

Abstract
Atypical antipsychotic (AA) medications including risperidone (RIS) and olanzapine (OLAN) are FDA approved for the treatment of psychiatric disorders including schizophrenia, bipolar disorder and depression. Clinical side effects of AA medications include obesity, insulin resistance, dyslipidemia, hypertension and increased cardiovascular disease risk. Despite the known pharmacology of these AA medications, however, the mechanisms contributing to adverse metabolic side-effects are not well understood. To evaluate drug-associated effects on the heart, we assessed changes in the cardiac proteomic signature in mice administered for 4 weeks with clinically relevant exposure of RIS or OLAN. Using proteomic and gene enrichment analysis, we identified differentially expressed (DE) proteins in both RIS- and OLAN-treated mouse hearts (p <0.05), including proteins comprising mitochondrial respiratory complex I and pathways involved in mitochondrial function and oxidative phosphorylation. A subset of DE proteins identified were further validated by both western blotting and quantitative real-time PCR. Histological evaluation of hearts indicated that AA-associated aberrant cardiac gene expression occurs prior to the onset of gross pathomorphological changes. Additionally, RIS treatment altered cardiac mitochondrial oxygen consumption and whole body energy expenditure. Our study provides insight into the mechanisms underlying increased patient risk for adverse cardiac outcomes with chronic treatment of AA medications.
Keywords: Antipsychotics, risperidone, olanzapine, heart, energy metabolism, mitochondria
Graphical abstract
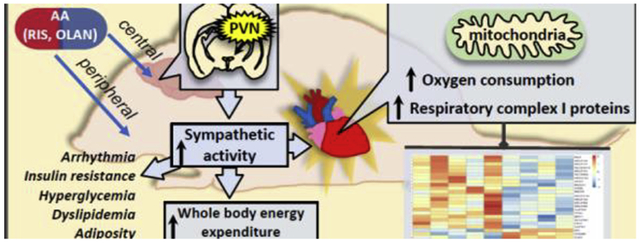
1. Introduction
Cardiovascular disease is the leading cause of death worldwide (World Health Organization, 2018), and cardiovascular risk is increased in patients with psychiatric disorders. Specifically, individuals with severe mental illness (schizophrenia or bipolar disorder) are twice as likely to die from cardiovascular disease compared with the general population (Daumit et al., 2008; Foley and Mackinnon, 2014; Iwamoto et al., 2005). It is not clear how much of this risk is due to genetics and the underlying mental health disorder, per se, versus environmental factors including psychiatric medications. Indeed, schizophrenia is considered an independent risk factor for metabolic syndrome and cardiovascular disease (Subashini et al., 2011; Thakore et al., 2002), and the metabolic syndrome is worsened with atypical antipsychotic medications (AA) (De Hert et al., 2011; Meyer et al., 2008). AA drugs, including risperidone (RIS) and olanzapine (OLAN), are FDA approved for the treatment of psychiatric disorders including schizophrenia and bipolar disorder and are increasingly prescribed off-label for diverse indications including attention deficit hyperactivity disorder, insomnia, post-traumatic stress disorder, autism and dementia. AA drugs are pharmacologically diverse, potently targeting dopamine and serotonergic signaling and also antagonizing other G protein coupled receptors, such as muscarinic, α-adrenergic and histaminergic receptors, to varying degrees. AA medications are associated with significant endocrine and metabolic side effects, including rapid onset weight gain, dyslipidemia, insulin resistance and hyperglycemia across the drug class (Calarge et al., 2009; Gude et al., 2018; Komossa et al., 2011; Lambert et al., 2015; Patel et al., 2009; Picca et al., 2018; Stoner, 2017). These metabolic side effects are observed to varying degrees with most AA medications despite differing receptor pharmacology. For example, OLAN has greater clinical liability for weight gain than RIS, and has higher affinity for histamine H1 and muscarinic receptors than RIS, whereas RIS has higher affinity for α-adrenergic receptors (Sullivan et al., 2015). Recently, it was reported that AA-associated weight gain/adiposity is linked to altered serotonergic (5HT2c) signaling (Lord et al., 2017; Reynolds and Kirk, 2010). However, the underlying pharmacological mechanisms associated with other AA-associated side effects remain elusive. Indeed, it is likely that the metabolic side effects are mediated by both central and peripheral mechanisms including altered autonomic nervous system effects (Hattori et al., 2018; Suda et al., 2018) as well as altered lipid metabolism, lipid trafficking and AMPK signaling (Vantaggiato et al., 2019). Using preclinical models, we and others have demonstrated the potent systemic effects of AAs, showing that even a single dose is sufficient to induce changes in gene expression in bone, induce whole body insulin resistance, increase plasma prolactin concentrations and alter lipid profiles in both rat and mouse models (Albaugh et al., 2012; Houseknecht et al., 2007; Motyl et al., 2012, 2015; Shimizu et al., 2015).
Clinical and preclinical literature report a strong link between AA therapy with adverse cardiac events such as a prolonged heart rate-corrected QT interval, associated with increased patient risk for ventricular arrhythmias and sudden cardiac death (Drici et al., 1998; Gopal et al., 2013; Haddad and Anderson, 2002; Harrigan et al., 2004; Li et al., 2013; Ray et al., 2009; Stöllberger et al., 2005; Straus et al., 2004; Vieweg, 2003; Witchel et al., 2003; Wu et al., 2015; Yerrabolu et al., 2000). AA have been shown to cause cardiometabolic abnormalities in children and adolescents and perspective clinical studies indicate improvement of cardiometabolic parameters with discontinuation of long term treatment (Calarge et al., 2014). Furthermore, AAs, including RIS, are associated with a significantly increased risk for acute myocardial infarction, stroke, myocarditis and cardiomyopathies (Citrome et al., 2013; Lin et al., 2014; Stoner, 2017).
Despite compelling evidence that AA medications increase cardiovascular risk, there is very little information on drug-associated effects on cardiac biology in terms of genetic and proteomic signatures as well as onset of drug-associated effects. We and others have reported that some of the AA-associated metabolic and endocrine effects can be rapid in onset and occur prior to drug associated weight gain/adiposity (Alfaro et al., 2002; Assié et al., 2008; Chintoh et al., 2008, 2009; Houseknecht et al., 2007; Motyl et al., 2012, 2015; Skrede et al., 2012). Therefore the primary aim of this study was to examine the effects of antipsychotic treatment on the cardiac proteomic signature following sub-chronic dosing to identify drug-associated mechanisms relating to cardiac function. We hypothesized that AA medications may accelerate the development of cardiovascular disease by targeting multiple physiological/pharmacological pathways. Given the complexity of the known pharmacology of these medications, we chose to use a pre-clinical model to identify relevant underlying mechanisms and pathways. Our approach combined assessment of whole body energetics, and cardiac proteome analysis to explore the pharmacology of AA medication-induced effects. We provide the first evidence that clinically relevant plasma concentrations of RIS or OLAN are associated with alterations in cardiac mitochondrial pathways prior to presentation of overt cardiac pathophysiology, suggesting that AA medications may promote the early onset of cardiovascular disease, at least in part, via impaired mitochondrial function.
2. Material and methods
2.1. In vivo studies:
The work reported here is derived from multiple cohorts of mice, and includes both sexes, as well as clinically relevant doses and dosing schedules conducted across multiple institutions. All measurements performed in each cohort are shown. Eight-week-old male and female C57BL/6J mice (The Jackson Laboratory, Bar Harbor ME) were fed a standard chow diet and administered once daily with vehicle (VEH; 0.1% acetic acid, oral PO), RIS (Sigma-Aldrich, St. Louis MO; 0.75 or 1 mg/kg, PO) or OLAN (Sigma-Aldrich, St. Louis MO; 5 mg/kg, PO) for 4 weeks (28 days). Our dosing strategy was chosen based on pharmacokinetic studies previously conducted in our laboratory in male and female mice (May et al., 2019; Motyl et al., 2017) designed to achieve peak plasma concentrations of drug that fall within the range of plasma drug exposure observed in patients (Aman et al., 2007; Mauri et al., 2014). Furthermore the drug dose employed in these studies falls within the low range of doses typically used in mice (1–6 mg/kg BW) (Gao et al., 2006). Mice were euthanized by IP injection of 2.5% avertin (in PBS) followed by cardiac perfusion. Blood samples were collected and centrifuged in EDTA tubes for plasma collection and immediately frozen −20°C. Brains and whole hearts were extracted following perfusion. Brains were fixed in 4% PFA and whole hearts were either snap frozen in liquid nitrogen for RNA and protein analysis, fixed in 4% paraformaldehyde and paraffin embedded or collected for mitochondrial isolation. All animal experiments were carried out in compliance with the federal animal welfare laws and policies using protocols approved by the Institutional Animal Care and Use Committee at the University of New England, Biddeford, ME and Maine Medical Research Institute (MMCRI), Scarborough, ME following National Institutes of Health guide for the care and use of Laboratory animals (National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals, 2011).
2.2. Indirect calorimetry:
Eight-week-old female and male C57BL/6J mice (The Jackson Laboratory, Bar Harbor ME) were fed a standard chow diet and water ad libitum. Mice were administered a once daily (8:00 to 9:00 PM) dose with vehicle (VEH; 0.1% acetic acid, oral (p.o.) gavage or a low, clinically relevant dose of risperidone (Sigma-Aldrich, St. Louis MO; 0.75 mg/kg, p.o.) for 4 weeks (28 days). Mice were maintained on a 14 hr light/10 hr dark schedule. During the third week of the study, indirect calorimetry measurements were performed while mice were singly housed in Promethiom metabolic cages (Sable Systems International, North Las Vegas, NV) located in the Physiology Core at MMCRI. Data acquisition and instrument control were performed using Meta Screen version 1.7.2.3, and the raw data obtained were processed with ExpeData version 1.5.4 (Sable Systems International, North Las Vegas, NV) using an analysis script detailing all aspects of data transformation (Motyl et al., 2015). Daytime, nighttime and 24-hour measurements were calculated. Temporal energy expenditure and respiratory quotient data was visualized and analyzed in GraphPad Prism. Female mice from these studies were euthanized and fresh whole hearts were removed and used for mitochondrial oxygen consumption measurements (described below).
2.3. RNA and protein isolation:
Whole hearts were homogenized under liquid nitrogen and total RNA was isolated by TriReagent:Chloroform extraction and RNA quality was assessed by Nanodrop (Thermo Fisher Scientific, Waltham, MA) and protein was isolated by extracting from precipitate pellet with lysis buffer containing 30 mM Tris-base, 7 M urea, 2 M thiourea, 4% (w/v) CHAPS, pH 8.5. Protein concentration was determined by ovalbumin using EZQ Protein Quantitation Kit (Molecular Probes, Eugene OR).
2.4. Proteomic methodology:
Whole hearts from male mice treated with daily dose of 1 mg/kg RIS (n=5), 5 mg/kg OLAN (n=4) or VEH (n=4) were homogenized in HTNG lysis buffer (20 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 10% glycerol, 1% Triton- X 100, 1 mM EDTA, protease- inhibitor cocktail (Calbiochem, Burlington MA). Protein concentration of the supernatant was determined by BCA (Pierce, Appleton WI). 40 μg of protein was used from each sample and Sequential Window Acquisition of all THeoretical spectra (SWATH) analysis of samples were performed as previously described (May et al., 2019; Peterson et al., 2018). In brief, tryptic digests of protein samples were performed using the ProteoExtract digestion kit (Calbiochem, Burlington MA). Tryptic peptides were then separated on an Ultimate RSLC system 3000 (ThermoFisher/Dionex, Waltham MA) nanoscale liquid chromatograph and infused onto a 5600 TripleTOF mass spectrometer (Sciex, Framingham MA). SWATH was used to profile all proteins in each sample using a data-independent acquisition method as previously described (May et al., 2019; Peterson et al., 2018). In brief, a mouse-specific ion library comprising 4489 proteins was constructed using ProteinPilot software (Sciex, Framingham MA). For identification of peptides, multiple fragment ion chromatograms were retrieved from the spectral library for each peptide of interest. These spectra were compared with the extracted fragment ion traces for the corresponding isolation window to identify the transitions that best identify the target peptide. Detailed proteomic analysis methods are available at PeptideAtlas (Identifier: PASS01349). SWATH analysis was performed using PeakView software, and MarkerView software was utilized for principal component analysis and T-test comparisons. Principal Component Analysis (PCA) normalized using MLR normalization scheme was performed to demonstrate close clustering for each of our three experimental groups (RIS, OLAN and VEH) (Suppl Fig. 1) (Gillet et al., 2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis (Gillet et al., 2012).
2.5. Proteomic analysis:
The proteomic data was analyzed comparing heart RIS or OLAN to heart control after log transformation to achieve normality, although fold changes were given on the original scale. A differentially expressed (DE) protein list with a p-value threshold of 0.05 was established for pathway analysis, with false discovery rate (FDR) values estimates for each protein using the p.adjust package in R using the BH (Benjamini Hochberg) method. Qiagen’s Ingenuity Pathway Analysis (IPA) was run on each significant gene lists using the mouse SWATH full library as the background with raw and Benjamini-Hochberg adjusted p-values reported for each result. Further analysis of enriched terms was performed using PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System (Mi et al., 2010) and Visual Annotation Display (VLAD) for visualizing Gene Ontology (GO) annotations (Richardson and Bult, 2015). The technical replicates were averaged to get values for the biological replicates, which were then used to produce heatmaps for overlapping significant gene lists. Heatmaps were created using the pheatmap package in R.
2.6. Western blowing:
The protein sample was mixed with Laemmli sample buffer at a ratio of 1:4 (v/v) and boiled for 5 min. Equal amounts of sample proteins (20 μg) were subjected to SDS- PAGE and transferred to PVDF membranes. Membranes were blocked for 1 h at room temperature in Tris- buffered saline (TBS) containing 0.05% Tween- 20 (TBS- T) and 5% nonfat dry milk. Membranes were then incubated overnight at 4°C with primary antibodies against the following proteins: NDUFB9 (sc-398869; Santa Cruz, Dallas TX), NDUFS3 (sc-374282; Santa Cruz, Dallas TX) and CAMK2D (MAB4176; R&D Systems, Minneapolis MN). After overnight incubation, the membranes were washed three times (5 min each) in TBS- T, and incubated for 2 h with CyDye- conjugated anti- mouse or anti-chicken IgG (Jackson ImmunoResearch Laboratories, West Grove PA) in TBS- T with 5% nonfat dry milk at room temperature. Membranes were then washed three times in TBS- T, and immunoreactive bands were visualized using Typhoon FLA 9500 scanner (GE Healthcare, Piscataway NJ). The signal intensity of scanned blotting was analyzed using ImageQuant TL v.8.1 (GE Healthcare, Piscataway NJ) and GAPDH (GTX85118 and GTX28245; GeneTex, Irvine, CA) was used as an internal control for equal protein loading.
2.7. Quantitative real-time PCR (qPCR):
The first cDNA strand was synthesized from 500 ng total RNA in a volume of 20 μl using the RT2 first strand kit (Qiagen, Valencia, CA) with genomic DNA elimination step, according to the manufacturer’s protocol. In the second step, we used oligo-dT primers to perform reverse transcription. The quantity of cDNA was measured on Nanodrop One spectrophotometer (ThermoFisher Scientific, Madison WI) at 260/280 nm absorbance. Newly synthesized cDNA was diluted 10-fold prior to determination of its quality and quantity and stored at −20°C until use for the qPCR study using StepOnePlus Real-Time PCR system (Applied Biosystems, Forest City, CA). Relative levels of gene expression were normalized to GAPDH expression with the ΔΔCt method (VanGuilder et al., 2008). Other housekeeping genes, such as Hprt, were evaluated yielding similar results. Primers used for gene expression studies found in Suppl Table 1.
2.8. Ex-vivo oxygen consumption studies:
Whole hearts were extracted from female mice dosed daily by oral gavage for 4 weeks (28 days) with VEH or 0.75 mg/kg RIS and homogenized in isolation buffer (70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, 0.5% BSA, pH 7.2) to isolate mitochondria. Mitochondria were loaded in equal amounts by normalizing to total protein concentration on the XF24 extracellular flux analyzer (Agilent Technologies, Santa Clara CA). Respiratory states were measured following injections of Succinate (10 mM) in the presence of Rotenone (2 μM), followed by 4.5 mM ADP, oligomycin (2 μM), FCCP (4 μM) and Antimycin A (2 μM). Each experiment was performed with n= 2 hearts per group for 3 independent experiments.
2.9. Histology:
Whole fixed hearts from male mice treated for 4 weeks (28 days) with daily dose of 1 mg/kg RIS, 5mg/kg OLAN or VEH were paraffin embedded and longitudinal sections were stained with hematoxylin and eosin and Masson’s Trichrome.
2.10. Plasma free fatty acid composition and analysis:
Plasma samples were extracted and solvent partitioned using the Folch method. A Thermo EASY 1000 nLC autosampler/pumping system interfaced to a Thermo LTQ-Orbitrap mass spectrometer was used for the analyses. All samples were run in triplicate by both positive ion and negative ion mass spectrometry performed in separate analyses, using a Thermo Scientific LTQ Orbitrap mass spectrometer. Full mass spectra were acquired at approximately 1 sec intervals from m/z 150 −2,000 with 2 μscans, a spray voltage at approximately 1,800 V, resolution 30,000 and maximum inject time 200 msec. TraceFindertm software (Thermo Scientific) was used for peak identification and peak area integration. Target compound lists of expected analytes were used to find peaks of interest with ion windows of 0.005 ppm mass accuracy for the expected ions. Target peaks were adjusted relative to the retention time of its assigned internal standard. Analyte concentrations were calculated by dividing its peak area by that of its internal standard and multiplying the result by the concentration of the internal standard in the plasma sample. The concentrations of each individual analyte and total concentration of each compound class and the CV for the 3 repeats of each sample are reported.
2.11. c-FOS expression:
Brains were collected from female mice 2 hours post oral dose of VEH or 0.75 mg/kg RIS or from male mice after 4 weeks of daily oral dosing 24 h following the last dose of VEH or 1mg/kg RIS and fixed in 4% PFA. Brains were transferred to 30% sucrose then frozen in OCT in 2-methylbutane (Alfa Aesar, Tewksbury MA) on dry ice. Coronal sections of PVN were collected at 30 microns using a Leica CM1950 cryostat (Wetzlar, Germany) and placed in PBS for free floating immunostaining. All staining steps were carried out on a gentle shaker. Tissue was rinsed with 0.1% PBS-Tx then incubated for 1 hour at room temperature in blocking solution consisting of 5% normal goat serum (ab7481; Abcam, Cambridge MA) and 0.1% PBS-Tx. Sections were incubated with mouse anti-c-FOS (NBP2-50037; RRID AB_2665387; Novus Biologicals, Littleton, CO) diluted 1:1000 in blocking solution overnight at 4°C. Sections were then rinsed with 0.1% PBS-Tx and incubated with goat anti-mouse IgG conjugated to Alexa Fluor 488 (ab150117; RRID AB_2688012; Abcam, Cambridge MA) for 2 hours at room temperature. Sections were rinsed with 0.1% PBS-Tx then mounted with Fluoromount-G containing DAPI (0100-20; Southern Biotechnology, Birmingham AL). Z-stacked images were captured on a Leica DM2500 epifluorescence microscope with Leica DFC365X camera. c-FOS(+) cells were quantified by number of positive cells average over 5 × 0.005 mm2 regions of the PVN for 3 sections per sample using ImageJ image processing and analysis software.
2.12. Statistical analysis:
All data are expressed as the mean ± standard error of the mean (SEM) unless specified. All statistical analysis was performed using student’s t-test or ANOVA with post-hoc analyses as indicated unless specified. All statistical analysis was performed using Prism 8 statistical software (GraphPad Software, Inc., La Jolla, CA) or SigmaPlot (Systat Software, Inc., San Jose, CA).
3. Results
3.1. Animal Health: Risperidone treatment increases fat mass in male mice with no change in body weight gain
The animals utilized in this study were healthy and the drugs and vehicles used were well tolerated as indicated by overall animal health, activity, feeding behavior and growth rates. The doses of RIS and OLAN used in these studies are in the low to moderate range of doses used in the published literature, and plasma drug concentrations fall into the low clinical range as previously reported. In contrast to the rapid onset of hyperphagia and obesity commonly observed in patients administered these AA medications, we did not observe notable difference in body weight gain among our male or female groups administered daily low doses of RIS (or OLAN-treated male mice) compared to our control groups after 4 weeks. However, there was a significant increase in fat mass (14%) with RIS treatment (p=0.04), and a trend for a reduction in percentage of lean mass (p=0.06) (Table 1). Daytime food and water consumption were also significantly increased in RIS-treated males compared to VEH (Table 2). In female mice treated with RIS, there was no effect of treatment on body weight, lean mass and fat mass (Suppl Table 2), with no change in food or water consumption (Suppl Table 3), confirming our previous findings using a similar, low dose of RIS in mice of similar age (Motyl et al., 2015). It is well known that AA drugs cause significant weight gain and increased adiposity with some gender differences in human patients. However, in pre-clinical species, literature reports indicate that the effects on body weight gain and adiposity are variable and are associated with significant sex and dose effects (Albaugh et al., 2012; Auger et al., 2014, 2018; Cope et al., 2009; Klingerman et al., 2014; Li et al., 2013). The dose of RIS used in these studies results in significant hyperprolactinemia, bone loss, increased adiposity and immunosuppression in mice (May et al., 2019; Motyl et al., 2012; Motyl et al. 2014; Motyl et al. 2017) but is lower than doses reported to significantly alter body weight 1.4 mg/kg (Cope et al., 2005, 2009; Li et al., 2013) Furthermore, studies published by others have also shown a lack of weight gain in female mice treated with RIS (0.5 mg/kg BW) for 4 weeks and male rats treated with RIS for treated for 3 weeks (Auger et al., 2018; Ota et al., 2002, 2005). Overall, the data reported here is consistent with the pre-clinical literature which shows that weight gain response to RIS and OLAN can be variable and dose-dependent, whereas other metabolic and endocrine effects occur at lower dose ranges.
Table 1.
Body composition of male C57BL/6J mice treated with VEH or 0.75 mg/kg RIS.
| Vehicle | Risperidone | p-value | |
|---|---|---|---|
| Body Mass (g) | 26.4 ± 0.8 | 25.8 ± 0.8 | 0.63 |
| Lean Mass (g) | 22.0 ± 0.6 | 21.5 ± 0.6 | 0.59 |
| Fat Mass (g) | 2.6 ± 0.2 | 3.0 ± 0.2 | 0.23 |
| Lean Mass (%) | 89.3 ± 0.5 | 87.9 ± 0.4 | 0.06 |
| Fat Mass (%) | 10.6 ± 0.4 | 12.1 ± 0.5 | 0.04 |
Values represent mean ± SEM. p-values calculated by Student’s t-test.
Table 2.
Energy metabolism measurements in male C57BL/6J mice treated with VEH or 0.75 mg/kg RIS at the beginning of the dark phase.
| Vehicle (n=4) | Risperidone (n=4) | p-value | |
|---|---|---|---|
| Daytime O2 Consumption (ml/min) | 1.094 ± 0.103 | 1.636 ± 0.029 | 0.002 |
| Nighttime O2 Consumption (ml/min) | 1.761 ± 0.091 | 2.064 ± 0.080 | 0.047 |
| 24-hour O2 Consumption (ml/min) | 1.499 ± 0.056 | 1.850 ± 0.033 | 0.002 |
| Daytime CO2 Expulsion (ml/min) | 1.201 ± 0.181 | 1.221 ± 0.074 | 0.924 |
| Nighttime CO2 Expulsion (ml/min) | 1.505 ± 0.075 | 1.790 ± 0.058 | 0.024 |
| 24-hour CO2 Expulsion (ml/min) | 1.231 ± 0.048 | 1.531 ± 0.027 | 0.002 |
| Daytime Average EE (Kcal/hr) | 0.356 ± 0.007 | 0.471 ± 0.009 | <0.001 |
| Nighttime Average EE (Kcal/hr) | 0.516 ± 0.027 | 0.607 ± 0.023 | 0.041 |
| 24-hour Average EE (Kcal/hr) | 0.436 ± 0.016 | 0.539 ± 0.010 | 0.002 |
| Daytime Resting EE (Kcal/30 min) | 0.354 ± 0.016 | 0.385 ± 0.009 | 0.132 |
| Nighttime Resting EE (Kcal/30 min) | 0.345 ± 0.018 | 0.434 ± 0.005 | 0.003 |
| 24-hour Resting EE (Kcal/30 min) | 0.350 ± 0.011 | 0.410 ± 0.003 | 0.002 |
| Daytime Active EE (Kcal/30 min) | 0.498 ± 0.011 | 0.588 ± 0.012 | 0.001 |
| Nighttime Active EE (Kcal/30 min) | 0.577 ± 0.037 | 0.702 ± 0.020 | 0.024 |
| 24-hour Active EE (Kcal/30 min) | 0.538 ± 0.023 | 0.645 ± 0.011 | 0.006 |
| Daytime RQ | 0.770 ± 0.009 | 0.776 ± 0.008 | 0.636 |
| Nighttime RQ | 0.852 ± 0.007 | 0.864 ± 0.007 | 0.264 |
| 24-hour RQ | 0.811 ± 0.007 | 0.820 ± 0.006 | 0.336 |
| Daytime Resting RQ | 0.790 ± 0.024 | 0.810 ± 0.009 | 0.468 |
| Nighttime Resting RQ | 0.841 ± 0.018 | 0.826 ± 0.023 | 0.635 |
| 24-hour Resting RQ | 0.815 ± 0.020 | 0.818 ± 0.013 | 0.922 |
| Daytime Active RQ | 0.827 ± 0.009 | 0.839 ± 0.015 | 0.524 |
| Nighttime Active RQ | 0.854 ± 0.013 | 0.880 ± 0.017 | 0.266 |
| 24-hour Active RQ | 0.840 ± 0.004 | 0.860 ± 0.015 | 0.260 |
| Daytime Food Consumption (g) | 0.514 ± 0.101 | 1.053 ± 0.115 | 0.013 |
| Nighttime Food Consumption (g) | 2.713 ± 0.523 | 2.829 ± 0.171 | 0.840 |
| 24-hour Food Consumption (g) | 3.227 ± 0.624 | 3.882 ± 0.280 | 0.375 |
| Daytime Water Consumption (ml) | 0.729 ± 0.062 | 1.109 ± 0.103 | 0.019 |
| Nighttime Water Consumption (ml) | 2.635 ± 0.164 | 2.661 ± 0.178 | 0.916 |
| 24-hour Water Consumption (ml) | 3.363 ± 0.203 | 3.770 ± 0.270 | 0.274 |
Values represent mean ± SEM. p-values calculated by Student’s t-test.
Abbreviations: EE, energy expenditure; RQ, respiratory quotient.
3.2. Risperidone treatment alters the cardiac proteome
We performed protein profiling using an unbiased SWATH proteomic approach to characterize the unique protein signature of whole hearts from male mice following 4-week treatment of RIS. Using SWATH analysis, we identified 111 proteins that were differentially expressed (DE) in RIS-treated hearts compared to control hearts (p< 0.05), of which 80 were significantly upregulated and 31 downregulated (Suppl File 1). Using Ingenuity Pathway Analysis (IPA) software from our DE protein dataset, we identified 18 canonical pathways that are significantly enriched in RIS hearts. Of these results, the top five canonical pathways were identified as sirtuin signaling pathway, xenobiotic metabolism signaling, oxidative phosphorylation, superpathway of cholesterol biosynthesis and mitochondrial dysfunction (Table 3; Suppl File 1). We identified 6 proteins that comprise the mitochondrial complex I, including NDUFS3, NDUFB9, NDUFA8, NDUFA9, NDUFA10 and NDUFB8 within these top canonical pathways in RIS hearts (p<0.05) (Table 3).
Table 3.
Canonical pathways are altered in the male mouse heart with chronic RIS exposure.
| Ingenuity Canonical Pathways | −log(p-value) | Ratio | Molecules |
|---|---|---|---|
| Sirtuin Signaling Pathway | 2.5 | 0.08 | NDUFB9,NDUFA9,SLC25A4,NDUFS7,NQO1,NDUFA10,NDUFB8,LDHA,NDUFS3,HMGCS2,NDUFA8 |
| Xenobiotic Metabolism Signaling | 2.15 | 0.09 | RAP2B,UGT2B28,SRA1,CAMK2D,UGT1A6,NQO1,MGST3,GSTO1 |
| Oxidative Phosphorylation | 2.12 | 0.1 | NDUFB9,NDUFA9,NDUFS7,NDUFA10,NDUFB8,NDUFS3,NDUFA8 |
| Superpathway of Cholesterol Biosynthesis | 1.96 | 0.2 | IDI1,HMGCS2,CYP51A1 |
| Mitochondrial Dysfunction | 1.71 | 0.08 | NDUFB9,NDUFA9,NDUFS7,NDUFA10,NDUFB8,ACO1,NDUFS3,NDUFA8 |
IPA analysis of significant protein expression in RIS hearts identified 18 significantly altered canonical pathways. The five top canonical pathways altered with RIS exposure include metabolism signaling, mitochondrial dysfunction and oxidative phosphorylation with RIS exposure demonstrated by −log(p-value) by Fisher’s exact test and ratio of proteins in DE dataset to total proteins mapped to the canonical pathway. P-value < 0.05 = −log(p-value) > 1.3.
Over 500 enriched terms corresponding to disease and biological function in RIS-treated hearts were found using IPA software (p<0.05). Of the top 50 significantly enriched disease and biological function terms that were identified, many correspond to terms relating to mitochondria and energy metabolism, including mitochondrial complex I deficiency, assembly respiratory chain complex, quantity of NADH, accumulation of mitochondria and mitochondrial disorder (Table 4, Suppl File 1). Recently we reported that proteins involved in the immune response are significantly altered in hearts of male mice treated with RIS (May et al., 2019). In the present study, IPA analysis identified several categories of immune proteins involved in migration of inflammatory leukocytes, quantity of t-lymphocytes, which play a pathophysiological role in cardiovascular diseases and the development of heart failure (Bansal et al., 2017).
Table 4.
Selected results from the top 50 IPA function and disease categories of RIS hearts filtered for tissue specificity and metabolic functions.
| Categories | Diseases or Functions Annotation | p-value | Molecules |
|---|---|---|---|
| Cell Signaling, Post-Translational Modification, Protein Synthesis | Assembly of Respiratory chain complex I | 0.0001 | NDUFA10,NDUFA8,NDUFA9,NDUFB8,NDUFB9,NDUFS7 |
| Cellular Assembly and Organization | Accumulation of mitochondria | 0.0006 | MAP1S,TARDBP,TFRC |
| Cellular Movement, Immune Cell Trafficking | Migration of inflammatory leukocytes | 0.001 | PLAUR,PPIA |
| Molecular Transport, Nucleic Acid Metabolism, Small Molecule Biochemistry | Quantity of NADH | 0.003 | NQO1,SLC27A1 |
| Inflammatory Response | Inflammation of lesion | 0.006 | CD14, GJA1 |
| Cellular Development, Cellular Growth and Proliferation, Connective Tissue Development and Function, Hematological System Development and Function, Lymphoid Tissue Structure and Development, Tissue Development | Proliferation of peripheral T lymphocyte | 0.006 | TFRC, CSNK1A1 |
| Metabolic Disease | Mitochondrial disorder | 0.007 | DLD,HMGCS2,NDUFA10,NDUFA9,NDUFB9,NDUFS3,NDUFS7,SLC25A4 |
| Cardiovascular System Development and Function, Organ Morphology | Dilation of heart ventricle | 0.009 | CAMK2D,CAST,LMNA |
| Cell-mediated Immune Response, Lymphoid Tissue Structure and Development | Frequency of T lymphocytes | 0.009 | PTPN6, ZEB2, NOTCH2 |
| Cardiovascular Disease, Hereditary Disorder, Organismal Injury and Abnormalities, Skeletal and Muscular Disorders | Arrhythmogenic right ventricular dysplasia familial 9 | 0.01 | LMNA,MYH7 |
| Cellular Movement, Hematological System Development and Function, Immune Cell Trafficking, Inflammatory Response | Chemotaxis of peripheral blood monocytes | 0.01 | PLAUR,PPIA |
| Cellular Assembly and Organization | Sliding of myofilaments | 0.01 | MYH7,MYL6 |
Using IPA, over 500 disease and function categories were identified from DE proteins in RIS hearts. Top results relating to cardiovascular disease, metabolic disease and inflammation were identified in RIS hearts. Significantly down-regulated proteins are highlighted in blue
To further characterize our DE proteins, we used PANTHER Software and VLAD software to categorize our DE proteins by protein class, cellular component, molecular function and biological processes (Suppl File 2; Suppl File 3). We found 34 DE proteins, or ~30% of total DE genes, in RIS hearts involved in metabolic processes (Suppl Fig. 2; Suppl File 3). Overrepresentation tests for biological processes using PANTHER software were performed to identify 5 significantly overrepresented GO terms relating to biological processes in our significant DE protein dataset all relate to metabolic processes (Fisher test, FDR<0.05) (Table 5; Suppl File 2). Using VLAD to categorize our DE proteins by cellular component, we found that ~23% and 5% of our total DE protein dataset were localized to the mitochondrion and the mitochondrial respiratory chain complex I, respectively (p<0.0001, FDR q<0.0001) (Suppl File 3). Top significant GO terms categorized by biological process using VLAD software from our dataset are related to oxidative-reduction process (p<0.0001, FDR q<0.0001), mitochondrial electron transport NADH to ubiquinone (p<0.0001, FDR q<0.0001), and cellular respiration (p<0.001, FDR q<0.0001) (Suppl file 4). A heat map of all proteins of the mitochondrion that were significantly altered in RIS hearts compared to VEH hearts is shown in Figure 1.
Table 5.
Significantly overrepresented biological processes in RIS hearts.
| Overrepresentation test PANTHER GO-Slim Biological Process | # of proteins | Fold Enrichment | p-value | FDR |
|---|---|---|---|---|
| small molecule metabolic process (GO:0044281) | 6 | 8.61 | 8.91E-05 | 3.97E-02 |
| carboxylic acid metabolic process (GO:0019752) | 8 | 6.61 | 3.63E-05 | 3.23E-02 |
| oxoacid metabolic process (GO:0043436) | 8 | 6.35 | 4.78E-05 | 2.84E-02 |
| organic acid metabolic process (GO:0006082) | 8 | 5.45 | 1.35E-04 | 4.81E-02 |
| cellular metabolic process (GO:0044237) | 24 | 3.18 | 4.25E-07 | 7.58E-04 |
Using PANTHER analysis, all 5 of the significantly overrepresented biological processes identified were related to metabolic process identified by gene ontology (GO) terms. Fisher test, False Discovery Rate, FDR <0.05
Figure 1. Heat map of differential changes in mitochondrial protein expression in RIS and VEH hearts.
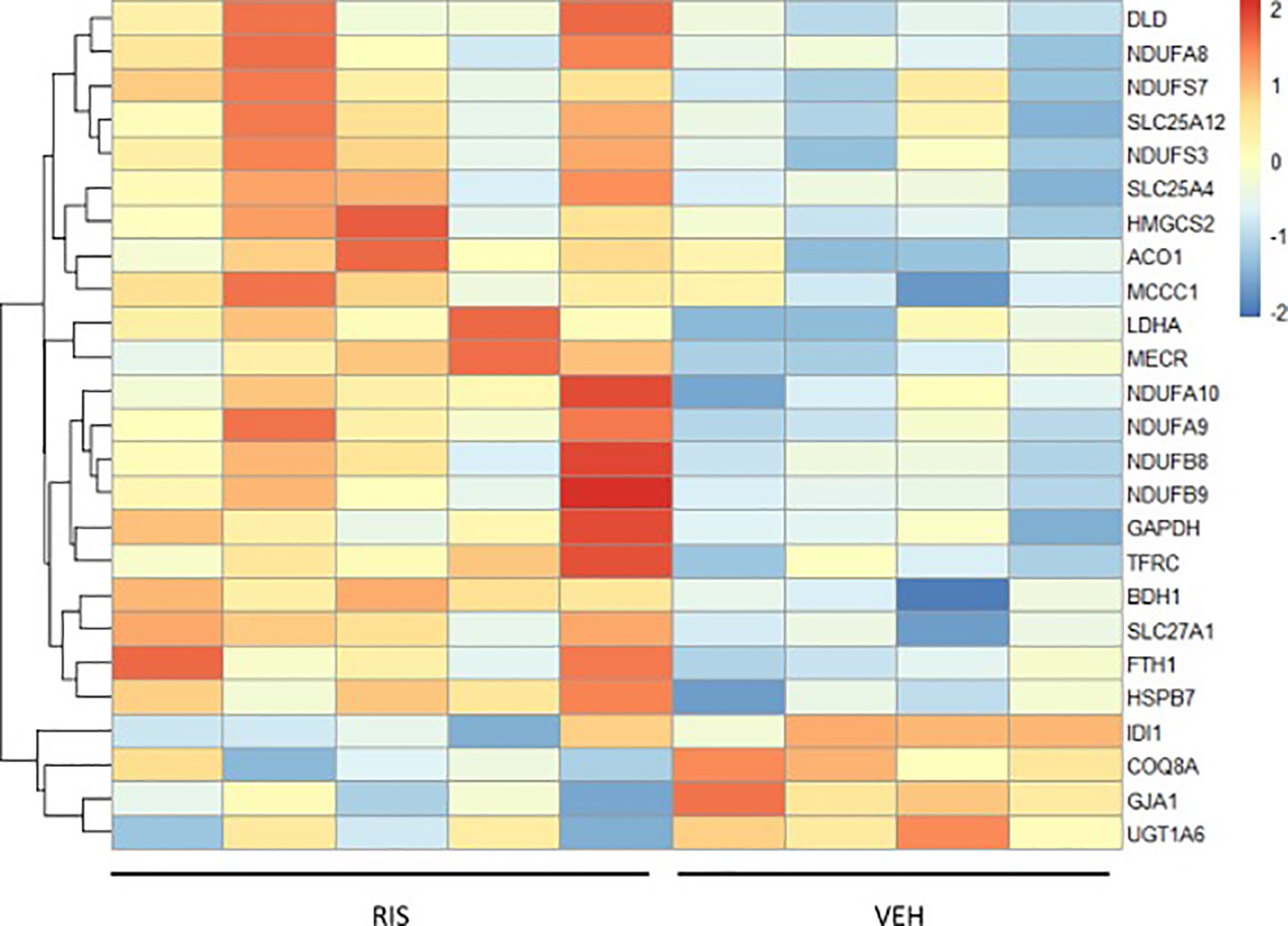
Each mitochondrial protein level is normalized to the mean of the protein across all male mouse heart samples. Red shows samples above mean protein expression and blue is below mean protein expression. (VEH/RIS n=4/n=5)
To validate a subset of proteins from our proteomic results we performed western blot analysis for mitochondrial complex I proteins NDUFS3 (iron-sulfur containing component), NDUFB9 (accessory subunit of NADH dehydrogenase) and calcium/calmodulin-dependent protein kinase type II subunit delta (CAMK2D) in both male and female treated mice (Figure 2A). Quantification of western blots combined from hearts of both male and female mice showed a significant increase of ~446% of CAMK2D (p<0.05) (Figure 2B), ~271% of NDUFS3 (p<0.05) (Figure 2C), and ~378% of NDUFB9 (p<0.05) (Figure 2D) in RIS-treated groups compared to VEH.
Figure 2: NDUFS3, NDUFB9 and CAMK2D protein expression increased in hearts of male and female mice treated with RIS.
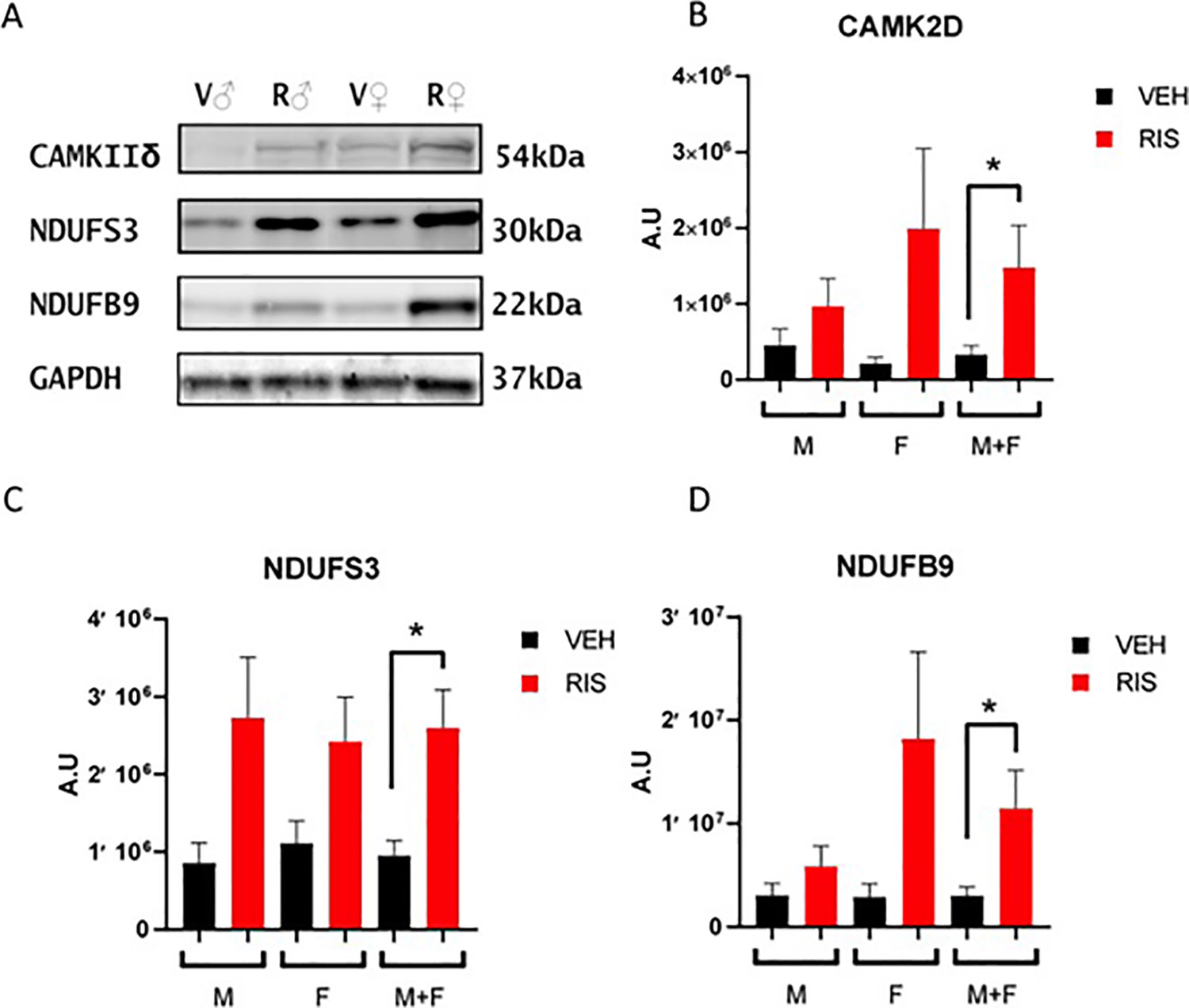
A) Western blot of RIS (R) and VEH (V) treated male (♂) and female (♀) mice for CAMK2D, NDUFS3 and NDUFB9. GAPDH was used as a loading control. Quantification of western blots for B) CAMK2D C) NDUFS3 and D) NDUFB9 for male (M) and female (F) and combined male and female (M+F) mice treated with RIS or VEH. Data is shown as mean +/− SEM. A.U.= arbitrary units (male RIS/VEH n=9/n=12; female RIS/VEH n=6/n=6.) (* p<0.05, student’s t-test)
3.3. Gene expression is altered in hearts of male and female mice treated with risperidone
To determine whether the action of RIS on protein modulation occurs at the transcriptional level, we performed qPCR targeting a subset of 15 genes either involved in the top canonical pathways identified from our gene enrichment results of our proteomic data, including xenobiotic metabolism signaling, oxidative phosphorylation and mitochondrial dysfunction, or showed the largest increase in protein levels in RIS hearts (Suppl Table 4). Results show significant fold change (FC) increase in the expression of genes identified from our proteomic results in the hearts of both male and female RIS mice compared to VEH hearts (p<0.05). Of 15 genes paneled, 5 of our target genes are significantly upregulated at the transcription level in RIS male hearts, 3-hydroxy-3-methylglutaryl CoA synthase 2 (Hmgcs2) (13.56 FC, p<0.001), calcium/calmodulin dependent protein kinase II delta (Camk2d) (2.64 FC, p=0.023), microsomal glutathione S-transferase 3 (Mgst3) (4.72 FC, p=0.04), and the subunit of respiratory complex I, NADH:ubiquinone oxidoreductase subunit B9 (Ndufb9) (1.88 FC, p<0.001). In female RIS hearts, 4 genes had significant fold change increase compared to control hearts with p-values less than 0.05; Camk2d (22.91 FC, p<0.001), and the NADH:ubiquinone oxidoreductase subunits of respiratory complex I Ndufs (p=0.013), Ndufs7 (p=0.041) and Ndufa9 (p=0.032) with fold changes of 4.83, 3.79 and 6.7, respectively. Camk2d expression was significantly increased in both RIS male and female mice hearts (2.64 and 22.91 FC vs VEH, respectively).
3.4. The AA drug, olanzapine, alters the cardiac proteome.
To determine whether drug-associated effects on the heart are specific to RIS, or are shared by other AA medications, we evaluated the effects of OLAN, an AA drug known to cause significant weight gain, dyslipidemia and hyperglycemia, clinically. We performed proteomic analysis on hearts from male mice treated with clinically relevant doses of OLAN (5 mg/kg) daily for 4 weeks compared to vehicle controls. Our dosing paradigm was based on pharmacokinetic studies which examined peak plasma concentrations of OLAN at 1 and 3 hours following dosing (data not shown). As with our RIS-treated animals, though increased adiposity was noted, we did not see a significant increase in weight in our OLAN group versus our control group at this dose (Suppl Fig. 3). As shown in Figure 3A, 19 of the 69 proteins significantly upregulated in OLAN hearts compared to VEH hearts (p<0.05) were also upregulated in our RIS hearts. Mitochondrial respiratory complex I proteins, NDUFS3, NDUFB9, NDUFA9, NDUFA8 and NDUFA10 were significantly increased in both RIS and OLAN hearts (Figure 3B, Suppl File 5). Similarly, 5 of the 27 proteins downregulated in OLAN hearts were also downregulated in RIS (Figure 3A). As validation for our proteomic results, we next performed western blotting to evaluate protein expression levels of NDUFB9 and NDUFS3 in our OLAN and VEH treated hearts. Compared to VEH, quantification of our western blots show significant increase in protein levels for both NDUFB9 (p=0.029) and NDUFS3 (p<0.001) in the hearts of mice treated with OLAN (Figure 3C). Using IPA, we identified significant canonical pathways relating to mitochondrial function, such as oxidative phosphorylation and mitochondrial dysfunction (Table 6; Suppl File 4) and disease and function terms such as mitochondrial complex I deficiency, mitochondrial disorder and accumulation of mitochondria that were also enriched in our OLAN hearts (Table 7; Suppl File 4).
Figure 3. Hearts of male mice treated with OLAN share altered expression of mitochondrial proteins with RIS hearts.
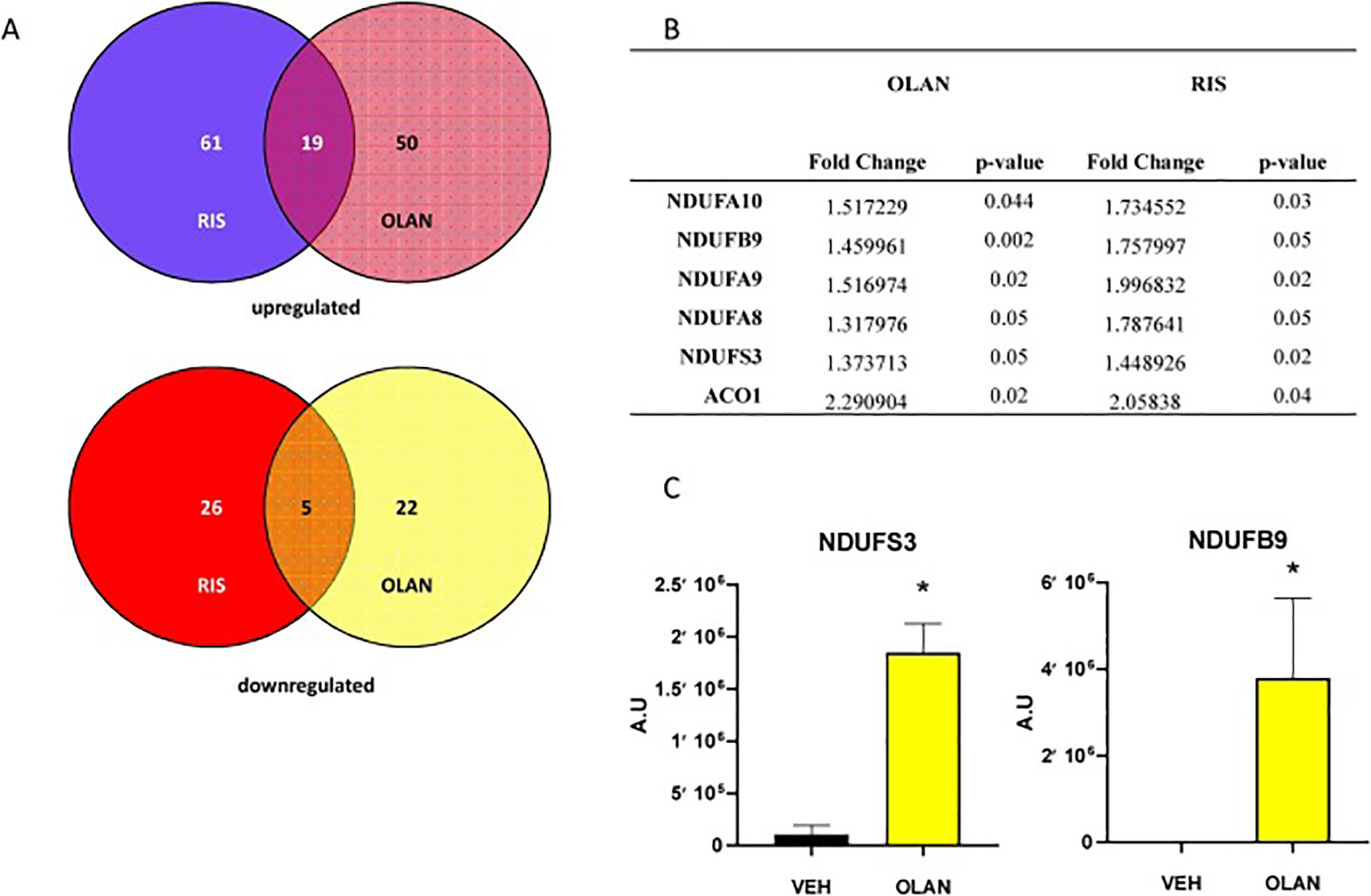
A) Venn diagrams showing shared proteins that are significantly upregulated (top) or downregulated (bottom) in RIS and OLAN treated male mice hearts (p<0.05). 24 proteins shared between RIS and OLAN treated hearts compared to control hearts. B) List of 6 protein relating to mitochondrial function that are significantly altered in both OLAN and RIS hearts, (fold change and p-value). C) Quantification of western blots of VEH (black) and OLAN (yellow) treated mice for NDUFS3 and NDUFB9. Data is shown as mean +/− SEM. A.U.= arbitrary units (VEH/OLAN n=4/n=4) (NDUFB9 *p=0.029; NDUFS3 p<0.001, ANOVA, Tukey test and Holm-Sidak post hoc).
Table 6.
Selected IPA canonical pathways significantly altered in hearts of OLAN treated male mice overlapping with RIS.
| Ingenuity Canonical Pathways | −log(p-value) | Ratio | Molecules |
|---|---|---|---|
| Sirtuin Signaling Pathway | 3.54 | 0.091 | TOMM40,NDUFB9,NDUFA9,SLC25A4,TIMM9,CYC1,SMARCA5,NDUFA10,PCK1,NDUFS3,NDUFA2,NDUFA8 |
| Oxidative Phosphorylation | 2.43 | 0.01 | NDUFB9,NDUFA9,CYC1,NDUFA10,NDUFS3,NDUFA2,NDUFA8 |
| Mitochondrial Dysfunction | 2.03 | 0.08 | NDUFB9,NDUFA9,CYC1,NDUFA10,ACO1,NDUFS3,NDUFA2,NDUFA8 |
IPA analysis of significant protein expression in OLAN hearts identified significantly altered canonical pathways. The top canonical pathways altered with RIS exposure include metabolism signaling, mitochondrial dysfunction and oxidative phosphorylation with RIS exposure demonstrated by −log(p-value) by Fisher’s exact test and ratio of proteins in DE dataset to total proteins mapped to the canonical pathway. P-value < 0.05 = −log(p-value) > 1.3.
Table 7.
Selected 5 of top 25 IPA disease and function categories of OLAN treated hearts, filtered for mitochondrial function
| Categories | Diseases or Functions Annotation | p-value | Molecules |
|---|---|---|---|
| Cell Signaling, Post-Translational Modification, Protein Synthesis | Assembly of Respiratory chain complex I | 0.0008 | NDUFA10,NDUFA2,NDUFA8,NDUFA9,NDUFB9 |
| Developmental Disorder, Hereditary Disorder, Metabolic Disease, Organismal Injury and Abnormalities | Mitochondrial complex I deficiency | 0.001 | NDUFA10,NDUFA2,NDUFA9,NDUFB9,NDUFS3 |
| Developmental Disorder, Hereditary Disorder, Metabolic Disease, Organismal Injury and Abnormalities | Mitochondrial respiratory chain deficiency | 0.0021 | CYC1,NDUFA10,NDUFA2,NDUFA9,NDUFB9,NDUFS3 |
| Metabolic Disease | Mitochondrial disorder | 0.0033 | CYC1,NDUFA10,NDUFA2,NDUFA9,NDUFB9,NDUFS3,OPA3,SLC25A4 |
| Cellular Assembly and Organization | Accumulation of mitochondria | 0.011 | MAP1S,TFRC |
Using IPA, over 500 disease and function categories were identified from DE proteins in OLAN hearts. Top terms relating to mitochondrial disorder were identified in OLAN hearts.
3.5. Mitochondrial function is altered in the hearts of risperidone-treated mice.
Metabolic state is directly associated with oxygen consumption and measuring cellular oxygen uptake rates serves as an important tool in assessing overall mitochondrial function. As we observed upregulation of mitochondrial proteins with RIS and OLAN therapy, we next assessed changes in cardiac mitochondrial function in a subset of mice treated with RIS. Oxygen consumption studies measuring respiratory states were performed on isolated cardiac mitochondria from female mice treated 4 weeks with RIS or VEH (Figure 4). Several parameters of respiration, including basal respiration, ATP-linked respiration, proton leak, maximal respiration capacity can be assessed by measuring oxygen consumption rates (OCR) in the presence of complex-specific inhibitors. OCR was measured in RIS-treated or VEH-treated isolated cardiac mitochondria. Basal respiration was measured in response to succinate in the presence of rotenone, a complex I inhibitor, and state 3 respiration was induced with ADP injection followed by state 4 oligomycin and then uncoupling with FCCP, and an inhibitor of complex III, antimycin A. Respiration and oxidative phosphorylation are well coupled in mitochondria of both RIS and VEH treated mice, with Respiratory Control Ratios (RCR) of ~6 (RCR >3). Oxygen consumption rates are increased by ~50% in RIS hearts compared to VEH hearts during inhibition of each of the complexes in the electron transport chain. These data demonstrate functional effects of chronic AA drug treatment on cardiac mitochondrial function and bioenergetics.
Figure 4. Cardiac mitochondrial function is altered with RIS treatment.
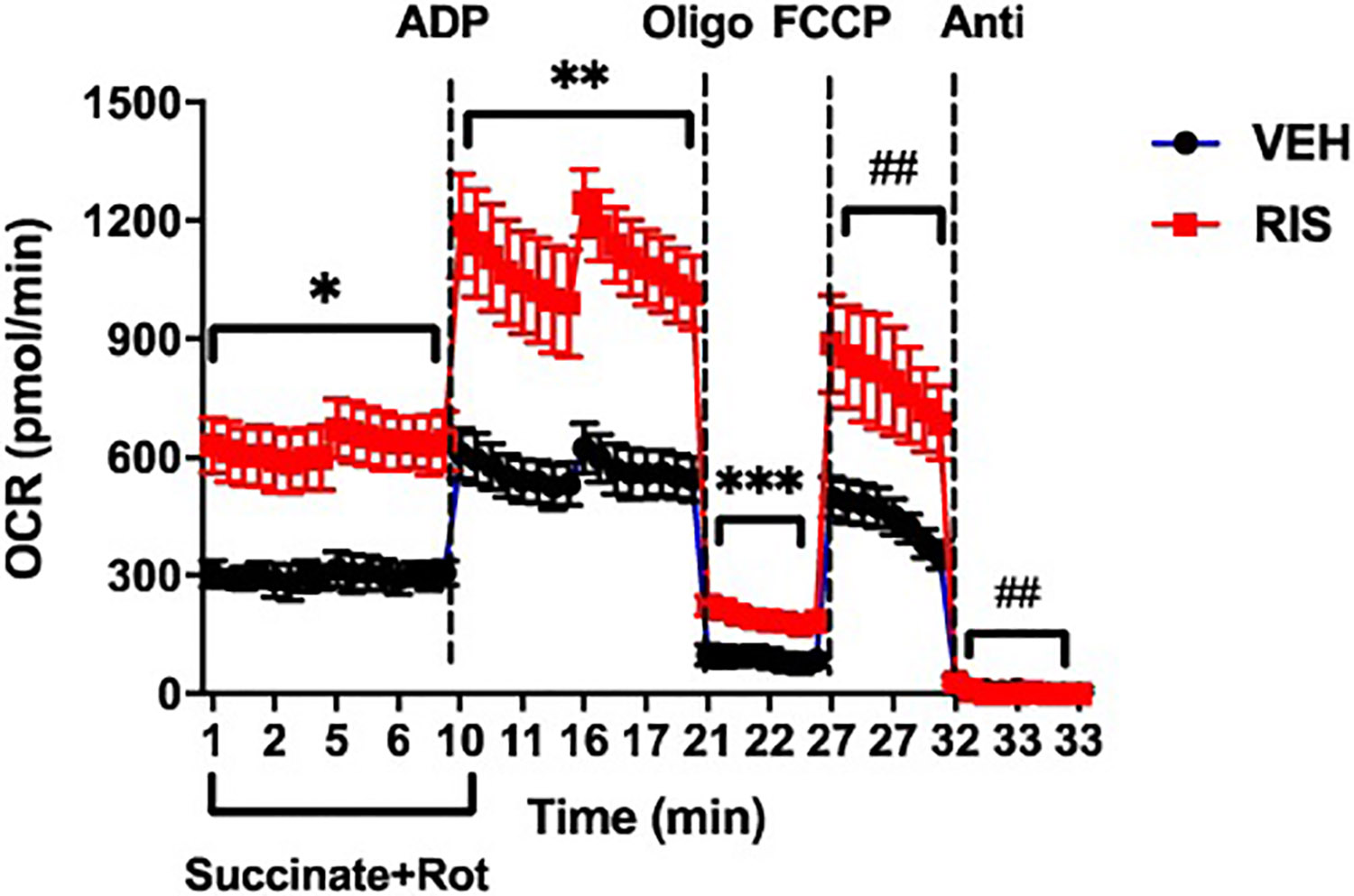
Oxygen consumption rates (OCR) were measured from isolated cardiac mitochondria of VEH (black) or RIS (red) treated female mice. Basal respiration was measured in response to substrate Succinate (10 mM) in the presence of Rotenone (2 μM), an inhibitor of complex I. This was followed by addition of 4.5 mM ADP for state 3, or maximal respiration. Injection of oligomycin (2 μM), an inhibitor of ATPase (complex V) measures state 4 to derive ATP-linked respiration and proton leak. Uncoupled respiration from ATP was obtained by adding FCCP (4 μM). Antimycin A (Anti, 2 μM) was used to block complex III of the electron transport chain. Respiration and oxidative phosphorylation are well coupled in RIS and VEH hearts, with a respiratory control ratio (RCR) of ~6. The data shown is representative of three independent experiments using 2 mice each for RIS treatment group and VEH treatment group. Respiratory states are significantly increased by ~50% in mitochondria of RIS hearts compared to VEH. Data is shown as mean +/− SEM (* p=9.3 × 10−20, ** p= 2.3 × 10−17, *** p= 3.4 × 10−08, # p= 2.9 × 10−10, ## p=0.05, student’s t-test)
3.6. Four week exposure to risperidone does not induce fibrosis or pathomorphological changes in the heart.
The dosing paradigm used in this study was sub-chronic in nature (4 weeks) and we wanted to determine if there were any signs of drug associated pathology in hearts of animals treated with AA medications. We evaluated cardiac histology by Hemotoxylin & Eosin staining and Masson trichrome staining (for collagen deposition) to determine if there were morphological or phenotypic changes in the male mouse heart following 4 weeks of RIS treatment. We did not observe qualitative treatment differences in collagen deposition (Figure 5A), nor did we observe overt pathological morphology in cardiac tissue (Figure 5B). We observed similar findings of indistinguishable histological differences in H&E and Masson’s Trichrome staining between hearts from mice following 4 weeks of treatment with OLAN and control hearts (data not shown). These findings indicate that RIS and OLAN do not induce any gross physical changes or fibrotic scarring in the heart following 4 weeks of drug exposure in mice.
Figure 5. RIS does not alter cardiac morphology or induce scarring after 4 weeks of treatment in male mice.
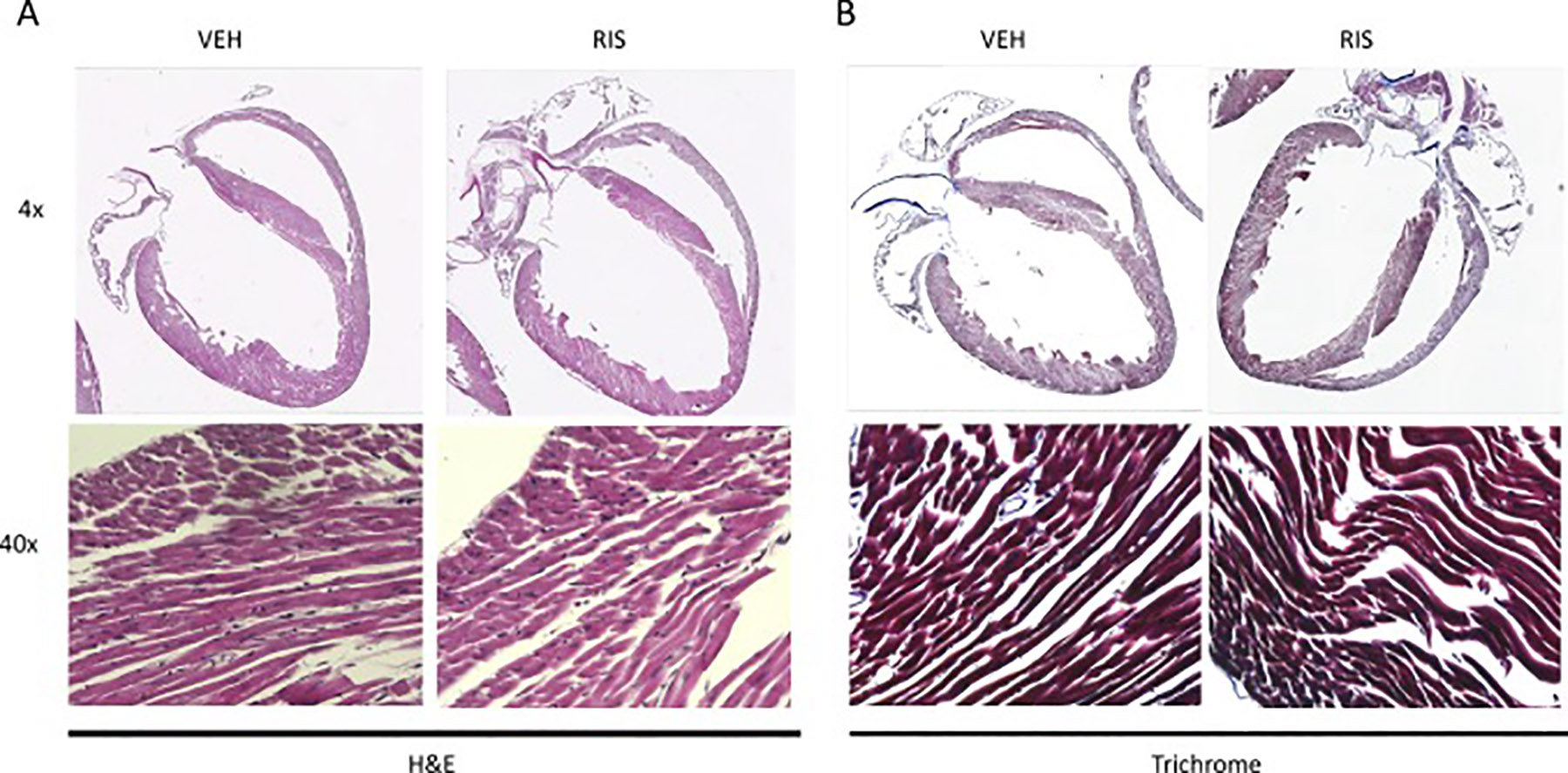
Histological sections of RIS and VEH treated male hearts. Representative images of RIS and VEH heart sections stained with A) H&E and B) Masson Trichrome. (RIS/VEH n=4/n=4)
3.7. Risperidone temporally alters whole body energy expenditure in male and female mice, consistent with plasma drug exposure
As we observed increased adiposity and altered cardiac gene expression and bioenergetics in our RIS-treated mice, we next wanted to determine whether RIS exposure alters whole body energy expenditure. In our male mice treated with RIS, we observed a significant increase in daytime (p<0.001) and nighttime energy expenditure (p=0.041) (Table 2). We observed a significant increase in resting energy expenditure at night (p=0.003) and active energy expenditure in both the day (p=0.001) and night (p=0.024) with no changes in respiratory quotient (Table 2). In our RIS-treated female mice, there was a trend toward higher energy expenditure at night (p=0.057), despite a significant decrease in daytime and total resting energy expenditure (Suppl Table 3). Furthermore, nighttime respiratory quotient was lower in RIS-treated mice (Suppl Table 3), indicating a shift toward lipid metabolism. Consistent with previous studies reporting fuel switching and decreased free fatty acid concentrations in plasma with AA treatment (Albaugh et al., 2012; Kaddurah-Daouk et al., 2007; Klingerman et al., 2014), we show ~20% decrease in plasma free fatty acid concentrations in male mice following 4 week exposure of RIS (p=0.03) (Suppl Figure 4).
To address potential drug-associated mechanism of this effect, we examined the timing of drug associated effects on energy expenditure and respiratory quotient to determine the role of pharmacokinetics on altered energy expenditure (Figure 6). Indeed, immediately after dosing with RIS, energy expenditure was significantly elevated (Figure 6A, B) with no change in respiratory quotient (Figure 6C, D) in our RIS-treated male mice compared to VEH. These changes normalized after approximately 2 hours, consistent with changes in plasma drug exposure (Aguirre et al., 2018; May et al., 2019). Similarly, as previously reported, we observed a significant increase in energy expenditure (Figure 7 A, B) with significant suppression of respiratory quotient (Figure 7C, D) 2 hours post-dose in female mice treated with RIS (Motyl et al., 2015). This increased temporal energy expenditure observed are consistent with peak plasma concentrations of RIS and the active metabolite 9-OH RIS as we have previously reported in this model (Motyl et al., 2015).
Figure 6. Risperidone increased energy expenditure in male mice immediately after dosing.
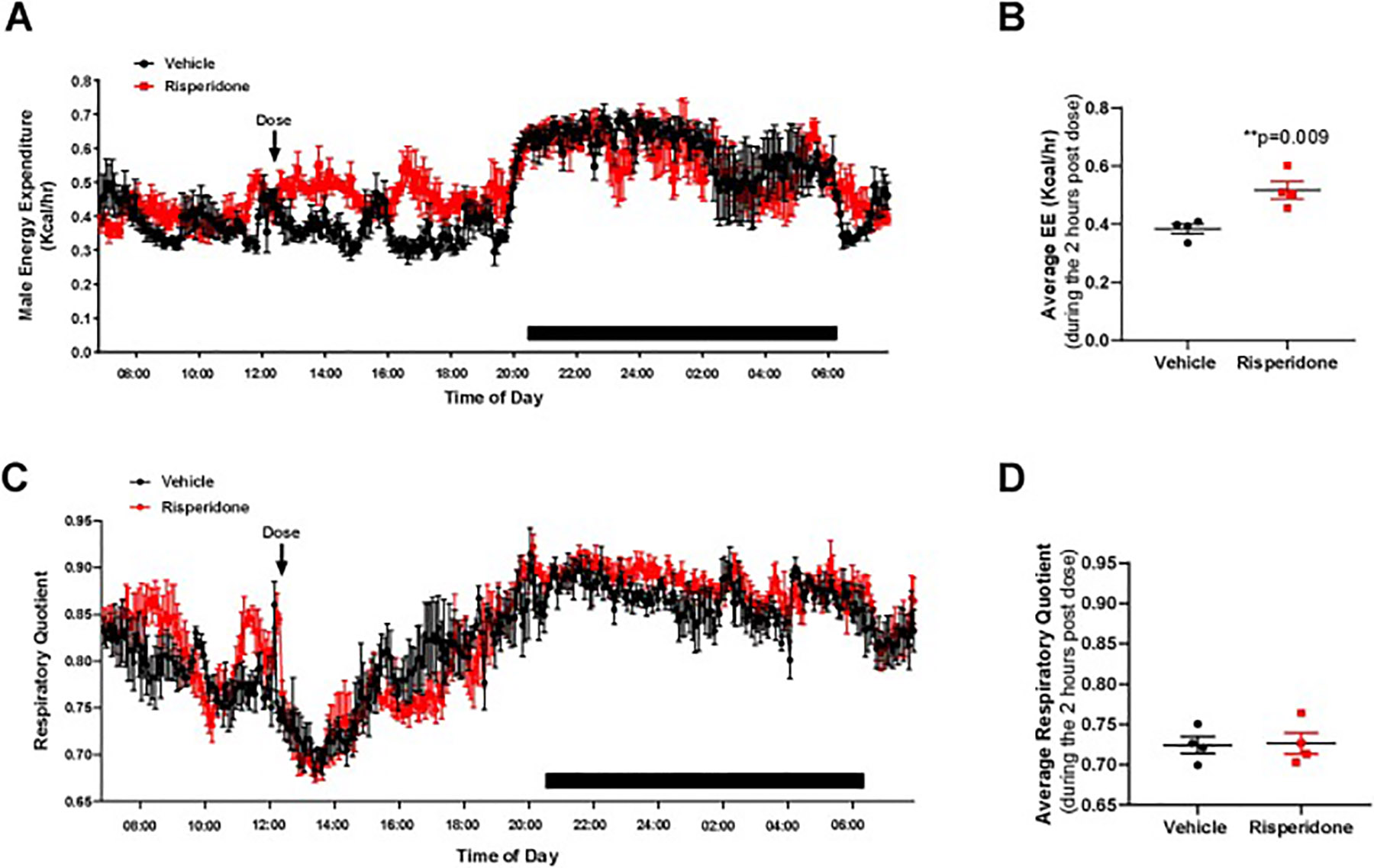
Male C57BL/6J mice were treated daily from 8 to 12 week of age with vehicle (black) or 0.75 mg/kg risperidone (red) p.o. at 12:15 PM. During the third week of treatment, mice were placed in metabolic cages measuring energy expenditure (A) and respiratory quotient (C) at 5 minute intervals. Points represent mean ± SEM for N=4 mice measured continuously over 4 days. Data shown is from one representative day. Because of changes immediately after dosing, we also calculated the average energy expenditure (B) and respiratory quotient (D) during the 2 hours after dosing. Points represent average RQ and EE for each of 4 mice per group during the time interval 2 hours after doing. **p<0.01 by Student’s t-test.
Figure 7. Risperidone increased energy expenditure and reduces respiratory quotient immediately after dosing in female mice.
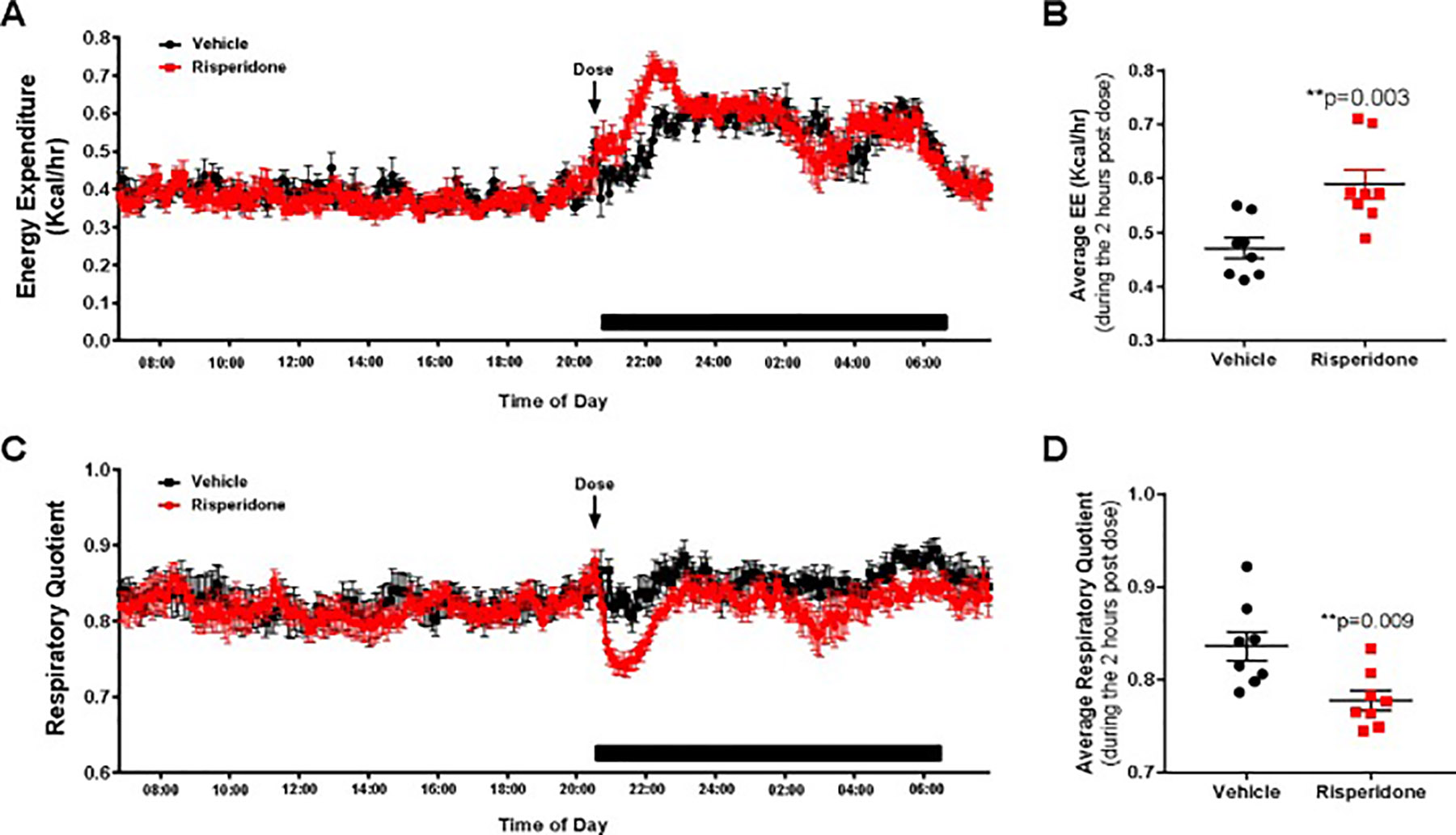
Female C57BL/6J mice were treated daily from 8 to 12 weeks of age with VEH (black) or 0.75 mg/kg RIS (red) PO at the beginning of the dark cycle. During the third week of treatment, mice were placed in metabolic cages measuring energy expenditure (A) and respiratory quotient (C) at 5 minute intervals. Points represent mean ± SEM for n=8 mice measured continuously over 4 days. Data shown is from one representative day. Because of changes immediately after dosing, we also calculated the average energy expenditure (B) and respiratory quotient (D) during the 2 hours after dosing. Points represent average RQ and EE for each of 8 mice per group during the time interval 2 hours after dosing. **p<0.01 by Student’s t-test.
Finally, in order to determine if changes in energy expenditure were associated with changes in the level of physical activity, we measured time and speed of wheel running and walking in the cage (Suppl. Table 5). We observed no significant changes in walking or wheel activity in our RIS-treated male groups compared to VEH. Additionally, RIS-treated male mice spent significantly less time sleeping and standing still in the daytime than VEH-treated mice. At nighttime, female mice treated with RIS ran a longer distance on wheels, and spend a greater amount of time on the wheels then vehicle treated mice, but the wheel speed did not differ between groups (Suppl. Table 6). During the two-hour time period of time that energy expenditure was high, however, there was no difference in wheel running or walking in the cage (not shown), indicating that activity could not explain the increased energy expenditure.
3.8. Risperidone increases c-FOS expression in the hypothalamic paraventricular nucleus
Altered autonomic nervous system (ANS) activity is a hallmark of schizophrenia and it is believed that AA therapy worsens ANS-associated metabolic syndrome (Hattori et al., 2018; Iwamoto et 2012; Nielsen et al., 1988; Scigliano and Ronchetti, 2013). We hypothesized that AA-associated increased sympathetic activity could alter cardiac gene expression, similar to AA-associated effects on bone (Calarge et al., 2013; Houseknecht et al., 2017; Motyl et al., 2015). Our observations of altered whole body energetics and mitochondrial function in the heart prompted us to evaluate RIS exposure on the activity of the paraventricular nucleus (PVN), a region of the brain involved in the regulation of sympathetic activity and energy balance. Increased sympathetic (PVN) activation is also involved in the pathogenesis of congestive heart failure and hypertension (Leimbach et al., 1986; Patel et al., 1993). We quantified the expression of the proto-oncogene c-FOS, a marker of neuronal activity, using immunocytochemistry in histological sections of the parvocellular region of the PVN, following both acute and chronic exposure to RIS or VEH. Images in Figure 8A, show increased c-FOS expression in the PVN of female mice 2 hours following a single dose of RIS (top right) and at 24 hours fallowing the last dose of male mice treated daily for 4 weeks (bottom right) compared to their respective VEH groups (top left, bottom left). In quantification of c-FOS positive nuclei images, we identified a ~500% increase in c-FOS expression in the PVN of RIS-treated mice compared to control (p=0.0014) following 2 hours of exposure (Figure 8B) and a significant increase of ~325% in c-FOS expression in our chronically-treated mice (p=0.019) (Figure 8C). These findings indicate increased PVN in response to RIS, consistent with activation of pathways involved in regulation of energy metabolism, including activation of sympathetic activity. Finally, these effects coincide with both acute plasma drug exposure and the temporal increase in whole body energy expenditure.
Figure 8. Acute and chronic RIS exposure alters activity in regions of the brain regulating sympathetic activity.
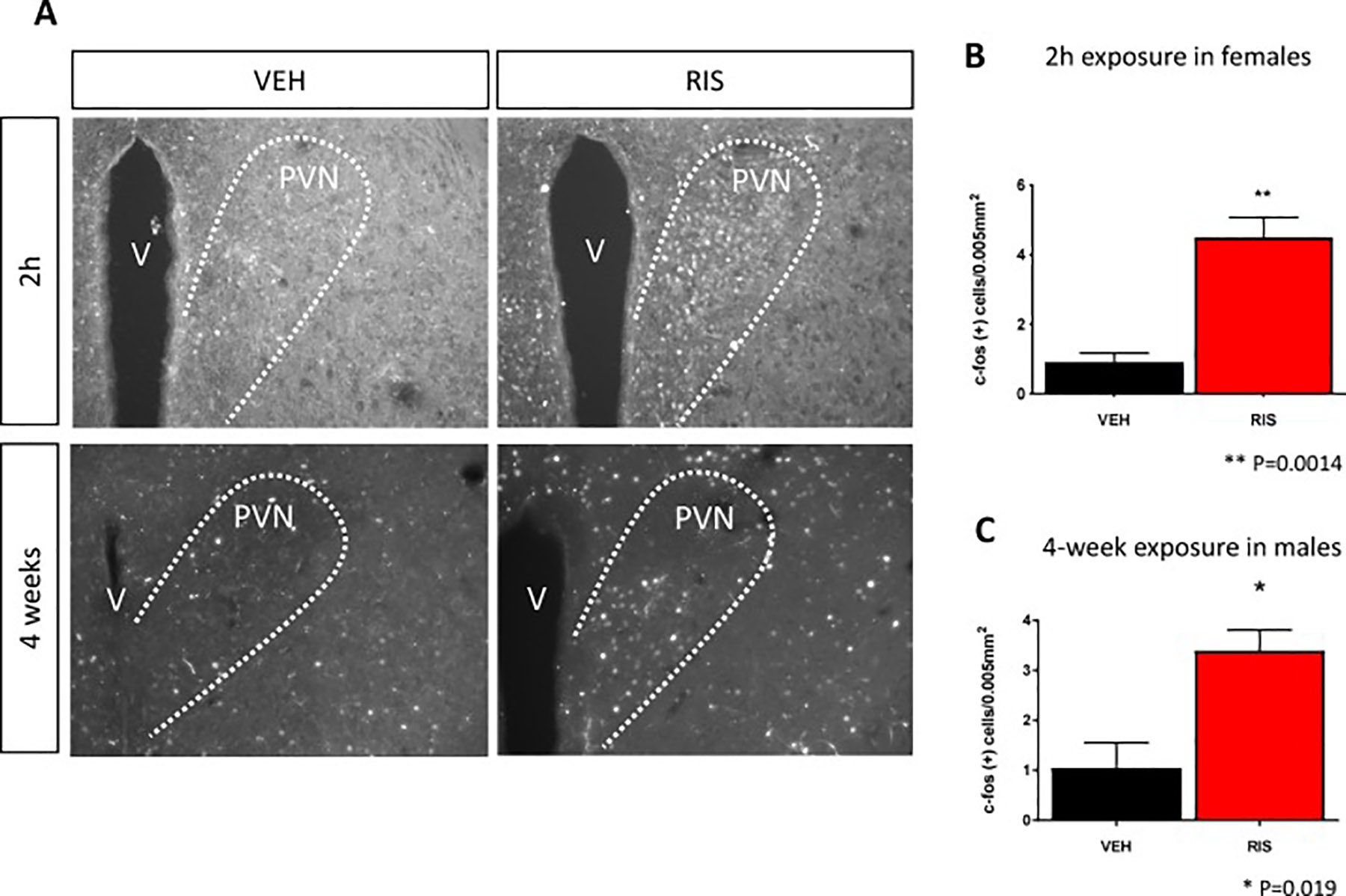
A) Images of c-FOS staining, a marker of neuronal activity, in regions of the brain involved in sympathetic output; the paraventricular nucleus (PVN) in our acute (top panel) and chronic models (bottom panel) of RIS exposure; female mice 2 hours following either a single dose of RIS (right) or VEH (left) and male mice following 4 weeks of daily exposure to RIS (right) or VEH (left), respectively. Quantification of c-FOS expression in PVN shows significant increase in c-FOS expression in B) acute and C) chronic models of RIS exposure (red) compared to VEH (black). Data is shown as mean +/− SEM. (v=ventricle) (** p=0.0014, * p=0.019 student’s t-test Acute RIS/VEH n=3/n=4; Chronic RIS/VEH n=3/n=4).
4. Discussion and conclusions
Atypical antipsychotic medications, including RIS and OLAN, are associated with significant endocrine and metabolic side effects which reduce the quality of life and increase cardiovascular disease risk and mortality (Gopal et al., 2013; Salvo et al., 2016). These risks include hyperglycemia, dyslipidemia, hypertension, hyperphagia, and obesity. Antipsychotic medications are a prominent cause of obesity in children and youth with mental health disorders, and obesity is a known risk for development of cardiovascular disease. It should be noted that many of these adverse effects (e.g. arrhythmia, hyperglycemia, dyslipidemia, fuel switching in rodents) are rapid in onset, occurring within hours or days upon initiation of dosing and prior to increased weight gain and adiposity, reflecting complex and diverse underlying pharmacology (Alfaro et al., 2002; Assié et al., 2008; Chintoh et al., 2008, 2009; Houseknecht et al., 2007; Motyl et al., 2012, 2015; Skrede et al., 2012). Importantly, despite the well documented AA-associated increase in cardiometabolic risk, the direct effects of AA drugs on the heart have not been fully elucidated. In this study, we present novel evidence that sub-chronic treatment with antipsychotic medications (risperidone or olanzapine) results in significant changes in gene expression and protein signatures in the heart, including pathways that regulate mitochondrial function, energy metabolism and immune function.
The heart is the most metabolically active organ in the body, requiring high energy demand for normal functionality. Therefore, heart function is closely dependent upon mitochondrial function. Impaired energy metabolism, caused by perturbations in mitochondrial activity, is a key factor in various pathological cardiac conditions including progression of heart failure (Sharov et al., 2000; Zhou and Tian, 2018). Antipsychotic drugs are associated with cardiac arrhythmia and sudden cardiac death in older adults and antipsychotic associated arrhythmias have been associated with altered QT interval (Drici et al., 1998; Haddad and Anderson, 2002; Harrigan et al., 2004; Lee et al., 2013; Stöllberger et al., 2005; Straus et al., 2004; Vieweg, 2003; Witchel et al., 2003; Yerrabolu et al., 2000). The underlying pharmacology for these effects remain elusive, however altered autonomic function appears to play a role. A body of literature report that antipsychotic drugs elicit both direct and indirect effects on the cardiovascular system by antagonism of adrenergic and cholinergic receptors (direct) and via central mediation of autonomic function (Khasawneh and Shankar, 2014; Leung et al., 2012). Cardiometabolic risk, including AA-associated effects on cardiac bioenergetics and mitochondrial function, however, are less understood. Given the rapid and potent effects of AA medications on multiple pharmacological and physiological pathways, and the rapid onset of drug-associated insulin resistance and dyslipidemia, we hypothesized that AA medications may act acutely/semi-acutely on the heart to accelerate the development of cardiovascular disease. Previous experimental and clinical studies have shown inhibitory effects of AA drugs, including RIS, on complex I activity and mitochondrial bioenergetics (Balijepalli et al., 2001; Casademont et al., 2007; Elmorsy et al., 2017; Garabadu et al., 2015; Modica-Napolitano et al., 2003). None of these studies, however, examine the effect of AA therapy on mitochondrial activity specifically in the heart. In our study, we examined the effect of sub-chronic RIS treatment on cardiac bioenergetics. We observed increased oxygen consumption in cardiac mitochondria of RIS-treated mice, indicating RIS-induced alterations in the energy metabolism of the heart. Increased cardiac oxygen consumption has been associated with decreased cardiac efficiency in diabetic mice (How et al., 2006). Therefore, our observations of increased oxygen consumption in our RIS-treated hearts may be indicative of or contribute to aberrant cardiac function. To our knowledge, this is the first study to show that AA medications significantly alter cardiac mitochondrial function prior to presentation of an overt cardiac pathophysiological phenotype. These effects, combined with other AA-associated metabolic factors observed clinically (e.g. insulin resistance, hyperglycemia and dyslipidemia) only further potentiate the risk for cardiovascular disease.
Using an unbiased proteomic approach, we identified a subset of proteins involved in mitochondrial function and oxidative phosphorylation altered in RIS hearts. In contrast to previous studies in other tissues, we identified an increase in mitochondrial proteins that comprise complex I of the electron transport chain with RIS treatment, such as NDUFS3 and NDUFB9. We confirmed these findings with proteomic analysis and validation studies using a second AA medication, OLAN, showing that mitochondrial complex I proteins are also increased in the heart following 4 weeks of drug treatment. Recent clinical studies have shown that acute treatment with antipsychotics improves cardiorespiratory coupling in schizophrenic patients under psychotic states (Aguirre et al., 2018). Though the increase we observe in cardiac bioenergetics with RIS exposure may be explained by increased complex I protein expression and assembly, future studies are needed to further investigate functional changes to complex I activity with RIS exposure, such as NADH oxidation and reactive oxygen species (ROS) production. Although outside the scope of the current study, we cannot exclude the possible contributions of altered mitochondrial dynamics, such as mitochondrial biogenesis, fission and fusion processes and mitophagy on the observed alterations in cardiomyocyte energy metabolism with RIS treatment. Assessing mitochondrial dynamics, mitochondrial density and morphology may provide further insight on RIS action on cardiac mitochondria.
To our knowledge, our study elucidates for the first time significant changes in the cardiometabolic protein expression profile and cardiac mitochondrial function in mice treated with AA medications, in the absence of gross morphological changes in heart tissue. Therefore, our findings indicate an early metabolic consequence of prolonged RIS exposure prior to the onset of pathological cardiac phenotype. Though not examined in this study, future studies investigating the effects of prolonged RIS treatment on cardiac output and efficiency will provide further evidence of the early consequences of RIS on heart function. We have also shown significant increase in CAMK2D at the transcriptional and protein level in our RIS-treated hearts. CAMK2D plays a regulatory role in initiating the inflammatory response and increased CAMK2D expression has been shown to be involved in the pathogenesis of cardiac fibrosis, dilated cardiomyopathy and heart failure (Sag et al., 2009; Toko et al., 2010; Willeford et al., 2018; Zhang et al., 2003). Moreover, we have identified a number of significantly altered inflammatory proteins in RIS hearts (May et al., 2019) that are reported to play a key role in the onset of cardiovascular diseases and heart failure (Ramos et al., 2017). Given the detrimental effects of mitochondrial dysfunction on cardiac structure and function and involvement of chronic inflammation in heart disease, our findings of RIS-induced immune response in the heart may provide an early indication of the progression towards pathogenesis. Prospective studies examining the presence of cytokines and inflammatory cells in RIS hearts may provide further evidence of the contribution of chronic inflammation induced by RIS in the progression of cardiac disease.
In our previous work, we have shown that RIS increases sympathetic output to bone (Motyl et al., 2012) contributing to AA-associated bone loss as well as increased markers of thermogenesis in brown adipose tissue (BAT). Sympathetic overreactivity, regulated by the paraventricular nucleus (PVN) of the hypothalamus, has been linked with insulin resistance, hypercholesterolemia, hypertriglyceridemia, and central adiposity (Thorp and Schlaich, 2015). Furthermore, prolonged activation of cardiac SNS through PVN activation is known to cause deleterious effects on cardiac structure and function and potentiate the development of cardiac arrhythmias, heart failure and increased cardiometabolic risk, well-known adverse side-effects of AA therapy (Zhang and Anderson, 2014). Scigliano and Ronchetti (Scigliano and Ronchetti, 2013) proposed that AA-induced metabolic and cardiovascular effects are due to disease (schizophrenia) and AA-associated autonomic dysfunction. As cardiac energy metabolism and altered mitochondrial function are also observed in patients with diabetic cardiomyopathy (Duncan, 2011; Wassef et al., 2018), we hypothesized that increased sympathetic tone may serve as a potential mechanism for cardiometabolic effects observed in with RIS treatment. Here, we confirm that RIS treatment causes an acute increase in c-FOS expression in the parvocellular region of the PVN in mice, consistent with previous findings (Kiss, 2018) of c-FOS induction in the rat PVN by both RIS and OLAN (Kiss et al., 2010; Sebens et al., 1998). Given the role of the PVN in regulating SNS, these results suggest an over-activation of SNS by RIS. Indeed, this acute effect coincides temporally with altered whole body energy expenditure and peak plasma drug concentrations. The drug associated increase in c-fos activity in the PVN was also observed 24 hours following the final dose of RIS (4 week treatment), a time when there is no longer any RIS or 9-OH RIS (active metabolite) present in plasma. These prolonged effects on c-FOS activation in the brain have also been observed in rat studies 24 hours after cessation of chronic haloperidol treatment (Sebens et al., 1996). Furthermore, lasting effects on cognitive performance have been identified as long as 4 days following cessation of chronic RIS treatment (Terry et al., 2003). Markers of SNS overactivity, such as increased plasma concentrations of norepinephrine, have been observed with RIS treatment (Elman et al., 2002; See et al., 1999), and prolonged SNS activation has been associated with metabolic syndrome and heart disease (Thorp and Schlaich, 2015). However, the direct causality of SNS on the observed mitochondrial and metabolic effects with chronic RIS treatment, specifically of the heart, remains unknown. Prospective studies elucidating the contributions of SNS and the upstream regulators of RIS-induced cardiac mitochondrial dysfunction would provide further insight on the pharmacological and molecular pathways that promote these negative cardiometabolic side effects. In addition to SNS overactivation, it is likely that other direct and indirect mechanisms contributing to our observed effects of RIS and OLAN on the heart (Ballon et al., 2014; Vantaggiato et al., 2019). As this is a first report of drug-associated changes in cardiac proteome and gene expression, future studies will be needed to further elucidate the specific contributions of peripheral and central drug associated effects on cardiac function, including, but not limited to, insulin resistance, altered lipid metabolism and neurotransmitter suppression (Ballon et al., 2014; Vantaggiato et al., 2019).
The adverse effects we observe with RIS treatment in the heart and whole body energy metabolism were independent of weight gain and may, in part, be explained through activation of SNS and increased thermogenesis as previously reported (Motyl et al., 2012, 2015). Furthermore, the dose of RIS used in this study falls results in plasma drug exposure in the low clinical range, and is also lower than reported to cause significant weight gain and adiposity in the rodent literature (Cope et al., 2005, 2009; Li et al., 2013). Data demonstrating gender-specific adverse metabolic effects in clinical and pre-clinical studies are inconsistent, as contributions overall to increased risk and severity is multi-faceted, including baseline weight, age at first treatment, duration, dosage, and the type of AA medication itself used for therapy, to name a few (Basson et al., 2001; Gebhardt et al., 2009). Though the role of sex hormones, such as estradiol, have been implicated in the regulation of cardiac mitochondrial function and energy regulation (Rattanasopa et al., 2015; Skrede et al., 2017), studies in mice have shown no significant differences in cardiac mitochondrial respiratory function between males and females (Khalifa et al., 2017). In our study, we attempted to assess potential sex differences of the effects of RIS on energy metabolism and cardiac bioenergetics through the inclusion of both sexes in our study. Although subtle differences in daytime vs. nighttime and resting vs active energy expenditure and activity were observed among sexes, we did see a consistently significant increase in energy expenditure in both our male and female mice treated with RIS, occurring 2 hours post-dose. These findings indicate that the temporal effects of RIS on whole body energy expenditure are independent of sex. We also observed increased mitochondrial protein expression, NDUFS3 and NDUFB9, in both sexes.
In summary, our study provides novel insight into the effects of AA medications on cardiac gene expression, the cardiac proteomic signature, and cardiac mitochondrial respiration following sub-chronic treatment of mice. We provide the first evidence that clinically relevant concentrations AA medications are associated with alterations in expression of genes and proteins in cardiac mitochondrial pathways, suggesting that AA medications may accelerate cardiovascular disease progression, at least in part, via impaired mitochondrial function. Future studies evaluating the effects of altered cardiac gene expression on the development of cardiovascular disease will deepen our understanding of the pharmacology underlying increased cardiac risk in patients prescribed AA medications.
Supplementary Material
7. Acknowledgements
Histological imaging was supported by the Histology and Imaging Core with the financial support provided by the NIGMS grant P20GM103643 (Dr. Ian Meng). We thank Ms. Barbara Conley for proteomic sample preparation and Ms. Theresa McKenzie and Ms. Annika Treyball for assistance with mouse work. This research utilized the services of the Histopathology and Histomorphometry Core supported by NIH/NIGMS P20GM121301 (Dr. Lucy Liaw). Proteomics and Lipidomics Core receives funding support from National Institutes of Health grants P30GM103392 (Drs. Robert Friesel/Donald St. Germain), P20GM121301 (Dr. Lucy Liaw), and U54GM115516 (Dr. Clifford Rosen). Metabolic cage analyses utilized services from the MMCRI Physiology Core, which is supported by NIH/NIGMS P30GM106391 (Dr. Robert Friesel), P20G121301 (Dr. Lucy Liaw) and U54GM115516 (Dr. Clifford Rosen).
6. Funding: This work was supported by the National Institutes of Health NIDDK award number DK095143 to KLH; NIAMS award number AR067858 and NIGMS award number GM121301 to KJM.
Abbreviations:
- AA
Atypical antipsychotic
- DE
Differentially expressed
- FDR
False discovery rate
- GO
Gene Ontology
- PVN
Hypothalamic paraventricular nucleus
- IPA
Ingenuity Pathway Analysis
- OLAN
Olanzapine
- OCR
Oxygen consumption rates
- PANTHER
Protein Analysis Through Evolutionary Relationships
- qPCR
quantitative real-time PCR
- RIS
Risperidone
- RCR
Respiratory Control Ratios
- SWATH
Sequential window acquisition of all theoretical spectra
- VLAD
Visual Annotation Display
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data statement
The proteomic datasets analyzed for this study can be found in the PeptideAtlas (Identifier: PASS01349) http://www.peptideatlas.org/PASS/PASS01349. The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Declaration of interest: none
9. References:
- Aguirre RR, Mustafa MZ, Dumenigo A, Schulz S, Voss A, Goubran B, Dumenigo R, and Sanchez-Gonzalez MA (2018). Influence of Acute Antipsychotic Treatment on Cardiorespiratory Coupling and Heart Rate Variability. Cureus 10, e2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh VL, Vary TC, Ilkayeva O, Wenner BR, Maresca KP, Joyal JL, Breazeale S, Elich TD, Lang CH, and Lynch CJ (2012). Atypical Antipsychotics Rapidly and Inappropriately Switch Peripheral Fuel Utilization to Lipids, Impairing Metabolic Flexibility in Rodents. Schizophr Bull 38, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro CL, Wudarsky M, Nicolson R, Gochman P, Sporn A, Lenane M, and Rapoport JL (2002). Correlation of antipsychotic and prolactin concentrations in children and adolescents acutely treated with haloperidol, clozapine, or olanzapine. J Child Adolesc Psychopharmacol 12, 83–91. [DOI] [PubMed] [Google Scholar]
- Aman MG, Vinks AA, Remmerie B, Mannaert E, Ramadan Y, Masty J, Lindsay RL, and Malone K (2007). Plasma Pharmacokinetic Characteristics of Risperidone and Their Relationship to Saliva Concentrations in C’ildrei with Psychiatric or Neurodevelopmental Disorders. Clin Ther 29, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assié M-B, Carilla-Durand E, Bardin L, Maraval M, Aliaga M, Malfètes N, Barbara M, and Newman-Tancredi A (2008). The antipsychotics clozapine and olanzapine increase plasma glucose and corticosterone levels in rats: Comparison with aripiprazole, ziprasidone, bifeprunox and F15063. European Journal of Pharmacology 592, 160–166. [DOI] [PubMed] [Google Scholar]
- Auger F, Duriez P, Martin-Nizard F, Durieux N, Bordet R, and Pétrault O (2014). Long-Term Risperidone Treatment Induces Visceral Adiposity Associated with Hepatic Steatosis in Mice: A Magnetic Resonance Approach. Schizophr Res Treatment 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger F, Martin F, Pétrault O, Samaillie J, Hennebelle T, Trabelsi M-S, Bailleul F, Staels B, Bordet R, Duriez P, et al. (2018). Risperidone-induced metabolic dysfunction is attenuated by Curcuma longa extract administration in mice. Metabolic Brain Disease; New York 33, 63–77. [DOI] [PubMed] [Google Scholar]
- Balijepalli S, Kenchappa RS, Boyd MR, and Ravindranath V (2001). Protein thiol oxidation by haloperidol results in inhibition of mitochondrial complex I in brain regions: comparison with atypical antipsychotics. Neurochem. Int 38, 425–435. [DOI] [PubMed] [Google Scholar]
- Ballon JS, Pajvani U, Freyberg Z, Leibel RL, and Lieberman JA (2014). Molecular pathophysiology of metabolic effects of antipsychotic medications. Trends Endocrinol. Metab 25, 593–600. [DOI] [PubMed] [Google Scholar]
- Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, and Prabhu SD (2017). Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ Heart Fail 10, e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson BR, Kinon BJ, Taylor CC, Szymanski KA, Gilmore JA, and Tollefson GD (2001). Factors influencing acute weight change in patients with schizophrenia treated with olanzapine, haloperidol, or risperidone. J Clin Psychiatry 62, 231–238. [DOI] [PubMed] [Google Scholar]
- Calarge CA, Acion L, Kuperman S, Tansey M, and Schlechte JA (2009). Weight gain and metabolic abnormalities during extended risperidone treatment in children and adolescents. J Child Adolesc Psychopharmacol 19, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarge CA, Ivins SD, Motyl KJ, Shibli-Rahhal AA, Bliziotes MM, and Schlechte JA (2013). Possible mechanisms for the skeletal effects of antipsychotics in children and adolescents. Ther Adv Psychopharmacol 3, 278–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarge CA, Nicol G, Schlechte JA, and Burns TL (2014) Cardiometabolic outcomes in children and adolescents following discontinuation of long-term risperidone treatment. J Child Adolesc Psychopharmacol 24, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casademont J, Garrabou G, Miró O, López S, Pons A, Bernardo M, and Cardellach F (2007). Neuroleptic treatment effect on mitochondril electron transport chain: peripheral blood mononuclear cells analysis in psychotic patients. J Clin Psychopharmacol 27, 284–288. [DOI] [PubMed] [Google Scholar]
- Chintoh AF, Mann SW, Lam L, Lam C, Cohn TA, Fletcher PJ, Nobrega JN, Giacca A, and Remington G (2008). Insulin Resistance and Decreased Glucose-Stimulated Insulin Secretion After Acute Olanzapine Administration. Journal of Clinical Psychopharmacology 28, 494. [DOI] [PubMed] [Google Scholar]
- Chintoh AF, Mann SW, Lam L, Giacca A, Fletcher P, Nobrega J, and Remington G (2009). Insulin resistance and secretion in vivo: Effects of different antipsychotics in an animal model. Schizophrenia Research 108, 127–133. [DOI] [PubMed] [Google Scholar]
- Citrome L, Collins JM, Nordstrom BL, Rosen EJ, Baker R, Nadkarni A, and Kalsekar I (2013). Incidence of cardiovascular outcomes and diabetes mellitus among users of second-generation antipsychotics. J Clin Psychiatry 74, 1199–1206. [DOI] [PubMed] [Google Scholar]
- Cope MB, Nagy TR, Fernández JR, Geary N, Casey DE, and Allison DB (2005). Antipsychotic drug-induced weight gain: development of an animal model. Int J Obes (Lond) 29, 607–614. [DOI] [PubMed] [Google Scholar]
- Cope MB, Li X, Jumbo-Lucioni P, DiCostanzo CA, Jamison WG, Kesterson RA, Allison DB, and Nagy TR (2009). Risperidone alters food intake, core body temperature, and locomotor activity in mice. Physiol. Behav 96, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daumit GL, Goff DC, Meyer JM, Davis VG, Nasrallah HA, McEvoy JP, Rosenheck R, Davis SM, Hsiao JK, Stroup TS, et al. (2008). Antipsychotic effects on estimated 10-year coronary heart disease risk in the CATIE schizophrenia study. Schizophr. Res 105, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Hert M, Dobbelaere M, Sheridan EM, Cohen D, and Correll CU (2011). Metabolic and endocrine adverse effects of second-generation antipsychotics in children and adolescents: A systematic review of randomized, placebo controlled trials and guidelines for clinical practice. Eur. Psychiatry 26, 144–158. [DOI] [PubMed] [Google Scholar]
- Drici MD, Wang WX, Liu XK, Woosley RL, and Flockhart DA (1998). Prolongation of QT interval in isolated feline hearts by antipsychotic drugs. J Clin Psychopharmacol 18, 477–481. [DOI] [PubMed] [Google Scholar]
- Duncan JG (2011). Mitochondrial dysfunction in diabetic cardiomyopathy. Bi chim. Biophys. Acta 1813, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elman I, Goldstein DS, Green AI, Eisenhofer G, Folio CJ, Holmes CS, Pickar D, and Breier A (2002). Effects of risperidone on the peripheral noradrenegic system in patients with schizophrenia: a comparison with clozapine and placebo. Neuropsychopharmacology 27, 293–300. [DOI] [PubMed] [Google Scholar]
- Elmorsy E, Al-Ghafari A, Aggour AM, Mosad SM, Khan R, and Amer S (2017). Effect of antipsychotics on mitochondrial bioenergetics of rat ovarian theca cells. Toxicol. Lett 272, 94–100. [DOI] [PubMed] [Google Scholar]
- Foley DL, and Mackinnon A (2014). A systematic review of antipsychotic drug effects on human gene expression related to risk factors for cardiovascular disease. Pharmacogenomics J. 14, 446–451. [DOI] [PubMed] [Google Scholar]
- Gao X-M, Cooper T, Suckow RF, and Tamminga CA (2006). Multidose risperidone treatment evaluated in a rodent model of tardive dyskinesia. Neuropsychopharmacology 31, 1864–1868. [DOI] [PubMed] [Google Scholar]
- Garabadu D, Ahmad A, and Krishnamurthy S (2015). Risperidone Attenuates Modified Stress-Re-stress Paradigm-Induced Mitochondrial Dysfunction and Apoptosis in Rats Exhibiting Post-traumatic Stress Disorder-Like Symptoms. J. Mol. Neurosci 56, 299–312. [DOI] [PubMed] [Google Scholar]
- Gebhardt S, Haberhausen M, Heinzel-Gutenbrunner M, Gebhardt N, Remschmidt H, Krieg J-C, Hebebrand J, and Theisen FM (2009). Antipsychotic-induced body weight gain: predictors and a systematic categorization of the long-term weight course. J Psychiatr Res 43, 620–626. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell Proteomics 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal S, Hough D, Karcher K, Nuamah I, Palumbo J, Berlin JA, Baseman A, Xu Y, and Kent J (2013). Risk of Cardiovascular Morbidity With Risperidone or Paliperidone Treatment: Analysis of 64 Randomized, Double-Blind Trials. Journal of Clinical Psychopharmacology 33, 157. [DOI] [PubMed] [Google Scholar]
- Gude NA, Broughton KM, Firouzi F, and Sussman MA (2018). Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat Rev Cardiol 15, 523–542. [DOI] [PubMed] [Google Scholar]
- Haddad PM, and Anderson IM (2002). Antipsychotic-related QTc prolongation, torsade de pointes and sudden death. Drugs 62, 1649–1671. [DOI] [PubMed] [Google Scholar]
- Harrigan EP, Miceli JJ, Anziano R, Watsky E, Reeves KR, Cutler NR, Sramek J, Shiovitz T, and Middle M (2004). A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. J Clin Psychopharmacol 24, 62–69. [DOI] [PubMed] [Google Scholar]
- Hattori S, Kishida I, Suda A, Miyauchi M, Shiraishi Y, Fujibayashi M, Tsujita N, Ishii C, Ishii N, Moritani T, et al. (2018). Effects of four atypical antipsychotics on autonomic nervous system activity in schizophrenia. Schizophr. Res 193, 134–138. [DOI] [PubMed] [Google Scholar]
- Houseknecht KL, Robertson AS, Zavadoski W, Gibbs EM, Johnson DE, and Rollema H (2007). Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: implications for adverse metabolic effects. Neuropsychopharmacology 32, 289–297. [DOI] [PubMed] [Google Scholar]
- Houseknecht KL, Bouchard CC, and Black CA (2017). Elucidating the Mechanism(s) Underlying Antipsychotic and Antidepressant-Mediated Fractures. J Ment Health Clin Psychol 1, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- How O-J, Aasum E, Severson DL, Chan WYA, Essop MF, and Larsen TS (2006). Increased Myocardial Oxygen Consumption Reduces Cardiac Efficiency in Diabetic Mice. Diabetes 55, 466–473. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, and Kato T (2005). Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum. Mol. Genet 14, 241–253. [DOI] [PubMed] [Google Scholar]
- Iwamoto Y, Kawanishi C, Kishida I, Furuno T, Fujibayashi M, Ishii C, Ishii N, Moritani T, Taguri M, and Hirayasu Y (2012). Dose-dependent effect of antipsychotic drugs on autonomic nervous system activity in schizophrenia. BMC Psychiatry 12, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R, McEvoy J, Baillie RA, Lee D, Yao JK, Doraiswamy PM, and Krishnan KRR (2007). Metabolomic mapping of atypical antipsychotic effects in schizophrenia. Mol. Psychiatry 12, 934–945. [DOI] [PubMed] [Google Scholar]
- Khalifa ARM, Abdel- Rahman EA, Mahmoud AM, Ali MH, Noureldin M, Saber SH, Mohsen M, and Ali SS (2017). Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiological Reports 5, e13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasawneh FT, and Shankar GS (2014). Minimizing Cardiovascular Adverse Effects of Atypical Antipsychotic Drugs in Patients with Schizophrenia. [DOI] [PMC free article] [PubMed]
- Kiss A (2018). c-Fos expression in the hypothalamic paraventricular nucleus after a single treatment with a typical haloperidol and nine atypical antipsychotics: a pilot study. Endocr Regul 52, 93–100. [DOI] [PubMed] [Google Scholar]
- Kiss A, Bundzikova J, Pirnik Z, and Mikkelsen JD (2010). Different antipsychotics elicit different effects on magnocellular oxytocinergic and vasopressinergic neurons as revealed by Fos immunohistochemistry. Journal of Neuroscience Research 88, 677–685. [DOI] [PubMed] [Google Scholar]
- Klingerman CM, Stipanovic ME, Bader M, and Lynch CJ (2014). Second-Generation Antipsychotics Cause a Rapid Switch to Fat Oxidation That Is Required for Survival in C57BL/6J Mice. Schizophr Bull 40, 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komossa K, Rummel-Kluge C, Schwarz S, Schmid F, Hunger H, Kissling W, and Leucht S (2011). Risperidone versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev CD006626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz M, and Robbins J (2004). Impact of beta-myosin heavy chain expression on cardiac function during stress. J. Am. Coll. Cardiol 44, 2390–2397. [DOI] [PubMed] [Google Scholar]
- Lambert EA, Straznicky NE, Dixon JB, and Lambert GW (2015). Should the sympathetic nervous system be a target to improve cardiometabolic risk in obesity? Am. J. Physiol. Heart Circ. Physiol 309, H244–258. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kim HR, Han RX, Oqani RK, and Jin DI (2013). Cardiovascular risk assessment of atypical antipsychotic drugs in a zebrafish model. J Appl Toxicol 33, 466–470. [DOI] [PubMed] [Google Scholar]
- Leimbach WN, Wallin BG, Victor RG, Aylward PE, Sundlöf G, and Mark AL (1986). Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 73, 913–919. [DOI] [PubMed] [Google Scholar]
- Leung JYT, Barr AM, Proyshyn RM, Honer WG, and Pang CCY (2012). Cardiovascular side-effects of antipsychotic drugs: The role of the autonomic nervous system. Pharmacology & Therapeutics 135, 113–122. [DOI] [PubMed] [Google Scholar]
- Li X, Johnson MS, Smith DL, Li Y, Kesterson RA, Allison DB, and Nagy TR (2013). Effects of risperidone on energy balance in female C57BL/6J mice. Obesity (Silver Spring) 21, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-T, Chen C-C, Tsang H-Y, Lee C-S, Yang P, Cheng K-D, Li D-J, Wang C-J, Hsieh Y-C, and Yang W-C (2014). Association between antipsychotic use and risk of acute myocardial infarction: a nationwide case-crossover study. Circulation 130, 235–243. [DOI] [PubMed] [Google Scholar]
- Lord CC, Wyler SC, Wan R, Castorena CM, Ahmed N, Mathew D, Lee S, Liu C, and Elmquist JK (2017). The atypical antipsychotic olanzapine causes weight gain by targeting serotonin receptor 2C. J Clin Invest 127, 3402–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, et al. (1997). Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest 100, 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri MC, Paletta S, Maffini M, Colasanti A, Dragogna F, Di Pace C, and Altamura AC (2014). Clinical pharmacology of atypical antipsychotics: an update. EXCLI J 13, 1163–1191. [PMC free article] [PubMed] [Google Scholar]
- May M, Beauchemin M, Vary C, Barlow D, and Houseknecht KL (2019). The antipsychotic medication, risperidone, causes global immunosuppression in healthy mice. PLOS ONE 14, e0218937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JM, Davis VG, Goff DC, McEvoy JP, Nasrallah HA, Davis SM, Rosenheck RA, Daumit GL, Hsiao J, Swartz MS, et al. (2008). Change in metabolic syndrome parameters with antipsychotic treatment in the CATIE Schizophrenia Trial: prospective data from phase 1. Schizophr. Res 101, 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, and Thomas PD (2010). PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38, D204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modica-Napolitano JS, Lagace CJ, Brennan WA, and Aprille JR (2003). Differential effects of typical and atypical neuroleptics on mitochondrial function in vitro. Arch. Pharm. Res 26, 951–959. [DOI] [PubMed] [Google Scholar]
- Motyl KJ, Dick-de-Paula I, Maloney AE, Lotinun S, Bornstein S, de Paula FJA, Baron R, Houseknecht KL, and Rosen CJ (2012). Trabecular bone loss after administration of the second-generation antipsychotic risperidone is independent of weight gain. Bone 50, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl KJ, DeMambro VE, Barlow D, Olshan D, Nagano K, Baron R, Rosen CJ, and Houseknecht KL (2015). Propranolol Attenuates Risperidone-Induced Trabecular Bone Loss in Female Mice. Endocrinology 156, 2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyl KJ, Beauchemin M, Barlow D, Le PT, Nagano K, Treyball A, Contractor A, Baron R, Rosen CJ, and Houseknecht KL (2017). A novel role for dopamine signaling in the pathogenesis of bone loss from the atypical antipsychotic drug risperidone in female mice. Bone 103, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals (2011). Guide for the Care and Use of Laboratory Animals (Washington (DC): National Academies Press (US)). [PubMed] [Google Scholar]
- Nielsen BM, Mehlsen J, and Behnke K (1988). Altered balance in the autonomic nervous system in schizophrenic patients. Clin Physiol 8, 193–199. [DOI] [PubMed] [Google Scholar]
- Ota M, Mori K, Nakashima A, Kaneko YS, Fujiwara K, Itoh M, Nagasaka A, and Ota A (2002). Peripheral injection of risperidone, an atypical antipsychotic, alters the bodyweight gain of rats. Clin. Exp. Pharmacol. Physiol 29, 980–989. [DOI] [PubMed] [Google Scholar]
- Ota M, Mori K, Nakashima A, Kaneko YS, Takahashi H, and Ota A (2005). Resistance to excessive bodyweight gain in risperidone-injected rats. Clin. Exp. Pharmacol. Physiol 32, 279–287. [DOI] [PubMed] [Google Scholar]
- Patel JK, Buckley PF, Woolson S, Hamer RM, McEvoy JP, Perkins DO, Lieberman JA, and CAFE Investigators (2009). Metabolic profiles of second-generation antipsychotics in early psychosis: findings from the CAFE study. Schizophr. Res 111, 9–16. [DOI] [PubMed] [Google Scholar]
- Patel KP, Zhang PL, and Krukoff TL (1993). Alterations in brain hexokinase activity associated with heart failure in rats. Am. J. Physiol 265, R923–928. [DOI] [PubMed] [Google Scholar]
- Peterson SM, Turner JE, Harrington A, Davis-Knowlton J, Lindner V, Gridley T, Vary CPH, and Liaw L (2018). Notch2 and Proteomic Signatures in Mouse Neointimal Lesion Formation. Arterioscler. Thromb. Vasc. Biol 38, 1576–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picca A, Mankowski RT, Burman JL, Donisi L, Kim J-S, Marzetti E, and Leeuwenburgh C (2018). Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol 15, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, Friedrich M, Pinnecker J, Abeßer M, Heinze KG, et al. (2017). Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. U.S.A 114, E2420–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattanasopa C, Phungphong S, Wattanapermpool J, and Bupha-Intr T (2015). Significant role of estrogen in maintaining cardiac mitochondrial functions. J. Steroid Biochem. Mol. Biol 147, 1–9. [DOI] [PubMed] [Google Scholar]
- Ray WA, Chung CP, Murray KT, Hall K, and Stein CM (2009). Atypical Antipsychotic Drugs and the risk of Sudden Cardiac Death. N Engl J Med 360, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser PJ, Portman MA, Ning X-H, and Moravec CS (2001). Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. American Journal of Physiology-Heart and Circulatory Physiology 280, H1814–H1820. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, and Kirk SL (2010). Metabolic side effects of antipsychotic drug treatment-pharmacological mechanisms. Pharmacol. Ther 125, 169–179. [DOI] [PubMed] [Google Scholar]
- Richardson JE, and Bult CJ (2015). Visual annotation display (VLAD): a tool for finding functional themes in lists of genes. Mamm. Genome 26, 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, Opiela M-K, Backs J, Olson EN, Brown JH, et al. (2009). Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail 2, 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvo F, Pariente A, Shakir S, Robinson P, Arnaud M, Thomas S, Raschi E, Fourrier-Réglat A, Moore N, Sturkenboom M, et al. (2016). Sudden cardiac and sudden unexpected death related to antipsychotics: A meta-analysis of observational studies. Clin. Pharmacol. Ther 99, 306–314. [DOI] [PubMed] [Google Scholar]
- Scigliano G, and Ronchetti G (2013). Antipsychotic-induced metabolic and cardiovascular side effects in schizophrenia: a novel mechanistic hypothesis. CNS Drugs 27, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebens JB, Koch T, and Korf J (1996). Lack of cross-tolerance between haloperidol and clozapine towards Fos-protein induction in rat forebrain regions. Eur. J. Pharmacol 315, 269–275. [DOI] [PubMed] [Google Scholar]
- Sebens JB, Koch T, Ter Horst GJ, and Korf J (1998). Olanzapine-induced Fos expression in the rat forebrain; cross-tolerance with haloperidol and clozapine. Eur. J. Pharmacol 353, 13–21. [DOI] [PubMed] [Google Scholar]
- See RE, Fido AA, Maurice M, Ibrahim MM, and Salama GMS (1999). Risperidone-induced increase of plasma norepinephrine is not correlated with symptom improvement in chronic schizophrenia. Biological Psychiatry 45, 1653–1656. [DOI] [PubMed] [Google Scholar]
- Sharov VG, Todor AV, Silverman N, Goldstein S, and Sabbah HN (2000). Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell. Cardiol 32, 2361–2367. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Hashiguchi M, Shiga T, Tamura H, and Mochizuki M (2015). Meta-Analysis: Effects of Probiotic Supplementation on Lipid Profiles in Normal to Mildly Hypercholesterolemic Individuals. PLoS ONE 10, e0139795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrede S, Fernø J, Vázquez MJ, Fjær S, Pavlin T, Lunder N, Vidal-Puig A, Diéguez C, Berge RK, López M, et al. (2012). Olanzapine, but not aripiprazole, weight-independently elevates serum triglycerides and activates lipogenic gene expression in female rats. Int J Neuropsychopharmacol 15, 163–179. [DOI] [PubMed] [Google Scholar]
- Skrede S, González-García I, Martins L, Berge RK, Nogueiras R, Tena-Sempere M, Mellgren G, Steen VM, López M, and Fernø J (2017). Lack of Ovarian Secretions Reverts the Anabolic Action of Olanzapine in Female Rats. Int. J. Neuropsychopharmacol 20, 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöllberger C, Huber JO, and Finsterer J (2005). Antipsychotic drugs and QT prolongation. Int Clin Psychopharmacol 20, 243–251. [DOI] [PubMed] [Google Scholar]
- Stoner SC (2017). Management of serious cardiac adverse effects of antipsychotic medications. Ment Health Clin 7, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus SMJM, Bleumink GS, Dieleman JP, van der Lei J, ‘t Jong GW, Kingma JH, Sturkenboom MCJM, and Stricker BHC (2004). Antipsychotics and the risk of sudden cardiac death. Arch. Intern. Med 164, 1293–1297. [DOI] [PubMed] [Google Scholar]
- Subashini R, Deepa M, Padmavati R, Thara R, and Mohan V (2011). Prevalence of diabetes, obesity, and metabolic syndrome in subjects with and without schizophrenia (CURES-104). J Postgrad Med 57, 272–277. [DOI] [PubMed] [Google Scholar]
- Suda A, Hattori S, Kishida I, Miyauchi M, Shiraishi Y, Fujibayashi M, Tsujita N, Ishii C, Ishii N, Moritani T, et al. (2018). Effects of long-acting injectable antipsychotics versus oral antipsychotics on autonomic nervous system activity in schizophrenic patients. Neuropsychiatr Dis Treat 14, 2361–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LC, Clarke WP, and Berg KA (2015). Atypical Antipsychotics and Inverse Agonism at 5-HT2 Receptors. Curr Pharm Des 21, 3732–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry AV, Hill WD, Parikh V, Waller JL, Evans DR, and Mahadik SP (2003). Differential effects of haloperidol, risperidone, and clozapine exposure on cholinergic markers and spatial learning performance in rats. Neuropsychopharmacology 28, 300–309. [DOI] [PubMed] [Google Scholar]
- Thakore JH, Mann JN, Vlahos I, Martin A, and Reznek R (2002). Increased visceral fat distribution in drug-naive and drug-free patients with schizophrenia. Int. J. Obes. Relat. Metab. Disord 26, 137–141. [DOI] [PubMed] [Google Scholar]
- Thorp AA, and Schlaich MP (2015). Relevance of Sympathetic Nervous System Activation in Obesity and Metabolic Syndrome. [DOI] [PMC free article] [PubMed]
- Toko H, Takahashi H, Kayama Y, Oka T, Minamino T, Okada S, Morimoto S, Zhan D-Y, Terasaki F, Anderson ME, et al. (2010). Ca2+/calmodulin-dependent kinase IIdelta causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation 122, 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGuilder HD, Vrana KE, and Freeman WM (2008). Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques 44, 619–626. [DOI] [PubMed] [Google Scholar]
- Vantaggiato C, Panzeri E, Citterio A, Orso G, and Pozzi M (2019). Antipsychotics Promote Metabolic Disorders Disrupting Cellular Lipid Metabolism and Trafficking. Trends Endocrinol. Metab 30, 189–210. [DOI] [PubMed] [Google Scholar]
- Vieweg WV (2003). New Generation Antipsychotic Drugs and QTc Interval Prolongation. Prim Care Companion J Clin Psychiatry 5, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassef MAE, Tork OM, Rashed LA, Ibrahim W, Morsi H, and Rabie DMM (2018). Mitochondrial Dysfunction in Diabetic Cardiomyopathy: Effect of Mesenchymal Stem Cell with PPAR-γ Agonist or Exendin-4. Exp. Clin. Endocrinol. Diabetes 126, 27–38. [DOI] [PubMed] [Google Scholar]
- Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, and Heller Brown J (2018). CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witchel HJ, Hancox JC, and Nutt DJ (2003). Psychotropic drugs, cardiac arrhythmia, and sudden death. J Clin Psychopharmacol 23, 58–77. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2018). World health statistics 2018: monitoring health for the SDGs, sustainable development goals. [Google Scholar]
- Wu C-S, Tsai Y-T, and Tsai H-J (2015). Antipsychotic drugs and the risk of ventricular arrhythmia and/or sudden cardiac death: a nation-wide case-crossover study. J Am Heart Assoc 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerrabolu M, Prabhudesai S, Tawam M, Winter L, and Kamalesh M (2000). Effect of risperidone on QT interval and QT dispersion in the elderly. Heart Dis 2, 10–12. [PubMed] [Google Scholar]
- Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Bers DM, and Brown JH (2003). The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res 92, 912–919. [DOI] [PubMed] [Google Scholar]
- Zhou B, and Tian R (2018). Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 128, 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.