

MSC spheroids self-organize in a hierarchical manner, resulting in a spatial patterning of the function of individual cells.
Abstract
Organoids that recapitulate the functional hallmarks of anatomic structures comprise cell populations able to self-organize cohesively in 3D. However, the rules underlying organoid formation in vitro remain poorly understood because a correlative analysis of individual cell fate and spatial organization has been challenging. Here, we use a novel microfluidics platform to investigate the mechanisms determining the formation of organoids by human mesenchymal stromal cells that recapitulate the early steps of condensation initiating bone repair in vivo. We find that heterogeneous mesenchymal stromal cells self-organize in 3D in a developmentally hierarchical manner. We demonstrate a link between structural organization and local regulation of specific molecular signaling pathways such as NF-κB and actin polymerization, which modulate osteo-endocrine functions. This study emphasizes the importance of resolving spatial heterogeneities within cellular aggregates to link organization and functional properties, enabling a better understanding of the mechanisms controlling organoid formation, relevant to organogenesis and tissue repair.
INTRODUCTION
In recent years, organoids have emerged as powerful tools for basic research, drug screening, and tissue engineering. The organoids formed in vitro show many features of the structural organization and the functional hallmarks of adult or embryonic anatomical structures (1). In addition, the formation of organoids alleviates the need to perform animal studies and provides an attractive platform for robust quantitative studies on the mechanisms regulating organ homeostasis and tissue repair in vivo (1). The formation of organoids usually starts with populations of stem cells. They are therefore expected to be heterogeneous because pluripotent stem cells [induced pluripotent stem cells (pscs) or embryonic stem cells] have been shown to dynamically and stochastically fluctuate from ground to differentiated state (2). In the same vein, LGR5+ intestinal stem cells are reported to contain several distinct populations (3). As such, the formation of organoids involves the inherent capacity of these heterogeneous populations to self-sort and self-pattern to form an organized three-dimensional (3D) architecture (4). However, the rules underlying organoid formation as well as the contribution of intrinsic population heterogeneity to the organoid self-assembly remain poorly understood (5). Consequently, there is a need for novel quantitative approaches at the single-cell level to reliably understand the mechanisms of spatial tissue patterning in 3D organoids, for which microfluidic and quantitative image analysis methods are well suited.
In this work, we use mesenchymal progenitors, alternatively named mesenchymal stromal cells (MSCs), which constitute a self-renewing population with the ability to differentiate into adipocytes, chondrocytes, and osteoblasts (5). Although human MSCs (HMSCs) express high levels of undifferentiation markers (e.g., CD105, CD44, CD73), they constitute a heterogeneous population of cells that exhibit considerable variation in their biophysical properties and epigenetic status, as well as the basal level of expression of genes related to differentiation, immunoregulation, and angiogenesis (6, 7). Nonetheless, their aggregation leads to the formation of highly cohesive 3D spherical structures [which we designate hereafter as mesenchymal bodies (MBs)] with improved biological activities in comparison to 2D cultures (8). However, little is known on how HMSCs self-organize or whether the intrinsic heterogeneity of the population regulates MB formation and individual cell functions in 3D.
The self-aggregation of HMSCs into MBs can recapitulate the early stages of mesenchymal condensation, and it promotes the secretion of paracrine molecules taking part in the process of ossification (9). During mesenchymal condensation in vivo, mesenchymal progenitors self-aggregate and form dense cell-cell contacts that lead to the initiation of bone organogenesis through endochondral (necessitating a chondrogenic intermediate) and intramembranous (direct osteogenic differentiation) ossification (10). In addition, the formation of these 3D MBs in vivo is associated with the secretion of important paracrine molecules such as prostaglandin E2 (PGE2) and vascular endothelial growth factor (VEGF), which participate in the recruitment of endogenous osteoblasts, osteoclasts, and blood vessels, leading to the initiation/restoration of bone homeostasis (11, 12). In these two ossification processes, the induction of nuclear factor κB (NF-κB) target genes, such as cyclooxygenase-2 (COX-2), and their downstream products (e.g., PGE2 and VEGF) plays a critical role as developmental regulators of ossification and bone healing (13). However, while mesenchymal condensation is critical for bone organogenesis, there is still a limited understanding on how the cellular spatial organization within 3D MBs regulates the individual cells’ endocrine functions (14).
In the present work, we interrogate the influence of phenotypic heterogeneity within a population of stem cells on the mechanisms of self-assembly and functional patterning within 3D organoids using HMSCs as a model of heterogeneous progenitor cell population. This is performed using a novel microfluidic platform for high-density formation of mensenchymal bodies, combined with the analysis of individual cells by quantitative image analysis. Our study reveals that the progenitor cell population self-assembles in a developmentally hierarchical manner. We also find that the structural arrangement in mensenchymal bodies is linked with the functional patterning in 3D, through a modulation of the activity of regulatory molecular signaling at a local scale. This study demonstrates the interplay between cell size and differentiation status, which mediates cellular spatial rearrangement in 3D, leading to the regionalized activation of unique biological functions while forming aggregates.
RESULTS
Self-organization of HMSCs in 3D MBs
HMSCs are known to constitute a heterogeneous population (6, 7). In this study, fetal HMSCs were derived from the Wharton’s jelly of the umbilical cord (UC). UC-derived HMSCs are considered to be more primitive than HMSCs derived from adult bone marrow because of their higher proliferative capacity, their ability to form colony-forming unit–fibroblast, as well as their lower degree of basal commitment (15). To examine the cellular diversity within the population, HMSCs were first characterized by their expression of membrane markers. Most of the HMSC population consistently expresses CD73, CD90, CD105, and CD146, but not CD31 (an endothelial cell marker), CD34 (a hematopoietic cell marker), CD14 (an immune cell marker), or human leukocyte antigen–DR (HLA-DR) (a type of major histocompatibility complex II) (Fig. 1, A to F, and fig. S1, A to C). However, a deeper analysis of the flow cytometric data shows that the HMSC population contains cells of heterogeneous size [coefficient of variation (CV) = 33 to 37%] (Fig. 1, G and I), having a broad distribution in the expression of CD146 (Fig. 1F). Of note, the CD146 level of expression was linked to the size of the cells: The highest levels of CD146 were found for the largest cells (Fig. 1, H and J). Similar correlations with cell size were also observed for CD73, CD90, and CD105 (fig. S1, D to F). In addition, upon specific induction, the HMSC population used in this study successfully adopted an adipogenic (Fig. 1K), an osteogenic (Fig. 1L), or a chondrogenic (Fig. 1M) phenotype, demonstrating their mesenchymal progenitor identity.
Fig. 1. Characterization of the hMSC population.
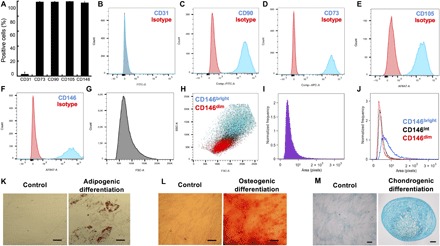
(A) Percentage of positive cells for CD31, CD73, CD90, CD105, and CD146 (n = 3). Representative histograms of the distribution of the CD31− (B), CD73− (C), CD90− (D), CD105− (E), and CD146− (F) level of expression are shown. (G) Representative histogram of the forward scatter (FSC) distribution. (H) Correlation between cell size [FSC and side scatter (SSC)] and the level of CD146 expression. (I) Representative histogram of the cell projected area distribution. (J) Representative histogram of the size distribution of the CD146dim, CD146int, and CD146bright (ImageSteam analysis). (K) Representative images of hMSCs differentiated toward adipogenic lineage (Oil Red O staining). (L) Representative images of UC-hMSCs differentiated toward osteogenic lineage in (Alizarin Red S staining). (M) Representative images of UC-hMSCs differentiated toward chondrogenic lineage (Alcian Blue staining in 2D and cryosectioned micromass cultures). Scale bars, 50 μm. The images were acquired using a binocular. FITC-A, fluorescein isothiocyanate–A; APC-A, allophycocyanin-A.
To interrogate contribution of cellular heterogeneity (i.e., in terms of size and levels of CD marker expression) in the self-organization of HMSCs in 3D, MBs were formed at high density on an integrated microfluidic chip. This was done by encapsulating cells into microfluidic droplets at a density of 380 cells per droplet, with a CV of 24% (fig. S2, A and B). The drops were then immobilized in 250 capillary anchors in a culture chamber, as previously described (Fig. 2, A and B) (16). The loading time for the microfluidic device was about 5 min, after which the typical time for complete formation of MBs was about 4 hours (movie S1), as obtained by measuring the time evolution of the projected area (Fig. 2, C and D) and circularity of individual MBs (Fig. 2E and movie S2). The protocol resulted in the formation of a single MB per anchor (fig. S2C) with an average diameter of 158 μm (Fig. 2F), when starting with a seeding concentration of 6 × 106 cells ml−1. The diameter of the aggregates can easily be tuned by modulating the concentration of cells in the seeding solution (fig. S2, A and B). In addition, the complete protocol yielded the reproducible formation of a high-density array of fully viable MBs ready for long-term culture (for the images of the individual fluorescent channel, see Fig. 2G and fig. S2D), as described previously (16). Of interest, the CV of the MB diameter distribution was lower than the CV of the individual cell size and of the cell number in droplets (CV MB diameter = 13.3%, CV cell number per drop = 24%, and CV cell size = 35%), which demonstrates that the production of MBs leads to more homogeneous size conditions, compared with the broad heterogeneity in the cell population.
Fig. 2. Formation of MBs on chip.
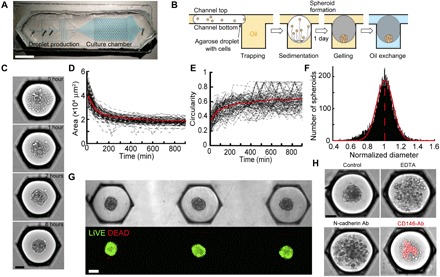
(A) Chip design. Scale bar, 1 cm. (B) Schematized side view of an anchor through the MB formation and culture protocol. (C) Representative time lapse of an MB formation. Scale bar, 100 μm. (D and E) Measurement of the time evolution of the projected area (D) and circularity of each aggregate (E). n = 120 MBs. (F) Distribution of the MB diameter normalized by the mean of each chip (n = 10,072 MBs). (G) Top: Representative images of MBs after agarose gelation and oil-to-medium phase change. Bottom: The same MBs are stained with LIVE/DEAD. Scale bar, 100 μm. (H) Representative images of MBs formed in the presence of EDTA, an N-cadherin, or a CD146-conjugated blocking antibody (Ab) (the red color shows the position of the CD146 brightest cells, and the dilution of the antibody was 1/100 and remain in the droplet for the whole experiment). Scale bar, 100 μm. The images were acquired using a wide-field microscope.
To gain insight into the cellular components required to initiate the self-organization of HMSCs in 3D, the MB formation was disrupted by altering cell-cell interactions. This was first performed by adding EDTA, a chelating agent of the calcium involved in the formation of cadherin junctions, to the droplet contents. Doing so disrupted the MB formation, as shown in Fig. 2H, where the projected area of the cells increased and the circularity decreased in the presence of EDTA compared with the controls, as previously reported (17). The role of N-cadherins among different types of cadherins was further specified by adding a blocking antibody in the droplets before MB formation. This also led to a disruption of the MB formation, demonstrating that N-cadherin homodimeric interactions are mandatory to initiate the process of HMSC aggregation. CD146 [melanoma cell adhesion molecule (M-CAM)] plays important dual roles: as an adhesion molecule (that binds to Laminin 411) (18) and a marker of the commitment of HMSCs (19). We, thus, interrogate its contribution to MB formation. The addition of a CD146-conjugated blocking antibody also disrupted the formation of the MB (Fig. 2H), demonstrating that cell-cell interactions involving CD146 are also required during MB formation, as reported with other cell types (18). Of note, the brightest signal from the CD146-stained cells was located in the core of the cellular aggregates (Fig. 2H), suggesting that HMSCs self-organize relatively to their degree of commitment.
We found that the population of HMSCs constituted of cells of broad size and expressing different levels of undifferentiated markers [i.e., CD90, CD73, CD105, and CD146 are known to be down-regulated upon differentiation; (20)] and that the cells are capable of self-organizing cohesively in 3D. To better understand how the heterogeneous cells organized within the MBs, we measured how the different cell types composing the population self-assembled spatially in 3D by investigating the role of CD146. For this purpose, the CD146dim and CD146bright cells were separated from the whole HMSC population by flow cytometry (Fig. 3, A and B). The cells were then reseeded on a chip for the MB formation after fluorescently labeling the brighter and/or the dimmer CD146 populations. Image analysis revealed that the CD146bright cells were mostly located in the center of the cellular aggregates, while CD146dim cells were found at the boundaries of the MBs (Fig. 3, C to E, figs. S3A and S5A for confocal images, and movie S1). This organization was stable for a 3-day culture (fig. S3B).
Fig. 3. CD146 expression and cellular commitment regulate the structural organization of MBs.
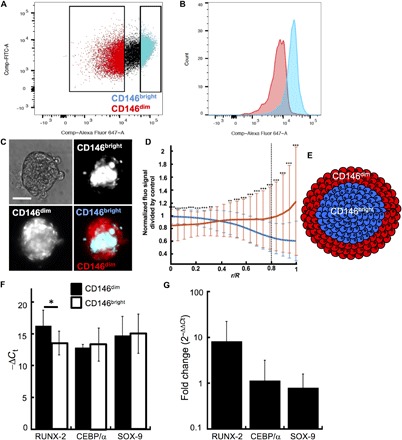
(A) Representative dot plot of the hMSC population separation based on the level of CD146: The CD146dim constitutes 20% of the population expressing the highest levels of CD146; the CD146bright constitutes the 20% of the population expressing the lowest levels of CD146. (B) Fluorescence signal distribution in the CD146dim and CD146bright populations after cell sorting. (C) After cell sorting, the CD146bright or the CD146dim was stained with Vybrant Dil (red) or Vybrant DiO (green), remixed together and allowed to form MBs. Representative images of the CD146bright and CD146dim within the MBs. Scale bar, 100 μm (n = 185 MBs). (D) The position of the CD146bright and CD146dim was quantified by correlating the fluorescence signal of the different stained cells as a function of their radial position within the MBs, after the staining of individual population with Vybrant Dil (CD146bright) = 500 and CD146dim (MBs; CD146bright = 85). Error bars show the SD. (E) Schematized representation of the structural organization of MBs. (F and G) RT-qPCR analysis of the relative RUNX-2, CEBP/α, and SOX-9 expression to glyceraldehyde-3-phosphate dehydrogenase (GADPH) [−ΔCt (cycle threshold) (D) and relative RNA expression (E)] in the CD146bright and CD146dim populations (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001.
As we found that the CD146bright cells were larger than the CD146dim cells, the cells from the HMSC population were also separated on the basis of their relative size (a parameter that also discriminates the CD90−, CD105−, and CD73−bright from the CD90−, CD105−, and CD73−dim cells; fig. S1, D to F). After reseeding on the chip, the MBs were composed of large cells in the core, while the smallest cells were located at the boundaries, as expected from the previous experiments (fig. S3A). Moreover, we found that the speed of self-assembly of each population is not related to the rearrangement of CD146dim and CD146bright cells in 3D, because the mixing of dissociated cells or the fusion of aggregates made each population give rise to the same structural organization (21). It is well established that CD146bright defines the most undifferentiated HMSCs (20). The heterogeneity in level of commitment between the two subpopulations was therefore checked by reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis to quantify differences in the expression of differentiation markers. The analysis showed that the CD146dim cells expressed higher levels of osteogenic differentiation markers (i.e., RUNX-2) than the CD146bright cells (Fig. 3, F and G).
The level of RUNX-2 expression was also quantified at the protein level using immunocytochemistry and image analysis of the MBs on the microfluidic device by developing a layer-by-layer description of the MBs. This mapping was constructed by estimating the boundaries of each cell in the image from a Voronoi diagram, built around the positions of the cell nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 4A) (22). These estimates were then used to associate the fluorescence signal from each cell with one of the concentric layers (Fig. 4B). Such a mapping provides better resolution for discriminating the spatial heterogeneity of protein expression than simply assigning a fluorescence signal to a defined radial coordinate (fig. S4). Moreover, the reliability of the measurements by quantitative image analysis was confirmed by performing several control experiments. In particular, we verified (i) the specificity of the fluorescence labeling, (ii) the absence of limitation for antibody diffusion, and (iii) the absence of the light path alteration in the 3D structure (fig. S5 and Materials and Methods). Consistent with the qPCR data, we found that HMSCs located at the boundaries of the MBs expressed higher levels of the protein RUNX-2 than the cells located in the core (see Fig. 4, C and D, and fig. S7 for individual experiments).
Fig. 4. Quantitative mapping of RUNX-2 expression and structural organization in MBs.
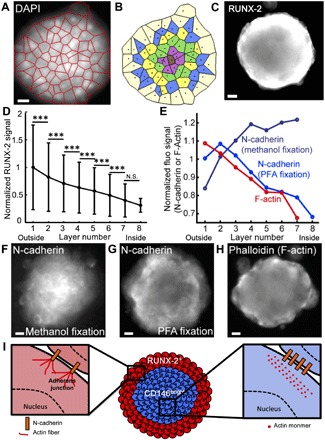
(A and B) The detection of nuclei within MBs enables the construction of a Voronoi diagram (A) that allows the identification of concentric cell layers (B) within the MBs. (C and D) Representative image (C) and quantitative analysis (D) (error bars represent the SD) of RUNX-2 staining within the cell layers of the MB (Nchips = 3 and nMBs = 458). N.S., nonsignificant. (E to H) Quantitative analysis (E) and representative images (F to H) of N-cadherin staining after methanol/acetone (F) (Nchips = 3 and nMBs = 405), after PFA/Triton X-100 fixation and permeabilization (G) (Nchips = 3 and nMBs = 649), and F-actin staining with phalloidin (H) (Nchips = 3 and nMBs = 421). Scale bars, 20 μm. The images were acquired using a wide-field microscope. ***P < 0.001. (I) Schematized representation of the structural organization of MBs.
Thus, as CD146 defines the most undifferentiated and clonogenic cells as well as regulates the trilineage differentiation potential of HMSCs, the results indicate that HMSCs self-organize within MBs based on their initial commitment. The most undifferentiated and largest cells are found in the core (r/R < 0.8), while more differentiated cells positioned in the outer layers of the MBs (r/R > 0.8) (Fig. 4, C to E). In addition, these data reveal that HMSCs are conditioned a priori to occupy a specific location within the MBs.
The commitment of HMSCs is known to regulate their level of CD146 expression and the type of cell-cell adhesion molecules (23), which plays a fundamental role in the structural cohesion of the MBs (Fig. 2H). For this reason, we interrogated the organization of cell-cell junctions after the MB formation through measurements of the N-cadherin and F-actin fluorescence signal distribution. Two different protocols were used to discriminate several forms of N-cadherin interactions. First, paraformaldehyde (PFA) fixation and Triton X-100 permeabilization were used, because they were reported to retain in place only the detergent-insoluble forms of N-cadherin. Alternatively, ice-cold methanol/acetone fixation and permeabilization enabled the detection of all forms of N-cadherins (26). The results show a higher density of total N-cadherins in the core of the MBs (Fig. 4, E and F), while a higher density of F-actin was found in the cell layers located near the edge of the MBs (Fig. 4, E and H). The pattern of F-actin distribution was not related to the agarose gel surrounding the MBs (fig. S5B). These results are consistent with the theories of cell sorting in spheroids that postulate that more adhesive cells (i.e., expressing more N-cadherin or CD146) should be located in the core, while more contractile cells (i.e., containing denser F-actin) are located at the edge of the MBs (24). Moreover, our observations are in accordance with recent results demonstrating that HMSCs establishing higher N-cadherin interactions show reduced osteogenic commitment than HMSCs making fewer N-cadherin contacts, potentially through the modulation of Yap/Taz signaling and cell contractility (23).
In contrast, the most triton-insoluble forms of N-cadherins were located at the boundaries of the HMSC aggregates (Fig. 4, E and G), at the same position as the cells containing the denser F-actin. These results demonstrate that different types of cellular interactions were formed between the core and the edges of the MBs, which correlated with the degree of cell commitment that apparently stabilize the adherens junctions (Fig. 4I) (25).
Mapping the biological functions in MBs
We found above that the degree of commitment was linked with the pattern of HMSC self-organization in MBs (i.e., formation of adherens junctions), which may also regulate their paracrine functions (26). We therefore interrogated the functional consequences of the cellular organization in MBs by investigating the distribution of VEGF- and PGE2-producing cells.
The specific production of COX-2, VEGF, and two other molecules regulating bone homeostasis such as tumor necrosis factor–inducible gene 6 (TSG-6) (27) and stanniocalcin 1 (STC-1) (28) was evaluated by RT-qPCR analysis. An increased transcription (20- to 60-fold) of these molecules was measured in 3D in comparison to the monolayer culture (Fig. 5, A and B). Consistent with this observation, while a very limited level of secreted PGE2 and VEGF was measured by enzyme-linked immunosorbent assay (ELISA) in 2D culture, they were significantly increased (by about 15-fold) upon the aggregation of HMSCs in 3D (Fig. 5C). In addition, to interrogate the specific role of COX-2 [the only inducible enzyme catalyzing the conversion of arachidonic acid into prostanoids; (29)] in PGE2 and VEGF production, indomethacin (a pan-COX inhibitor) was added to the culture medium. Indomethacin abrogated the production of PGE2, and it significantly decreased VEGF secretion (Fig. 5C), which suggests an intricate link between COX-2 expression and the secretion of these two molecules (30, 31).
Fig. 5. Spatial patterning of the biological functions of hMSCs in 3D.
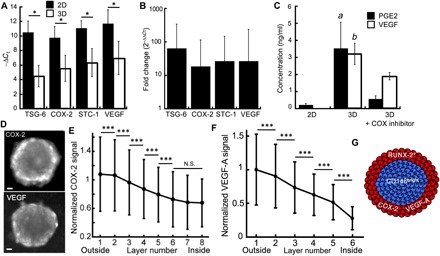
(A and B) RT-qPCR analysis of the relative TSG-6, COX-2, STC-1, and VEGF expression to GADPH (−ΔCt) (A) and relative RNA expression (B) in the 3D and 2D populations (n3D = 3 and n2D = 3). (C) Quantification by ELISA of the PGE-2 and VEGF secreted by hMSCs cultivated in 2D, as MBs or as MBs treated with indomethacin (nchips = 3 and n2D = 3). (D and E) Representative image (D) and quantitative analysis (E) of COX-2 (Nchips = 13 and nMBs = 2936) and (F) VEGF-A (Nchips = 3 and nMBs = 413) staining within the cell layers of the MBs (error bars represent the SD). Scale bars, 50 μm. The images were acquired using a wide-field microscope *P < 0.05; ***P < 0.001; a and b: P < 0.05. (G) Schematized representation of the structural organization of MBs.
To further interrogate the link between the COX-2– and the VEGF-producing cells, their location was analyzed by quantitative image analysis at a layer-by-layer resolution. These measurements showed significantly higher levels of COX-2 in the first two layers, compared with the successive layers of the MBs (Fig. 5, D and E), with a continuous decrease of about 40% of the COX-2 signal between the edge and the core. This pattern of COX-2 distribution was not affected by the MB diameter (fig. S7B). Similar observations were made with VEGF (Fig. 5, D and F), demonstrating that cells at the boundaries of the MBs expressed both COX-2 and VEGF (Fig. 5G). Taken with the measurements of Fig. 5C, these results imply that COX-2 acts as an upstream regulator of PGE2 and VEGF secretion. Conversely, oxygen deprivation was unlikely to occur within the center of the MBs because no hypoxic area was detected through the whole MBs (fig. S5). Consequently, it is unlikely that hypoxia-inducible factor–1α (HIF-1α) signaling mediates the increase in VEGF expression at the boundaries of the MBs. Note that finding the link between these three molecules requires the 3D format, because the molecules are not detected in 2D. Here, the combination of population-scale measurements (Fig. 5C) and cell layer analysis (Fig. 5, E and F) provides strong evidence for this pathway.
Because variations of COX-2 and adherens junction distribution are colocalized within the MBs (Figs. 4, E to G, and 5, D and E), the results point to a link between the quality of cell-cell interactions and the spatial distribution of the COX-2high cells in 3D. The mechanisms leading to the spatial patterning of COX-2 expression in the MBs were therefore explored using inhibitors of the signaling pathways related to anti-inflammatory molecule production and of the molecular pathways regulating the structural organization (table S3): (i) 4-N-[2-(4-phenoxyphenyl)ethyl]quinazoline-4,6-diamine (QNZ) that inhibits NF-κB, a critical transcription factor regulating the level of COX-2 expression (32); (ii) N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) that inhibits the canonical Notch pathway, modulating cell-cell interactions and several differentiation pathways; (iii) Y-27632 (Y27) that inhibits ROCK involved in the bundling of F-actin (i.e., formation of stress fibers) to assess the role of actomyosin organization; and (iv) cytochalasin D (CytoD) that inhibits the polymerization of actin monomers.
While the addition of DAPT had virtually no effect on the ability of the cells to form MBs, Y27 led to MBs with more rounded cells, and both QNZ and CytoD strongly interfered with the MB formation process (Fig. 6, A to C). The results indicate that NF-κB activation and the promotion of actin polymerization are critical signaling steps initiating the process of MB formation by HMSCs.
Fig. 6. Molecular mechanisms regulating the local level of COX-2 expression in MBS.
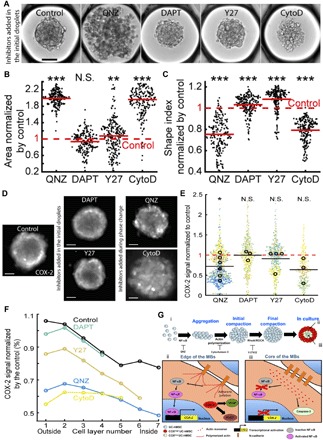
(A) Representative images of MBs formed 1 day after the droplet loading. Scale bar, 100 μm. Inhibitors are added to the culture medium before the MB formation. (B and C) Quantitative analysis of the aggregates projected area (B) and shape index (C) in the presence of the different inhibitors. Red lines represent the mean value for each condition. (D and E) Representative images (D) (contrast is adjusted individually for a better visualization of the pattern; scale bar, 100 μm; the images were acquired using a wide-field microscope) and quantitative analysis (E) of the COX-2 fluorescence signal intensity normalized by the control value with the different inhibitors. For these longer culturing times, QNZ and CytoD are only added during the phase change to allow the MB formation. Small dots represent one MB. Large dots represent the average normalized COX-2 fluorescence signal per chip. Each color corresponds to a specific chip. *P < 0.05. (F) Estimation of inhibitor effect in the cell layers with the COX-2 signal normalized by the control value. Control: Nchips = 11 and nMBs = 2,204; QNZ: Nchips = 6 and nMBs = 1215; DAPT: Nchips = 3 and nMBs = 658; Y27: Nchips = 4 and nMBs = 709; CytoD: Nchips = 3 and nMBs = 459. *P < 0.05; **P < 0.01; ***P < 0.001. (G) Proposed mechanisms regulating the MB formation and the patterning of their biological functions. (i) Regulation of the formation of MBs. (ii and iii) Spatial patterning of hMSC biological properties within MBs.
To assess the role of NF-κB and actin polymerization in the pattern and the level of COX-2 expression in the MBs, QNZ and CytoD were added 1 day after the cell seeding, once the MBs were completely formed. In contrast, Y27 and DAPT were included in the initial droplets and maintained in the culture medium for the whole culture period. Typical images showing the COX-2 signal in these different conditions are shown in Fig. 6D (see also fig. S7 for quantification of the individual experiments). Of note, none of the inhibitors had an effect on Casp3 activation, indicating that they do not induce apoptosis at the concentration used in this study (fig. S8). The levels of COX-2 expression in MBs, after 3 days in culture, were significantly reduced with QNZ, also decreasing after the addition of CytoD (Fig. 6E). By contrast, Y27 and DAPT had no effect on the levels of COX-2 expression. As a consequence, the results demonstrate that a sustained NF-κB activity after the MB formation is required to promote COX-2 expression. Moreover, the induction of actin polymerization in MBs constitutes a mandatory step to initiate COX-2 production.
To get a deeper understanding on the local regulation of these signaling pathways, we analyzed at the single-cell resolution the distribution of COX-2 within the MBs. The spatial mapping revealed that the COX-2 fluorescence intensity was mostly attenuated at the edge of the MBs treated with CytoD and QNZ, while more limited change in the pattern of its expression was observed in the presence of Y27 and even less so with DAPT (Fig. 6F). Consequently, the results revealed a strong link between cell phenotype, the capability to form functional adherens junctions, and the local regulation of NF-κB and actin polymerization leading to the increased expression of PGE2 and VEGF that are mediated by COX-2 in 3D (Fig. 6G). Together, the results indicate that in 3D cell aggregates, the spatial organization has some implications on the specific activation of signaling pathways, resulting in local functional heterogeneity.
DISCUSSION
Understanding the mechanisms of the formation and the spatial tissue patterning within organoids requires a characterization at single-cell level in 3D. In this study, we used a novel microfluidic and epifluorescence imaging technology to obtain a precise quantitative mapping of the structure, the position, and the link with individual cell functions within MBs. The image analysis provided quantitative data that were resolved on the scale of the individual cells, yielding measurements on 700,000 cells in situ within over 10,000 MBs.
While the microfluidic technology developed here is very efficient for high-density size-controlled MB formation, the method is prone to some limitations. Chief among them, the cultivation in nanoliter-scale drops may subject the cells to nutrient deprivation and by-product accumulation under static culture conditions. This limits the duration of the culture to a few days, depending on the cell type and droplet size. To overcome this limitation, it is possible to continuously perfuse the chip with fresh culture medium after performing the oil-aqueous phase exchange, as we demonstrated previously (16). Alternatively, it is also possible to maintain the cells in liquid droplets (without using a hydrogel) by resupplying culture medium through the fusion of additional drops at later times. This operation requires, however, a new design of the anchors and additional microfluidic steps (21).
The second major drawback of the method emerges from the large distance between the MBs and the microscope objective, which requires the use of very large working distance objectives. This compounds the difficulty of applying different confocal techniques by limiting the fluorescence intensity of the images, which, in turn, reduces the throughput when 3D image stacks are required. Although we have shown above that wide-field imaging can be used to obtain spatial mappings of spheroid structure and cell functions, true single-cell measurements will need to overcome the limitations on imaging in the future.
A Voronoi segmentation was used to categorize the cells into concentric layers, starting from the edge of the MBs and ending with the cells in the central region (22), which allowed us to measure variations in the structural organization and in the protein expressions on a layer-by-layer basis within the 3D cultures. The MBs were found to organize into a core region of undifferentiated cells, surrounded by a shell of committed cells. This hierarchical organization results from the spatial segregation of an initially heterogeneous population, as is generally the case for populations of pluripotent and somatic stem cells (2, 3, 33). The process of aggregation of HMSCs obtained within a few hours takes place through different stages (Fig. 6G): The first steps of the aggregation of MBs are mediated by N-cadherin interactions. In parallel, NF-κB signaling is activated, promoting cell survival by preventing anoikis of suspended cells (34, 35). At later stages, the formation of polymerized F-actin and, to a lesser extent, stress fibers mediates the MB compaction, mainly at the edge of the MBs where the cellular commitment helps the stabilization of adherens junctions. The formation of adherens junctions facilitates the cohesion of the 3D structure, probably through the enhanced α- and β-catenin availability in the CD146dim/RUNX-2+ cells (36, 37, 38), which are recruited in the CCC complexes of the adherens junctions to promote the stable coupling of the F-actin to the N-cadherin (39), which become more insoluble to Triton X-100 than unbounded N-cadherins.
A functional phenotype that correlates with this hierarchical segregation is an increase in endocrine activity of the cells located at the boundaries of the MBs. COX-2 expression is increased in the outer layers of the MBs, which also contain more functional adherens junctions as well as a sustained NF-κB activity in this region. The promoter of COX-2 contains RUNX-2 and NF-κB cis-acting elements (40). While RUNX-2 is required for COX-2 expression in mesenchymal cells, its level of expression does not regulate the levels of COX-2 (40). The increased COX-2 expression is, in turn, due to the unbundled form of F-actin (i.e., a more relaxed form of actin, in comparison to the dense stress fibers observed in 2D) near the edge of the MBs, which was reported to sustain NF-κB activity (41) and to down-regulate COX-2 transcriptional repressors (42). Therefore, NF-κB has a high activity in the outer layers of the MB, where it locally promotes COX-2 expression.
These results show that the 3D culture format may provide some insights to understand the mesenchymal cell behavior in vivo, because we found that the expression of key bone regulatory molecules is spatially regulated as a function of the structural organization of the MBs. The 3D structure obtained here recalls some of the conditions found at the initial steps of intramembranous ossification that occurs after mesenchymal condensation (i.e., no chondrogenic intermediate was found in the MBs). In the developing calvaria, the most undifferentiated mesenchymal cells (e.g., Sca-1+/RUNX-2− cells) are located in the intrasutural mesenchyme, which is surrounded by an osteogenic front containing more committed cells (e.g., Sca-1−/RUNX-2+ cells) (43, 44). Similarly, we observed that undifferentiated HMSCs (i.e., CD146bright/RUNX-2− HMSCs) were surrounded by osteogenically committed cells (i.e., CD146dim/RUNX-2+ HMSCs), which also coexpressed pro-osteogenic molecules, namely, COX-2 and its downstream targets, PGE2 and VEGF. While the link between COX-2 and PGE2 is well established, there is also evidence that COX-2 can induce the production of VEGF in different cell types, e.g., colon cancer cells (45), prostate cancer cells (46), sarcoma (47), pancreatic cancer cells (48), retinal Müller cells (49), gastric fibroblasts (50), skin or lung fibroblasts (51). In these cases, the mechanism for VEGF production through COX-2 induction is thought to be linked to PGE-2, either in an autocrine/paracrine manner (52) or in an intracrine manner (53).
Beyond HMSCs, spatial organization related to the level of differentiation and cell size has been documented in growing embryoids and organoids, with more committed cells being positioned in the outer layers (54, 55, 56). Our results show that a similar hierarchical structure can also be obtained through the aggregation of a mixed population of adult progenitors. This suggests that cell sorting, based on the size and commitment, plays a dominant role in organizing stem cell aggregates. This data-driven approach of combining high-throughput 3D culture and multiscale cytometry (16) on complex biological models can be applied further for getting a better understanding of the equilibria that determine the structure and the function of cells within multicellular tumor spheroids, embryoid bodies, or organoids.
MATERIALS AND METHODS
Human UC-derived MSC culture
HMSCs derived from the Wharton’s jelly of the UC (HMSCs) [American Type Culture Collection (ATCC) PCS-500-010, LGC, Molsheim, France] were obtained at passage 2. Four different lots of HMSCs were used in this study (lot nos. 60971574, 63739206, 63516504, and 63739206). While the lots were not selected a priori, we found consistent results for COX-2 and CD146 distribution within MBs. HMSCs from the different lots were certified for being CD29, CD44, CD73, CD90, CD105, and CD166 positive (more than 98% of the population is positive for these markers) and CD14, CD31, CD34, and CD45 negative (less than 0.6% of the population is positive for these markers) and to differentiate into adipocytes, chondrocytes, and osteocytes (ATCC, certificates of analysis). HMSCs were maintained in T175 cm2 flasks (Corning, France) and cultivated in a standard CO2 incubator (Binder, Tuttlingen, Germany). The culture medium was composed of α–modified Eagle’s medium (α-MEM) (Gibco, Life Technologies, Saint Aubin, France) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco) and 1% (v/v) penicilin-streptomycin (Gibco). The cells were seeded at 5 × 103 cells/cm2, subcultivated every week, and the medium was refreshed every 2 days. HMSCs at passage 2 were first expanded until passage 4 [for about five to six population doublings (PDs)], then cryopreserved in 90% (v/v) FBS/10% (v/v) dimethyl sulfoxide (DMSO), and stored in a liquid nitrogen tank. The experiments were carried out with HMSCs at passages 4 to 11 (about 24 to 35 PDs, after passage 2).
Surface marker staining, analysis, and sorting by flow cytometry
HMSCs were harvested by scrapping or trypsinization from T175 cm2 flasks. Then, the cells were incubated in staining buffer [2% FBS in phosphate-buffered saline (PBS)], stained with a mouse anti-human CD146–Alexa Fluor 647 (clone P1-H12, BD Biosciences), a mouse anti-human CD31–Alexa Fluor 488 (BD Biosciences, San Jose, CA) antibody, a mouse anti-human CD105–Alexa Fluor 647 (BD Biosciences, San Jose, CA), a mouse anti-human CD90–fluorescein isothiocyanate (FITC) and a mouse anti-human CD73–allophycocyanin (APC) (Miltenyi Biotec, Germany), a CD14-APC (Miltenyi Biotec), a CD34-FITC (BioLegend), and an HLA-DR–APC (BD Biosciences).
The percentages of CD73-, CD90-, CD105-, CD146-, CD31-, CD34-, and HLA-DR–positive cells were analyzed using a FACS LSRFortessa (BD Biosciences, San Jose, CA) or an ImageStream (Amnis) flow cytometer. To validate the specificity of the antibody staining, the distributions of fluorescently labeled cells were compared to cells stained with isotype controls: mouse immunoglobulin G1 (IgG1), k-PE-Cy5 (clone MOPC-21, BD Biosciences), and mouse IgG2a K isotype control FITC (BD Biosciences, San Jose, CA). Alternatively, HMSCs were sorted on the basis of their level of expression of CD146 or their size [forward scatter (FSC) and side scatter (SSC)] using a FACSAria III (BD Biosciences, San Jose, CA).
Adipogenic, osteogenic, and chondrogenic differentiation
To induce adipogenic differentiation, UC-HMSCs were seeded at 1 × 104 cells/cm2 in culture medium. The day after, the culture medium was switched to StemPro Adipogenesis Differentiation medium (Life Technologies) supplemented with 10 μM rosiglitazone (Sigma-Aldrich) for 2 weeks. To visualize the differentiated adipocytes, the cells were stained with Oil Red O (Sigma-Aldrich). As a control, UC-HMSCs were maintained in culture medium for 2 weeks and stained with Oil Red O, as above.
To induce osteogenic differentiation, UC-HMSCs were seeded at 5 × 103 cells/cm2 in culture medium. The day after, the culture medium was switched to StemPro Osteogenesis Differentiation medium (Life Technologies) supplemented with 2-nm bone morphogenetic protein 2 (BMP-2) (Sigma-Aldrich) for 2 weeks. To visualize the differentiated osteoblasts, the cells were stained with Alizarin Red S (Sigma-Aldrich). As a control, UC-HMSCs were maintained in culture medium for 2 weeks and stained with Alizarin Red S, as above.
To induce chondrogenic differentiation, UC-HMSCs were seeded at 1 × 106 cells/ml in a 15-ml conical tube to promote micromass culture. The medium consisted of StemPro Chondrogenic Differentiation medium (Life Technologies). After 3 weeks in culture, the pellets were fixed and cryosectioned and then stained for Alcian Blue 8GX (Sigma-Aldrich). As a control, UC-HMSCs were maintained in 2D using culture medium for 3 weeks and stained with Alcian Blue, as above.
The color images were acquired using a binocular (SMZ18, Nikon) equipped with a camera (D7500, Nikon).
Microfabrication
Standard dry-film soft lithography was used for the flow-focusing device (top of the chip) fabrication, while a specific method for the fabrication of the anchors (bottom of the chip) was developed. For the first part, up to five layers of dry-film photoresist consisting of 50-μm Eternal Laminar E8020, 33-μm Eternal Laminar E8013 (Eternal Materials, Taiwan), and 15-μm Alpho NIT215 (Nichigo-Morton, Japan) negative films were successively laminated using an office laminator (PEAK pro PS320) at a temperature of 100°C until the desired channel height, either 135, 150, 165, or 200 μm, was reached. The photoresist film was then exposed to ultraviolet (Lightningcure, Hamamatsu, Japan) through a photomask of the junction, the channels, and the culture chamber boundaries. The masters were revealed after washing in a 1% (w/w) K2CO3 solution (Sigma-Aldrich). For the anchor fabrication, the molds were designed with RhinoCAM software (MecSoft Corporation, LA) and were fabricated by micromilling a brass plate (CNCMini-Mill/GX, Minitech Machinery, Norcross). The topography of the molds and masters was measured using an optical profilometer (Veeco Wyco NT1100, Veeco, Mannheim, Germany).
For the fabrication of the top of the chip, poly(dimethylsiloxane) [PDMS; SYLGARD 184, Dow Corning, 1:10 (w/w) ratio of curing agent to bulk material] was poured over the master and cured for 2 hours at 70°C. For the fabrication of the bottom of the chip, the molds for the anchors were covered with PDMS. Then, a glass slide was immersed into uncured PDMS, above the anchors. The mold was lastly heated on a hot plate at 180°C for 15 min. The top and the bottom of the chip were sealed after plasma treatment (Harrick, Ithaca). The chips were filled three times with Novec Surface Modifier (3M, Paris, France), a fluoropolymer coating agent, for 30 min at 110°C on a hot plate.
Formation of MBs on the chip
HMSCs were harvested with TrypLE at 60 to 70% confluence, and a solution containing 6 × 105 cells in 70 μl of medium was mixed with 30 μl of a 3% (w/v) liquid low-melting agarose solution (i.e., stored at 37°C) (Sigma-Aldrich, Saint Quentin Fallavier, France) diluted in culture medium containing gentamicin (50 μg/ml; Sigma-Aldrich) (1:3, v/v), resulting in a 100-μl solution of 6 × 106 cells/ml in 0.9% (w/v) agarose.
HMSCs and agarose were loaded into a 100-μl glass syringe (SGE, Analytical Science, France), while Fluorinert FC-40 oil (3M, Paris, France) containing 1% (w/w) PEG-di-Krytox surfactant (RAN Biotechnologies, Beverly, USA) was loaded into a 1- and 2.5-ml glass syringes (SGE, Analytical Science). Droplets of cell-liquid agarose were generated in the FC-40 containing PEG-di-Krytox, at the flow-focusing junction, by controlling the flow rates using syringe pumps (neMESYS Low-Pressure Syringe Pump, Cetoni GmbH, Korbussen, Germany) (table S1). After complete loading, the chips were immersed in PBS, and the cells were allowed to settle down and to organize as MBs overnight in the CO2 incubator. Then, the agarose was gelled at 4°C for 30 min, after which the PEG-di-Krytox was extensively washed in flushing pure FC-40 in the culture chamber. After washing, cell culture medium was injected to replace the FC-40. All flow rates are indicated in table S1. Further operations were allowed by gelling the agarose in the droplets, such that the resulting beads were retained mechanically in the traps rather than by capillary forces (Fig. 2G). This step allowed the exchange of the oil surrounding the droplets by an aqueous solution, for example, to bring fresh medium for long-term culture, chemical stimuli, or the different solutions required for cell staining.
Live analysis of the MB formation
For the live imaging of the MB formation, the chips were immersed in PBS and then were incubated for 24 hours in a microscope incubator equipped with temperature, CO2, and hygrometry controllers (Okolab, Pozzuoli, Italy). The cells were imaged every 20 min.
Immunocytochemistry
2D cultures or MBs were washed in PBS and incubated with a 5 μM NucView 488 caspase-3 substrate (Interchim, Montluçon, France) diluted in PBS. After washing with PBS, HMSCs were fixed with a 4% (w/v) PFA (Alpha Aesar, Heysham, UK) for 30 min and permeabilized with 0.2 to 0.5% (v/v) Triton X-100 (Sigma-Aldrich) for 5 min. The samples were blocked with 5% (v/v) FBS in PBS for 30 min and incubated with a rabbit polyclonal anti–COX-2 primary antibody (ab15191, Abcam, Cambridge, UK) diluted at 1:100 in 1% (v/v) FBS for 4 hours. After washing with PBS, the samples were incubated with an Alexa Fluor 594–conjugated goat polyclonal anti-rabbit IgG secondary antibody (A-11012, Life Technologies, Saint Aubin, France) diluted at 1:100 in 1% (v/v) FBS for 90 min. Last, the cells were counterstained with 0.2 μM DAPI for 5 min (Sigma-Aldrich) and then washed with PBS.
The same protocol was used for the staining of VEGF-A–expressing cells using a rabbit anti-human VEGF-A monoclonal antibody (ab52917, Abcam, Cambridge, UK), which was revealed using the same secondary antibody as above. RUNX-2–positive cells were similarly stained using a mouse anti-human RUNX-2 monoclonal antibody (ab76956, Abcam, Cambridge, UK), which was revealed using an Alexa Fluor 488 goat anti-mouse IgG2a secondary antibody (A-21131, Life Technologies, Saint Aubin, France), both diluted at 1:100 in 1% (v/v) FBS.
Detection of hypoxia within the MBs
To measure potential induction of hypoxia within the core of the MBs, the cells were stained with Image-iT Red Hypoxia Reagent (Invitrogen) for 3 hours and then imaged using a fluorescence microscope. As a positive control, the chips containing the MBs were immersed into PBS, incubated overnight in an incubator set at 37°C under 3% O2/5% CO2, and lastly imaged as above.
Inhibition of molecular pathways regulating properties of HMSCs in MBs
To interrogate the contribution of signaling related to anti-inflammatory molecule production (COX-2 and NF-κB) or molecular pathways regulated by the cell structural organization (Notch, ROCK, and F-actin), several small molecules inducing their inhibition were added to the culture medium (table S1). For all the conditions, the final concentration of DMSO was below 0.1% (v/v) in the culture medium.
Viability assay
The cell viability was assessed using LIVE/DEAD staining kit (Molecular Probes, Life Technologies). The MBs were incubated for 30 min in PBS containing 1 μM calcein AM and 2 μM ethidium homodimer-1, in flushing 100 μl of the solution. The samples were then washed with PBS and imaged under a motorized fluorescence microscope (Nikon, France).
N-cadherin immunostaining
For the detection of the functional forms of N-cadherins (i.e., the N-cadherins closely linked to the actin network, which are PFA insoluble), the MBs were fixed with a 4% (w/v) PFA (Alpha Aesar, Heysham, UK) for 30 min and permeabilized with 0.2 to 0.5% (v/v) Triton X-100 (Sigma-Aldrich) for 5 min. Alternatively, the aggregates were incubated for 5 min with 100% cold methanol followed by 1 min with cold acetone, for the detection of total N-cadherins (i.e., the PFA-soluble and PFA-insoluble forms).
Then, the samples were blocked with 5% (v/v) FBS in PBS for 30 min and incubated with a rabbit polyclonal anti–N-cadherin primary antibody (ab18203, Abcam, Cambridge, UK) diluted at 1:100 in 1% (v/v) FBS for 4 hours. After washing with PBS, the samples were incubated with an Alexa Fluor 594–conjugated goat polyclonal anti-rabbit IgG secondary antibody (A-11012, Life Technologies, Saint Aubin, France) diluted at 1:100 in 1% (v/v) FBS for 90 min. Last, the cells were counterstained with 0.2 μM DAPI for 5 min (Sigma-Aldrich) and then washed with PBS.
F-actin staining
For the quantification of the polymerized form of actin (F-actin), the MBs were first fixed with a 4% (w/v) PFA (Alpha Aesar, Heysham, UK) for 30 min and permeabilized with 0.2 to 0.5% (v/v) Triton X-100 (Sigma-Aldrich) for 5 min. The samples were then blocked with a 5% (v/v) FBS solution and incubated for 90 min in a 1:100 phalloidin–Alexa Fluor 594 (Life Technologies) diluted in a 1% (v/v) FBS solution. The cells were then counterstained with 0.2 μM DAPI for 5 min (Sigma-Aldrich) and then washed with PBS.
Validation of the of the fluorescence signal patterns
To ensure the specificity of the antibody to COX-2 and N-cadherin, control UC-HMSCs were permeabilized, fixed, and incubated only with the secondary antibody (Alexa Fluor 594–conjugated goat polyclonal anti-rabbit IgG), as above. The absence of fluorescence signal indicated the specific staining for intracellular COX-2 and N-cadherin.
Next, to validate that the distribution of the fluorescence intensity was not related to any antibody diffusion limitation, the MBs were fixed and permeabilized, as above. For this assay, the MBs were not subjected to any blocking buffer. The cells were incubated for 90 min with the Alexa Fluor 594–conjugated goat polyclonal anti-rabbit IgG secondary antibody (A-11012, Life Technologies, Saint Aubin, France) diluted at 1:100 in 1% (v/v) FBS. Then, the cells were counterstained for DAPI, as above. Last, the MBs were collected from the chip, deposed on a glass slide, and imaged.
For the analysis of COX-2 expression by flow cytometry, the total MBs were recovered from the chip. The MBs were then trypsinized and triturated to obtain single-cell suspension. UC-HMSCs were stained for COX-2, as above. The percentage of COX-2–positive cells was quantified on 5 × 103 dissociated UC-HMSCs using a Guava easyCyte Flow Cytometer (Merck Millipore, Guyancourt, France). The results were compared to the fluorescence intensity distribution obtained by image analysis.
Clearing MBs derived from HMSCs
To interrogate the influence of the MB opacity in the COX-2 and N-cadherin fluorescence signals, the samples were treated by the Clear(T2) method after immunostaining (57). Briefly, the MBs were incubated for 10 min in 25% (v/v) formamide/10% (w/v) polyethylene glycol (PEG) (Sigma-Aldrich), then for 5 min in 50% (v/v) formamide/20% (w/v) PEG, and lastly for 60 min in 50% (v/v) formamide/20% (w/v) PEG, before their imaging. The fluorescence signal distribution was compared with the noncleared samples.
Cryosections
The MBs were collected from the chip and then fixed using PFA, as above. The MBs were incubated overnight in a 30% sucrose solution at 4°C. Then, the sucrose solution was exchanged to O.C.T. medium (optimal cutting temperature; Tissue-Tek) in inclusion molds, which were slowly cooled down using dry ice in ethanol. The molds were then placed at −80°C. On the day of the experiments, the O.C.T. blocks were cut at 7 μm using a cryostat (CM3050 S, Leica). The cryosections were placed on glass slides (SuperFrost Plus Adhesion, Thermo Fisher Scientific), dried at 37°C, and rehydrated using PBS. The cryosections were permeabilized and stained for COX-2, as above. The slides were lastly mounted in mounting medium containing DAPI (Fluoromount-G, Invitrogen).
Microscopy
All the images used for the quantitative analysis were taken using a motorized wide-field microscope (Ti, Eclipse, Nikon), equipped with a CMOS (complementary metal-oxide semiconductor) camera (ORCA-Flash4.0, Hamamatsu) and a fluorescence light-emitting diode source (Spectra X, Lumencor). The images were taken with a 10× objective with a 4-mm working distance (extra-long working distance) and a 0.45 numerical aperture (NA) (Plan Apo λ, Nikon).
For control experiments, images were taken using a motorized (Ti2, Nikon) confocal spinning disc microscope equipped with lasers (W1, Yokogawa) and the same camera and objective as above. Alternatively, the samples were imaged with a multiphoton microscope (TCS SP8 NLO, MP, Leica). The objective was an HCX PL APO CS 10×, 0.40 NA, working distance of 2.2 mm (Leica).
All immunostained samples were counterstained with DAPI, and most of the images (i.e., for N-cadherin, COX-2, VEGF-A, and F-actin) were taken using red light excitation that is known to penetrate deeper into the 3D objects than dyes emitting at lower weight length (e.g., DAPI, FITC). For wide-field microscopy, the focal plane was defined as the area containing the maximal number of DAPI-stained nuclei covering the focal area, while z stacks were taken for the whole in-focus planes containing DAPI-stained nuclei using spinning discs and two-photon confocal microscopy.
Wide-field imaging is sensitive for the emission of fluorescence from inside and outside the focal plane (i.e., from the out-of-focus upper and bottom planes of the spheroids) (58). Consistently, more DAPI signal from nuclei is emitted from the core than in the edges of MBs using epifluorescence microscopy (fig. S5I). We confirmed that our interpretation of the signal distribution from epifluorescence images was consistent with confocal and two-photon microscopy by comparing with images taken from the median z plane and the maximal z projection (fig. S5, N to P).
Consequently, the results unambiguously demonstrate that even if there are more cells in the z plane of the middle area of the MBs, the contribution of the out-of-focus signal from N-cadherin, COX-2, VEGF-A, and F-actin staining in this area of the MBs is minimal using wide-field imaging. Because of the higher throughput of wide-field microscopy, this method was chosen to quantitatively analyze the distribution of these immunolabeled proteins within MBs.
Competitive ELISA for PGE2
The culture supernatants of six-well plates were collected, while the total medium content of the chip was recovered by flushing the culture chamber with pure oil. A PGE2 human ELISA kit (ab133055, Abcam, Cambridge, UK) was used for the quantification of PGE2 concentration in the culture supernatant, following the manufacturer’s instructions. Briefly, a polynomial standard curve of PGE2 concentration derived from the serial dilution of a PGE2 standard solution was generated (r2 > 0.9). The absorbance was measured using a plate reader (Chameleon, Hidex, Finland).
ELISA for VEGF-A
A VEGF-A human ELISA kit (Ab119566, Abcam, Cambridge, UK) was used for the quantification of VEGF-A concentration in the culture supernatant of 2D cultures or from the chips. A linear standard curve of VEGF-A concentration derived from the serial dilution of a VEGF-A standard solution was generated (r2 > 0.9). The absorbance was measured using a plate reader (Chameleon, Hidex, Finland).
RT-qPCR analysis
The total MBs of a 3-day culture period were harvested from the chips, as described above. Alternatively, cells cultured on regular six-well plates were recovered using trypsin after the same cultivation time; CD146dim and CD146bright isolated cells were immediately treated for RNA extraction after sorting. The total RNA of 1 × 104 cells were extracted and converted to complementary DNA (cDNA) using SuperScript III CellsDirect cDNA Synthesis System (18080200, Invitrogen, Life Technologies), following the manufacturer’s instructions. After cell lysis, a comparable quality of the extracted RNA was observed using a bleach agarose gel, and similar RNA purity was obtained by measurement of the optical density at 260 and 280 nm using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE) between total RNA preparations from 2D and on-chip cultures.
The cDNA was amplified using a GoTaq qPCR Master Mix (Promega, Charbonnieres, France) or a FastStart Universal SYBR Green Master Mix (containing Rox) (Roche) and primers (Life Technologies, Saint Aubin, France or Eurofins Scientific, France) at the specified melting temperature (Tm) (table S2) using a MiniOpticon (Bio-Rad) or a QuantStudio 3 (Thermo Fisher Scientific) thermocycler. As a negative control, water and total RNA served as template for PCR. To validate the specificity of the PCR, the amplicons were analyzed by dissociation curve and subsequent loading on a 2.5% (w/v) agarose gel and migration at 100 V for 40 min. The PCR products were revealed by ethidium bromide (Sigma-Aldrich) staining, and the gels were imaged using a transilluminator. The analysis of the samples not subjected to reverse transcription (RT−) indicated negligible genomic DNA contamination (i.e., <0.1%), while no amplification signal was observed for the water template (no template control). The amount of TSG-6, COX-2, STC-1, VEGF-A, RUNX-2, CEBP-α, and SOX-9 transcripts was normalized to the endogenous reference [glyceraldehyde-3-phosphate dehydrogenase (GADPH)], and the relative expression to a calibrator (2D cultures) was given by 2−ΔΔCt calculation. At least five biological replicates of 2D and on-chip cultures were analyzed by at least duplicate measurements. The standard curves for GADPH, TSG-6, COX-2, and STC-1 were performed using a five serial dilution of the cDNA templates and indicated almost 100% PCR efficiency.
Image acquisition and analysis
The image analysis allowed us to perform a multiscale analysis (16) of the MBs. For each chip, single images of the anchors were acquired automatically with the motorized stage of the microscope. The analysis was conducted on a montage of the detected anchors using a custom MATLAB code (r2016a, MathWorks, Natick, MA). Two distinct routines were used: one with bright-field detection and one for the fluorescence experiments.
For the bright field-detection described previously (16), the cells were detected in each anchor as pixels with high values of the intensity gradient. This allowed for each cell aggregate to compute morphological parameters such as the projected area A and the shape index SI that quantifies the circularity of an object
where P is the perimeter. Shape index values range from 0 to 1, with 1 being assigned for perfect disk.
The MB detection with fluorescence staining (DAPI/Casp3/COX-2, DAPI/phalloidin, DAPI/N-cadherin, or LIVE/DEAD) was performed as described previously (16). First, morphological data were extracted at the MB level, such as the equivalent diameter of the MBs or the shape index. Also, the mean fluorescence signal of each MB was defined as the subtraction of the local background from the mean raw intensity.
At the cellular level, two different methods were used, both relying on the detection of the nuclei centers with the DAPI fluorescence signal. On the one hand, each cell location could be assigned to a normalized distance from the MB center (r/R) to correlate a nuclear fluorescence signal with a position in the MB, as previously described (16). On the other hand, the cell shapes inside the MBs were approximated by constructing Voronoi diagrams on the detected nuclei centers. Basically, the edges of the Voronoi cells are formed by the perpendicular bisectors of the segments between the neighboring cell centers. These Voronoi cells were used to quantify the cellular cytoplasmic signal (COX-2, F-actin and N-cadherin, VEGF and RUNX-2). In detail, to account for the variability of the cytoplasmic signal across the entire cell (nucleus included), the fluorescence signal of a single cell was defined as the mean signal of the 10% highest pixels of the corresponding Voronoi cell.
Image processing was also used to get quantitative data on 2D cultures, as previously described (16). Last, different normalization procedures were chosen in this paper. When an effect was quantified compared with a control condition, the test values were divided by the mean control value, and the significance was tested against 1. For some other data, the values were simply normalized by the corresponding mean at the chip level to discard the interchip variation from the analysis.
Statistical analysis
*P < 0.05; **P < 0.01; ***P < 0.001; NS, nonsignificant. Details of each statistical test and P values can be found in table S4.
Supplementary Material
Acknowledgments
C. Frot is gratefully acknowledged for the help with the microfabrication, and F. Soares da Silva is gratefully acknowledged for the help in flow cytometry. The group of Biomaterials and Microfluidics (BMCF) of the Center for Innovation and Technological Research as well as the Center for Translational Science (CRT)–Cytometry and Biomarkers Unit of Technology and Service (CB UTechS is also acknowledged for the access to the microfabrication and flow cytometry platform at the Institut Pasteur). Funding: The research leading to these results received funding from the European Research Council (ERC) grant agreement 278248 Multicell. Author contributions: S.S., C.N.B., and A.C. conceived the experiments. S.S. performed the experiments. R.F.-X.T. wrote the image processing code and performed the image analysis. R.F.-X.T., S.S., G.A., and A.B. performed the image and data analyses. S.S., C.N.B., and A.C. discussed the results and wrote the manuscript. All authors discussed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaw7853/DC1
Table S1. Flow rate for cell loading and phase exchange.
Table S2. Primer sequences.
Table S3. Small molecules for the inhibition of molecular pathways regulating hMSC behavior; the concentrations used in this study are adapted from (59, 60).
Table S4. Statistical tests and P values.
Fig. S1. Characterization of the hMSC population.
Fig. S2. Formation of MBs on chip.
Fig. S3. Structural organization in MBs.
Fig. S4. Intra-MB analysis of the COX-2 signal with concentric cell layers.
Fig. S5. Validation of the fluorescence signal patterns.
Fig. S6. Hypoxia analysis within MBs.
Fig. S7. Intra-MB fluorescence signal distribution in individual chip.
Fig. S8. Ratio of Casp3+ cells per MB formed with QNZ (nMBs = 1216), DAPT (nMBs = 658), Y27 (nMBs = 709), and CytoD (nMBs = 458).
Movie S1. Time lapse of MB formation.
Movie S2. Morphology of MB in 3D.
Movie S3. Representative z stack (from the bottom to the median plan of the MBs) acquired using a spinning disc confocal microcopy showing the distribution of de CD146dim (Vibrant Dil, green) and CD146bright (Vibrant DiO, red) within MBs.
REFERENCES AND NOTES
- 1.Rossi G., Manfrin A., Lutolf M. P., Progress and potential in organoid research. Nat. Rev. Genet. 19, 671–687 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Hough S. R., Thornton M., Mason E., Mar J. C., Wells C. A., Pera M. F., Single-cell gene expression profiles define self-renewing, pluripotent, and lineage primed states of human pluripotent stem cells. Stem Cell Rep. 2, 881–895 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim T.-H., Saadatpour A., Guo G., Saxena M., Cavazza A., Desai N., Jadhav U., Jiang L., Rivera M. N., Orkin S. H., Yuan G.-C., Shivdasani R. A., Single-cell transcript profiles reveal multilineage priming in early progenitors derived from Lgr5+ intestinal stem cells. Cell Rep. 16, 2053–2060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner D. A., Baillie-Johnson P., Martinez Arias A., Organoids and the genetically encoded self-assembly of embryonic stem cells. Bioessays 38, 181–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dominici M., Le Blanc K., Mueller I., Slaper-Cortenbach I., Marini F., Krause D., Deans R., Keating A., Prockop D., Horwitz E., Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Freeman B. T., Jung J. P., Ogle B. M., Single-cell RNA-seq of bone marrow-derived mesenchymal stem cells reveals unique profiles of lineage priming. PLOS ONE 10, e0136199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee W. C., Shi H., Poon Z., Nyan L. M., Kaushik T., Shivashankar G. V., Chan J. K. Y., Lim C. T., Han J., Van Vliet K. J., Multivariate biophysical markers predictive of mesenchymal stromal cell multipotency. Proc. Natl. Acad. Sci. U.S.A. 111, E4409–E4418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartosh T. J., Ylöstalo J. H., Mohammadipoor A., Bazhanov N., Coble K., Claypool K., Lee R. H., Choi H., Prockop D. J., Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc. Natl. Acad. Sci. U.S.A. 107, 13724–13729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhumiratana S., Eton R. E., Oungoulian S. R., Wan L. Q., Ateshian G. A., Vunjak-Novakovic G., Large, stratified, and mechanically functional human cartilage grown in vitro by mesenchymal condensation. Proc. Natl. Acad. Sci. U.S.A. 111, 6940–6945 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zelzer E., Olsen B. R., The genetic basis for skeletal diseases. Nature 423, 343–348 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Zelzer E., Mamluk R., Ferrara N., Johnson R. S., Schipani E., Olsen B. R., VEGFA is necessary for chondrocyte survival during bone development. Development 131, 2161–2171 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Biddulph D. M., Dozier M. M., Capehart A. A., Inhibition of prostaglandin synthesis reduces cyclic AMP levels and inhibits chondrogenesis in cultured chick limb mesenchyme. Methods Cell Sci. 22, 9–16 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Huang C., Xue M., Chen H., Jiao J., Herschman H. R., O’Keefe R. J., Zhang X., The spatiotemporal role of COX-2 in osteogenic and chondrogenic differentiation of periosteum-derived mesenchymal progenitors in fracture repair. PLOS ONE 9, e100079 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mammoto T., Mammoto A., Torisawa Y.-s., Tat T., Gibbs A., Derda R., Mannix R., de Bruijn M., Yung C. W., Huh D., Ingber D. E., Mechanochemical control of mesenchymal condensation and embryonic tooth organ formation. Dev. Cell 21, 758–769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh J.-Y., Fu Y.-S., Chang S.-J., Tsuang Y.-H., Wang H.-W., Functional module analysis reveals differential osteogenic and stemness potentials in human mesenchymal stem cells from bone marrow and wharton’s jelly of umbilical cord. Stem Cells Dev. 19, 1895–1910 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Sart S., Tomasi R. F.-X., Amselem G., Baroud C. N., Multiscale cytometry and regulation of 3D cell cultures on a chip. Nat. Commun. 8, 469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sart S., Tsai A.-C., Li Y., Ma T., Three-dimensional aggregates of mesenchymal stem cells: Cellular mechanisms, biological properties, and applications. Tissue Eng. Part B Rev. 20, 365–380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z., Yan X., CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 330, 150–162 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Sacchetti B., Funari A., Michienzi S., Di Cesare S., Piersanti S., Saggio I., Tagliafico E., Ferrari S., Robey P. G., Riminucci M., Bianco P., Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131, 324–336 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Delorme B., Ringe J., Gallay N., Le Vern Y., Kerboeuf D., Jorgensen C., Rosset P., Sensebé L., Layrolle P., Häupl T., Charbord P., Specific plasma membrane protein phenotype of culture-amplified and native human bone marrow mesenchymal stem cells. Blood 111, 2631–2635 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Tomasi R. F.-X., Sart S., Champetier T., Baroud C. N., Studying 3D cell cultures in a microfluidic droplet array under multiple time-resolved conditions. Biorxiv, 407759 (2018). [Google Scholar]
- 22.Chang H., Yang Q., Parvin B., Segmentation of heterogeneous blob objects through voting and level set formulation. Pattern Recogn. Lett. 28, 1781–1787 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cosgrove B. D., Mui K. L., Driscoll T. P., Caliari S. R., Mehta K. D., Assoian R. K., Burdick J. A., Mauck R. L., N-cadherin adhesive interactions modulate matrix mechanosensing and fate commitment of mesenchymal stem cells. Nat. Mater. 15, 1297–1306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMillen P., Holley S. A., Integration of cell-cell and cell-ECM adhesion in vertebrate morphogenesis. Curr. Opin. Cell Biol. 36, 48–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinz B., Pittet P., Smith-Clerc J., Chaponnier C., Meister J.-J., Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol. Biol. Cell 15, 4310–4320 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boregowda S. V., Booker C. N., Phinney D. G., Mesenchymal stem cells: The moniker fits the science. Stem Cells 36, 7–10 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Mahoney D. J., Mikecz K., Ali T., Mabilleau G., Benayahu D., Plaas A., Milner C. M., Day A. J., Sabokbar A., TSG-6 regulates bone remodeling through inhibition of osteoblastogenesis and osteoclast activation. J. Biol. Chem. 283, 25952–25962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshiko Y., Aubin J. E., Maeda N., Stanniocalcin 1 (STC1) protein and mRNA are developmentally regulated during embryonic mouse osteogenesis: The potential of STC1 as an autocrine/paracrine factor for osteoblast development and bone formation. J. Histochem. Cytochem. 50, 483–492 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Arikawa T., Omura K., Morita I., Regulation of bone morphogenetic protein-2 expression by endogenous prostaglandin E2 in human mesenchymal stem cells. J. Cell. Physiol. 200, 400–406 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Kuwano T., Nakao S., Yamamoto H., Tsuneyoshi M., Yamamoto T., Kuwano M., Ono M., Cyclooxygenase 2 is a key enzyme for inflammatory cytokine-induced angiogenesis. FASEB J. 18, 300–310 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y. M., Azahri N. S. M., Yu D. C. W., Woll P. J., Effects of COX-2 inhibition on expression of vascular endothelial growth factor and interleukin-8 in lung cancer cells. BMC Cancer 8, 218 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Németh K., Leelahavanichkul A., Yuen P. S. T., Mayer B., Parmelee A., Doi K., Robey P. G., Leelahavanichkul K., Koller B. H., Brown J. M., Hu X., Jelinek I., Star R. A., Mezey É., Bone marrow stromal cells attenuate sepsis via prostaglandin E2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 15, 42–49 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singer Z. S., Yong J., Tischler J., Hackett J. A., Altinok A., Surani M. A., Cai L., Elowitz M. B., Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol. Cell 55, 319–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toruner M., Fernandez-Zapico M., Sha J. J., Pham L., Urrutia R., Egan L. J., Antianoikis effect of nuclear factor-κB through up-regulated expression of osteoprotegerin, BCL-2, and IAP-1. J. Biol. Chem. 281, 8686–8696 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Kipp A. P., Deubel S., Arnér E. S. J., Johansson K., Time- and cell-resolved dynamics of redox-sensitive Nrf2, HIF and NF-κb activities in 3D spheroids enriched for cancer stem cells. Redox Biol. 12, 403–409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guntur A. R., Rosen C. J., Naski M. C., N-cadherin adherens junctions mediate osteogenesis through PI3K signaling. Bone 50, 54–62 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou S., TGF-β regulates β-catenin signaling and osteoblast differentiation in human mesenchymal stem cells. J. Cell. Biochem. 112, 1651–1660 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D., Yang J.-Y., Shin C. S., Overexpression of α-catenin increases osteoblastic differentiation in mouse mesenchymal C3H10T1/2 cells. Biochem. Biophys. Res. Commun. 382, 745–750 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Gumbiner B. M., Regulation of cadherin adhesive activity. J. Cell Biol. 148, 399–404 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Susperregui A. R. G., Gamell C., Rodríguez-Carballo E., Ortuño M. J., Bartrons R., Rosa J. L., Ventura F., Noncanonical BMP signaling regulates cyclooxygenase-2 transcription. Mol. Endocrinol. 25, 1006–1017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sero J. E., Sailem H. Z., Ardy R. C., Almuttaqi H., Zhang T., Bakal C., Cell shape and the microenvironment regulate nuclear translocation of NF-κB in breast epithelial and tumor cells. Mol. Syst. Biol. 11, 790 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Y., Chen H., Li H., Wu Y., 3D culture increases pluripotent gene expression in mesenchymal stem cells through relaxation of cytoskeleton tension. J. Cell. Mol. Med. 21, 1073–1084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takarada T., Nakazato R., Tsuchikane A., Fujikawa K., Iezaki T., Yoneda Y., Hinoi E., Genetic analysis of Runx2 function during intramembranous ossification. Development 143, 211–218 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Lana-Elola E., Rice R., Grigoriadis A. E., Rice D. P. C., Cell fate specification during calvarial bone and suture development. Dev. Biol. 311, 335–346 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Tsujii M., Kawano S., Tsuji S., Sawaoka H., Hori M., DuBois R. N., Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 93, 705–716 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Liu X. H., Kirschenbaum A., Yao S., Lee R., Holland J. F., Levine A. C., Inhibition of cyclooxygenase-2 suppresses angiogenesis and the growth of prostate cancer in vivo. J. Urol. 164, 820–825 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Yoshida S., Amano H., Hayashi I., Kitasato H., Kamata M., Inukai M., Yoshimura H., Majima M., COX-2/VEGF-dependent facilitation of tumor-associated angiogenesis and tumor growth in vivo. Lab. Invest. 83, 1385–1394 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Eibl G., Bruemmer D., Okada Y., Duffy J. P., Law R. E., Reber H. A., Hines O. J., PGE2 is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 306, 887–897 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Yanni S. E., McCollum G. W., Penn J. S., Genetic deletion of COX-2 diminishes VEGF production in mouse retinal Müller cells. Exp. Eye Res. 91, 34–41 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hatazawa R., Tanigami M., Izumi N., Kamei K., Tanaka A., Takeuchi K., Prostaglandin E2 stimulates VEGF expression in primary rat gastric fibroblasts through EP4 receptors. Inflammopharmacology 15, 214–217 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Nakanishi M., Sato T., Li Y., Nelson A. J., Farid M., Michalski J., Kanaji N., Wang X., Basma H., Patil A., Goraya J., Liu X., Togo S., Toews M. L., Holz O., Muller K.-C., Magnussen H., Rennard S. I., Prostaglandin E2 stimulates the production of vascular endothelial growth factor through the E-prostanoid-2 receptor in cultured human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 46, 217–223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X., Klein R. D., Prostaglandin E2 induces vascular endothelial growth factor secretion in prostate cancer cells through EP2 receptor-mediated cAMP pathway. Mol. Carcinog. 46, 912–923 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Madrigal-Martínez A., Cazaña F. J. L., Fernández-Martínez y. A. B., Role of intracellular prostaglandin E2 in cancer-related phenotypes in PC3 cells. Int. J. Biochem. Cell Biol. 59, 52–61 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Yang D.-H., Cai K. Q., Roland I. H., Smith E. R., Xu X.-X., Disabled-2 is an epithelial surface positioning gene. J. Biol. Chem. 282, 13114–13122 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Rula M. E., Cai K. Q., Moore R., Yang D.-H., Staub C. M., Capo-Chichi C. D., Jablonski S. A., Howe P. H., Smith E. R., Xu X.-X., Cell autonomous sorting and surface positioning in the formation of primitive endoderm in embryoid bodies. Genesis 45, 327–338 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Hubatsch L., Peglion F., Reich J. D., Rodrigues N. T. L., Hirani N., Illukkumbura R., Goehring N. W., A cell-size threshold limits cell polarity and asymmetric division potential. Nat. Phys. 15, 1078–1085 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boutin M. E., Hoffman-Kim D., Application and assessment of optical clearing methods for imaging of tissue-engineered neural stem cell spheres. Tissue Eng. Part C Methods 21, 292–302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanderson M. J., Smith I., Parker I., Bootman M. D., Fluorescence microscopy. Cold Spring Harb. Protoc. 2014, pdb.top071795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai A.-C., Liu Y., Yuan X., Ma T., Compaction, fusion, and functional activation of three-dimensional human mesenchymal stem cell aggregate. Tissue Eng. Part A 21, 1705–1719 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartosh T. J., Ylöstalo J. H., Bazhanov N., Kuhlman J., Prockop D. J., Dynamic compaction of human mesenchymal stem/precursor cells into spheres self-activates caspase-dependent IL1 signaling to enhance secretion of modulators of inflammation and immunity (PGE2, TSG6, and STC1). Stem Cells 31, 2443–2456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/10/eaaw7853/DC1
Table S1. Flow rate for cell loading and phase exchange.
Table S2. Primer sequences.
Table S3. Small molecules for the inhibition of molecular pathways regulating hMSC behavior; the concentrations used in this study are adapted from (59, 60).
Table S4. Statistical tests and P values.
Fig. S1. Characterization of the hMSC population.
Fig. S2. Formation of MBs on chip.
Fig. S3. Structural organization in MBs.
Fig. S4. Intra-MB analysis of the COX-2 signal with concentric cell layers.
Fig. S5. Validation of the fluorescence signal patterns.
Fig. S6. Hypoxia analysis within MBs.
Fig. S7. Intra-MB fluorescence signal distribution in individual chip.
Fig. S8. Ratio of Casp3+ cells per MB formed with QNZ (nMBs = 1216), DAPT (nMBs = 658), Y27 (nMBs = 709), and CytoD (nMBs = 458).
Movie S1. Time lapse of MB formation.
Movie S2. Morphology of MB in 3D.
Movie S3. Representative z stack (from the bottom to the median plan of the MBs) acquired using a spinning disc confocal microcopy showing the distribution of de CD146dim (Vibrant Dil, green) and CD146bright (Vibrant DiO, red) within MBs.