

Abstract
Ganglion cells (RGCs) are the sole output neurons of the retinal circuity. Here, we investigated whether and how dopamine D2 receptors modulate the excitability of dissociated rat RGCs. Application of the selective D2 receptor agonist quinpirole inhibited outward K+ currents, which were mainly mediated by glybenclamide- and 4-aminopyridine-sensitive channels, but not the tetraethylammonium-sensitive channel. In addition, quinpirole selectively enhanced Nav1.6 voltage-gated Na+ currents. The intracellular cAMP/protein kinase A, Ca2+/calmodulin-dependent protein kinase II, and mitogen-activated protein kinase/extracellular signal-regulated kinase signaling pathways were responsible for the effects of quinpirole on K+ and Na+ currents, while phospholipase C/protein kinase C signaling was not involved. Under current-clamp conditions, the number of action potentials evoked by positive current injection was increased by quinpirole. Our results suggest that D2 receptor activation increases RGC excitability by suppressing outward K+ currents and enhancing Nav1.6 currents, which may affect retinal visual information processing.
Electronic supplementary material
The online version of this article (10.1007/s12264-019-00431-3) contains supplementary material, which is available to authorized users.
Keywords: Retinal ganglion cell, Dopamine D2 receptor, Outward K+ current, Nav1.6 voltage-gated Na+ current, Excitability
Introduction
By activating distinct G-protein-coupled receptors, dopamine (DA) has been demonstrated to be involved in diverse functions in the central nervous system [1–6]. Five DA receptor (DAR) subtypes (D1–D5) have been identified according to their biochemical and pharmacological characteristics. On the basis of their effects on adenylate cyclase activity, DARs are further classified into D1 (subtypes D1 and D5) and D2 receptors (subtypes D2, D3, and D4). Activation of D1 receptors positively regulates adenylate cyclase activity by coupling with the Gs protein, while activation of D2 receptors negatively regulates adenylate cyclase activity through the Gi/o protein [3, 7, 8].
DA, released mainly from dopaminergic amacrine cells in retina, plays an important role in modulating retinal neuronal functions [9–12]. As the sole output neurons of the retina, ganglion cells (RGCs) integrate retinal visual signals. Therefore, changes in the excitability of RGCs may modulate visual information processing in the retina. It has been reported that DA might change RGC spiking by regulating retinal neuronal circuits [13–16]. In addition, DA might also modulate RGC activity directly. For example, DA inhibits the discharges of dissociated RGCs by modulating voltage-dependent Na+ channels [17–20]. RGCs express DARs [18, 21, 22], while also expressing various voltage-gated ion channels [23–27]. There is evidence that stimulation of D1 receptors reduces outward K+ current amplitudes in rat RGC preparations and hyperpolarization-activated cation currents in retinal slices [28, 29]. Our recent study showed that temporal summation of excitatory postsynaptic potentials in rat RGCs is enhanced by D1 receptor activation, mainly by affecting inward-rectifier K+ (Kir) currents [30]. These studies indicate that RGC excitability is influenced by D1 receptor stimulation. Moreover, activation of D2 receptors modulates neuronal excitability and synaptic integration in different brain regions, thereby participating in the regulation of reward-related behaviors, working memory, and locomotion [2]. D2 receptor-induced modulation of neuronal excitability is largely a result of changes in the functions of voltage-gated ion channels [31–35]. In the present study, we investigated the effects of D2 receptors on RGC excitability and the underlying mechanisms by whole-cell patch-clamp recording in acutely-dissociated rat RGCs.
Materials and Methods
Animals
All experimental procedures were performed in compliance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals. The experimental protocol was approved by the Animal Care Committee of the Institutes of Brain Science, Fudan University. Male Sprague-Dawley rats (70 g–100 g, 4 weeks–5 weeks old) were purchased from the SLAC Laboratory Animal Co., Ltd (Shanghai, China). Animals were housed on a 12-h light/dark cycle. To the best of our ability, we minimized the number of animals used in this study, and ameliorated their suffering.
Retrograde Labeling of RGCs
RGCs were retrogradely labeled following the procedures previously described in detail [29, 37]. RGCs were labeled with 1% cholera toxin B subunit (CTB; List Biological Laboratories, Campbell, CA) or 4% rhodamine-B-isothiocyanate (RITC; List Biological Laboratories).
Preparation of Isolated RGCs
Isolated RGCs were obtained by digesting retinal tissue with papain and mechanically dissociated as previously described [29, 38].
Immunohistochemistry and Immunocytochemistry
Immunohistochemistry was performed in vertical retinal slices and immunocytochemistry in isolated RGCs, following the procedures described previously [29, 36, 39]. The primary antibodies used were anti-dopamine D2 receptor antibody (1:200 dilution; AB1558, Chemicon International Inc., Temecula, CA) and polyclonal goat anti-CTB (1:4000, List Biological Laboratories). Images were captured using a Leica SP2 confocal laser-scanning microscope (Nussloch, Germany).
Whole-Cell Patch-Clamp Recordings
Standard whole-cell patch-clamp techniques were applied to record membrane currents and potentials in RITC-labeled RGCs using a patch amplifier (Axopatch700B; Molecular Devices, Novato, CA) with a Digidata 1440A data acquisition board and pClamp 10.2 software [37, 38, 40]. The sampling rate was set at 10 kHz, and signals were filtered at 1 kHz. The recording pipettes (3 MΩ–6 MΩ) were filled with internal solutions. The bath solution used for K+ current recordings consisted of (in mmol/L) NaCl 140, KCl 5, CaCl2 2, MgCl2 1, HEPES 10, and glucose 20, with 0.5 μmol/L tetrodotoxin (TTX) and 100 μmol/L CdCl2 (pH 7.4 with NaOH, 300 mOsm/L–310 mOsm/L with sucrose); the pipette solution contained (in mmol/L) KCl 140, NaCl 9, MgCl2 1, EGTA 0.2, ATP-Mg 2, GTP-Na 0.25, and HEPES 10 (pH 7.2 with KOH, 290 mOsm/L–300 mOsm/L with sucrose). To record Na+ currents, the bath solution contained (in mmol/L) NaCl 130, CaCl2 2, MgCl2 1, HEPES 10, tetraethylammonium (TEA)-Cl 15, 4-aminopyridine (4-AP) 10, and glucose 10 (pH 7.4 with NaOH, 300 mOsm/L–310 mOsm/L with sucrose); the pipette solution contained (in mmol/L) CsCl 130, NaCl 10, HEPES 5, EGTA 8, TEA-Cl 10, ATP-Mg 2, and GTP-Na 1 (pH 7.2 adjusted with CsOH, 290 mOsm/L–300 mOsm/L with sucrose). To record evoked action potentials, the bath solution consisted of (in mmol/L) NaCl 135, KCl 3, CaCl2 2, MgCl2 1, HEPES 10, glucose 11, and sucrose 10 (pH 7.4 with NaOH, 300 mOsm/L–310 mOsm/L with sucrose), while the pipettes were filled with an internal solution containing (in mmol/L) potassium D-gluconate 120, EGTA 1, HEPES 10, ATP-Mg 4, GTP-Na 0.3, phosphocreatine 10, CaCl2 0.1, and MgCl2 1 (pH 7.2 with KOH, 280 mOsm/L–290 mOsm/L with sucrose). All experiments were performed at room temperature (22 °C–25 °C).
Reagents and Drug Application
Quinpirole, sulpiride, 4-AP, TEA, Rp-cAMP, bisindolylmaleimide IV (Bis IV), KN-62, ICA121431, 4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126), 4,9-anhydrotetrodotoxin (AHTTX), phrixotoxin-3, and TTX were from Tocris (Tocris Bioscience, Ellisville, MO), while others were from Sigma (Sigma-Aldrich, Inc., St. Louis, MO). The drug-containing solution was delivered by a stepper motor-based rapid solution exchanger (RSC-160, Bio-Logic, Claix, France) [29, 38].
Data Analysis
Data were analyzed using Clampfit 10.2 (Molecular Devices, Foster City, CA), SigmaPlot 10.0 (version 10.0, Systat Software Inc., San Jose, CA), and Igor 4.0 software (WaveMetrics, Lake Oswego, OR). A Boltzmann function was used to fit the activation and inactivation curves. Data are shown as the mean ± SEM. Statistical analysis was performed by either one-way ANOVA with Bonferroni’s post-hoc test (multiple comparisons) or the paired t test. A P value < 0.05 was considered significant.
Results
Activation of D2 Receptors Selectively Suppresses Glybenclamide- and 4-AP-Sensitive Outward K+ Currents
It has been shown that D2 receptors are expressed in cells of the retinal ganglion cell layer [22] and D2 receptor agonists and antagonists modulate the responses of rat RGCs [19, 41], suggesting that D2 receptors are expressed in rat RGCs. We first detected the expression of D2 receptors on isolated RGCs labelled retrogradely with CTB using double immunofluorescent labeling (Fig. S1A). We further confirmed the expression of D2 receptors on RGCs in retinal vertical slices in which labeled D2 receptors co-localized with RGCs retrogradely labeled by CTB (Fig. S1B). To avoid possible non-specific binding of the D2 receptor antibody, we did negative control experiment by replacing the D2 receptor antibody with phosphate-buffered saline. This experiment demonstrated the specificity of the D2 receptor antibody used in the present study (Fig. S1B). These results confirmed that D2 receptors are indeed expressed in rat retinal RGCs.
Outward K+ currents in rat RGCs were recorded and the effects of D2 receptor activation on these currents were examined. The protocol for inducing the K+ currents is shown in Fig. 1A. RGCs were held at −70 mV and stepped to +30 mV in increments of 10 mV. Bath application of quinpirole (10 µmol/L), a selective D2 receptor agonist, markedly reduced the K+ current amplitudes (Fig. 1A). The current-voltage curves showed that quinpirole inhibited the K+ currents in a voltage-dependent manner (Fig. 1B, C). The time course of changes in K+ current amplitudes at +30 mV with or without quinpirole clearly showed that stable recordings could be obtained in the control group (Fig. 1D). Extracellular application of quinpirole (10 μmol/L) progressively reduced the current amplitudes and washout with normal solution brought the currents to the control level. Moreover, the suppressive effect of quinpirole on the K+ currents was dose-dependent (Fig. 1E, F). To confirm the quinpirole effect was through activating D2 receptors, sulpiride (10 μmol/L), a selective D2 receptor antagonist, was first applied to RGCs, which did not change the K+ currents (100.4% ± 1.4% of control, n = 11, P = 0.803); then, co-application of quinpirole failed to suppress the K+ currents (101.4% ± 3.2% of control, n = 11, P = 0.684) (Fig. 1G). These results indicated that the effect of quinpirole is indeed through activating D2 receptors.
Fig. 1.
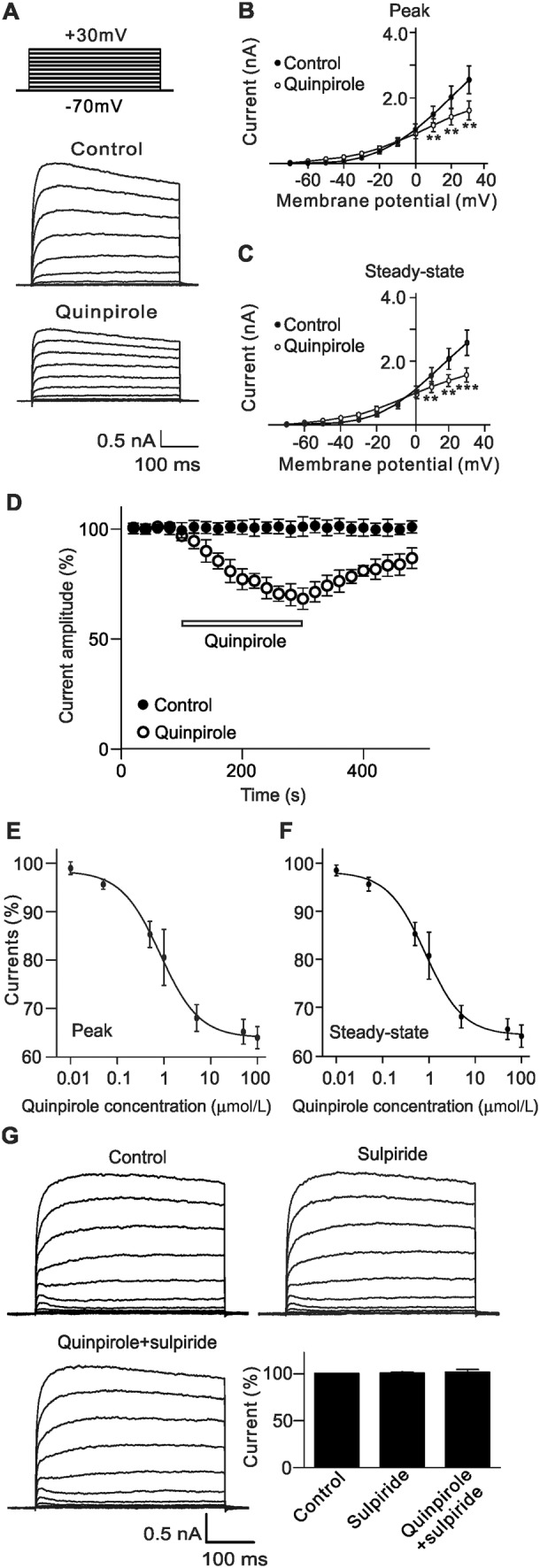
Suppression of outward K+ currents by activating D2 receptors in rat RGCs. A Representative outward K+ currents recorded from an RGC, showing that extracellular application of quinpirole (10 μmol/L), a selective D2 receptor agonist, significantly suppressed the current amplitudes. The cell was held at −70 mV and the currents were evoked by a series of voltage pulses (from −70 mV to +30 mV in increments of 10 mV). B, CI-V curves showing that quinpirole voltage-dependently suppressed average peak (B) and steady-state (C) current amplitudes (n = 12). **P < 0.01, ***P < 0.001 vs control. D Time course of quinpirole-induced suppression of K+ currents at +30 mV. Note that the current amplitudes were almost unchanged during a period of 8 min without quinpirole (Control), while quinpirole significantly reduced the current amplitudes. E, F Concentration-dependent suppression of peak (E) and steady-state (F) current amplitudes by quinpirole (n = 7–11 for each quinpirole concentration). G Sample current traces showing that sulpiride (10 μmol/L), a selective D2 receptor antagonist, blocked the quinpirole-induced suppression of K+ currents recorded in a rat RGC. Bar chart summarizing the changes of K+ current amplitudes at +30 mV under different conditions (n = 11). All data are normalized to control and presented as the mean ± SEM.
RGCs are known to express various K+ channels, including large-conductance Ca2+-activated K+ channels (BKCa) that are sensitive to TEA, delayed rectifying K+ channels that are blocked by 4-AP, and ATP-sensitive K+ channels (KATP) that are sensitive to glybenclamide (Gb) [24, 26, 29]. We then set out to identify which K+ channels were modulated by activating D2 receptors. Fig. 2A shows that the outward K+ currents recorded from RGCs pre-treated with 4-AP (5 mmol/L) and TEA (10 mmol/L) were still clearly reduced by perfusion with quinpirole. At +30 mV, the average current amplitude was reduced to 68.8% ± 5.2% of control (n = 8, P = 0.002; Fig. 2B). Similarly, when TEA- and Gb-sensitive currents were blocked by pre-treatment with TEA (10 mmol/L) and Gb (10 μmol/L), the quinpirole-induced suppression of K+ currents still occurred (80.2% ± 3.8% of control, n = 10, P = 0.002) (Fig. 2C, D). In contrast, after blockade of Gb- and 4-AP-sensitive K+ currents, the remaining currents were not influenced by quinpirole application (97.6% ± 1.3% of control, n = 9, P = 0.099; Fig. 2E, F), suggesting that D2 receptors selectively modulate the Gb- and 4-AP-sensitive K+ channels.
Fig. 2.
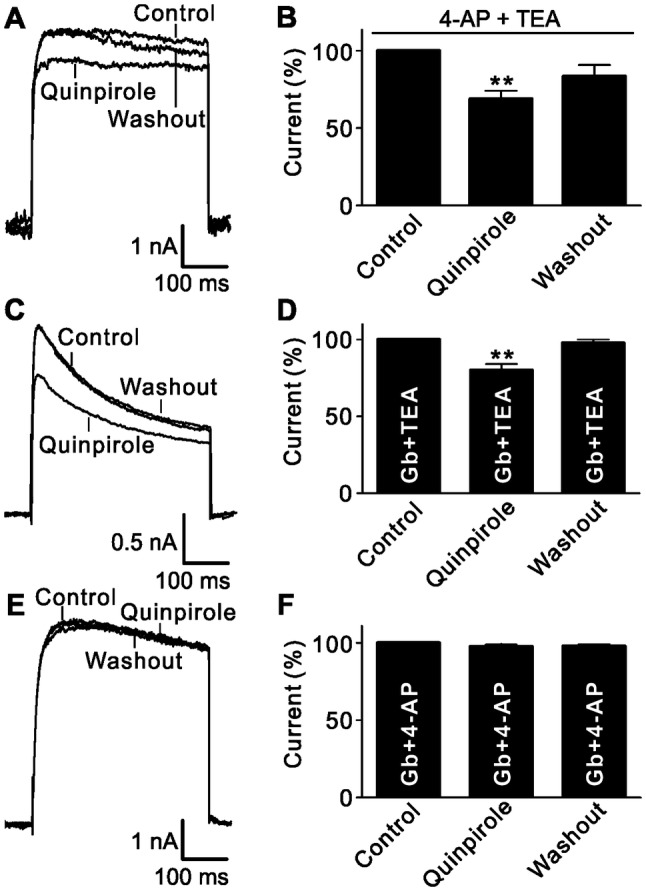
Quinpirole selectively suppresses Gb- and 4-AP-sensitive K+ current components. A Representative current traces recorded from an RGC, showing that extracellular application of 10 µmol/L quinpirole still suppressed the current amplitude after the 4-AP- and TEA-sensitive current components were blocked. B Bar chart summarizing the changes of K+ current amplitudes at +30 mV (n = 8, **P < 0.01 vs control). C Representative current recordings from an RGC, showing that extracellular application of quinpirole (10 μmol/L) significantly reduced the current amplitude in the presence of Gb (10 μmol/L) and TEA (10 mmol/L). D Bar chart summarizing the changes of K+ current amplitudes at +30 mV under different conditions (n = 10, **P < 0.01 vs control). E Sample current traces from an RGC showing that quinpirole (10 μmol/L) did not further reduce the current amplitude after the Gb- and 4-AP-sensitive components were blocked. F Bar chart summarizing the changes of K+ current amplitudes at +30 mV (n = 9).
cAMP/PKA, CaMKII, and MAPK/ERK Signaling Pathways are Involved in D2 Receptor-Mediated Effects
We then further investigated the possible signaling pathways involved in the D2 receptor-mediated effect on K+ currents. There is evidence showing that D2 receptors are linked to many intracellular signaling pathways [32, 42, 43]. We first examined the possible involvement of the forskolin-stimulated cAMP/protein kinase A (PKA) signaling pathway. Perfusion with Rp-cAMP (2 μmol/L), a cAMP/PKA pathway inhibitor [29, 38], for at least 3 min did not change the K+ current amplitude recorded from RGCs (100.7% ± 4.6% of control, n = 13, P = 0.926; Fig. 3A), and the current remained unchanged when quinpirole was added (107.3% ± 6.7% of control, n = 13, P = 0.403; Fig. 3B). Similarly, when RGCs were intracellularly dialyzed with KN-62 (10 μmol/L), a Ca2+/calmodulin-dependent protein kinase II (CaMKII) pathway blocker, or U0126 (10 μmol/L), a specific inhibitor of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, through the recording pipette [38], extracellular perfusion with quinpirole no longer suppressed outward K+ currents (94.5% ± 3.1% of control for KN-62, n = 11, P = 0.247; 96.8% ± 2.1% of control for U0126, n = 12, P = 0.06; Fig. 3C–F). Although activation of D2 receptors may also be coupled to the phospholipase C (PLC)/protein kinase C (PKC) pathway, experiments involving this pathway presented different results. After blocking PKC by intracellular dialysis with the PKC inhibitor Bis IV (10 μmol/L) [29, 38], the K+ currents recorded from RGCs were still reduced by quinpirole (68.9% ± 5.7% of control, n = 14, P = 0.004; Fig. 3G, H).
Fig. 3.
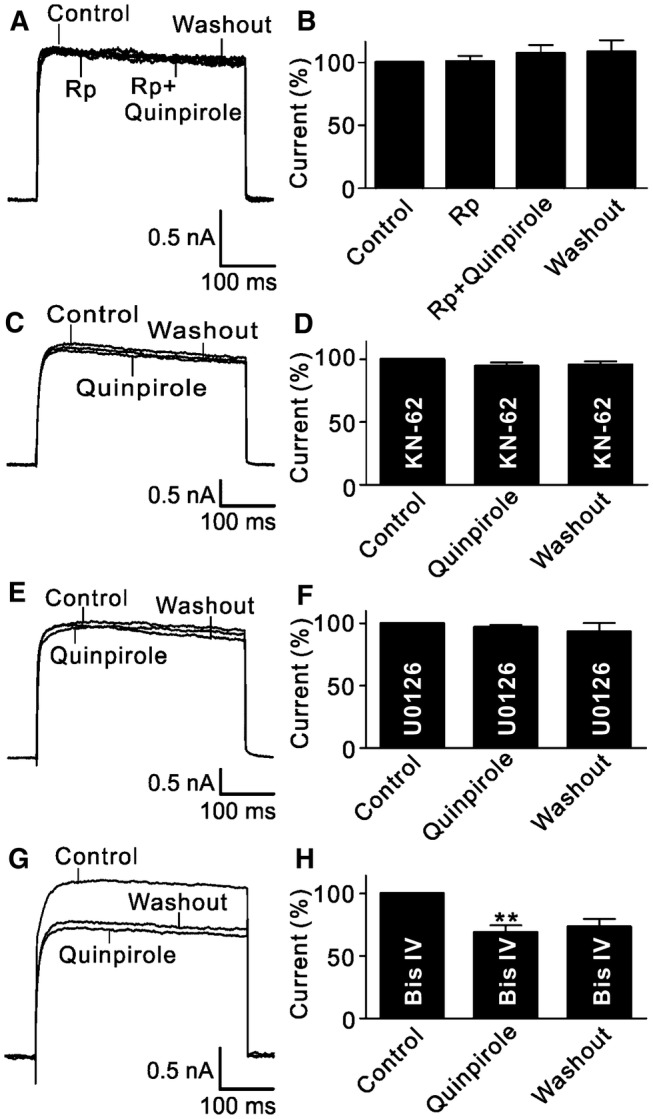
Signaling pathways involved in the quinpirole-induced suppression of outward K+ channels. A Representative current traces recorded from an RGC, showing that quinpirole did not change the outward K+ current amplitude when the cell was perfused with Rp-cAMP (2 μmol/L). B Bar chart summarizing the changes in K+ current amplitudes at +30 mV under different conditions (n = 13). C, D Sample current traces showing that the CaMKII signaling inhibitor KN-62 (10 μmol/L) blocked the quinpirole-induced suppression of outward K+ currents (C), and summary data are shown in (D) (n = 11). E Sample current traces showing that U0126 (10 μmol/L), a MAPK/ERK signaling inhibitor, blocked the quinpirole-induced suppression of outward K+ currents. F Bar chart summarizing the changes of outward K+ current amplitudes at +30 mV (n = 12). G Representative current traces recorded from an RGC, showing that the PKC inhibitor Bis IV (10 μmol/L) failed to block the quinpirole-induced suppression of outward K+ currents. H Summary bar graphs for the effect of Bis IV on the quinpirole-induced suppression of outward K+ currents (n = 14, **P <0.01 vs control).
Activation of D2 Receptors Selectively Increases Nav1.6 Channel Currents
RGCs express voltage-gated Na+ channels [27, 44, 45], so we further investigated whether activation of D2 receptors modulates Na+ currents in acutely isolated rat RGCs. Na+ currents were induced by 50-ms depolarizing voltage pulses from a holding potential of −70 mV to +30 mV in steps of 10 mV. Bath application of quinpirole remarkably enhanced the current amplitudes (Fig. 4A, B). The current-voltage curves showed that quinpirole voltage-dependently enhanced the current amplitudes (Fig. 4C). For example, the current amplitude at −20 mV was significantly increased to 130.2% ± 5.2% of control by quinpirole (n = 12, P < 0.001). The time course of quinpirole-induced changes in Na+ current amplitudes at −20 mV showed that they remained unchanged during 8 min of recording; extracellular application of quinpirole (10 μmol/L) significantly enhanced the current amplitudes; and the currents returned to the control level after washout (Fig. 4D). The quinpirole effect on Na+ currents was through activating D2 receptors. In the presence of sulpiride, addition of quinpirole failed to change the Na+ current amplitude (104.5% ± 2.6% of control, n = 6, P = 0.129; Fig. 4E, F).
Fig. 4.
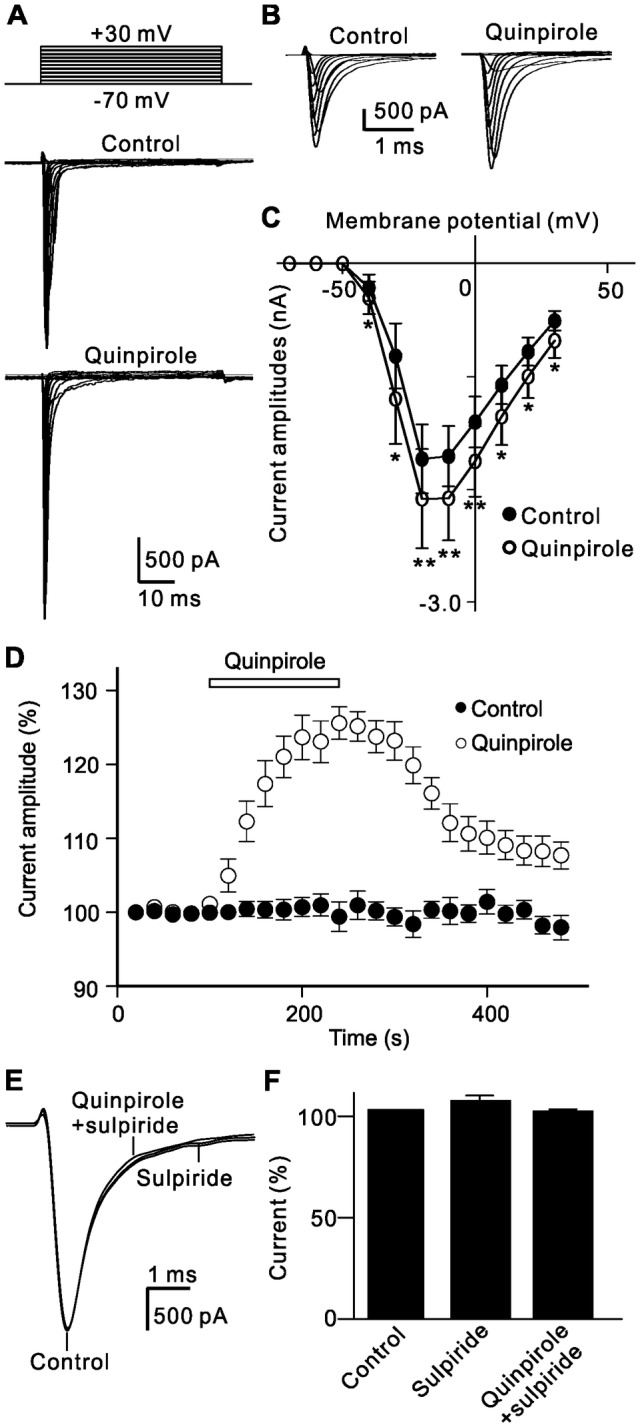
D2 receptor activation-induced enhancement of Na+ channel currents in rat RGCs. A Representative Na+ currents recorded from an RGC showing that extracellular application of quinpirole (10 μmol/L) significantly increased the current amplitudes. The cell was held at −70 mV and the currents were induced by a series of depolarizing voltage pulses from −70 mV to +30 mV at steps of 10 mV. B Expanded peak current traces before and after quinpirole application as in A. CI-V curves showing that quinpirole voltage-dependently enhanced Na+ currents (n = 12; *P < 0.05, **P < 0.01 vs control). D Time course of quinpirole-induced enhancement of Na+ currents at −20 mV. Note that the current amplitudes were almost unchanged during a period of 8 min without quinpirole (Control), while quinpirole significantly increased the current amplitudes. E Sample current traces showing that the quinpirole-induced increase in Na+ currents was blocked by sulpiride (10 µmol/L). F Bar chart summarizing the changes of Na+ current amplitudes at −20 mV under different conditions (n = 6).
We also studied changes in the activation and inactivation curves of Na+ channels. The steady-state activation curve of Na+ channels was not significantly shifted after quinpirole application; the Vh value obtained from Boltzmann function fitting was −31.4 mV ± 0.98 mV (n = 10), which was comparable to that of control (−29.2 mV ± 0.89 mV) (n = 10, P = 0.521; Fig. 5A). The inactivation curves also fitted a Boltzmann function. The cells were first given a 200-ms pre-pulse from a holding potential of −70 mV to different potentials, and then depolarized to −10 mV. The inactivation curves of the channels before and after quinpirole application were almost same (Vh values: −47.1 ± 0.57 mV in control; −48.3 ± 0.63 mV in quinpirole group, n = 12, P = 0.881; Fig. 5B, C).
Fig. 5.
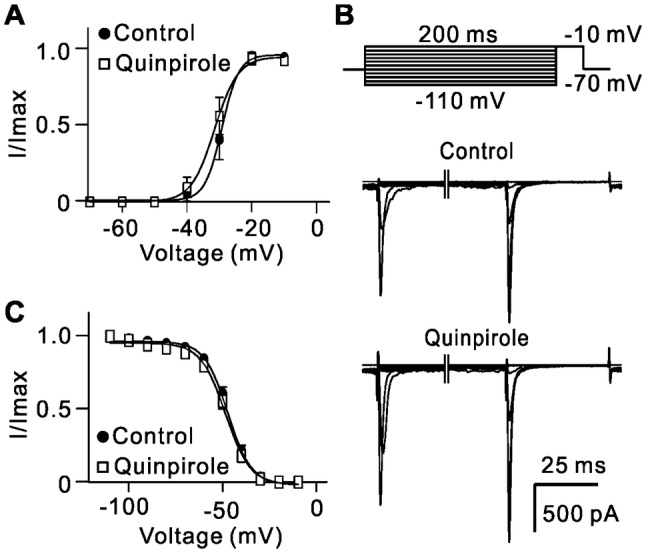
Activation of D2 receptors has no effect on activation and inactivation curves of Na+ currents. A Activation curves of Na+ currents before and after quinpirole (10 µmol/L) application. Normalized points are fitted with a Boltzmann function (n = 12). B Representative Na+ currents in an RGC evoked when the cells were first hyperpolarized to different potentials from a holding potential of −70 mV and then depolarized to −10 mV before and after application of quinpirole (10 µmol/L). C Inactivation curves of Na+ currents before and after application of quinpirole. Normalized points are fitted with a Boltzmann function (n = 12).
There are two components in Na+ channel currents, TTX-sensitive and TTX-resistant, both of which are expressed in sensory neurons of the mammalian nervous system [23]. Perfusion with 500 nmol/L TTX almost completely inhibited Na+ currents in isolated RGCs (Fig. 6A), suggesting that only TTX-sensitive Na+ channels are expressed in rat RGCs. Several Na+ channel subunits, including Nav1.1, Nav1.2, and Nav1.6, are expressed in RGCs [25, 27]. Our results showed that bath application of 10 nmol/L phrixotoxin-3 (Ph3), a Nav1.2 blocker, reduced the Na+ current amplitude to 96.5% ± 0.7% of control (n = 6, P = 0.007), and addition of 1 μmol/L ICA121431 (ICA), a Nav1.1 blocker, reduced the currents to 78.0% ± 2.9% of control (n = 6, P = 0.005), and further to 13.6% ± 2.4% of control (n = 6, P = 0.002) when 100 nmol/L 4,9-anhydrotetrodotoxin (AHTTX), a Nav1.6 blocker [46, 47], was added (Fig. 6B, C). These results demonstrated that Nav1.6 is the dominant Na+ channel in rat RGCs although other subunits are also expressed.
Fig. 6.
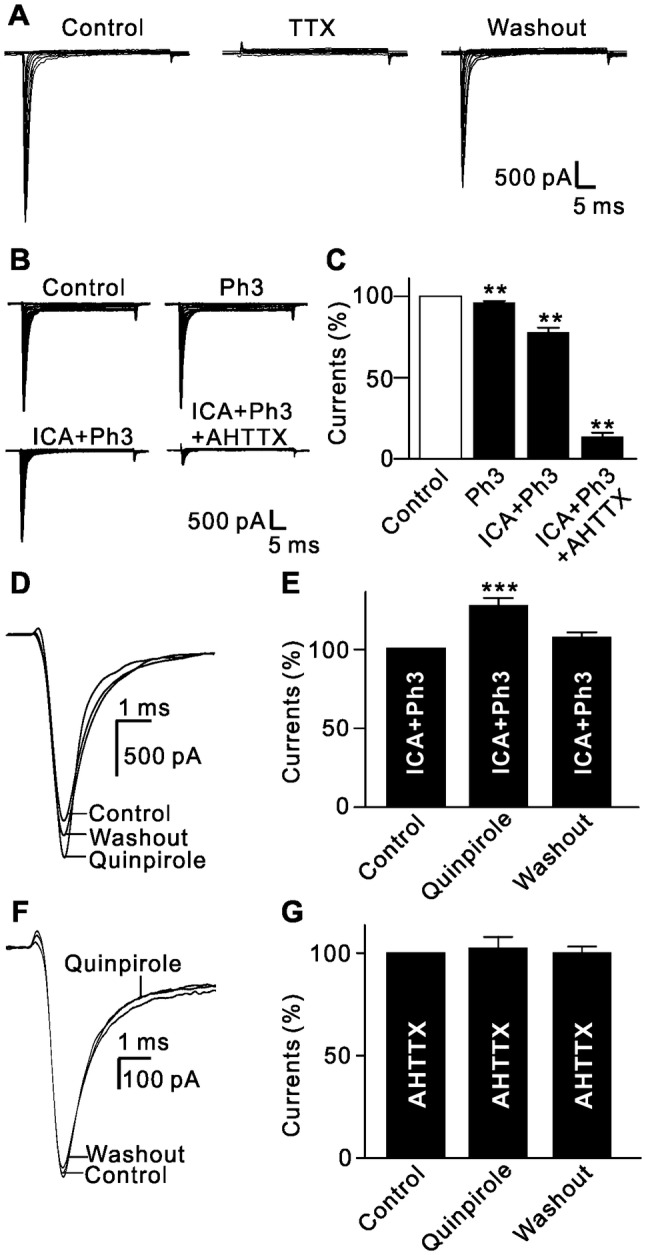
Activation of D2 receptors selectively enhances Nav1.6 voltage-gated Na+ currents. A Representative current recordings from an RGC, showing that TTX (500 nmol/L) completely and reversibly suppressed Na+ currents. B Representative Na+ currents recorded from an RGC, showing the effects of successive addition of the Nav1.2 blocker phrixotoxin-3 (Ph3, 10 nmol/L), the Nav1.1 blocker ICA121431 (ICA, 1 µmol/L), and the Nav1.6 blocker 4,9-anhydrotetrodotoxin (4,9-AHTTX, 100 nmol/L). C Bar chart summarizing the changes in Na+ current amplitudes at −20 mV under different conditions (n = 6/group, **P < 0.01 vs control). D Sample current traces showing that extracellular application of quinpirole (10 µmol/L) still enhanced current amplitudes after blocking the Nav1.1 and Nav1.2 channels. E Bar chart summarizing the changes in the Na+ current amplitudes at −20 mV (n = 10, ***P < 0.001 vs control). F Sample current traces showing that extracellular application of quinpirole (10 µmol/L) failed to change Na+ currents after blocking the Nav1.6 channels. G Bar chart summarizing the changes in the Na+ current amplitudes at −20 mV (n = 11).
We further determined which subunit(s) of the Na+ channels are modulated by activation of D2 receptors. In the presence of ICA and Ph3, bath application of quinpirole still enhanced the Na+ currents (125.6% ± 4.3% of control, n = 10, P < 0.001) (Fig. 6D, E). The increase was comparable to that of quinpirole in the absence of ICA and Ph3 (130.2% ± 5.2% of control, P = 0.351; Fig. 5A–C). In contrast, bath application of quinpirole had no effect on the currents when the cells were pretreated with AHTTX (102.5% ± 5.4% of control, n = 11, P = 0.567; Fig. 6F, G), suggesting that activation of D2 receptors selectively enhances Na+ currents mediated by the Nav1.6 subunit in rat RGCs.
Involvement of Signaling Pathways in the Quinpirole-Induced Enhancement of Na+ Currents
The intracellular signaling pathways mediating the effect of D2 receptor activation on Nav1.6 currents were similar to those on outward K+ currents. Quinpirole failed to enhance the current amplitude when the cells were pre-treated with Rp-cAMP (110.1% ± 5.7% of control, n = 9, P = 0.121; Fig. 7A, B). Moreover, when the CaMKII and MAPK/ERK pathways were blocked by intracellular dialysis with KN-62 or U0126 through the recording pipette, quinpirole perfusion had no significant effect on the current amplitudes (106.6% ± 4.9% of control for KN-62, n = 12, P = 0.25; 102.4% ± 2.5% of control for U0126, n = 11, P = 0.447; Fig. 7C–F). In addition, when PKC was inhibited by Bis IV, quinpirole continued to enhance the currents (117.8% ± 3.1% of control, n = 10, P < 0.01; Fig. 7G, H).
Fig. 7.
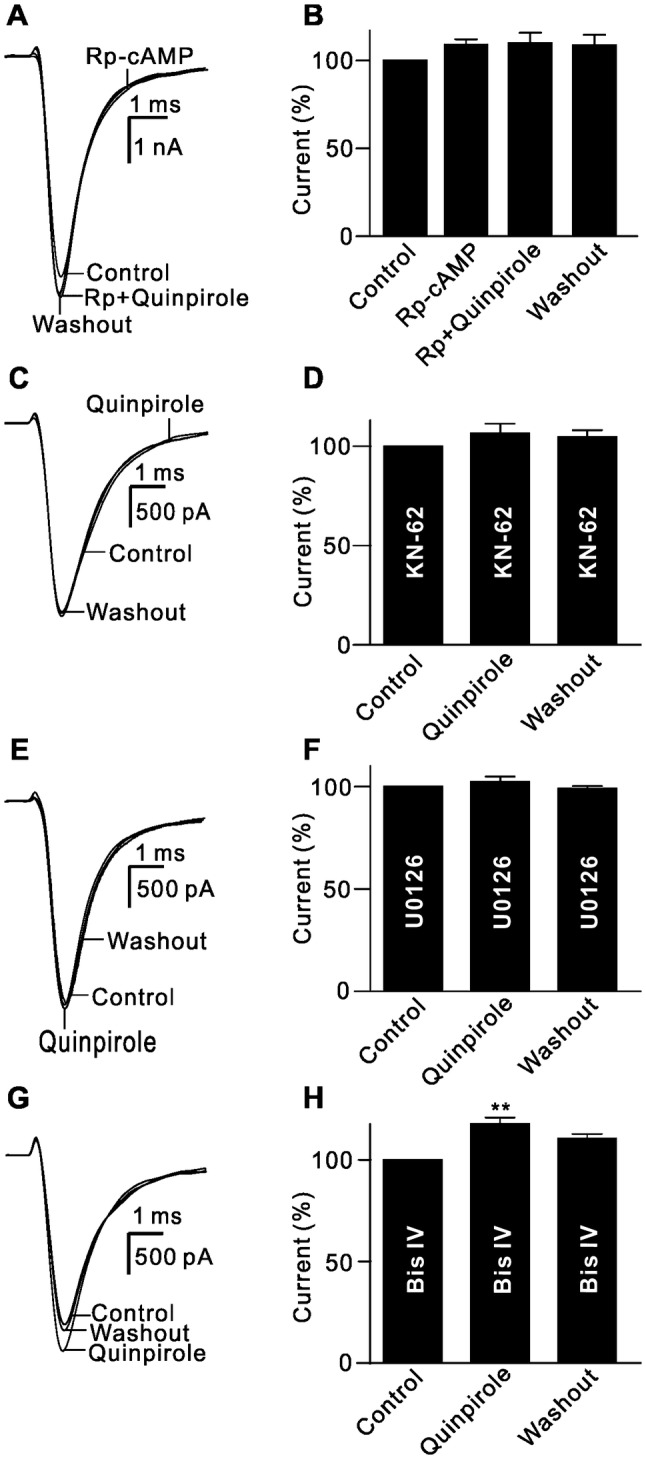
Signaling pathways involved in the quinpirole-induced enhancement of Na+ channels. A Sample current traces showing that Rp-cAMP (2 µmol/L) blocked the quinpirole-induced enhancement of Na+ currents. B Bar chart summarizing the changes in Na+ current amplitudes at −20 mV under different conditions (n = 9). C, D Sample current traces showing that extracellular application of quinpirole (10 µmol/L) failed to change Na+ currents when the cell was pre-treated with 10 µmol/L KN-62 (C), and summary data are shown in (D) (n = 10). E, F Sample current traces showing that U0126 (10 µmol/L) blocked the quinpirole-induced enhancement of Na+ currents (E), and cumulative data are shown in (F) (n = 11). G Sample current traces showing that extracellular application of quinpirole (10 µmol/L) enhanced Na+ currents when the cell was pre-treated by 10 µmol/L Bis IV. H Bar chart summarizing the changes of the Na+ current amplitudes at −20 mV under different conditions (n = 10, **P < 0.01 vs control).
D2 Receptor Activation Increases the Number of Evoked Action Potentials in RGCs
DA may modulate voltage-gated ion channels, thereby influencing the excitability of RGCs [17, 18, 28, 30]. The above results showed that activation of D2 receptors suppressed outward K+ current and enhanced Na+ current. We then tested whether D2 receptor activation changes RGC excitability by recording the numbers of action potentials induced by current injection before (control) and after perfusion of quinpirole (Fig. 8A). The numbers of action potentials increased with increasing positive current injection (Fig. 8B). On average, the firing frequency gradually increased from 0 Hz to 10.5 Hz ± 1.5 Hz with 0 pA to +100 pA current injections in controls (n = 15). After quinpirole application, the firing frequency at 0 pA current injection was 2.0 Hz ± 1.5 Hz (n = 15, P = 0.178), and 6.5 Hz ± 1.5 Hz to 15.5 Hz ± 1.7 Hz at 10 pA to 100 pA current injection (n = 15, P <0.05 for all; Fig. 8B).
Fig. 8.
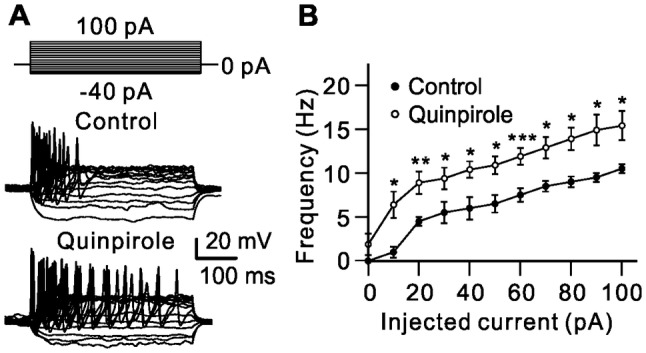
Activation of D2 receptors increases the numbers of action potential evoked by current injection in RGCs. A Representative recordings from an RGC, showing that extracellular application of quinpirole (10 μmol/L) increased the numbers of action potentials evoked by a series of 400-ms current injections from −40 pA to +100 pA in increments of 10 pA. B Plot of average firing frequency of evoked action potentials versus different injected currents (n = 15, *P < 0.05, **P < 0.01, ***P < 0.001 vs control).
Discussion
Modulation of K+ Channels in RGCs by D2 Receptors
Although previous studies have shown that specific staining for D2 receptors is observed in cells of the ganglion cell layer in rat retinas [22], and that pharmacological interference with D2 receptors modulates the responses of RGCs [19, 41], our present work clearly showed that D2 receptors are indeed expressed in rat RGCs, as evidenced by double immunofluorescent labeling in RGCs retrogradely labeled with CTB (Fig. S1).
In the present study we found that one of the major effects of D2 receptor stimulation in rat RGCs is to significantly suppress the Gb-sensitive KATP and the transient outward K+ channels (KA) and delayed rectifying K+ channels [48, 49], but not the TEA-sensitive BKCa channel (Fig. 2). Consistently, previous studies have shown that activation of the D4 subtype of D2-class receptors reversibly inhibits K+ currents in the neurohypophysial nerve terminals of rats [50]. In addition, D2 receptor activation reduces small-conductance Ca2+-activated K+ channel currents in subthalamic nucleus neurons [34]. In contrast to the data presented here, extensive evidence has shown that activation of D2 receptors increases the activity of several K+ channels in neurons of different brain regions [51–54]. For example, D2 receptor-modulated changes in the excitability of nucleus accumbens neurons are dynamically regulated and integrated by KA, Kir, and leak K+ currents [52]. In B5 neurons of the buccal ganglion in the freshwater pond snail Helisoma trivolvis, DA increases 4-AP- and TEA-sensitive K+ currents through D2 receptors, resulting in a strong hyperpolarization of membrane potential [54]. Activation of D2 receptors increases the activity of slowly inactivating K+ channels and enhances the action potential discharge in neurons of the ventrobasal thalamus [51], and decreases the spontaneous firing of rat substantia nigra pars compacta (SNc) dopaminergic neurons by directly activating G protein-dependent inward rectifying K+ channels [53].
It is of interest that modulation of KATP due to D2 receptor stimulation revealed a contradictory situation. Rather than a reduction in KATP currents in rat RGCs (Fig. 2), an increase in KATP currents has been reported in isolated rat striatal neurons and in primary rat lactotrophs [55, 56]. The absence of an effect on KATP has also been reported in neurons of the rat SNc. DA acting on D2 receptors activates K+ currents and this is not mediated by KATP [57]. In addition, it is noteworthy that the outward current components inhibited by D4 receptor activation are KA and BKCa channels in the neurohypophysial nerve terminals of rats [50]. A possible explanation is that the distinct K+ channel subtypes may be affected by different subtypes of D2 receptor. Which subtype(s) of D2 receptors are expressed in rat RGCs remains to be explored.
It should be noted that activation of both D1 and D2 receptors suppresses similar subtypes of outward K+ channel in rat RGCs [29] (and this study). We speculate that outward K+ currents in rat RGCs are modulated by a variety of signaling pathways coupled to distinct G protein subunits (see below).
D2 Receptor-Mediated Modulation of Na+ Currents in RGCs
Voltage-gated Na+ channels ignite neuronal action potentials. Nine distinct Na+ channels (Nav1.1–Nav1.9) have been identified, among which Nav1.1–Nav1.3 are expressed in neurons and play roles in electrogenesis [58–61]. Although the Nav1.1, Nav1.2, and Nav1.6 Na+ channel proteins have been identified in RGCs by immunohistochemistry [25, 27, 45], our data demonstrated that these Na+ channel subtypes indeed functionally exist in rat RGCs, and that the Nav1.6 channel predominates (Fig. 6). Furthermore, TTX completely blocked these Na+ currents, which is consistent with all these Na+ channels being TTX-sensitive.
An important finding in this study was that activation of D2 receptors enhanced Na+ currents in rat RGCs. We further demonstrated that Nav1.6 channels were selectively modulated by D2 receptors. Although previous studies have demonstrated that DA acting on D2 receptors modulates Na+ channels in various neurons [32, 33, 36, 62, 63], the present work is the first to show the selective modulation of Nav1.6 channels due to D2 receptor stimulation. In agreement with our findings, quinpirole induces a robust increase of TTX-sensitive Na+ influx in primary cultures of striatal neurons, and the effect is abolished by sulpiride, suggestive of involvement of D2 receptors [31]. Enhancement of Na+ currents by D2 receptor stimulation has also been reported in freshly-dissociated rat nucleus accumbens neurons [32] and in neurons of layer V entorhinal cortex in slice preparations [63]. However, it should be emphasized that a reduction in Na+ current amplitudes by D2 receptors has been reported in cochlear spiral ganglion neurons [36], and in striatal cholinergic interneurons [33]. One possibility for this inconformity is that D2 receptors are coupled to different subunits of G proteins in different neurons. For example, this is Gi/o in nucleus accumbens neurons [32], but Gβγ in striatal cholinergic interneuron [33]. On the other hand, Na+ channels are composed of α and β subunits [60, 61]. It has been reported that Nav1.6 and β1 or β2 are clustered in RGCs [64, 65]. Whether activation of D2 receptors modulates either α or β subunit in different cells remains to be addressed.
Since D2 receptor activation did not significantly shift the activation and inactivation curves, the enhancement of Na+ currents due to D2 receptor stimulation may be mediated through changing the Na+ channel conductance, but not through changing the activation and inactivation probability. In other words, D2 receptor stimulation did not change the activation and inactivation gates of the channels.
Mechanisms Underlying D2 Receptor-Mediated Effects on K+ and Na+ Channels in RGCs
It has been reported that activation of D2 receptors may be coupled to several signaling pathways [32, 42, 43, 66]. The data presented in this study demonstrated that similar signaling pathways (cAMP/PKA, CaMKII, and MAPK/ERK) mediated the modulation of outward K+ currents and Na+ currents in rat RGCs. Since D2 receptors negatively modulate the activity of adenylate cyclase, application of quinpirole should suppress cAMP/PKA signaling activity, then affect the outward K+ and Na+ currents. Consistently, the enhancement of Na+ currents or modulation of K+ channels in nucleus accumbens neurons due to D2 receptor stimulation results from suppressing tonic activity of cAMP/PKA signaling via the activation of Gi/o proteins; blockade of PKA activity mimics the D2 receptor-mediated effects [32, 52]. It has also been reported that Ca2+ influx through N-methyl-D-aspartate (NMDA) receptors is modulated by PKA; this process is inhibited by D2 receptors, thus reducing corticostriatal glutamate release. These results further demonstrate the suppression of tonic activity of cAMP/PKA signaling by D2 receptors [67].
Activation of D2 receptors may modulate the activity of CaMKII and MAPK/ERK signaling in cell lines and neurons [42, 43, 66, 68]. In this study, we found that the CaMKII and MAPK/ERK pathways were indeed involved in the quinpirole effects on K+ and Na+ channels because quinpirole no longer changed the current amplitudes when these two signaling pathways were blocked. How CaMKII and MAPK/ERK modulate outward K+ and Na+ currents remains to be explored.
Regulation of PLC/PKC is another classical signaling pathway due to D2 receptor stimulation [33, 69]. However, our results clearly showed that the PLC/PKC pathway did not participate in the quinpirole-induced modulation of K+ and Na+ currents (Figs. 3 and 7). In agreement with our data, the effects of D2 receptor activation in the rat striatum are not mediated by the PLC/PKC pathway since quinpirole does not affect the basal level of PLC or the levels of phosphoinositols (inositol monophosphate, inositol biphosphate, and inositol triphosphate), which are well characterized consequence of PLC stimulation [70]. On the other hand, the PLC/PKC pathway has been demonstrated to be involved in the effects of D2 receptor activation. For example, D2 receptor agonists reduce L-type Ca2+ currents and excitability in striatal medium spiny neurons through the PLCβ1/IP3/calcineurin pathway [69]. Inhibition of Na+ channels by D2 receptors in striatal cholinergic interneurons is mediated by PLC/PKC signaling [33]. The reason for this inconformity may be that D2 receptors are linked to different subunits of G proteins in different cells. In striatal cholinergic interneurons, Gβγ signaling that activates the PLC/PKC cascade mediates the D2 receptor-induced inhibition of Na+ currents [33], whereas the enhancement of Na+ currents in the present study was not mediated by this signaling.
Modulation of RGC Excitability by D2 Receptors
RGC excitability can be modulated by two elements, synaptic inputs and the intrinsic properties of the ion channels. K+ channels play a critical role in setting up the membrane potential [71], while neuronal voltage-gated Na+ channels ignite action potentials and determine the firing frequency of the cells. D2 receptor-induced inhibition of outward K+ currents and enhancement of Na+ currents would increase RGC excitability. In line with this, quinpirole indeed increased the numbers of action potential discharges evoked by current injection (Fig. 8). It is noteworthy that both increases and decreases of cell excitability due to D2 receptor activation have been reported. Activation of D2 receptors causes increased firing in thalamocortical relay neurons [51], and enhances hippocampal-accumbens neuron excitability [72]. In contrast, stimulation of D2 receptors decreases evoked firing in medium spiny neurons of the nucleus accumbens [52], and decreases the spontaneous firing activity in rat SNc neurons [53] and in the striatal cholinergic interneurons of mice [33]. We speculate that different subunits of G protein mediate these effects as noted above.
Increasing evidence shows that the modulatory effect of D2 receptors on cell excitability is mediated by increasing or inhibiting Kir currents [51–53]. However, we previously showed that Kir channels are mainly expressed in RGC dendrites [73]. In this study, we could not record Kir channels since most of the RGC dendrites were lost during cell dissociation. Whether D2 receptor activation also affects Kir channels in RGCs remains to be further explored.
Since both D1 and D2 receptors are extensively expressed in retinal cells, in the intact retina, intrinsic DA may regulate the activity of RGCs through two pathways. First, DA acts on the D1/D2 receptors expressed in bipolar and amacrine cells to regulate the balance of inhibitory and excitatory inputs to RGCs, thus affecting RGC activity [13–16]. Second, DA activates D1/D2 receptors expressed in RGCs, directly influencing RGC excitability. It is of interest that both increased and reduced RGC excitability induced by DA have been reported, although the effects of DA are all mediated by activating D1 receptors [18, 28]. In retinal slices, DA enhances RGC excitability through inhibiting Ih [28], while in dissociated RGCs DA reduces RGC excitability through inhibiting voltage-gated Na+ current [18]. In addition, in a previous study, we showed that the temporal summation of excitatory postsynaptic potentials in rat RGCs is enhanced by D1 receptor activation and that this is mediated by affecting Kir currents [30]. The reason for this inconsistency may be that the dissociated RGCs lost their intact dendrites. Therefore, the net effect of DA on RGC activity in the intact retina depends on the integration of presynaptic and postsynaptic effects. On the other hand, the density and affinity of D1 and D2 receptors in RGCs are also important factors. We speculate that DA may enhance visual contrast sensitivity by changing the center/surround balance of RGCs in the intact retina [12, 16]. The detailed mechanisms need to be explored.
In conclusion, we provide compelling evidence that activation of D2 receptors increases rat RGC excitability through suppressing outward K+ currents and selectively enhancing Nav1.6 currents, and subsequently modulating visual integrative processing. In addition, DA and its receptors have been shown to associate with retinal disorders such as diabetic retinopathy, retinitis pigmentosa, and NMDA-induced retinal injury [41, 74–76]. Modulation of RGC excitability by D2 receptors may provide a potential strategy for RGC neuroprotection.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Dr. Xiong-Li Yang for helpful discussion and critical comments on the manuscript. This work was supported by grants from the National Natural Science Foundation of China (31671078, 81790642, and 31872765), the Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZJ Lab.
Conflict of interest
The authors declare no potential conflict of interest.
Footnotes
Ning Yin and Yu-Long Yang have contributed equally to this work.
References
- 1.Barros M, Mello EL, Huston JP, Tomaz C. Behavioral effects of buspirone in the marmoset employing a predator confrontation test of fear and anxiety. Pharmacol Biochem Behav. 2001;68:255–262. doi: 10.1016/s0091-3057(00)00447-0. [DOI] [PubMed] [Google Scholar]
- 2.Bissiere S, Humeau Y, Luthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–592. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- 3.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: From structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 4.Paspalas CD, Goldman-Rakic PS. Presynaptic D1 dopamine receptors in primate prefrontal cortex: Target-specific expression in the glutamatergic synapse. J Neurosci. 2005;25:1260–1267. doi: 10.1523/JNEUROSCI.3436-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V-VI rat prefrontal cortex neurons in vitro: Modulation of dendritic-somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang ZY, Liang SX, Yu SS, Xie T, Wang BC, Wang JK, et al. Distinct roles of dopamine receptors in the lateral thalamus in a rat model of decisional impulsivity. Neurosci Bull. 2017;33:413–422. doi: 10.1007/s12264-017-0146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enjalbert A, Bockaert J. Pharmacological characterization of the D2 dopamine receptor negatively coupled with adenylate-cyclase in rat anterior pituitary. Mol Pharmacol. 1983;23:576–584. [PubMed] [Google Scholar]
- 8.Sibley DR, Monsma FJ., Jr Molecular-biology of dopamine-receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 9.Beaulieu JM, Espinoza S, Gainetdinov RR. Dopamine receptors - IUPHAR Review 13. Br J Pharmacol. 2015;172:1–23. doi: 10.1111/bph.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson CR, Ruan GX, Aseem F, Abey J, Gamble K, Stanwood G, et al. Retinal dopamine mediates multiple dimensions of light-adapted vision. J Neurosci. 2012;32:9359–9368. doi: 10.1523/JNEUROSCI.0711-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavoie J, Illiano P, Sotnikova TD, Gainetdinov RR, Beaulieu JM, Hebert M. The electroretinogram as a biomarker of central dopamine and serotonin: Potential relevance to psychiatric disorders. Biol Psychiat. 2014;75:479–486. doi: 10.1016/j.biopsych.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 12.Witkovsky P. Dopamine and retinal function. Doc Ophthalmol. 2004;108:17–40. doi: 10.1023/b:doop.0000019487.88486.0a. [DOI] [PubMed] [Google Scholar]
- 13.Gustincich S, Feigenspan A, Wu DK, Koopman LJ, Raviola E. Control of dopamine release in the retina: A transgenic approach to neural networks. Neuron. 1997;18:723–736. doi: 10.1016/s0896-6273(00)80313-x. [DOI] [PubMed] [Google Scholar]
- 14.Hokoc JN, Mariani AP. Tyrosine–hydroxylase immunoreactivity in the rhesus-monkey retina reveals synapses from bipolar cells to dopaminergic amacrine cells. J Neurosci. 1987;7:2785–2793. doi: 10.1523/JNEUROSCI.07-09-02785.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pourcho RG. Dopaminergic amacrine cells in the cat retina. Brain Res. 1982;252:101–109. doi: 10.1016/0006-8993(82)90982-9. [DOI] [PubMed] [Google Scholar]
- 16.Thier P, Alder V. Action of iontophoretically applied dopamine on cat retinal ganglion cells. Brain Res. 1984;292:109–121. doi: 10.1016/0006-8993(84)90895-3. [DOI] [PubMed] [Google Scholar]
- 17.Hayashida Y, Ishida AT. Dopamine receptor activation can reduce voltage-gated Na+ current by modulating both entry into and recovery from inactivation. J Neurophysiol. 2004;92:3134–3141. doi: 10.1152/jn.00526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashida Y, Rodriguez CV, Ogata G, Partida GJ, Oi H, Stradleigh TW, et al. Inhibition of adult rat retinal ganglion cells by D-1-type dopamine receptor activation. J Neurosci. 2009;29:15001–15016. doi: 10.1523/JNEUROSCI.3827-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogata G, Stradleigh TW, Partida GJ, Ishida AT. Dopamine and full-field illumination activate D1 and D2-D5-type receptors in adult rat retinal ganglion cells. J Comp Neurol. 2012;520:4032–4049. doi: 10.1002/cne.23159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaquero CF, Pignatelli A, Partida GJ, Ishida AT. A dopamine- and protein kinase A-dependent mechanism for network adaptation in retinal ganglion cells. J Neurosci. 2001;21:8624–8635. doi: 10.1523/JNEUROSCI.21-21-08624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran VT, Dickman M. Differential localization of dopamine D1-receptors and D2 receptors in rat retina. Invest Ophthalmol Vis Sci. 1992;33:1620–1626. [PubMed] [Google Scholar]
- 22.Wagner HJ, Luo BG, Ariano MA, Sibley DR, Stell WK. Localization of D2 dopamine-receptors in vertebrate retinae with anti-peptide antibodies. J Comp Neurol. 1993;331:469–481. doi: 10.1002/cne.903310404. [DOI] [PubMed] [Google Scholar]
- 23.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 24.Ettaiche M, Heurteaux C, Blondeau N, Borsotto M, Tinel N, Lazdunski M. ATP-sensitive potassium channels (K-ATP) in retina: a key role for delayed ischemic tolerance. Brain Res. 2001;890:118–129. doi: 10.1016/s0006-8993(00)03152-8. [DOI] [PubMed] [Google Scholar]
- 25.Fjell J, DibHajj S, Fried K, Black JA, Waxman SG. Differential expression of sodium channel genes in retinal ganglion cells. Mol Brain Res. 1997;50:197–204. doi: 10.1016/s0169-328x(97)00187-3. [DOI] [PubMed] [Google Scholar]
- 26.Koeberle P D, Wang Y, Schlichter L C. Kv1.1 and Kv1.3 channels contribute to the degeneration of retinal ganglion cells after optic nerve transection in vivo. Cell Death & Differentiation. 2009;17(1):134–144. doi: 10.1038/cdd.2009.113. [DOI] [PubMed] [Google Scholar]
- 27.Van Wart A, Matthews G. Expression of sodium channels Na(v)1.2 and Na(v)1.6 during postnatal development of the retina. Neurosci Lett 2006, 403: 315–317. [DOI] [PMC free article] [PubMed]
- 28.Chen L, Yang XL. Hyperpolarization–activated cation current is involved in modulation of the excitability of rat retinal ganglion cells by dopamine. Neuroscience. 2007;150:299–308. doi: 10.1016/j.neuroscience.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Li Q, Wu N, Cui P, Gao F, Qian WJ, Miao Y, et al. Suppression of outward K(+) currents by activating dopamine D1 receptors in rat retinal ganglion cells through PKA and CaMKII signaling pathways. Brain Res. 2016;1635:95–104. doi: 10.1016/j.brainres.2016.01.039. [DOI] [PubMed] [Google Scholar]
- 30.Cui P, Li XY, Zhao Y, Li Q, Gao F, Li LZ, et al. Activation of dopamine D1 receptors enhances the temporal summation and excitability of rat retinal ganglion cells. Neuroscience. 2017;355:71–83. doi: 10.1016/j.neuroscience.2017.04.046. [DOI] [PubMed] [Google Scholar]
- 31.Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, et al. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3:226–230. doi: 10.1038/72929. [DOI] [PubMed] [Google Scholar]
- 32.Hu XT, Dong Y, Zhang XF, White FJ. Dopamine D2 receptor-activated Ca2+ signaling modulates voltage-sensitive sodium currents in rat nucleus accumbens neurons. J Neurophysiol. 2005;93:1406–1417. doi: 10.1152/jn.00771.2004. [DOI] [PubMed] [Google Scholar]
- 33.Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, et al. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramanathan Sankari, Tkatch Tatiana, Atherton Jeremy F., Wilson Charles J., Bevan Mark D. D2-Like Dopamine Receptors Modulate SKCa Channel Function in Subthalamic Nucleus Neurons Through Inhibition of Cav2.2 Channels. Journal of Neurophysiology. 2008;99(2):442–459. doi: 10.1152/jn.00998.2007. [DOI] [PubMed] [Google Scholar]
- 35.Chen XY, Xue B, Wang J, Liu HX, Shi LM, Xie JX. Potassium channels: a potential therapeutic target for Parkinson’s disease. Neurosci Bull. 2018;34:341–348. doi: 10.1007/s12264-017-0177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valdes-Baizabal C, Soto E, Vega R. Dopaminergic modulation of the voltage-gated sodium current in the cochlear afferent neurons of the rat. PLoS One. 2015;10:e0120808. doi: 10.1371/journal.pone.0120808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong LD, Gao F, Wang XH, Miao Y, Wang SY, Wu Y, et al. GluA2 trafficking is involved in apoptosis of retinal ganglion cells induced by activation of EphB/EphrinB reverse signaling in a rat chronic ocular hypertension model. J Neurosci. 2015;35:5409–5421. doi: 10.1523/JNEUROSCI.4376-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian WJ, Yin N, Gao F, Miao Y, Li Q, Li F, et al. Cannabinoid CB1 and CB2 receptors differentially modulate L- and T-type Ca2+ channels in rat retinal ganglion cells. Neuropharmacology. 2017;124:143–156. doi: 10.1016/j.neuropharm.2017.04.027. [DOI] [PubMed] [Google Scholar]
- 39.Gao F, Li F, Miao Y, Xu LJ, Zhao Y, Li Q, et al. Involvement of the MEK-ERK/p38-CREB/c-fos signaling pathway in Kir channel inhibition-induced rat retinal Müller cell gliosis. Sci Rep. 2017;7:1480. doi: 10.1038/s41598-017-01557-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li LZ, Yin N, Li XY, Miao Y, Cheng S, Li F, et al. Rac1 modulates excitatory synaptic transmission in mouse retinal ganglion cells. Neurosci Bull. 2019;35:673–687. doi: 10.1007/s12264-019-00353-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen R. Effects of dopamine d2-like receptor antagonists on light responses of ganglion cells in wild-type and P23H rat retinas. PLoS One. 2015;10:e0146154. doi: 10.1371/journal.pone.0146154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bofill-Cardona E, Kudlacek O, Yang Q, Ahorn H, Freissmuth M, Nanoff C. Binding of calmodulin to the D-2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J Biol Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- 43.Liu XY, Chu XP, Mao LM, Wang M, Lan HX, Li MH, et al. Modulation of D2R-NR2B interactions in response to cocaine. Neuron. 2006;52:897–909. doi: 10.1016/j.neuron.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 44.Fohlmeister JF. Voltage gating by molecular subunits of Na+ and K+ ion channels: higher-dimensional cubic kinetics, rate constants, and temperature. J Neurophysiol. 2015;113:3759–3777. doi: 10.1152/jn.00551.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mojumder DK, Frishman LJ, Otteson DC, Sherry DM. Voltage-gated sodium channel alpha-subunits Na(v)1.1, Na(v)1.2, and Na(v)1.6 in the distal mammalian retina. Mol Vis 2007, 13: 2163–2182. [PubMed]
- 46.Bosmans F, Rash L, Zhu SY, Diochot S, Lazdunski M, Escoubas P, et al. Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol Pharmacol. 2006;69:419–429. doi: 10.1124/mol.105.015941. [DOI] [PubMed] [Google Scholar]
- 47.Rosker Christian, Lohberger Birgit, Hofer Doris, Steinecker Bibiane, Quasthoff Stefan, Schreibmayer Wolfgang. The TTX metabolite 4,9-anhydro-TTX is a highly specific blocker of the Nav1.6 voltage-dependent sodium channel. American Journal of Physiology-Cell Physiology. 2007;293(2):C783–C789. doi: 10.1152/ajpcell.00070.2007. [DOI] [PubMed] [Google Scholar]
- 48.Saito M, Murai Y, Sato H, Bae YC, Akaike T, Takada M, et al. Two opposing roles of 4-AP-sensitive K+ current in initiation and invasion of spikes in rat mesencephalic trigeminal neurons. J Neurophysiol. 2006;96:1887–1901. doi: 10.1152/jn.00176.2006. [DOI] [PubMed] [Google Scholar]
- 49.Wu RL, Barish ME. Modulation of a slowly inactivating potassium current, I-D, by metabotropic glutamate receptor activation in cultured hippocampal pyramidal neurons. J Neurosci. 1999;19:6825–6837. doi: 10.1523/JNEUROSCI.19-16-06825.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilke RA, Hsu SF, Jackson MB. Dopamine D4 receptor mediated inhibition of potassium current in neurohypophysial nerve terminals. J Pharmacol Exp Ther. 1998;284:542–548. [PubMed] [Google Scholar]
- 51.Govindaiah G, Wang Y, Cox CL. Dopamine enhances the excitability of somatosensory thalamocortical neurons. Neuroscience. 2010;170:981–991. doi: 10.1016/j.neuroscience.2010.08.043. [DOI] [PubMed] [Google Scholar]
- 52.Perez MF, White FJ, Hu XT. Dopamine D2 receptor modulation of K+ channel activity regulates excitability of nucleus accumbens neurons at different membrane potentials. J Neurophysiol. 2006;96:2217–2228. doi: 10.1152/jn.00254.2006. [DOI] [PubMed] [Google Scholar]
- 53.Uchida S, Akaike N, Nabekura J. Dopamine activates inward rectifier K+ channel in acutely dissociated rat substantia nigra neurones. Neuropharmacology. 2000;39:191–201. doi: 10.1016/s0028-3908(99)00111-2. [DOI] [PubMed] [Google Scholar]
- 54.Zhong LR, Artinian L, Rehder V. Dopamine suppresses neuronal activity of helisoma B5 neurons via a D2-Like receptor, activating PLC and K channels. Neuroscience. 2013;228:109–119. doi: 10.1016/j.neuroscience.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 55.Lin YJ, Greif GJ, Freedman JE. Multiple sulfonylurea-sensitive potassium channels - a novel subtype modulated by dopamine. Mol Pharmacol. 1993;44:907–910. [PubMed] [Google Scholar]
- 56.Einhorn LC, Gregerson KA, Oxford GS. D2 dopamine receptor activation of potassium channels inidentified rat lactotrophs - whole-cell and single-channel recording. J Neurosci. 1991;11:3727–3737. doi: 10.1523/JNEUROSCI.11-12-03727.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hicks GA, Henderson G. Lack of evidence for coupling of the dopamine D2 receptor to an adenosine triphosphate-sensitive potassium (ATP-K+) channel in dopaminergic neurones of the rat substantia nigra. Neurosci Lett. 1992;141:213–217. doi: 10.1016/0304-3940(92)90897-g. [DOI] [PubMed] [Google Scholar]
- 58.Catterall William A., Goldin Alan L., Waxman Stephen G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacological Reviews. 2005;57(4):397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 59.Black JA, Waxman SG. Noncanonical roles of voltage-gated sodium channels. Neuron. 2013;80:280–291. doi: 10.1016/j.neuron.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 60.Catterall WA. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 61.Catterall WA. Forty years of sodium channels: structure, function, pharmacology, and epilepsy. Neurochem Res. 2017;42:2495–2504. doi: 10.1007/s11064-017-2314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Centonze D, Bracci E, Pisani A, Gubellini P, Bernardi G, Calabresi P. Activation of dopamine D1-like receptors excites LTS interneurons of the striatum. Eur J Neurosci. 2002;15:2049–2052. doi: 10.1046/j.1460-9568.2002.02052.x. [DOI] [PubMed] [Google Scholar]
- 63.Rosenkranz JA, Johnston D. State-dependent modulation of amygdala inputs by dopamine-induced enhancement of sodium currents in layer V entorhinal cortex. J Neurosci. 2007;27:7054–7069. doi: 10.1523/JNEUROSCI.1744-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fein AJ, Meadows LS, Chen C, Slat EA, Isom LL. Cloning and expression of a zebrafish SCN1B ortholog and identification of a species-specific splice variant. BMC Genomics. 2007;8:226. doi: 10.1186/1471-2164-8-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaplan Miriam R, Cho Min-Hee, Ullian Erik M, Isom Lori L, Levinson S.Rock, Barres Ben A. Differential Control of Clustering of the Sodium Channels Nav1.2 and Nav1.6 at Developing CNS Nodes of Ranvier. Neuron. 2001;30(1):105–119. doi: 10.1016/s0896-6273(01)00266-5. [DOI] [PubMed] [Google Scholar]
- 66.Cho DI, Zheng M, Kim KM. Current perspectives on the selective regulation of dopamine D2 and D3 receptors. Arch Pharm Res. 2010;33:1521–1538. doi: 10.1007/s12272-010-1005-8. [DOI] [PubMed] [Google Scholar]
- 67.Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cussac D, Newman-Tancredi A, Pasteau V, Millan MJ. Human dopamine D3 receptors mediate mitogen- activated protein kinase activation via a phosphatidylinositol 3-kinase and an atypical protein kinase C-dependent mechanism. Mol Pharmacol. 1999;56:1025–1030. doi: 10.1124/mol.56.5.1025. [DOI] [PubMed] [Google Scholar]
- 69.Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, et al. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gupta SK, Mishra RK. The effect of dopamine-D1 and dopamine-D2 receptor agonists on inositol phosphate turnover in rat striatal slices. Biochem Int. 1990;22:887–894. [PubMed] [Google Scholar]
- 71.Hille B. Potassium channels and chloride channels. In: Hille B, editor. Ion Channels of Excitable Membrane. Sunderland: Sinauer Associates, Inc.; 2001. pp. 131–167. [Google Scholar]
- 72.Yang CR, Mogenson GJ. Dopamine enhances terminal excitability of hippocampal accumbens neurons via D2 receptor: role of dopamine in presynaptic inhibition. J Neurosci. 1986;6:2470–2478. doi: 10.1523/JNEUROSCI.06-08-02470.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Q, Cui P, Miao Y, Gao F, Li XY, Qian WJ, et al. Activation of group I metabotropic glutamate receptors regulates the excitability of rat retinal ganglion cells by suppressing Kir and I (h) Brain Struct Funct. 2017;222:813–830. doi: 10.1007/s00429-016-1248-3. [DOI] [PubMed] [Google Scholar]
- 74.Aung MH, Park HN, Han MK, Obertone TS, Abey J, Aseem F, et al. Dopamine deficiency contributes to early visual dysfunction in a rodent model of type 1 diabetes. J Neurosci. 2014;34:726–736. doi: 10.1523/JNEUROSCI.3483-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kitaoka Y, Kumai T. Modulation of retinal dopaminergic cells by nitric oxide. A protective effect on NMDA-induced retinal injury. In Vivo 2004, 18: 311–315. [PubMed]
- 76.Nishimura C, Kuriyama K. Alterations in the retinal dopaminergic neuronal system in rats with streptozotocin-induced diabetes. J Neurochem. 1985;45:448–455. doi: 10.1111/j.1471-4159.1985.tb04008.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.