

Abstract
Background
Congenital myasthenic syndrome 22 (CMS22) is a rare autosomal recessive disorder due to isolated PREPL deficiency and characterized by neonatal hypotonia, muscular weakness, and feeding difficulties. Eight such cases have already been reported, while maternal uniparental disomy with a PREPL pathogenic mutation has never been involved.
Methods
Trio whole‐exome sequencing (WES), comparative genomic hybridization microarray (arry‐CGH), and Sanger sequencing were performed on a 6‐month‐old girl with severe neonatal hypotonia and feeding difficulties. Also, the phenotype and genotype of reported CMS22 patients were reviewed.
Results
In this female infant, we identified a novel homozygous frameshift mutation in PREPL (c.1282_1285delTTTG, p.Phe428Argfs*18) by trio‐WES. Sanger sequencing confirmed that her mother was heterozygous and her father was normal. Trio‐WES data showed that 96.70% (1668/1725) variants on chromosome 2 were homozygous and maternally inherited, suggesting maternal uniparental disomy of chromosome 2 [UPD(2)mat]. Array‐CGH did not show copy number variants (CNVs) but revealed complete UPD(2).
Conclusion
To date, nine patients with CMS22 have been reported including our patient, and we report the youngest and the first UPD(2)mat with PREPL novel homozygous pathogenic mutation case, which expand the mutation spectrum of PREPL gene.
Keywords: congenital myasthenic syndrome 22, uniparental disomy, PREPL gene, pyridostigmine treatment
Congenital myasthenic syndrome 22 (CMS22) is a rare autosomal recessive disorder due to isolated PREPL deficiency and characterized by neonatal hypotonia, muscular weakness and feeding difficulties. Eight cases have been reported, while maternal uniparental disomy with a PREPL pathogenic mutation has never been involved. Here, we report the first and the youngest CMS22 patient of UPD(2)mat with a novel homozygous frameshift mutation in PREPL, which expanded the mutation spectrum of the PREPL gene.

1. INTRODUCTION
Congenital myasthenic syndrome 22 (CMS22, OMIM 616,224) is a rare autosomal recessive disorder, characterized by severe neonatal hypotonia, muscular weakness, feeding difficulties, growth hormone deficiency, and childhood obesity (Engel, Shen, Selcen, & Sine, 2015). CMS22 is due to homozygous or compound heterozygous mutation in the PREPL gene (OMIM 609,557), which encodes a serine oligopeptidase involved in the filling of acetylcholine (ACh) into synaptic vesicles (Jaeken et al., 2006). Patients with CMS22 also present with facial weakness (ptosis and tented upper lip), motor developmental delay, and cognitive deficiency (Regal et al., 2018, 2014). The patient had favorable response to pyridostigmine in infancy and may improve spontaneously after 1 year old (Engel, 2018). Regal et al. (2014) reported that they treated three identified patients at 1, 11, and 12 years old with pyridostigmine. Only the infant improved, the two elder children did not respond to pyridostigmine. To date, eight patients with CMS22 have been reported previously, among which there are eight different truncating mutations and six gross deletions of PREPL gene (Laugwitz et al., 2018; Regal et al., 2018, 2014; Silva, Miyake, Tapia, & Matsumoto, 2018).
Uniparental disomy (UPD) is the inheritance of segmental or total a homologous pair of chromosomes from only one parent (Siegel & Slavotinek, 2005). The patient with UPD may be involved in autosomal‐recessive disorders, which unmasks the rare pathogenic variants (Carmichael, Shen, Nguyen, Hirschhorn, & Dauber, 2013; Labrijn‐Marks et al., 2019). However, maternal UPD of chromosome 2 (UPD(2)mat) with a homozygous pathogenic mutation in PREPL has not been reported before.
Here, we report the first case that UPD(2)mat renders a novel homozygous frameshift mutation in PREPL causing CMS22 features. Meanwhile, we review the clinical and genetic features of CMS22, which further delineate the phenotype and genotype of CMS22.
2. MATERIALS AND METHODS
2.1. Molecular analysis
Genomic DNA was extracted from the infant's peripheral blood and her parents using a whole blood genomic DNA extraction kit (Qiagen, German). DNA fragments were enriched using the Agilent SureSelect XT Human All Exon 50 Mb kit (Santa Clara, CA). Then DNA libraries were sequenced on the HiSeq2000/2500 sequencer according to the manufacturer's instructions (Illumina, San Diego, CA). The data analysis method followed the pipeline established in house (Yang et al., 2019). Human Phenotype Ontology (HPO) was converted and integrated into the PhenoPro pipeline, a method developed by our team (Li et al., 2019). The criteria of the molecular diagnosis followed the American College of Medical Genetics (ACMG) guidelines (Matthijs et al., 2016). The distribution of variant heterozygosity on each chromosome was calculated to scan the UPD event. UPD was detected using “B Allele Frequency” (BAF) (van Riet et al., 2018).
The variant of PREPL (NM_006036.4) was confirmed by Sanger sequencing using ABI 3,730 Genetic Analyzer (Applied Biosystems). Paired primers were designed by Primer3 website: primer‐F (5′‐TTAATGATACTTGGTGGCCTAAATAAA‐3′) and primer‐R (5′‐GCTTTCAGTAAATGGGAGCTGA‐3′).
Agilent SurePrint G3 comparative genomic hybridization (CGH) and SNP 4 × 180K microarray (Agilent Technologies, USA) was used to confirm the UPD(2) following the manufacturer's instructions. We used Agilent Cytogenomics software package for CNVs and loss of heterozygosity (LOH) calling and visualization.
2.2. Patient
The proband is a 6‐month‐old Chinese Han female infant who is the first child of healthy non‐consanguineous parents. She was born after an uneventful full‐term pregnancy with a birth weight of 2.8 kg. She presented with neonatal hypotonia and feeding difficulties, and was suspected of spinal muscular atrophy (SMA) in the local hospital. She was referred to our hospital because of poor nursing, weak crying, and delayed developmental milestones. Her motor examination demonstrated low muscle tone and muscle strength (4/5, MRC scale) for all major muscle groups in the upper and lower extremities. Needle EMG showed myopathic changes. The test results of liver function, thyroid function, blood ammonia, liquid chromatography‐tandem mass spectrometry analysis of blood and urine and SMA‐ multiplex ligation‐dependent probe amplification (MLPA) were all normal. She was diagnosed with CMS22 at the age of 6 months and treated with pyridostigmine (15 mg, three times a day, per orally) at the age of 8 months. At the last follow‐up when she was 9 months, she was better than before in feeding and could raise her head steadily, rollover independently, and stand with support.
Pretest counseling was performed in the clinic. Informed consent was signed by the parents. The criteria of genetic testing received approval from the ethics committees of the Children's Hospital, Fudan University (2015–130).
3. RESULTS
Trio‐WES data showed that 96.70% (1668/1725) variants on chromosome 2 had a BAF higher than 0.95 inherited from mother, revealing the LOH of the proband and suggesting UPD(2)mat (Figure 1a). Meanwhile, a novel homozygous frameshift mutation in PREPL (c.1282_1285delTTTG, p.Phe428Argfs*18) was detected in the proband, her mother was heterozygous and her father was normal (Figure 1b). This variant was confirmed by Sanger sequencing (Figure 1c). It has never been reported in the 1,000 Genome Project, the EXAC or the gnomAD database, and it is the only record in our in‐house database, which contains more than 30 000 exome sequencing data of patients. The frameshift variant may form truncated proteins, which lead to loss of the peptidase S9 prolyl oligopeptidase catalytic domain. Three of the eight reported truncating mutations (including nonsense and frameshift mutations) are located after this frameshift variant (Figure 2a). Therefore, this frameshift variant was classified as pathogenic.
Figure 1.
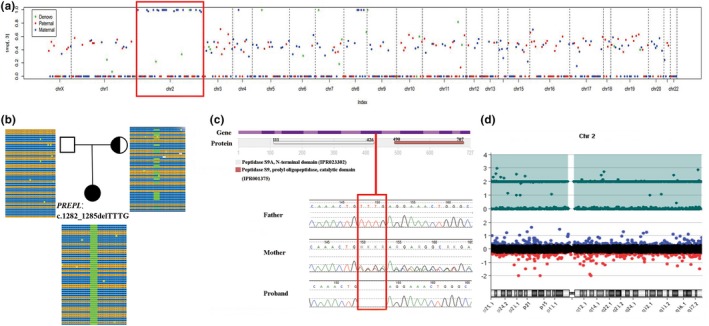
Results of genetic tests in our patient. (a) Trio‐WES identifies that 96.70% (1668/1725) variants on chromosome 2 are homozygous and inherited from the mother, suggesting that the proband has UPD(2)mat. (b) A novel homozygous frameshift variant on PREPL (c.1282_1285delTTTG) is detected in the proband. Her mother is heterozygous and her father is normal. (c) The variant is confirmed by Sanger sequencing. D Array‐CGH identifies UPD(2) by showing the LOH across the whole chromosome 2
Figure 2.
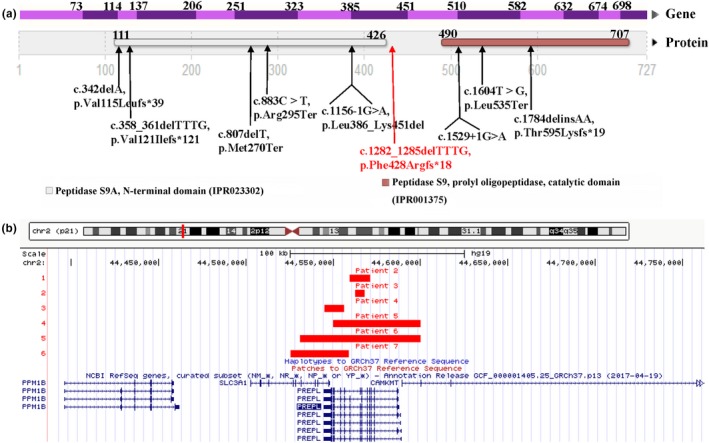
Summary of pathogenic variants and gross deletions in PREPL. (a) Nine truncating mutations on PREPL. The red shows the novel frameshift mutation in our patient. (b) The size and location of the six reported gross deletions in CMS22 patients
Trio‐WES did not identify other pathogenic or likely pathogenic variants associated with the clinical features or other inherited diseases. Data from array‐CGH showed complete UPD(2) by showing LOH across the entire chromosome 2, which supported the diagnosis of UPD(2). Array‐CGH did not reveal any other pathogenic CNVs (Figure 1d).
In combination of the clinical phenotype (neonatal hypotonia, muscular weakness, and feeding difficulties) and the molecular genetic finding, the proband was diagnosed with CMS22 caused by a PREPL novel homozygous pathogenic variation with UPD(2)mat.
4. DISCUSSION
The PREPL gene is one of the genes affected in contiguous gene deletion syndromes at 2p21 (Bartholdi et al., 2013). Isolated PREPL deficiency results in CMS22 (OMIM 616,224). PREPL is localized in the cytosol with highest expression in brain, kidney, and muscle, in decreasing order (Martens et al., 2006). It is an effector of clathrin‐associated adaptor protein 1 (AP‐1) to take part in the regulation of AP‐1 membrane binding (Radhakrishnan, Baltes, Creemers, & Schu, 2013), which is required for normal trafficking of the vesicular ACh transporter between the synaptic vesicle and the cytosol (Kim & Hersh, 2004). In a CMS22 patient, Régal et al found the absence of PREPL in frozen muscle fibers and at the endplates (EPs) in the neuromuscular junction, but EPs showed normal ACh receptor and ACh enzyme. However, microelectrode studies showed reduced miniature endplates potential (MEPP) and miniature endplates current (MEPC) amplitudes and decreased quantal release. Hence, Régal et al hypothesized that the absence of PREPL may affect function of AP‐1 and lead to reduced filling of the synaptic vesicles with ACh (Regal et al., 2014). Hence, we speculate that pyridostigmine, as a cholinesterase inhibitor, can inhibit the decomposition of ACh, and relieve the neuromuscular symptoms of patients. These may provide a potential explanation for the pyridostigmine treatment of CMS22.
Nine patients with CMS22 have been reported including our patient (Table 1). There were seven females and two males with the symptoms onset in neonatal period (9/9). However, the age of diagnosis varied from 6 months to 25 years. Our patient was the youngest one who was diagnosed with CMS22 at 6 months. The CMS22 patient may respond to pyridostigmine in the first year of life (Regal et al., 2014). Patient 7 were treated with pyridostigmine before 1 year old and turned out to have a positive response, while patient 8 started treatment at 14 months without clinical response. Our patient (patient 1) started pyridostigmine treatment at 8 months and had a positive response. Interestingly, other 6 patients without pyridostigmine treatment and patient 8 all showed symptom improvement after one year old. Although the rationale of spontaneous improvement of symptoms is not clear, it is noticeable that the positive response to pyridostigmine seemingly appeared when the patients were performed treatment before 1 year old (patient 1 and 7), and the earlier the treatment, the better the response (patient 7 with a strongly positive response, better than our patient). If treated after 1 year old, the effect of pyridostigmine treatment seems to be weak, and the symptom may start to relieve.
Table 1.
Clinical characteristic and molecular genetic features of nine CMS22 patients
| Feature/Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| PMID | This study | 28,726,805 (IPD1) | 28,726,805 (IPD2) | 28,726,805 (IPD3) | 28,726,805 (IPD4) | 28,726,805 (IPD5) | 24,610,330 | 29,913,539 | 29,483,676 | NA |
| Gender | F | F | F | M | F | M | F | F | F | 7F, 2M |
| Phenotype onset | Neonate | Neonate | Neonate | Neonate | Neonate | Neonate | Neonate | Neonate | Neonate | 9/9 Neonate |
| Age at diagnosis | 6 months | 15 years | 1 years | 2 years | 25 years | 3 years 6 months | NA | NA | 9 years | 6 months−25 years |
| Last evaluated age | 9 months | 17 years | 2 years 5 months | 2 years 10 months | 27 years | 3 years 8 months | 17 months | 3 years | 10 years | 9 months−27 years |
| Failure to thrive | + | + | + | − | − | − | NA | + | + | 5/8 |
| GH deficiency | NA | + | + | + | − | − | + | + | NA | 5/7 |
| PWS/SMA test | SMA (−) | PWS (−) | PWS (−) | PWS (−) | PWS (−) | NA | NA | NA | NA | 5/9 |
| Cystinuria | − | − | − | − | − | + | − | NA | NA | −:6/+:1/NA:2 |
| Neonatal hypotonia | + | + | + | + | + | + | + | + | + | 9/9 |
| Muscular weakness | + | − | + | − | + | + | + | + | + | 7/9 |
| Feeding difficulties | + | + | + | + | + | ‐ | + | + | + | 8/9 |
| Ptosis | − | − | + | + | + | + | + | − | + | 6/9 |
| Walking age | NA | 21 months | 23 months | 22 months | 19 months | 24 months | 17 months | 26 months | 27 months | 17−27 months |
| Childhood‐onset obesity | NA | + | − | − | + | − | NA | NA | + | 3/6 |
| Cognitive/behavioral problems | NA | − | +(DI 69) | + | +(IQ 64) | +(IQ 80) | NA | +(mild) | +(IQ 49) | 6/7 |
|
Pyridostigmine treatment |
Yes, started at 8 months with a positive response | NA | NA | NA | NA | NA | Yes, started at age of 11 weeks with a strongly positive response | Yes, started at 14 months without clinical response | NA | 3/9 |
| Improvement of symptoms (age) | + (9 months) | + (6.5 years) | + (13 months) | + (18 months) | + (1 year) | NA | + (11 weeks) | + (2 years) | + (3 years) | 8/8 |
| Zygosity | Homozygous du to UPD(2)mat | Homozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous | Homozygous | UPD(2)mat:1/Hom:2/Compound Het:6 |
| PREPL mutation | c.1282_1285delTTTG, p.Phe428Argfs*18 | NA | c.358_361delTTTG, p.Val121Ilefs*121 | c.1604T>G, p.Leu535Ter | c.883 C>T, p.Arg295Ter | c.1156−1G>A, p.Leu386_Lys451del | c.807delT, p.Met270Ter | c.1529 + 1G>A; c.1784delinsAA, p.Thr595Lysfs*19 | c.342delA, p.Val115Leufs*39) | Nine different mutations |
| PREPL deletion | NA | PREPL exon 5–10 | PREPL exon 7–8 | SLC3A1 exon10‐PREPL exon10 | PREPL exon12‐CAMKMT exon2 | SLC3A1 exon 7‐CAMKMT exon 2 | SLC3A1 exon5‐PREPL exon9 | NA | NA | Six different deletions |
Abbreviations: +, positive; −, negative; DI, developmental index; IQ, intelligence quotient; Het, heterozygous; Hom, homozygous; F, female; M, male; GH, growth hormone; NA, not applicable.
In total, nine mutations and six gross deletions in PREPL have been detected currently (Figure 2). The nine mutations are all truncating mutations which include four frameshifts (p.Val121Ilefs*121, p.Thr595Lysfs*19, p.Val115Leufs*39, and p.Phe428Argfs*18), three nonsense mutations (p.Leu535Ter,p.Arg295Ter, and p.Met270Ter), and two splicing mutations (c.1156‐1G>A and c.1529+1G>A). The eight reported mutations all locate in the protein domains (peptidase S9A N‐terminal domain or peptidase S9 prolyl oligopeptidase catalytic domain). The novel frameshift mutation at exon 8 detected in our study (p.Phe428Argfs*18) do not locate in the protein domains. But, it could lead to the loss of the peptidase S9 prolyl oligopeptidase catalytic domain (Figure 2a). In the six patients with gross deletions, patient 2 and 3 with PREPL single gene deletion, other 4 involve contiguous genes of SLC3A1 and CAMKMT (Figure 2b).
UPD can be associated with human diseases through three primary mechanisms: imprinting, homozygosity for an autosomal recessive trait, or mosaic aneuploidy (Siegel & Slavotinek, 2005). It is worth mentioning that, both UPD(2)mat and UPD(2)pat have been reported in individuals with normal phenotype (Bernasconi et al., 1996; Keller et al., 2009), suggesting that the imprinting is not the pathogenic mechanism for patients with UPD2 presenting phenotype.
The clinical features of CMS22 are similar to those of Prader‐Willi syndrome (PWS), such as neonatal hypotonia, feeding difficulties, the improvement of symptoms after 1 year old and childhood‐onset obesity. So, four of nine patients were firstly suspected of PWS. Our infantile patient showed neonatal hypotonia and feeding difficulties, and she was firstly suspected of SMA. Therefore, CMS22 should be carefully considered by the pediatrician when the common causes including PWS and SMA were negative. The timely genetic test is crucial to the early diagnosis of CMS22.
In summary, we report the first and the youngest case of CMS22 for UPD(2)mat with a novel homozygous frameshift variant in PREPL, which expands the mutation spectrum and further delineates the phenotype and genotype of CMS22.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
ACKNOWLEDGMENTS
The authors thank the clinicians' care for patients and our genetic laboratory team's contribution to this study. The work was supported by grants from the project of Shanghai Science and Technology Committee, grant number 13DZ2260600.
Zhang P, Wu B, Lu Y, et al. First maternal uniparental disomy for chromosome 2 with PREPL novel frameshift mutation of congenital myasthenic syndrome 22 in an infant. Mol Genet Genomic Med. 2020;8:e1144 10.1002/mgg3.1144
Ping Zhang and Bingbing Wu contributed equally to this work.
Funding information
Shanghai Science and Technology Committee, Grant numbers:13DZ2260600.
Contributor Information
Wenhao Zhou, Email: zhouwenhao@fudan.edu.cn.
Huijun Wang, Email: huijunwang@fudan.edu.cn.
REFERENCES
- Bartholdi, D. , Asadollahi, R. , Oneda, B. , Schmitt‐Mechelke, T. , Tonella, P. , Baumer, A. , & Rauch, A. (2013). Further delineation of genotype‐phenotype correlation in homozygous 2p21 deletion syndromes: First description of patients without cystinuria. American Journal of Medical Genetics. Part A, 161A(8), 1853–1859. 10.1002/ajmg.a.35994 [DOI] [PubMed] [Google Scholar]
- Bernasconi, F. , Karaguzel, A. , Celep, F. , Keser, I. , Luleci, G. , Dutly, F. , & Schinzel, A. A. (1996). Normal phenotype with maternal isodisomy in a female with two isochromosomes: I(2p) and i(2q). American Journal of Human Genetics, 59(5), 1114–1118. [PMC free article] [PubMed] [Google Scholar]
- Carmichael, H. , Shen, Y. , Nguyen, T. T. , Hirschhorn, J. N. , & Dauber, A. (2013). Whole exome sequencing in a patient with uniparental disomy of chromosome 2 and a complex phenotype. Clinical Genetics, 84(3), 213–222. 10.1111/cge.12064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, A. G. (2018). Congenital myasthenic syndromes in 2018. Current Neurology and Neuroscience Reports, 18(8), 46 10.1007/s11910-018-0852-4 [DOI] [PubMed] [Google Scholar]
- Engel, A. G. , Shen, X. M. , Selcen, D. , & Sine, S. M. (2015). Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. The Lancet Neurology, 14(5), 461 10.1016/S1474-4422(15)00010-1 [DOI] [PubMed] [Google Scholar]
- Jaeken, J. , Martens, K. , François, I. , Eyskens, F. , Lecointre, C. , Derua, R. , … Matthijs, G. (2006). Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia‐cystinuria syndrome. American Journal of Human Genetics, 78(1), 38–51. 10.1086/498852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, M. C. , McRae, A. F. , McGaughran, J. M. , Visscher, P. M. , Martin, N. G. , & Montgomery, G. W. (2009). Non‐pathological paternal isodisomy of chromosome 2 detected from a genome‐wide SNP scan. American Journal of Medical Genetics. Part A, 149A(8), 1823–1826. 10.1002/ajmg.a.32973 [DOI] [PubMed] [Google Scholar]
- Kim, M. H. , & Hersh, L. B. (2004). The vesicular acetylcholine transporter interacts with clathrin‐associated adaptor complexes AP‐1 and AP‐2. Journal of Biological Chemistry, 279(13), 12580–12587. 10.1074/jbc.M310681200 [DOI] [PubMed] [Google Scholar]
- Labrijn‐Marks, I. , Somers‐Bolman, G. M. , In ’t Groen, S. L. M. , Hoogeveen‐Westerveld, M. , Kroos, M. A. , Ala‐Mello, S. , … Halley, D. J. (2019). Segmental and total uniparental isodisomy (UPiD) as a disease mechanism in autosomal recessive lysosomal disorders: Evidence from SNP arrays. European Journal of Human Genetics, 27(6), 919–927. 10.1038/s41431-019-0348-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugwitz, L. , Redler, S. , Buchert, R. , Sturm, M. , Zeile, I. , Schara, U. , … Distelmaier, F. (2018). Isolated PREPL deficiency associated with congenital myasthenic syndrome‐22. Klinische Padiatrie, 230(5), 281–283. 10.1055/a-0605-3659 [DOI] [PubMed] [Google Scholar]
- Li, Z. , Zhang, F. , Wang, Y. , Qiu, Y. , Wu, Y. , Lu, Y. , … Tian, W. (2019). PhenoPro: A novel toolkit for assisting in the diagnosis of Mendelian disease. Bioinformatics, 10.1093/bioinformatics/btz100 [DOI] [PubMed] [Google Scholar]
- Martens, K. , Derua, R. , Meulemans, S. , Waelkens, E. , Jaeken, J. , Matthijs, G. , & Creemers, J. W. (2006). PREPL: A putative novel oligopeptidase propelled into the limelight. Biological Chemistry, 387(7), 879–883. 10.1515/BC.2006.111 [DOI] [PubMed] [Google Scholar]
- Matthijs, G. , Souche, E. , Alders, M. , Corveleyn, A. , Eck, S. , Feenstra, I. , … Bauer, P. (2016). Guidelines for diagnostic next‐generation sequencing. European Journal of Human Genetics, 24(10), 1515 10.1038/ejhg.2016.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan, K. , Baltes, J. , Creemers, J. W. , & Schu, P. (2013). Trans‐Golgi network morphology and sorting is regulated by prolyl‐oligopeptidase‐like protein PREPL and the AP‐1 complex subunit mu1A. Journal of Cell Science, 126(Pt 5), 1155–1163. 10.1242/jcs.116079 [DOI] [PubMed] [Google Scholar]
- Régal, L. , Mårtensson, E. , Maystadt, I. , Voermans, N. , Lederer, D. , Burlina, A. , … Creemers, J. W. M. (2018). PREPL deficiency: Delineation of the phenotype and development of a functional blood assay. Genetics in Medicine, 20(1), 109–118. 10.1038/gim.2017.74 [DOI] [PubMed] [Google Scholar]
- Regal, L. , Shen, X. M. , Selcen, D. , Verhille, C. , Meulemans, S. , Creemers, J. W. , & Engel, A. G. (2014). PREPL deficiency with or without cystinuria causes a novel myasthenic syndrome. Neurology, 82(14), 1254–1260. 10.1212/WNL.0000000000000295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, D. H. , & Slavotinek, A. (2005). Uniparental disomy. Pediatric Dermatology, 22(5), 482–487. 10.1111/j.1525-1470.2005.00122.x [DOI] [PubMed] [Google Scholar]
- Silva, S. , Miyake, N. , Tapia, C. , & Matsumoto, N. (2018). The second point mutation in PREPL: A case report and literature review. Journal of Human Genetics, 63(5), 677–681. 10.1038/s10038-018-0426-y [DOI] [PubMed] [Google Scholar]
- van Riet, J. , Krol, N. M. G. , Atmodimedjo, P. N. , Brosens, E. , van IJcken, W. F. J. , Jansen, M. P. H. M. , … van de Werken, H. J. G. (2018). SNPitty: An intuitive web application for interactive B‐Allele frequency and copy number visualization of next‐generation sequencing data. The Journal of Molecular Diagnostics, 20(2), 166–176. 10.1016/j.jmoldx.2017.11.011 [DOI] [PubMed] [Google Scholar]
- Yang, L. , Kong, Y. , Dong, X. , Hu, L. , Lin, Y. , Chen, X. , … Zhou, W. (2019). Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genetics in Medicine, 21(3), 564–571. 10.1038/s41436-018-0091-8 [DOI] [PMC free article] [PubMed] [Google Scholar]