Fig. 7C).
The functionality of thrombin generated from the mutant prothrombin variant was then investigated. The amidolytic activity of thrombin towards S-2238 was similar between wild-type (82.7 ± 6.5 mOD/min) and C138A/Δ15 (89.0 ± 6.4 mOD/min) (Fig. 7D). However, C138A/Δ15-thrombin had a significantly prolonged clot time (200.6 ± 5.3 s) compared to wild-type (67.7 ± 3.1 s) (Fig. 7E), mostly due to delayed clot initiation and 1.5-fold higher total turbidity change (Fig. 7F) compared with wild-type. The rate of clot formation was also slower (82.4 ± 6.3 mOD/min) compared with the wild-type (135.9 ± 12.3 mOD/min) (Fig. 7G). These data suggest that thrombin derived from C138A/Δ15 prothrombin has normal catalytic properties towards small peptidyl substrates, but impaired activity towards macromolecular substrates like fibrinogen.
Discussion
The zebrafish model has been developed as a useful tool for understanding coagulation, especially during early development. In contrast to mammals, in which a number of coagulation factors are necessary for embryonic and/or neonatal viability, zebrafish are able to survive the loss of many aspects of the canonical cascade at least until early adulthood. Loss of antithrombin, fibrinogen, FV, and FX are all compatible with development to adulthood22,26–28,37. Targeting of f10 in fish results in the absence of larval hemostasis, but is accompanied by extended survival28. These data suggest that there could be residual thrombin activity present. Previous studies have utilized transient knockdown and chemical inhibition to study the loss of prothrombin in zebrafish29,38–40, providing valuable insight into the conserved function of prothrombin and suggesting a potential role for the coagulation cascade in development. Unfortunately, these technologies are susceptible to toxicity and off target effects. Additionally, specific chemical inhibition targets the proteolytic activity of thrombin without addressing the potential for exosite mediated binding of other factors. We sought to leverage knockout technology to create a specific genetic knockout of f2. We produced a genomic deletion in exon 6 with the intent of creating a nonsense mutation. Unexpectedly, we found reduced expression of an alternatively spliced version of prothrombin lacking a conserved cysteine in the kringle 1 domain. In vitro biochemical studies demonstrated that this protein is synthesized and secreted at a low level, and the secreted protein has decreased activation and activity. In vivo, the Δ15 mutant results in the loss of the ability to form fibrin rich clots and lethality by 2 months of age due to spontaneous hemorrhage. However, thrombocyte attachment was unaffected, consistent with the dogma that platelet adherence is mediated primarily via von Willebrand factor and collagen binding41–43. Overall, our data suggest that this is likely a severe hypomorph, if not functionally a complete null. Given that f2Δ14/Δ14 phenocopies the previously described f10 knockout, it appears that any residual thrombin activity is likely to be physiologically negligible.
Surprisingly, no obvious developmental defects were observed, including grossly normal vascular development, with survival through early adulthood. Previous work using antisense technology has demonstrated a variety of developmental malformations including hemorrhage, and circulatory, brain, and tailbud malformations38. These defects were not observed across multiple clutches in our knockout. This inconsistency could be due generally to off target effects of antisense knockdown or genetic compensation that can occur in response to genomic editing44. Conversely, the discrepancy may be a result of the fibrinogen-independent residual activity of the mutant thrombin, as detected in the amidolytic assay. Notably, the victoria mutant, which mapped close to f2 and is likely a mutant allele, similarly affects induced thrombus formation without a described developmental defect45, consistent with our data.
Kringle 1 is an understudied domain of prothrombin, but it is thought to interact directly with FVa and regulate prothrombin activation11. For the first time, we demonstrate that a defect in the kringle 1 domain may lead to decreased levels of prothrombin in circulation as well as decreased thrombin generation, thus contributing to the observed coagulopathy. The impairment of overall thrombin activity occurs at multiple levels. First, presumed inefficiency of the cryptic splice site in mRNA maturation leads to a roughly 45% reduction in transcript levels due to predicted nonsense mediated decay of the mutant transcript making use of the canonical splice site. Notably, splicing to the cryptic site is not seen in wild-type fish and a cryptic AG splice acceptor is not present in the human transcript. Second, when introduced into human prothrombin, the deletion resulted in a substantial decrease in secretion. It is possible that a large amount of the mutant prothrombin misfolds, aggregates, and is degraded intracellularly. Additionally, low secretion may be indicative of a more involved role of the kringle 1 domain in regulating prothrombin secretion as has been previously suggested46. The C138A/Δ15 prothrombin derivative (controlling for the unpaired C138 resulting from Δ15) showed a 3-fold reduction in its rate of activation by prothrombinase when compared with the wild-type prothrombin. In addition, thrombin generated from the C138A/Δ15 variant had a decreased procoagulant function (i.e. macromolecular substrates) while maintaining a functional active site as demonstrated by the unaltered S-2238 hydrolysis (i.e. small synthetic substrates) when compared with wild-type thrombin. Although similar thrombin functionality towards S-2238 was expected since the mutations did not involve the protease domain of prothrombin (i.e. thrombin), the delineation of substrate specificity towards fibrinogen was unexpected. This variation in substrate specificity could be explained by incomplete cleavage leading to an accumulation of a meizothrombin intermediate47. However, we did not observe the associated increase in DAPA-thrombin fluorescence predicted by increased meizothrombin presence. Conversely, our findings propose the potential role of F1.2 in the substrate recognition and specificity of thrombin, whereby the disruption of the kringle 1 domain leads to reduced fibrinogen recognition and/or cleavage by the F1.2:thrombin complex. Therefore, our data suggest that F1.2 is more than just a peptide fragment that results in product-inhibition of thrombin48. Overall, the compounding reductions in transcription, secretion, activation, and activity show that this genomic deletion results in a severe hypomorph. At the same time our data propose a role for the kringle 1 domain in the activation kinetics and subsequent substrate recognition of thrombin.
The ability of zebrafish to survive severe hemostatic imbalance may be due to a combination of influences, including the absence of birthing trauma, limited hemostatic challenges in an aqueous laboratory environment, a relatively low systolic blood pressure, and possible species-specific genetic differences. Nevertheless, the phenotypes of all zebrafish hemostatic mutants eventually converge with their mammalian counterparts in adulthood. Overall, this study further demonstrates the conservation of the coagulation cascade in fish while leveraging unique physiologic differences to build our understanding of the complex biological functions of thrombin. Furthermore, understanding how fish tolerate such a severe bleeding diathesis could provide new insights into managing patients with congenital bleeding disorders or acquired hemorrhage, with future studies leveraging the model system to develop new diagnostic markers and therapies.
Methods
Animal care
Zebrafish were maintained according to protocols approved by the University of Michigan Animal Care and Use Committee. All wild-type fish were a hybrid line generated by crossing AB and TL fish acquired from the Zebrafish International Resource Center. A hybrid line was selected to minimize strain specific effects and for improved reproductive success49. Tg(cd41:egfp) fish were used for tracking of fluorescently labeled thrombocytes50. Tris-buffered tricaine methanosulfate (Western Chemical) was used for anesthesia during all live animal experimental manipulation and for euthanasia.
Sequence analysis
Prothrombin protein sequences (NP_000497.1, NP_034298.1, NP_989936.1, NP_998555.1, NP_001015797.1) were downloaded from the National Center for Biotechnology Information RefSeq database51. Homology predicted propeptides were removed and the prothrombin numbering scheme52 was used relative to the human prothrombin sequence unless otherwise noted. Zebrafish sequence was modified to include an incompletely annotated exon. Sequences were aligned using MUSCLE53 and shaded using the Boxshade server (https://embnet.vital-it.ch/software/BOX_form.html).
Targeted mutagenesis using TALENs
The f2 coding sequence was identified in the zebrafish reference genome (Gene ID: 325881)54. Factoring in technical design limitations and predicted efficacy, an optimal target site 5’ to the protease domain was selected in exon 6 for mutagenesis. TALEN constructs were created using fast ligation-based solid-phase high-throughput (FLASH) assembly targeted to exon 6 of the zebrafish genomic f2 locus and validated as described55,56. TALEN mRNA was transcribed from plasmids and injected into single cell embryos. The resulting chimeric founders were raised, outcrossed, and offspring screened for deleterious mutations. A single founder heterozygous for a 14 base pair deletion (f2Δ14) in the coding sequence was identified and crossed to wild-type fish to establish the mutant line.
Genotyping of mutant offspring
Whole embryos or adult fin biopsies were lysed in buffer (10 mM Tris-HCl, pH 8.0, 2 mM EDTA, 2% Triton X-100, 100 µg/mL proteinase K) for 2 hours at 55 °C followed by incubation at 95 °C for 5 minutes. One microliter of the resulting solution was used as template for gene specific polymerase chain reaction (Table S1) and analyzed by gel electrophoresis.
Histochemical analysis
For in situ hybridization, DIG-labeled riboprobes (DIG RNA-labeling kit, Roche) were synthesized using 2-day old wild type embryonic cDNA and gene specific primers with T7 overhangs (Table S1). Embryos were fixed overnight at 4 °C in 4% paraformaldehyde in phosphate buffered saline prior to dehydration. Permeabilization and staining were performed as described57. Stained samples were evaluated by phenotype prior to genotyping.
For hematoxylin and eosin staining, juvenile zebrafish (30–89 days) were fixed overnight in 4% phosphate buffered paraformaldehyde and embedded in paraffin. Sagittal sections (3 µm) were collected every 50 µm and stained.
Single molecule real-time sequencing of RNA
Three f2+/Δ14 larvae were homogenized at 5 dpf in lysis buffer using a 21-gauge syringe. Total RNA was purified from the lysate using the PureLink RNA Mini Kit (Life Technologies) followed by DNAse I treatment (Invitrogen) and cDNA synthesis using the Superscript III First Strand cDNA kit (Invitrogen). Primers (Table S1) were used to amplify a 918 base pair (bp) region surrounding the predicted deletion. This region included several non-deleterious single nucleotide polymorphisms (SNPs) in downstream exons known to be allelic with either the wild-type or mutant alleles. Purified products from the 3 samples were pooled and sent to the University of Michigan Sequencing Core for library preparation and single molecule real-time high throughput sequencing (SMRT, Pacific Biosciences). Circular consensus reads with at least 5x coverage were filtered for full-length single inserts. After filtering for quality, the resulting 3826 reads were sorted by haplotype and splice variation.
Quantitative real-time PCR
Total RNA was extracted from 7 days post fertilization larvae (dpf) using the RNeasy Mini Plus kit (Qiagen) and transcribed using oligo(dT)12–18 primer and Superscript III (Invitrogen). Three pools of three whole larvae were used per genotype. The resulting cDNA was used as template for qPCR (StepOnePlus, Applied Biosystems) using gene specific PrimeTime© probe-based qPCR Assays (Integrated DNA Technologies) (Table S1). The expression level of f2 was normalized to the actb2 gene and significance analyzed using the double delta Ct method as described58. Transcript flanking the deletion was amplified using allele unbiased primers and separated by capillary gel electrophoresis (Table S1). Molar ratios were calculated from standardized relative band intensities using QIAxcel ScreenGel software (Qiagen).
Laser-induced endothelial injury
Anesthetized zebrafish larvae were embedded in 0.8% low melting point agarose and oriented in the sagittal position on a glass coverslip. Larvae were then positioned under an Olympus IX73 inverted microscope with an attached pulsed nitrogen dye laser (Micropoint, Andor Technology). For venous injury, ninety-nine laser pulses were administered to the luminal surface of the endothelium on the ventral side of the posterior cardinal vein (PCV) 5 somites posterior to the anal pore of 3 dpf larvae as previously described59. For arterial injury, pulses were administered to the endothelial surface of the dorsal aorta 3 somites posterior to the anal pore of 5 dpf larvae59. Following injury, time to occlusion and/or time to thrombocyte attachment were monitored for 2 minutes. Larvae were manually extracted from agarose for genotyping.
Chemical treatment
Dabigatran etexilate, apixaban, and rivaroxaban were dissolved in DMSO and diluted in embryo water to final concentrations of 50, 100, and 250 µM, respectively. At 5 dpf larvae were treated for 24 hours prior to laser-mediated endothelial injury on day 6. For ε-aminocaproic acid (Sigma) treatment, zebrafish embryos were incubated in 100 mM at 1 dpf and the time to occlusion was evaluated at 3 dpf following laser-induced endothelial injury.
O-dianisidine staining
Anesthetized larvae were incubated in the dark at 4 dpf for 30 minutes in o-dianisidine staining solution as previously described60,61. Larvae were subsequently fixed overnight at 4 °C in 4% paraformaldehyde, and pigment was removed using bleaching solution (1% KOH, 3% H2O2).
Human prothrombin expression vector construction and injection
Plasmid pF2-bio-His was obtained from Addgene (plasmid #52179) and human F2 cDNA amplified using primers containing vector sequence homology (Table S1). pcDNA3.1 was digested with BstBI and KpnI, followed by gel purification of the linearized backbone. The NEBuilder kit was used to fuse the F2 cDNA with linearized vector and transformed into 10beta competent cells (NEB). Site directed mutagenesis was used for missense mutation introduction62 and NEBuilder end homology joining was used to create internal deletions. Replacement of residues 100 to 115 of human F2 with a glutamic acid resulted in a homologous 15 amino acid deletion in human prothrombin (Δ15). As the deletion includes the cysteine residue at position 114, the effect of introducing a free-cysteine at position 138 due to the loss of its binding partner was investigated by generating two additional vectors that included an alanine substitution without (C138A) or with the 15-residue deletion (C138A/Δ15). For in vivo rescue assays, plasmid DNA (90 pg) in 0.1 M KCl was injected into one-cell stage embryos generated from heterozygous f2Δ14 incrosses using pulled glass capillary needles.
Human prothrombin protein expression and isolation
F2 mutant vectors were transfected into human embryonic kidney 293 T (HEK293T) cells in 6-well plates using Lipofectamine 3000 (ThermoFisher Scientific; L3000008) according to manufacturer’s instructions. The media and cells were collected after 3 days. The cells were washed with DPBS and lysed with 50 µL RIPA buffer on ice for 15 minutes. Prothrombin concentration in the expression media and cell lysate were quantified by ELISA using a matched-pair antibody set (Affinity Biologicals Inc., Ancaster, Canada), according to manufacturer’s instructions.
To characterize the functional characteristics of the prothrombin derivative, the plasmid containing the cDNA for prothrombin C138A/Δ15 was transfected and selected in HEK293 cells to generate a stable cell line. Using sustained rolling incubation at 37 °C, expression was induced in D-MEM supplemented with vitamin K1, and collected every 2–3 days63. The media were then centrifuged at 2,000 × g to remove cell debris, and the supernatant was filtered through a 0.22 µm filter (Millipore). The media was loaded onto tandem XAD2 and Q-Sepharose fast flow columns pre-equilibrated with 0.02 M Tris, 0.15 M NaCl, pH 7.4 (TBS) and eluted with 0.02 M Tris, 0.5 M NaCl, pH 7.4 as described previously64.
Prothrombin activation and activity
Prothrombin activation was quantified as described previously64. Briefly, prothrombin (0.1 µM) was incubated with PCPS vesicles (50 µM; 75% phosphatidylcholine, 25% phosphatidylserine), FVa (20 nM), CaCl2 (5 mM), and dansylarginine N-(3-ethyl-1,5-pentanediyl)amide (1 µM, DAPA) in 0.02 M HEPES, 0.15 M NaCl, pH 7.4 with 0.01% Tween80 (HBST). The reactions were initiated by the addition of FXa (70 pM) and activation was monitored for fluorescence change at 1 minute intervals using a SpectraMax M2 plate reader (Molecular Devices). Excitation and emission spectra were 280 nm and 540 nm, respectively, with an emission cutoff filter set at 530 nm. The quantum yield of the thrombin-DAPA complex was determined by plotting the total signal change observed with respect to known concentrations of the thrombin-DAPA complex12.
To determine the resulting thrombin activity, prothrombin was activated to thrombin by prothrombinase as described above. After a 30-minute incubation, complete prothrombin activation was verified by SDS-PAGE (not shown). The resulting thrombin (5 nM) was simultaneously added to either a thrombin-specific substrate S-2238 (400 µM; DiaPharma, West Chester, OH) or fibrinogen (3 µM), all in the presence of 5 mM CaCl2 in HBST. Both reactions were monitored at 405 nm using SpectraMax M2 plate reader (Molecular Devices) to quantify the amidolytic (S-2238) or clotting (fibrinogen) activity of thrombin.
Statistical analysis
The occlusion data were analyzed using Mann-Whitney U or two-tailed Student t tests. Survival was evaluated by log-rank (Mantel-Cox) testing. Significance testing, graphs, and survival curves were made using GraphPad Prism (Graphpad Software, La Jolla, California). P-values (p < 0.05 or p < 0.0001) were used to evaluate statistical significance.
All data generated or analyzed during this study are included in this published article (and its Supplementary Information Files).
Supplementary Information
Acknowledgements
The authors thank the University of Michigan Sequencing and Microscopy & Image Analysis cores for services. This work was supported by National Institutes of Health grants R01 HL124232 and R01 HL125774 (J.A.S.), and T32 GM007863, T32 HL125242, and an American Heart Association Predoctoral Fellowship Award (S.J.G.), a Hemophilia of Georgia Clinical Scientist Development Grant, the National Hemophilia Foundation/Novo Nordisk Career Development Award, and the Bayer Hemophilia Awards Program (J.A.S.), and NIH R01 GM088040 (J.K.J.). J.A.S. is the Diane and Larry Johnson Family Scholar of Pediatrics and the Henry and Mala Dorfman Family Professor of Pediatric Hematology/Oncology. This work was also supported by Natural Sciences and Engineering Research Council of Canada (RGPIN-2017-05347 to P.Y.K. and RGPIN-2017-04692 to C.A.K). P.Y.K. and C.A.K. hold the Department of Medicine Early Career Award (McMaster University).
Author contributions
S.J.G. designed and performed research, analyzed data and wrote the manuscript; Z.H. designed and performed research, analyzed data and assisted with figure design. D.R. and J.K.J. designed and performed research; Y.L., X.Y., A.C.F., H.M. and A.P.F. performed research. P.Y.K. and C.A.K. designed research, analyzed data, and edited the manuscript; J.A.S. designed, performed, and supervised research, analyzed data, and wrote the manuscript. All authors reviewed the manuscript.
Competing interests
J.A.S. has been a consultant for Bayer, Shire, CSL Behring, Spark Therapeutics, and NovoNordisk. J.K.J. has financial interests in Beam Therapeutics, Editas Medicine, Excelsior Genomics, Pairwise Plants, Poseida Therapeutics, Transposagen Biopharmaceuticals, and Verve Therapeutics (f/k/a Endcadia). J.K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.K.J. is a member of the Board of Directors of the American Society of Gene and Cell Therapy. J.K.J. is a co-inventor on various patents and patent applications that describe gene editing and epigenetic editing technologies. Additional authors have no competing interests as defined by Nature Research, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Steven J. Grzegorski and Zhilian Hu.
Supplementary information
is available for this paper at 10.1038/s41598-020-60840-7.
References
- 1.Davie EW, Kulman JD. An overview of the structure and function of thrombin. Semin. Thromb. Hemost. 2006;32(Suppl 1):3–15. doi: 10.1055/s-2006-939550. [DOI] [PubMed] [Google Scholar]
- 2.Wolberg Alisa S. Determinants of fibrin formation, structure, and function. Current Opinion in Hematology. 2012;19(5):349–356. doi: 10.1097/MOH.0b013e32835673c2. [DOI] [PubMed] [Google Scholar]
- 3.COUGHLIN S. R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. Journal of Thrombosis and Haemostasis. 2005;3(8):1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 4.Rosendaal, F. R. et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb. Haemost. (1998). [PubMed]
- 5.Poort, S. R., Rosendaal, F. R., Reitsma, P. H. & Bertina, R. M. A Common Genetic Variation in the 3′-Untranslated Region of the Prothrombin Gene Is Associated With Elevated Plasma Prothrombin Levels and an Increase in Venous Thrombosis. [PubMed]
- 6.Shavit, J. A. & Ginsburg, D. Hemophilias and Other Disorders of Hemostasis. In Emery and Rimoins Principles and Practice of Medical Genetics (eds. Rimoin, D., Pyeritz, R. & Korf, B.) 1–33 (2013).
- 7.Peyvandi F, Duga S, Akhavan S, Mannucci PM. Rare coagulation deficiencies. Haemoph. 2002;8:308–321. doi: 10.1046/j.1365-2516.2002.00633.x. [DOI] [PubMed] [Google Scholar]
- 8.Blanchard Rita A., Furie Barbara C., Jorgensen Maria, Kruger Steven F., Furie Bruce. Acquired Vitamin K-Dependent Carboxylation Deficiency in Liver Disease. New England Journal of Medicine. 1981;305(5):242–248. doi: 10.1056/NEJM198107303050502. [DOI] [PubMed] [Google Scholar]
- 9.Bristol, A. et al. Biosynthesis of prothrombin: intracellular localization of the vitamin K-dependent carboxylase and the sites of gamma-carboxylation. Blood (1996). [PubMed]
- 10.Kotkow Karen J., Deitcher Steven R., Furie Bruce, Furie Barbara C. The Second Kringle Domain of Prothrombin Promotes Factor Va- mediated Prothrombin Activation by Prothrombinase. Journal of Biological Chemistry. 1995;270(9):4551–4557. doi: 10.1074/jbc.270.9.4551. [DOI] [PubMed] [Google Scholar]
- 11.Deguchi, H., Takeya, H., Gabazza, E. C., Nishioka, J. & Suzuki, K. Prothrombin kringle 1 domain interacts with factor Va during the assembly of prothrombinase complex. Biochem J (1997). [DOI] [PMC free article] [PubMed]
- 12.Friedmann AP, et al. Identification and characterization of a factor Va-binding site on human prothrombin fragment 2. Sci. Rep. 2019;9:2436. doi: 10.1038/s41598-019-38857-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun WY, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Med. Sci. 1998;95:7597–7602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue J., Wu Q., Westfield L. A., Tuley E. A., Lu D., Zhang Q., Shim K., Zheng X., Sadler J. E. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proceedings of the National Academy of Sciences. 1998;95(13):7603–7607. doi: 10.1073/pnas.95.13.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dewerchin M, et al. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb. Haemost. 2000;83:185–190. doi: 10.1055/s-0037-1613783. [DOI] [PubMed] [Google Scholar]
- 16.Cui J, O’Shea KS, Purkayastha A, Saunders TL, Ginsburg D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nat. 1996;384:66–8. doi: 10.1038/384066a0. [DOI] [PubMed] [Google Scholar]
- 17.Mullins Eric S., Kombrinck Keith W., Talmage Kathryn E., Shaw Maureen A., Witte David P., Ullman Joni M., Degen Sandra J., Sun William, Flick Matthew J., Degen Jay L. Genetic elimination of prothrombin in adult mice is not compatible with survival and results in spontaneous hemorrhagic events in both heart and brain. Blood. 2009;113(3):696–704. doi: 10.1182/blood-2008-07-169003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ansell Jack, Hirsh Jack, Poller Leon, Bussey Henry, Jacobson Alan, Hylek Elaine. The Pharmacology and Management of the Vitamin K Antagonists. Chest. 2004;126(3):204S–233S. doi: 10.1378/chest.126.3_suppl.204S. [DOI] [PubMed] [Google Scholar]
- 19.Kretz CA, Weyand AC, Shavit JA. Modeling Disorders of Blood Coagulation in the Zebrafish. Curr. Pathobiol. Rep. 2015;3:155–161. doi: 10.1007/s40139-015-0081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dulley K, Zon LI. Zebrafish: a model system for the study of human disease. Curr. Opin. Genet. Dev. 2000;10:252–256. doi: 10.1016/S0959-437X(00)00074-5. [DOI] [PubMed] [Google Scholar]
- 21.Vo AH, Swaroop A, Liu Y, Norris ZG, Shavit JA. Loss of fibrinogen in zebrafish results in symptoms consistent with human hypofibrinogenemia. PLoS One. 2013;8:e74682. doi: 10.1371/journal.pone.0074682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, et al. Targeted mutagenesis of zebrafish antithrombin III triggers disseminated intravascular coagulation and thrombosis, revealing insight into function. Blood. 2014;124:142–50. doi: 10.1182/blood-2014-03-561027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanumanthaiah R, Day K, Jagadeeswaran P. Comprehensive Analysis of Blood Coagulation Pathways in Teleostei: Evolution of Coagulation Factor Genes and Identification of Zebrafish Factor VIIi. Blood Cells, Mol. Dis. 2002;29:57–68. doi: 10.1006/bcmd.2002.0534. [DOI] [PubMed] [Google Scholar]
- 24.Jagadeeswaran P, Sheehan JP. Analysis of blood coagulation in the zebrafish. Blood Cells. Mol. Dis. 1999;25:239–249. doi: 10.1006/bcmd.1999.0249. [DOI] [PubMed] [Google Scholar]
- 25.Jagadeeswaran P, Paris R, Rao P. Laser-induced thrombosis in zebrafish larvae: a novel genetic screening method for thrombosis. Methods Mol. Med. 2006;129:187–95. doi: 10.1385/1-59745-213-0:187. [DOI] [PubMed] [Google Scholar]
- 26.Fish Richard J., Di Sanza Corinne, Neerman-Arbez Marguerite. Targeted mutation of zebrafish fga models human congenital afibrinogenemia. Blood. 2014;123(14):2278–2281. doi: 10.1182/blood-2013-12-547182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu, Z. et al. Loss of fibrinogen in zebrafish results in an asymptomatic embryonic hemostatic defect and synthetic lethality with thrombocytopenia. J. Thromb. Haemost. (2019). [DOI] [PMC free article] [PubMed]
- 28.Hu Z, et al. Genome editing of factor X in zebrafish reveals unexpected tolerance of severe defects in the common pathway. Blood. 2017;130:666–676. doi: 10.1182/blood-2017-02-765206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jagadeeswaran Pudur, Liu Yuan C. Developmental Expression of Thrombin in Zebrafish Embryos: A Novel Model to Study Hemostasis. Blood Cells, Molecules, and Diseases. 1997;23(2):147–156. doi: 10.1006/bcmd.1997.0132. [DOI] [PubMed] [Google Scholar]
- 30.Reilly Paul A., Lehr Thorsten, Haertter Sebastian, Connolly Stuart J., Yusuf Salim, Eikelboom John W., Ezekowitz Michael D., Nehmiz Gerhard, Wang Susan, Wallentin Lars. The Effect of Dabigatran Plasma Concentrations and Patient Characteristics on the Frequency of Ischemic Stroke and Major Bleeding in Atrial Fibrillation Patients. Journal of the American College of Cardiology. 2014;63(4):321–328. doi: 10.1016/j.jacc.2013.07.104. [DOI] [PubMed] [Google Scholar]
- 31.Gulilat Markus, Tang Anthony, Gryn Steven E., Leong-Sit Peter, Skanes Allan C., Alfonsi Jeffrey E., Dresser George K., Henderson Sara L., Rose Rhiannon V., Lizotte Daniel J., Teft Wendy A., Schwarz Ute I., Tirona Rommel G., Kim Richard B. Interpatient Variation in Rivaroxaban and Apixaban Plasma Concentrations in Routine Care. Canadian Journal of Cardiology. 2017;33(8):1036–1043. doi: 10.1016/j.cjca.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Monroe Dougald M., Hoffman Maureane. A mouse bleeding model to study oral anticoagulants. Thrombosis Research. 2014;133:S6–S8. doi: 10.1016/j.thromres.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Genmin, DeGuzman Francis R, Hollenbach Stanley J, Karbarz Mark J, Abe Keith, Lee Gail, Luan Peng, Hutchaleelaha Athiwat, Inagaki Mayuko, Conley Pamela B, Phillips David R, Sinha Uma. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nature Medicine. 2013;19(4):446–451. doi: 10.1038/nm.3102. [DOI] [PubMed] [Google Scholar]
- 34.Berghmans Stéphane, Butler Paul, Goldsmith Paul, Waldron Gareth, Gardner Iain, Golder Zoe, Richards Frances M., Kimber Gillian, Roach Alan, Alderton Wendy, Fleming Angeleen. Zebrafish based assays for the assessment of cardiac, visual and gut function — potential safety screens for early drug discovery. Journal of Pharmacological and Toxicological Methods. 2008;58(1):59–68. doi: 10.1016/j.vascn.2008.05.130. [DOI] [PubMed] [Google Scholar]
- 35.Koster, R. & Sassen, W. A. A molecular toolbox for genetic manipulation of zebrafish. Adv. Genomics Genet., 10.2147/agg.s57585 (2015).
- 36.Rupp R A, Snider L, Weintraub H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes & Development. 1994;8(11):1311–1323. doi: 10.1101/gad.8.11.1311. [DOI] [PubMed] [Google Scholar]
- 37.Weyand, A. et al. Analysis of factor V in zebrafish demonstrates minimal levels needed for hemostasis and risk stratifies human variants. BioRxiv, 10.1101/567453 (2019).
- 38.Day K. Knockdown of prothrombin in zebrafish. Blood Cells, Molecules, and Diseases. 2004;32(1):191–198. doi: 10.1016/j.bcmd.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Hanumanthaiah R, Thankavel B, Day K, Gregory M, Jagadeeswaran P. Developmental Expression of Vitamin K-Dependent Gamma-Carboxylase Activity in Zebrafish Embryos: Effect of Warfarin. Blood Cells, Mol. Dis. 2001;27:992–999. doi: 10.1006/bcmd.2001.0472. [DOI] [PubMed] [Google Scholar]
- 40.Day Kenneth R., Jagadeeswaran Pudur. Microarray analysis of prothrombin knockdown in zebrafish. Blood Cells, Molecules, and Diseases. 2009;43(2):202–210. doi: 10.1016/j.bcmd.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.REININGER A. J. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia. 2008;14:11–26. doi: 10.1111/j.1365-2516.2008.01848.x. [DOI] [PubMed] [Google Scholar]
- 42.Bryckaert Marijke, Rosa Jean-Philippe, Denis Cécile V., Lenting Peter J. Of von Willebrand factor and platelets. Cellular and Molecular Life Sciences. 2014;72(2):307–326. doi: 10.1007/s00018-014-1743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruggeri Zaverio M. Structure of von Willebrand factor and its function in platelet adhesion and thrombus formation. Best Practice & Research Clinical Haematology. 2001;14(2):257–279. doi: 10.1053/beha.2001.0133. [DOI] [PubMed] [Google Scholar]
- 44.Rossi Andrea, Kontarakis Zacharias, Gerri Claudia, Nolte Hendrik, Hölper Soraya, Krüger Marcus, Stainier Didier Y. R. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524(7564):230–233. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]
- 45.Gregory Michael, Hanumanthaiah Ravikumar, Jagadeeswaran Pudur. Genetic Analysis of Hemostasis and Thrombosis Using Vascular Occlusion. Blood Cells, Molecules, and Diseases. 2002;29(3):286–295. doi: 10.1006/bcmd.2002.0568. [DOI] [PubMed] [Google Scholar]
- 46.Wu W., Bancroft J. D., Suttie J. W. Structural features of the kringle domain determine the intracellular degradation of under- -carboxylated prothrombin: Studies of chimeric rat/human prothrombin. Proceedings of the National Academy of Sciences. 1997;94(25):13654–13660. doi: 10.1073/pnas.94.25.13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doyle, M. F. & Mann, K. G. Multiple active forms of thrombin. IV. Relative activities of meizothrombins. J. Biol. Chem. (1990). [PubMed]
- 48.Dasgupta Swapan Kumar, Thiagarajan Perumal. Inhibition of thrombin activity by prothrombin activation fragment 1.2. Journal of Thrombosis and Thrombolysis. 2007;24(2):157–162. doi: 10.1007/s11239-007-0018-8. [DOI] [PubMed] [Google Scholar]
- 49.Monson Christopher A., Sadler Kirsten C. Inbreeding Depression and Outbreeding Depression Are Evident in Wild-Type Zebrafish Lines. Zebrafish. 2010;7(2):189–197. doi: 10.1089/zeb.2009.0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin H-F, et al. Analysis of thrombocyte development in CD41-GFP transgenic zebrafish. Blood. 2005;106:3803–10. doi: 10.1182/blood-2005-01-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Leary Nuala A., Wright Mathew W., Brister J. Rodney, Ciufo Stacy, Haddad Diana, McVeigh Rich, Rajput Bhanu, Robbertse Barbara, Smith-White Brian, Ako-Adjei Danso, Astashyn Alexander, Badretdin Azat, Bao Yiming, Blinkova Olga, Brover Vyacheslav, Chetvernin Vyacheslav, Choi Jinna, Cox Eric, Ermolaeva Olga, Farrell Catherine M., Goldfarb Tamara, Gupta Tripti, Haft Daniel, Hatcher Eneida, Hlavina Wratko, Joardar Vinita S., Kodali Vamsi K., Li Wenjun, Maglott Donna, Masterson Patrick, McGarvey Kelly M., Murphy Michael R., O'Neill Kathleen, Pujar Shashikant, Rangwala Sanjida H., Rausch Daniel, Riddick Lillian D., Schoch Conrad, Shkeda Andrei, Storz Susan S., Sun Hanzhen, Thibaud-Nissen Francoise, Tolstoy Igor, Tully Raymond E., Vatsan Anjana R., Wallin Craig, Webb David, Wu Wendy, Landrum Melissa J., Kimchi Avi, Tatusova Tatiana, DiCuccio Michael, Kitts Paul, Murphy Terence D., Pruitt Kim D. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 2015;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jenny, N. S., Lundblad, R. L. & Mann, K. G. Thrombin. in Hemostasis and Thrombosis: Basic Principles and Clinical Practice (eds. Colman, R. W., Marder, V. J., Clowes, A. W., George, J. N. & Goldhaber, S. J.) 193–214 (2008).
- 53.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howe Kerstin, Clark Matthew D., Torroja Carlos F., Torrance James, Berthelot Camille, Muffato Matthieu, Collins John E., Humphray Sean, McLaren Karen, Matthews Lucy, McLaren Stuart, Sealy Ian, Caccamo Mario, Churcher Carol, Scott Carol, Barrett Jeffrey C., Koch Romke, Rauch Gerd-Jörg, White Simon, Chow William, Kilian Britt, Quintais Leonor T., Guerra-Assunção José A., Zhou Yi, Gu Yong, Yen Jennifer, Vogel Jan-Hinnerk, Eyre Tina, Redmond Seth, Banerjee Ruby, Chi Jianxiang, Fu Beiyuan, Langley Elizabeth, Maguire Sean F., Laird Gavin K., Lloyd David, Kenyon Emma, Donaldson Sarah, Sehra Harminder, Almeida-King Jeff, Loveland Jane, Trevanion Stephen, Jones Matt, Quail Mike, Willey Dave, Hunt Adrienne, Burton John, Sims Sarah, McLay Kirsten, Plumb Bob, Davis Joy, Clee Chris, Oliver Karen, Clark Richard, Riddle Clare, Elliott David, Threadgold Glen, Harden Glenn, Ware Darren, Begum Sharmin, Mortimore Beverley, Kerry Giselle, Heath Paul, Phillimore Benjamin, Tracey Alan, Corby Nicole, Dunn Matthew, Johnson Christopher, Wood Jonathan, Clark Susan, Pelan Sarah, Griffiths Guy, Smith Michelle, Glithero Rebecca, Howden Philip, Barker Nicholas, Lloyd Christine, Stevens Christopher, Harley Joanna, Holt Karen, Panagiotidis Georgios, Lovell Jamieson, Beasley Helen, Henderson Carl, Gordon Daria, Auger Katherine, Wright Deborah, Collins Joanna, Raisen Claire, Dyer Lauren, Leung Kenric, Robertson Lauren, Ambridge Kirsty, Leongamornlert Daniel, McGuire Sarah, Gilderthorp Ruth, Griffiths Coline, Manthravadi Deepa, Nichol Sarah, Barker Gary, Whitehead Siobhan, Kay Michael, Brown Jacqueline, Murnane Clare, Gray Emma, Humphries Matthew, Sycamore Neil, Barker Darren, Saunders David, Wallis Justene, Babbage Anne, Hammond Sian, Mashreghi-Mohammadi Maryam, Barr Lucy, Martin Sancha, Wray Paul, Ellington Andrew, Matthews Nicholas, Ellwood Matthew, Woodmansey Rebecca, Clark Graham, Cooper James D., Tromans Anthony, Grafham Darren, Skuce Carl, Pandian Richard, Andrews Robert, Harrison Elliot, Kimberley Andrew, Garnett Jane, Fosker Nigel, Hall Rebekah, Garner Patrick, Kelly Daniel, Bird Christine, Palmer Sophie, Gehring Ines, Berger Andrea, Dooley Christopher M., Ersan-Ürün Zübeyde, Eser Cigdem, Geiger Horst, Geisler Maria, Karotki Lena, Kirn Anette, Konantz Judith, Konantz Martina, Oberländer Martina, Rudolph-Geiger Silke, Teucke Mathias, Lanz Christa, Raddatz Günter, Osoegawa Kazutoyo, Zhu Baoli, Rapp Amanda, Widaa Sara, Langford Cordelia, Yang Fengtang, Schuster Stephan C., Carter Nigel P., Harrow Jennifer, Ning Zemin, Herrero Javier, Searle Steve M. J., Enright Anton, Geisler Robert, Plasterk Ronald H. A., Lee Charles, Westerfield Monte, de Jong Pieter J., Zon Leonard I., Postlethwait John H., Nüsslein-Volhard Christiane, Hubbard Tim J. P., Crollius Hugues Roest, Rogers Jane, Stemple Derek L. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reyon Deepak, Tsai Shengdar Q, Khayter Cyd, Foden Jennifer A, Sander Jeffry D, Joung J Keith. FLASH assembly of TALENs for high-throughput genome editing. Nature Biotechnology. 2012;30(5):460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foley Jonathan E, Maeder Morgan L, Pearlberg Joseph, Joung J Keith, Peterson Randall T, Yeh Jing-Ruey J. Targeted mutagenesis in zebrafish using customized zinc-finger nucleases. Nature Protocols. 2009;4(12):1855–1868. doi: 10.1038/nprot.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thisse Christine, Thisse Bernard. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature Protocols. 2007;3(1):59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 58.Schmittgen Thomas D, Livak Kenneth J. Analyzing real-time PCR data by the comparative CT method. Nature Protocols. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 59.Rost M.S., Grzegorski S.J., Shavit J.A. Methods in Cell Biology. 2016. Quantitative methods for studying hemostasis in zebrafish larvae; pp. 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iuchi Ichiro, Yamamoto Masamichi. Erythropoiesis in the developing rainbow trout,Salmo gairdneri irideus: histochemical and immunochemical detection of erythropoietic organs. Journal of Experimental Zoology. 1983;226(3):409–417. doi: 10.1002/jez.1402260311. [DOI] [PubMed] [Google Scholar]
- 61.Paffett-Lugassy, N. & Zon, L. Analysis of hematopoietic development in the zebrafish. Methods in molecular medicine 171–198 (2005). [DOI] [PubMed]
- 62.Liu Huanting, Naismith James H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnology. 2008;8(1):91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orcutt Steven J., Krishnaswamy Sriram. Binding of Substrate in Two Conformations to Human Prothrombinase Drives Consecutive Cleavage at Two Sites in Prothrombin. Journal of Biological Chemistry. 2004;279(52):54927–54936. doi: 10.1074/jbc.M410866200. [DOI] [PubMed] [Google Scholar]
- 64.Kim Paul Y., Nesheim Michael E. Further Evidence for Two Functional Forms of Prothrombinase Each Specific for Either of the Two Prothrombin Activation Cleavages. Journal of Biological Chemistry. 2007;282(45):32568–32581. doi: 10.1074/jbc.M701781200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.