

The Schlafen (SLFN) Family
The Schlafen (SLFN) family of genes, which is limited to mammals, was initially discovered in mice by Schwarz et al., investigating the T cell lineage, regulating differentiation and in some instances ablating growth [1]. In particular, overexpression of SLFN1 resulted in a cell cycle arrest at the G0/G1 stage. Therefore, the family had been named Schlafen, which is translated from German as “to sleep” [1]. Subsequent studies have classified the SLFN family into three distinct subgroups based on gene/protein size and domain homology [2,3]. Ten mouse and six human SLFN genes have been identified to date (Figure 1) [4]. All 10 mouse SLFN proteins possess a core region containing a unique “slfn box” motif with an unknown function. Subgroups II and III contain an extra domain at the C-terminus of the slfn box, conserved by the flanking five amino acid signature (Ser-Trp-Ala … -Asp-Leu) ‘SWA … DL’ which appears to be SLFN specific. This was discovered in early characterization of the SLFN family with SLFN3 and SLFN4 (members 3 and 4) possessing a 200 amino acid sequence not found in those in subgroup I [1]. Adjacent to the slfn box is the ‘ATPases associated with diverse cellular activities’ (AAA) domain. Based on protein homology studies, the AAA motif is thought to function similarly to classical AAA domains with the role of ATP/GTP binding in the course of DNA and RNA metabolism and therefore playing a fundamental role in the production and function of SLFN proteins [5,6]. Another protein was discovered with significant similarity to those in subgroup II, extending a further 400 amino acids towards the C-terminus terminal tail. The sequence was aligned by BLAST and the first 570 amino acids were homologous to SLFNs 3 and 4 while the remainder was unique to named SLFN8 leading to the classification of the final SLFN subgroup, III [2]. Additional homologous genes were identified as SLFN5, SLFN9, SLFN10 and SLFN14, whereby the last two coding exons correspond to this unique subgroup. NCBI conserved domain database (CDD) searches revealed significant homology of subgroup III specific extension to motifs typical in superfamily I of RNA/DNA helicases which are known to mediate DNA and RNA metabolism [2]. The same characterization method applies to the human-specific SLFN genes with the absence of any subgroup I members and structural difference in SLFN12L (SLFN12-LIKE). Notably, SLFN5 and SLFN14 are the only SLFN family genes present in both human and mouse. The predicted transcript of SLFN12L is of a similar length to those in subgroup III; however, it also encompasses sequences which are unaccounted for and based on this structural analysis, has not yet been assigned a subgroup. According to length and homology, SLFN14 belongs to subgroup III (Figure 1) [7]. The recently discovered crystal structure of rat SLFN13 (related to human SLFN13 and mouse SLFN8) N-domain gives a functional insight into its ability to cleave RNA acting as an endoribonuclease in inhibiting protein synthesis [8]. Importantly, SLFN13 is the closest paralogue for SLFN14 (44% identity and 61% similarity).
Figure 1.
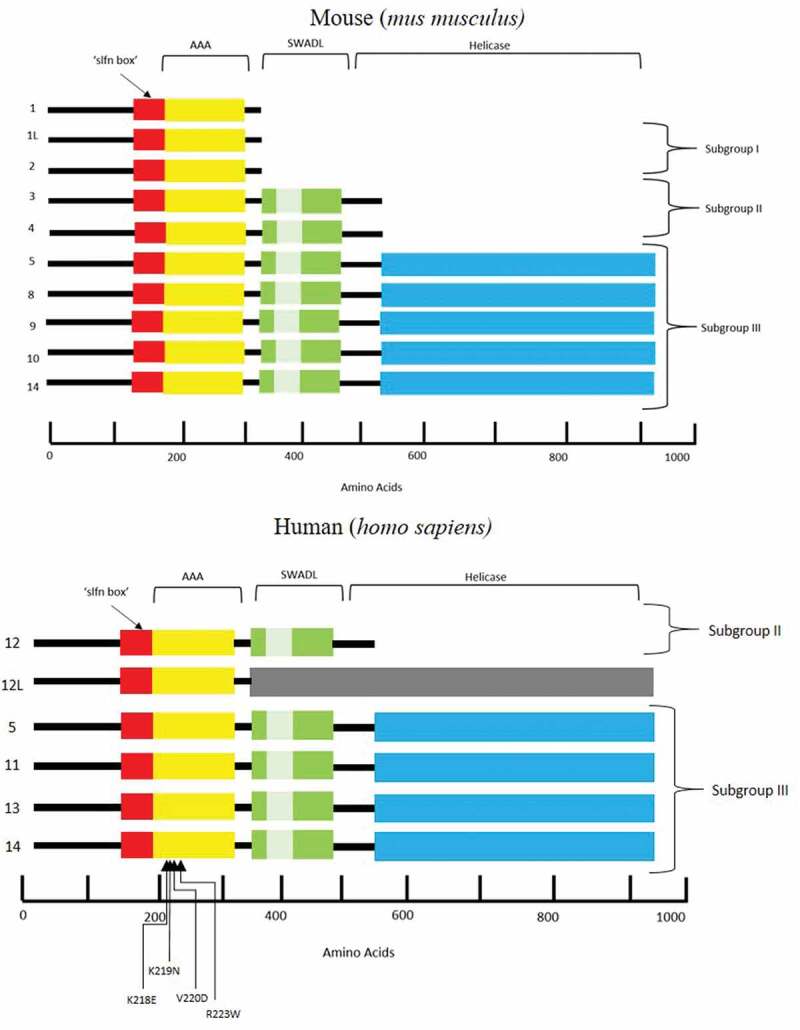
Schematic representation of the protein structure encoded by the SLFN family of genes, highlighting domains and regions in both mus musculus and homo sapiens.
‘slfn’ box is unique to SLFN proteins and function remains unknown. The AAA domain is responsible for DNA and RNA metabolism and the SWADL region, again believed to be SLFN specific, is a sequence flanked by SWA and DL amino acids. The helicase regions at the C-terminal end of the protein are known to mediate DNA and RNA metabolism. The involvement of mutations and thrombocytopenia within the AAA domain of the SLFN14 protein remains unclear.
SLFN14 in mice is located on chromosome 11 and comprised of five coding exons and in humans is on chromosome 17 with four coding exons [9]. SLFN genes are highly conserved throughout the mammalian species and are located in regions with other genes attributable to T-cell and macrophage development [1]. Early mouse knock-out studies investigating the role of SLFN1 in the immune response showed no phenotype, suggesting potential functional redundancy or minimal contribution of some members in the process [9]. The precise mechanisms of how some of the specific members of the SLFN family cause human disease is still unclear.
SLFN14 Mutations in Patients with Platelet Disorders and Bleeding
Platelet function disorders are associated with excessive bleeding and with the advent of next-generation sequencing (NGS) including whole exome sequencing (WES) the genetic impact of affected individuals can be evaluated. Initially, three extended families were investigated in the Genotyping and Phenotyping of Platelets Study, with three heterozygous single nucleotide variations in SLFN14 resulting in missense mutations of consecutive amino acids in the AAA domain [10]. The following year, an additional family was discovered with another missense mutation in the same region (R223W) and in 2019, Saes et al., reported a further patient with an alternative base change at nucleotide c.657 yet still resulting in the K219N mutation (Table I and Figure 1) [11,12]. All four mutations were inherited in an autosomal dominant fashion whereby amino acid changes are conserved across all mammalian species, except for V220D. The R223W variant is the only variant out of these families to be reported on gnomAD (genome aggregation database) with a frequency of 6.5e-6. This highlights variant rarity amongst different populations and confinement to the families described. All individuals had macrothrombocytopenia, characterized by a reduced platelet count (below 150x109/L) and enlarged platelets suggesting this is a typical feature of SLFN14 related thrombocytopenia. Patients from the three initial families showed a platelet function defect with impaired aggregation to ADP, protease-activated receptor-1 (PAR-1) and Collagen and reduced ATP secretion from dense granules after PAR-1 stimulation [10]. Platelet aggregation was not stated for the cases in family D or E but an unspecified secretion defect in the latter case was reported [12]. The proband in family A was 31 years of age at the time of measurement and had experienced severe cutaneous bruising, prolonged bleeding from wounds and menorrhagia [10]. These symptoms were consistent across the three generations and are all typical of moderate thrombocytopenia. The proband in family B was 35 years old and had a history of spontaneous epistaxis; her mother was also recruited but had not experienced any prior bleeding tendencies [10]. Family C index case was 3 years old at the time, displayed a very low platelet count and reported easy bruising since birth [10]. Patients with variant R223W also reported bleeding symptoms after dental surgery and mild menorrhagia, typical of thrombocytopenia [11]. Individual history for the patient in family E was not detailed in publication however the patient was recruited to the study based on bleeding tendency so, therefore, is likely to be similar to those with the same mutation [12].
Table I.
SLFN14 variants reported to date in patients with inherited macrothrombocytopenia, platelet-type bleeding disorder 20 (BDPLT20).
|
Patient |
Genomic Variant |
Protein effect |
Variant type |
Inheritance |
Platelet count (x109/L) |
Mean platelet volume (MPV fl) |
ISTH BAT Score |
Aggregation/Secretion Defect |
Reference |
| A; III 2 | c.659 T > A | p.V220D | Missense | Het | 140 | 9.1 | 5 | ADP, Collagen and PAR-1-activating peptide/ATP | [10] |
| A; III 3 | c.659 T > A | p.V220D | Missense | Het | 74 | 10.4 | 10 | ADP, Collagen and PAR-1-activating peptide/ATP | [10] |
| A; IV 2 | c.659 T > A | p.V220D | Missense | Het | 110 | 9.3 | 13 | ADP,Collagen and PAR-1-activating peptide/ATP | [10] |
| A; IV 4* | c.659 T > A | p.V220D | Missense | Het | 100 | 11.1 | 22 | ADP, Collagen and PAR-1-activating peptide/ATP | [10] |
| A; IV 5 | c.659 T > A | p.V220D | Missense | Het | 116 | 11.2 | 21 | ADP, Collagen and PAR-1-activating peptide/ATP | [10] |
| B; I 2 | c.657 A > T | p.K219N | Missense | Het | 83 | 11.9 | 13 | ADP, Collagen and PAR-1-activating peptide/ATP | [10] |
| B; II 3* | c.657 A > T | p.K219N | Missense | Het | 68 | 11.9 | 20 | ADP, Collagen and PAR-1-activating peptide/ATP | [10] |
| C; II 2* | c.652 A > G | p. K218E | Missense | Het | 89 | 13.0 | NA | ADP, Collagen and PAR-1-activating peptide/ATP | [10] |
| D; II 1 | c.667 C > T | p. R223W | Missense | Het | 87 | 12.1 | 5 | NA/NA | [11] |
| D; II 2 | c.667 C > T | p. R223W | Missense | Het | 91 | 21.0 | 2 | NA/NA | [11] |
| D; III 3* | c.667 C > T | p. R223W | Missense | Het | 79 | 12.3 | 9 | NA/NA | [11] |
| E; I 1* | c.657 A > C | p.K219N | Missense | Het | NR | NR | NR | NA/NR | [12] |
All patients identified with variants in the SLFN14 gene and affected by inherited bleeding. *:Proband in family case; Het: Heterozygous inheritance pattern; International Society on Thrombosis and Haemostasis Bleeding Assessment Tool (BAT) score; NA: Not Available for in vitro study; NR: Not Reported in publication. Thrombocytopenia was defined as platelet count <150x109/L
All patients in these reported families were heterozygous for the mutations and therefore phenotypically defined by platelet-type bleeding disorder 20 (BDPLT20) which is specific to heterozygous mutations in the SLFN14 gene. The absence of homozygous patients in the families suggests the mutant allele in each variant group has significant influence over the wild type. Indeed, heterozygous SLFN14 protein expression was reduced by 65%-80% when compared to controls and in platelet spreading assays there is a reduction of proplatelet extensions from developing megakaryocytes [10,11]. This apparent dominant negative effect on overall protein expression can impact the maturation of megakaryocytes, affecting differentiation and production of fully functional platelets [10,11]. In order to decipher the role of SLFN14 in platelet biogenesis, it has been demonstrated that SLFN14 acts as an endoribonuclease in controlling gene expression [13,14]. SLFN14 is strongly overexpressed in rabbit reticulocyte lysate where it is one of the major ribosome-associated proteins [13]. SLFN14 also binds to ribosomes in different model cell lines [14]. Upon binding, SLFN14 cleaves ribosomal RNA and ribosome-associated messenger RNA resulting in ribosomal degradation and disrupting translation [13,14]. Endoribonucleases mediate effective mechanisms in regulating gene expression; however, mutant variants of ribosome-related proteins, such as SLFN14, may cause errors in ribosome homeostasis and subsequent hematopoietic lineage dysfunction [15,16]. For this reason, we can hypothesize that SLFN14 contributes to improper platelet formation from the disrupted translation of their precursors, megakaryocytes, which then results in an inherited thrombocytopenia [14]. Given the structural similarity of SLFN13 to other subgroup III genes (such as SLFN14) and the recent report on SLFN13’s role as an endoribonuclease it seems plausible to hypothesise that SLFN14 may be impacting megakaryocyte differentiation and platelet synthesis by also acting as an endoribonuclease, inhibiting translation and preventing complete protein synthesis within the hematopoietic lineage [8].
It is only with the help of new genetic approaches over recent years, that inherited thrombocytopenias with previously unknown causes can now be attributed to single genetic defects. This not only allows for expanding knowledge within the field of genetics and hematological disorders but also provides patients with a clear diagnosis and a plan for tailored clinical management.
Main Findings
Mutations in SLFN14 cause inherited macrothrombocytopenia characterized by enlarged platelets and moderate to severe bleeding phenotypes.
Three out of the five families with SLFN14 genetic variants show reduced aggregation to agonists ADP, PAR-1 and Collagen, decreased ATP secretion and dominant inheritance pattern.
Endoribonucleolytic activity of SLFN14 contributes to platelet formation by regulating translation process during their maturation.
The exact role of SLFN14 in platelet biogenesis and how mutations within its AAA domain contribute to inherited thrombocytopenia remain to be explored.
Funding Statement
The work in these authors’ laboratory is supported by grants provided by the British Heart Foundation (PG/16/103/32650; FS/18/11/33443) to N.V.M and also supported by a National Institutes of Health Grant GM097014 to A.V.P.
Declaration of interest
The authors report no conflict of interest.
References
- 1.Schwarz DA, Katayama CD, Hedrick SM.. Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity 1998;9(5):657–668. [DOI] [PubMed] [Google Scholar]
- 2.Geserick P, Kaiser F, Klemm U, Kaufmann SHE, Zerrahn J. Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int Immunol 2004;16(10):1535–1548. doi: 10.1093/intimm/dxh155 [DOI] [PubMed] [Google Scholar]
- 3.Neumann B, Zhao L, Murphy K, Gonda TJ. Subcellular localization of the Schlafen protein family. Biochem Biophys Res Commun 2008;370(1):62–66. doi: 10.1016/j.bbrc.2008.03.032 [DOI] [PubMed] [Google Scholar]
- 4.Mavrommatis E, Arslan AD, Sassano A, Hua Y, Kroczynska B, Platanias LC. Expression and regulatory effects of murine Schlafen (Slfn) genes in malignant melanoma and renal cell carcinoma. J Biol Chem 2013;288(46):33006–33015. doi: 10.1074/jbc.M113.460741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 2005;6:519. doi: 10.1038/nrm1684 [DOI] [PubMed] [Google Scholar]
- 6.Lupas AN, Martin J. AAA proteins. Curr Opin Struct Biol 2002;12(6):746–753. [DOI] [PubMed] [Google Scholar]
- 7.Liu F, Zhou P, Wang Q, Zhang M, Li D. The Schlafen family: complex roles in different cell types and virus replication. Cell Biol Int 2018;42(1):2–8. doi: 10.1002/cbin.10778 [DOI] [PubMed] [Google Scholar]
- 8.Yang J-Y, Deng X-Y, Li Y-S, Ma X-C, Feng J-X, Yu B, Chen Y, Luo Y-L, Wang X, Chen M-L, et al. Structure of Schlafen13 reveals a new class of tRNA/rRNA- targeting RNase engaged in translational control. Nat Commun 2018;9(1):1165. doi: 10.1038/s41467-018-03544-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bustos O, Naik S, Ayers G, Casola C, Perez-Lamigueiro MA, Chippindale PT, Pritham EJ, de la Casa-Esperón E. Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene 2009;447(1):1–11. doi: 10.1016/j.gene.2009.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fletcher SJ, Johnson B, Lowe GC, Bem D, Drake S, Lordkipanidzé M, Guiú IS, Dawood B, Rivera J, Simpson MA, et al. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J Clin Invest 2015;125(9):3600–3605. doi: 10.1172/JCI80347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marconi C, Di Buduo CA, Barozzi S, Palombo F, Pardini S, Zaninetti C, Pippucci T, Noris P, Balduini A, Seri M, et al. SLFN14-related thrombocytopenia: identification within a large series of patients with inherited thrombocytopenia. Thromb Haemost 2016;115(5):1076–1079. doi: 10.1160/TH15-11-0884 [DOI] [PubMed] [Google Scholar]
- 12.Saes JL, Simons A, de Munnik SA, Nijziel MR, Blijlevens NMA, Jongmans MC, van der Reijden BA, Smit Y, Brons PP, van Heerde WL, et al. Whole exome sequencing in the diagnostic workup of patients with a bleeding diathesis. Haemophilia 2019;25(1):127–135. doi: 10.1111/hae.13638 [DOI] [PubMed] [Google Scholar]
- 13.Pisareva VP, Muslimov IA, Tcherepanov A, Pisarev AV. Characterization of novel ribosome-associated endoribonuclease SLFN14 from rabbit reticulocytes. Biochemistry 2015;54(21):3286–3301. doi: 10.1021/acs.biochem.5b00302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher SJ, Pisareva VP, Khan AO, Tcherepanov A, Morgan NV, Pisarev AV. Role of the novel endoribonuclease SLFN14 and its disease-causing mutations in ribosomal degradation. Rna 2018;24(7):939–949. doi: 10.1261/rna.066415.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ricciardi S, Miluzio A, Brina D, Clarke K, Bonomo M, Aiolfi R, Guidotti LG, Falciani F, Biffo S. Eukaryotic translation initiation factor 6 is a novel regulator of reactive oxygen species-dependent megakaryocyte maturation. J Thromb Haemost 2015;13(11):2108–2118. doi: 10.1111/jth.13150 [DOI] [PubMed] [Google Scholar]
- 16.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood 2010;115(16):3196–3205. doi: 10.1182/blood-2009-10-178129 [DOI] [PMC free article] [PubMed] [Google Scholar]