

Abstract
Mitomycin C (MC) is an anti-cancer agent which alkylates DNA to form monoadducts and interstrand crosslinks (ICLs). Decarbamoylmitomycin C (DMC) is an analog of MC lacking the carbamate on C10. The major DNA adducts isolated from treatment of culture cells with MC and DMC are N2-deoxyguanosine (dG) adducts and adopt an opposite stereochemical configuration at the dG-mitosene bond. In order to elucidate the molecular mechanisms of DMC-DNA alkylation, we have reacted short oligonucleotides, calf thymus and M. Luteus DNA with DMC using biomimetic conditions. These experiments revealed that DMC is able to form two stereoisomeric deoxyadenosine (dA) adducts with DNA under bifuntional reduction conditions and at low temperature. The dA-DMC adducts formed were detected and quantified by HPLC analysis after enzymatic digestion of the alkylated DNA substrates. Results revealed the following rules for DMC dA alkylation i) DMC dA adducts are formed at a 48 to 4-fold lower frequency than dG adducts ii) the 5’-phosphodiester linkage of the dA adducts is resistant to snake venom diesterase iii) End-chain dA residues are more reactive than internal ones in duplex DNA iv) Nucleophilic addition by dA occurs on both faces of DMC and the ratio of stereoisomeric dA adducts formed is dependent on the end bases located at the 3’ or 5’ position. A key finding was to discover that temperature plays a determinant role in the regioselectivity of duplex DNA alkylation by DMC: At 0°C, both dA and dG alkylation occur; but at 37 °C, DMC alkylates dG residues preferentially.
Keywords: antitumor agents, biomimetic synthesis, deoxyadenosine adducts, mitomycins, oligonucleotides
Graphical Abstract
Introduction
DNA-alkylation is possible at several different nucleophilic sites within the DNA molecule. In the case of simple alkylating agents, the reaction is generally not specific for a nucleobase and not specific for a given position within a nucleobase. For example, N-nitrosamines potentially can form adducts with all exocyclic oxygens and ring nitrogens with the exception of N1 site of dG.1 Similarly, Polycyclic Aromatic Hydrocarbons, (PAHs), can bind to dA, dG and dC (deoxycytosine) although dG is generally the favored site of DNA alkylation.1 In contrast, compounds with more complex structures tend to have higher selectivity in their binding with DNA than simple alkylating agents do. This is exhibited by their selective alkylation of a given nucleobase within DNA, by regioselective alkylation within that base and, in some cases, by a selective alkylation within some sequence contexts. Examples of such selective natural products include the antitumor natural products Leinamycin,2 which alkylates specifically at N7 of dG residues and Duocarmycin A,3 which preferentially alkylates N3 of dA at the 3’ end of three or more consecutive AT base pairs in DNA (for a review on sequence-selectivity of natural products see ref. 4).
Mitomycin C (MC) is a complex natural product which alkylates DNA selectively at several sites.5-8 MC has been widely used in cancer chemotherapy and other adjuvant therapies.9-13 A total of nine adducts have been identified from cancer cells treated with MC. Seven of them (including the major monoadduct, 1a, Figure 1, and the major interstrand cross-link, ICL 3a, Figure 1) result from direct alkylation of MC at the 2-amino group of dG.14-16 Decarbamoylmitomycin C (DMC) is an analog of MC lacking the carbamate on C10 and also generates monoadducts and ICLs in the presence of DNA (2b and 3b figure 1).17-19
Figure 1.

Major DNA adducts generated by MC and DMC.
In mammalian cells, MC targets CpG sequences preferentially for ICL formation whereas DMC favors GpC sequences.20 In addition, for MC, the stereochemical configuration at C1’’ of the major adducts is always R (e.g. 1a, 2a, 3a, figure 1) whereas DMC major crosslink (3b, Figure 1) and monoadduct (2b, Figure 1) present a S stereochemical configuration at C1’’ -In this manuscript we will refer to dG or dA adducts with a R stereochemistry at C1’’as α-adducts, while adducts with a C1’’-S configuration will be denoted as β-adducts-.
Although MC and DMC’s ability to form interstrand cross-links (ICLs) is believed to be the primary cause of their antitumor activity, DMC displays a higher cytotoxicity than MC in several cell lines.19, 21, 22 Our long term goal is to understand the molecular basis of DMC enhanced cytotoxicity and our hypothesis is that the opposite stereochemistry of the DNA-adducts formed by MC and DMC is the major reason for this phenomenon. To investigate the reactivity of DMC with DNA, short oligonucleotides have been reacted in the presence of reduced DMC and the resulting DNA adducts have been characterized. These experiments revealed the regioselectivity and stereoselectivity of the alkylation of DNA by DMC.20 Our results showed that, contrary to the current consensus, DMC and MC are able to generate crosslinks both at CpG and GpC sequences and that the crosslinking reaction is diastereospecific and diastereodivergent: Only the 1’’S-diastereomer of the original monoadduct can crosslink DNA at GpC sequences, and only the 1’’R-diastereomer can crosslink DNA at CpG sequences. This investigation led to another interesting discovery: We report here that DMC forms 2 minor stereoisomeric dA adducts by alkylating the exocyclic 6-amino group of deoxyadenosine residues in DNA. Although one of the stereoisomers is similar to a MC-dA adduct previously isolated (4a, Figure 2),23 the new isomeric DMC adducts reported here have never been previously detected. We also show that the formation of these adducts is favored when the reaction is performed at low temperature, both with DNA and with duplex oligonucleotides. No crosslinking activity arising from these monoadducts was observed. These DMC-dA adducts have not been detected in cellular nuclear DNA yet but it is possible that such dA adducts may form in the presence of other type of dA-rich nucleic acids such as dA residues in single-stranded teleomer repeats and/or dA end tails found in some RNAs. If that is the case, these adducts may contribute to DMC cytotoxicity.
Figure 2.

Products of MC and DMC dA alkylation.
Materials and Methods
Caution:
Mitomycin C and Decarbamoylmitomycin C are hazardous and should be handled carefully.24
Materials.
Mitomycin C was a generous gift from Prof. Maria Tomasz. Decarbamoylmitomycin C was synthesized from MC by a published procedure.16 Phosphodiesterase I (snake venom diesterase (SVD), Crotalus adamanteus venom, E.C. 3.1.4.1.) and alkaline phosphatase (Escherichia coli, EC 3.1.3.1) were obtained from Worthington Biochemical Corp (Freehold, NJ). Nuclease P1 (penicillium citrinum, EC 3.1.30.1), 2’-deoxyadenosine, M. lysodeicticus DNA (M. luteus DNA, type XI; 72% GC) and calf thymus DNA (type XV; 42% GC) were from Sigma Life Sciences (St. Louis, MO). Both were sonicated before use. Sep-Pak C-18 cartridges were purchased from Waters Corp (Milford, MA). Oligonucleotides were purchased from Midland Certified Reagent (Midland, TX).
Quantitative Analysis.
Quantitation of DNA and DMC-DNA adducts was based on UV spectrophotometry using the following molar extinction coefficients: DMC, 21800 (367 nm); M. luteus-DNA, 6000 (260 nm, mononucleotide unit); calf thymus DNA, 6500 (260 nm, mononucleotide); dT, 6600 (254 nm); dA, 14200 (254 nm); dG, 11500 (254 nm) DMC monoadducts (2a, 2b, 5a, 5b), 24000 (254 nm); The percent yields of DMC DNA-adducts designate the mole percent adducted dG (2a, 2b) or dA (5a and 5b) of total dG or dA nucleotide residues present in the oligonucleotides or DNA substrates. A full description of the method is available in previous publications.23, 25, 26
High Resolution Mass Spectroscopy (HRMS).
HRMS spectra were recorded by direct infusion using Bruker’s micrOTOF-II ESI instrument at Notre Dame University Mass Spectrometry Facility.
Circular Dichroism Spectra and UV Spectra.
CD spectra were obtained with a Jasco 1500 CD spectrometer. UV spectra were obtained using a Shimadzu UV 2600 UV-vis spectrometer.
Reproducibility.
Reactions were repeated at least twice independently and average deviation from the mean value is represented on graphs or in brackets in tables.
Oligonucleotide Alkylation by DMC and MC under Bifunctional Conditions
Oligonucleotides 6-24 (10 A260 unit scale; Tm: 57°C) were each mixed with an equal amount of their complementary strand (10 A260 unit scale) in water. The mixture was lyophilized. Oligonucleotides were then annealed by heating (90°C, 10 min) after the addition of 10 mM potassium phosphate buffer (pH 5.8, 355 μL) followed by slow cooling to 0°C. The reaction mixture was put under ice and deaerated via argon bubbling (30 min) while kept at 0°C. Excess Na2S2O4 (6.48 μmol in 20 μL of potassium phosphate buffer, 10 mM, pH 5.8) from a freshly prepared anaerobic solution was then added to the mixture quickly and immediately followed by addition of DMC (1.30 μmol). The reaction was allowed to stir for 10 min before another addition of Na2S2O4 (3.24 μmol in 10 μL of potassium phosphate buffer, 10 mM pH 5.8) was immediately followed by addition of DMC (1.30 μmol in 200μL). This process was repeated 3 more times until the reaction was treated with a total of 6.48 μmol of DMC. The reaction mixture was allowed to stir with argon bubbling for 1 hr and then opened to air, followed by gentle mixing until a consistent purple color was obtained. The mixture was then allowed to stir for 20 min under air and chromatographed on a 2.5*56 cm Sephadex G-25 column using 20 mM NH4HCO3 as eluent. Oligonucleotide containing fractions were lyophilized. A similar process was followed for oligonucleotides 31, 32, 33 and 34 (5’-TGCTTGCTTGCTTGCTTGCTTGCT-3’, TGCATGCATGCAAGCTAGCTAGCT-3’, 5’-TCGTTCGTTCGTTCGTTCGTTCGT-3’, TCGATCGATCGAACGTACGTACGT-3’, 10 A260 unit scale; Tm: 57°C). However, with these 24 MER oligonucleotides, the reaction was performed both at 0 and 37°C. A similar process was also followed with single stranded oligonucleotides (25-30). In this case, the reaction was performed on 10 A260 unit scale and the reagents (DMC and Na2S2O4) were scaled accordingly. No annealing was necessary and reactions were performed both at 0 and 37°C.
M. luteus and Calf Thymus DNA Alkylation by DMC under Na2S2O4 Activation.
A solution of sonicated calf thymus DNA or M. Luteus DNA (12 mM) and DMC (1mM) was deaerated in 10 mM potassium phosphate-11mM EDTA (pH 5.8, 1 mL). A deaerated solution of Na2S2O4 (2mM) was added. Na2S2O4 and DMC were added incrementally as described above until a total of 10 mM of Na2S2O4 and 5 mM of DMC were added. The drug-DNA complexes were extracted by adding 10mM potassium phosphate, pH 9.0, to adjust the pH to 8.0 and then adding 1 mL of phenol:CHCl3:isoamyl alcohol extraction solution (25:24:1 v/v). The solution was vortexed vigorously and allowed to settle before it was transferred into vials and centrifuged (13,000 rpm, 10 min). DNA was isolated by applying only the top aqueous layer on a Sephadex G-25 column.
Enzymatic Digestion of the DMC-DNA Complexes to Nucleosides and DMC-Nucleoside Adducts.
The lyophilized DMC-DNA complex (calf thymus or M. Luteus) was dissolved in 20 mM ammonium acetate (pH 5.5) (2.5 A260 units/mL). Nuclease P1 (1.0 unit/ A260 unit of complex) was added to the mixture followed by incubation for 4 hr at 37°C. The pH was adjusted to 8.2 by addition of 20 mM NaOH, and MgCl2 was added to a concentration of 0.9 mM. Addition of Snake Venom Diesterase (SVD) (2.25 units/A260 unit of complex) was followed by incubation at 37°C (2 hr). Alkaline phosphatase (AP) (1.6 units/ A260 unit of complex) was then added and the digest was incubated overnight at 37°C. Samples were lyophilized and redissolved for HPLC analysis.
Enzymatic Digestion of Alkylated Oligonucleotides.
(1) SVD, AP protocol: 1 A260 unit of oligonucleotide, 2.25 units of SVD and 1.6 units of AP in 100 mM Tris-2mM MgCl2 (pH 8.2) (200μL) were incubated for 4 hr at 45°C. (2) Nuclease P1/SVD/AP protocol: 1 A260 unit of oligonucleotide and 2 units of nuclease P1 were incubated at 37°C for 2 hr in 0.8 mL of 20 mM ammonium acetate (pH 5.5); 100 mM MgCl2 (20μL) was added, and the pH was adjusted to 8.2 by addition of 20 μL of 200 mM NaOH. SVD (2 units) and AP (2 units) were added and incubation was continued at 37°C for 2.5 h.
Analysis of DNA Adducts after Enzymatic Digestion of Alkylated Oligonucleotides and Calf Thymus or M. Luteus DNA.
Digestion mixtures were directly analyzed by HPLC using an Agilent 1200 HPLC system and a Kromasil C-18 reverse phase column (0.46*25 cm). The elution system was 6-18% acetonitrile in 30 mM potassium phosphate (pH 5.4), in 60 min, 1 mL/min flow rate.
Identification of 2a, 2b, 5a and 5b.
Adducts 2a and 2b were identified by their UV spectra, retention times and co-elution with authentic standards synthesized previously in our laboratories.27 Adducts 5a and 5b were identified by their UV, HRMS and Circular Dichroism spectra. A full description of their identification is provided in the results section.
NMR data:
Spectra were recorded in a mixture of D2O: pyridine-d5 (1:5). 1H NMR spectra were obtained at 300 MHz at room temperature and are referenced to tetramethylsylane. Chemical shifts are reported in parts per million and coupling constants are in hertz (Hz). The conventional numbering system is used for the mitosene moiety and numbered 1”-10”. The purine carbons are numbered 1-6 also as per convention. The sugar carbons are numbered 1’-5’ beginning at the anomeric carbon and proceeding via the carbon chain to the primary carbinol center. 2-D COSY NMR data were also collected and spectra are available in the supporting information.
dA trans adduct (5a): 1H NMR (C5D5N with 20% v/v D2O and 0.05% v/v TMS, 300 MHz): δ 2.15 (3H, s, CH3), 2.90 (1H, ddd, J = 14.1, 7.2, 2.0 Hz, H2’), 3.09 (1H, app quint, J = 18 Hz, H2’), 4.18 (1H, dd, J = 12.2, 3.0 Hz, H5’), 4.23 (1H, dd, J = 12.1, 3.0 Hz, H5’), 4.44 (1H, d, J = 4.5 Hz, H2”), 4.48 (1H, br s, H3’’), 4.66 (1H, dd, J = 6.2, 3.1 Hz, H4’), 4.96 (1H, br dd, J = 15.0, 5.0 Hz, H3”), 5.16 (2H, s, H10”), 5.19 (1H, d, J = 3.3 Hz, H3’), 6.90 (1H, t, J = 12.0 Hz, H1’), 8.65 (1H, s, H8 or H2). Note: There is one unobserved resonance corresponding to H1” and one unobserved resonance corresponding to either H8 or H2. Signals from exchanging protons (NH, OH) are not observed either.
dA cis adduct (5b): 1H NMR (C5D5N with 20% v/v D2O and 0.05% v/v TMS, 300 MHz): δ 2.16 (3H, s, CH3), 2.89 (1H, ddd, J = 13.3, 6.1, 3.0 Hz, H2’), 3.04 (1H, ddd, J = 14.0, 6.1, 1.0 Hz, H2’), 4.16 (1H, dd, J = 12.0, 3.0 Hz, H5’), 4.22 (1H, dd, J = 12.0, 3.0 Hz, H5’), 4.47 (1H, dd, J = 13.1, 7.2 Hz, H3”), 4.65 (1H, app. d, J = 3.1 Hz, H4’), 4.69 (1H, dd, J = 15.0, 7.2 Hz, H2”), 4.88 (1H, dd, J = 12.3, 8.0 Hz, H3’’), 5.17 (2H, s, H10”), 6.09 (1H, br s, NH), 6.89 (1H, t, J = 8.0 Hz, H1’), 8.60 (1H, s, H8 or H2), 9.34 (1H, s, NH). Note: There is one unobserved resonance corresponding to H1” and one unobserved resonance corresponding to either H8 or H2. Signals from exchanging protons (NH, OH) are not all observed either.
Formation and Isolation of dA Adducts 5a and 5b from Deoxyadenosine and DMC.
2’-deoxyadenosine (600 mg, 2.4 mmol) and DMC (180 mg, 0.62 mmol) were dissolved in 20 mL of water. PtO2 (60 mg) was added and the mixture was deaerated for 30 min with Argon after which H2 was bubbled through the solution for 5 min. The mixture was then filtered. dA adducts of DMC were isolated using a semi-preparative HPLC column: (Phenomenex, clarity, 10*250 mm, C-18). The elution system was 6-18% acetonitrile in 30 mM NH4OAc in 90 min at 4.0 mL/min flow rate. Fractions were collected manually and were desalted by passing through a sep-pak eluted with 10% acetonitrile in water. Lyophilization yielded 0.75 % (4.7 μmol, 2.37 mg) of adduct 5a (β, 7-diamino-1α-hydroxymitosene) and 0.8 % (5.12 μmol, 2.53 mg) of adduct 5b (β, 7-diamino-1β-hydroxymitosene).
Results
HPLC Analysis of the Digests from Oligonucleotides Treated with DMC and Sodium Dithionite Under Bifunctional Activation Revealed 2 New DMC Adducts (Figure 2).
Enzymatic digestions of reaction mixtures between oligonucleotides and DMC using AP (alkaline phosphatase), SVD (snake venom diesterase) and nuclease P1 yielded two novel adducts (5a and 5b, Figure 2). These two compounds eluted with retention times of 30 (5a) and 44 minutes (5b) (Figure 3 and Figure S1, supporting information) under our analysis conditions (see Materials and Methods). The UV spectra of both 5a and 5b show the presence of a deoxyadenosine chromophore (λmax 267 nm) and a 7-aminomitosene (λmax 313 nm) (Figure 4). Both spectra are similar to the spectrum of a previously isolated and characterized mitomycin C-N6-deoxyadenosine adduct.23 This prompted us to investigate if these new adducts were stereoisomeric N6-deoxyadenosine adducts of DMC.
Figure 3.

Representative HPLC chromatograms of enzymatic digests of DMC-oligonucleotide complexes formed under bifunctional conditions oligonucleotides 6 (a), 8 (b), 25 (c), 19 (d), 9 (e), 28 (f).
Figure 4.

UVspectra of 5a (a) and 5b (b), in water.
(1). 5a and 5b are dA adducts.
This was deduced because adducts 5a and 5b were detected in the reaction of DMC with duplex 6 (which contains only dA and dT) and with single stranded oligonucleotide 25 (which contains only dA) (Figures 3a and 3c). Additionally, 5a and 5b were directly synthesized from 2’-deoxyadenosine upon reductive activation of DMC using catalytic hydrogenation. Co-injection of samples of 5a and 5b (obtained from the direct reaction of 2’-deoxyadenosine and activated DMC) with the digests of the reactions between DMC and oligonucleotides 9 and 6 (Table 1) demonstrated the identity of the adducts formed with oligonucleotides (Figure S2 and S3, supporting information).
Table 1.
Short Oligonucleotides Reacted under Bifunctional Conditions.
| 6 | 5’-AAAAAAAAAAAA 3’-TTTTTTTTTTTT |
14 | 5’-AAAAAAAAAAAC 3’-TTTTTTTTTTTG |
22 | 5’-GCAAAAAAAAGC 3’-CGTTTTTTTTCG |
| 7 | 5’-AAAAAGCAAAAA 3’TTTTTCGTTTTT |
15 | 5’-AGAAAAAAAAAG 3’-TCTTTTTTTTTC |
23 | 5’-CGAAAAAAAACG 3’-GCTTTTTTTTGC |
| 8 | 5’-TATATATATATA 3’-ATATATATATAT |
16 | 5’-ACAAAAAAAAAG 3’-TGTTTTTTTTTC |
24 | 5’-GAAAAAAAAAAG 3’-CTTTTTTTTTTC |
| 9 | 5’-TTATTGCAATTA 3’-AATAACGTTAAT |
17 | 5’-ATATAGCTATAT 3’-TATATCGATATA |
25 | 5’-AAAAAAAAAAAA (single strand) |
| 10 | 5’-TATATGCATATA 3’-ATATACGTATAT |
18 | 5’-ATATACGTATAT 3’-TATATGCATATA |
26 | 5’-GAAAAAAAAAAG (single strand) |
| 11 | 5’-GAAAAAAAAAAA 3’-CTTTTTTTTTTT |
19 | 5’-ATTATTGCTATT 3’-TAATAACGATAA |
27 | 5’-TTAGG (single strand) |
| 12 | 5’-GAAAAAAAAAGA 3’-CTTTTTTTTTCT |
20 | 5’-ATTATCGTTATT 3’-TAATAGCAATAA |
28 | 5’-ATTATTGCTATT (single strand) |
| 13 | 5’-GAAAAAAAAACA 3’-CTTTTTTTTTGT |
21 | 5’-ATTATCGATAT 3’-TAATAGCTATAA |
29 | 5'-ATTATCGTTATT (single strand) |
| 30 | 5'-ATATAGCTATAT (single strand, 37° C) |
(2). Structural characterization of 4 and 5.
About 2.5 mg of adducts 5a and 5b were obtained from DMC and 2’-deoxyadenosine (Materials and Methods). HRMS data indicated that 5a and 5b had identical molecular formula. NMR data support the assignment of structures 5a and 5b for these adducts (supporting information, Figure S8-S11). Adducts 5a and 5b were further characterized by their circular dichroism (CD) spectra (Figure 5). The negative Cotton effect in the 500-600 nm region in the CD spectrum of adduct 5a shows that the mitosene has the 1”-α (trans) configuration and the positive Cotton effect in the same region in the CD spectrum of 5b indicates a 1”- β (cis) configuration.28
Figure 5.

CD spectra of 5a (blue) and 5b (red) in 0.1 M Tris·HCl; pH 7.4 (4.16*10−5 M).
SVD-Resistant Phosphodiester Linkage of adducts 5a and 5b.
When nuclease P1 was not added to the enzymatic digestion protocol (only SVD/AP enzymes were used), the digests from reactions between duplex 6 and DMC under bifunctional conditions yielded two other compounds, in addition to 5a and 5b. These unknown compounds (X and Y) were converted to 5a and 5b quantitatively upon addition of nuclease P1 (Figure S6, supporting information). This indicates that these two other compounds contain an SVD-resistant phosphodiester linkage which can be hydrolyzed by nuclease P1 and AP to a single DMC-nucleoside adduct. Although the sequence of these SVD-resistant dinucleoside phosphate adducts was not assessed in this work, the same phenomenon has been previously observed with an MC dA adduct.23 The authors proved that the 5’-phosphodiester linkage of this MC-dA adduct was resistant to SVD. The similarity of these previous findings with our own observations suggests a related structure for X and Y: 5’-dAp(5a) and 5’-dAp(5b), where the 5’-phosphodiester linkage is SVD resistant. The same SVD-resistance was observed with all oligonucleotides (6-34) studied. Our results in this regard bear some similarities with the previously reported observations on dA adducts formed by MC. These similarities are: i) similar UV spectra of dA dinucleoside phosphate adducts X and Y with previously isolated dA dinucleoside phosphate adducts of MC (Figure S7, supporting information) ii) disappearance of X and Y when nuclease P1 is used (Figure S6, supporting information) ii) earlier elution of dA dinucleoside phosphate adducts X and Y compared to the corresponding dA adducts 5a and 5b (Figure S6, supporting information).
dA Alkylation Frequency in Duplex versus Single Stranded Oligonucleotides (Table S1, supporting information, Figures 3, 6 and 7).
Figure 6.

Frequency of alkylation of oligonucleotides containing solely dA and dT. Comparison between duplexes 6: d(AAAAAAAAAAAA).(TTTTTTTTTTTT); 8: d(TATATATATATA).(TATATATATATA) and single stranded 25: 5’-AAAAAAAAAAAA.
Figure 7.

Frequency of alkylation of oligonucleotides containing dA, dT, dC, dG. Comparison between duplex 19: d(ATTATTGCTATT).(AATAGCAATAAT) and single stranded oligonucleotide 28: 5’-ATTATTGCTATT and between duplex 20 d(ATTATTCGTATT).(AATACGAATAAT) and single stranded oligonucleotide 29: 5’-ATTATTCGTATT.
The alkylated oligonucleotides were digested enzymatically and the digest was analyzed by HPLC. The frequency of the oligonucleotide-DMC adducts formed was determined by quantitative analysis of the nucleosides present in the enzymatic digest. With oligonucleotides containing solely dA and dT (oligonucleotides 6, 8 and 25), dA alkylation was mildly favored in duplex DNA (oligonucleotides 6 and 8) compared to single stranded DNA (oligonucleotide 25) (Figure 6). Unexpectedly, single-stranded oligonucleotides containing dG and dC bases, appeared to be more reactive toward dA alkylation than the corresponding duplexes (see frequency of alkylation of 19 versus 28 and 20 versus 29, Figure 7). Indeed, the highest yield for dA alkylation in all short oligonucleotides studied in Table 1 was obtained with single stranded oligonucleotide 29 (Figure 7, 0.48% of dA residues are alkylated for 29). This effect contrasts sharply with the reactivity of dG residues in alkylation reactions with DMC, which is strongly favored in duplex DNA versus single stranded DNA.25
dA Alkylation Frequency Compared to dG Alkylation Frequency in DNA (Table S1, supporting information, Table 2 and Figure 8).
Table 2.
Frequencies of DNA Adducts Detected in Calf Thymus DNA (42% CG), M. Luteus DNA (72% CG) and 24 base pairs Oligonucleotides (50% CG) under Bifunctional Conditions and at Different Temperature. ND: non-detected; NA: non available.
| DNA Substrate | Frequencies (%) of adducts detected | ||||
|---|---|---|---|---|---|
| dG | 5a | 5b | dG/ dA |
||
| Luteus 0°C | 1.54 (±0.03) |
0.61 (±0.03) |
0.10 (±0.01) |
2.17 | |
| Luteus RT | 0.73 (±0.05) |
0.23 (±0.05) |
ND | 3.17 | |
| Luteus 37°C | 0.97 (±0.08) |
ND | ND | NA | |
| CT-DNA 0°C | 1.87 (±0.008) |
0.02 (±0.002) |
ND | 93.5 | |
| CT-DNA RT | 1.83 (±0.03) |
0.03 (±0.02) |
ND | 61.0 | |
| CT-DNA 37°C | 0.90 (±0.03) |
ND | ND | NA | |
| 31 0°C | 5’-TGCTTGCTTGCTTGCTTGCTTGCT 3’-ACGAACGAACGAACGAACGAACGA |
1.52 (±0.08) |
0.16 (±0.06) |
ND | 9.50 |
| 31 37°C | 5’-TGCTTGCTTGCTTGCTTGCTTGCT 3’-ACGAACGAACGAACGAACGAACGA |
0.57 (±0.02) |
ND | ND | NA |
| 32 0°C | 5’-TCGTTCGTTCGTTCGTTCGTTCGT 3’-AGCAAGCAAGCAAGCAAGCAAGCA |
1.12 (±0.1) |
0.20 (±0.01) |
ND | 5.60 |
| 32 37°C | 5’-TCGTTCGTTCGTTCGTTCGTTCGT 3’-AGCAAGCAAGCAAGCAAGCAAGCA |
0.60 (±0.02) |
ND | ND | NA |
| 33 0°C | 5’-TGCATGCATGCAAGCTAGCTAGCT 3’-ACGTACGTACGTTCGATCGATCGA |
1.90 (±0.04) |
0.08 (±0.008) |
ND | 23.8 |
| 33 37°C | 5’-TGCATGCATGCAAGCTAGCTAGCT 3’-ACGTACGTACGTTCGATCGATCGA |
0.82 (±0.005) |
ND | ND | NA |
| 34 0°C | 5’-TCGATCGATCGAACGTACGTACGT 3’-AGCTAGCTAGCTTGCATGCATGCA |
1.99 (±0.09) |
0.28 (±0.07) |
ND | 7.10 |
|
34 37°C |
5’-TCGATCGATCGAACGTACGTACGT 3’-AGCTAGCTAGCTTGCATGCATGCA |
0.82 (±0.008) |
ND | ND | NA |
Figure 8.

dA Alkylation Frequency Compared to dG Alkylation Frequency (log10(dG/dA)) in duplex DNA, single stranded DNA CT DNA and M. Luteus DNA. 19: d(ATTATTGCTATT).(AATAGCAATAAT); 20 d(ATTATTCGTATT).(AATACGAATAAT) ; 27: 5’-TTAGG; 28: 5’-ATTATTGCTATT; 29: 5’-ATTATTCGTATT.
In all duplex oligonucleotides studied (12 MER 6 to 24 and 24 MER 31 to 34), the alkylation frequency of dA varied between 0.01 % and 0.39% (Table S1, supporting information and Table 2) and dA adducts were 4 to 48 times less abundant than dG adducts. In some cases, dA alkylation was not detected (oligonucleotides 22, 23, 24; Figure 9). Unexpectedly, in CT DNA and M. Luteus DNA, dA alkylation increases with the CG content of DNA (Table 2, Figure 8). The frequency of dA alkylation in M. Luteus (72% CG and 0.71% alkylation of A residues at 0°C) is considerably higher (36 times) than in CT DNA (42% CG and 0.02% alkylation of A residues at 0°C) (Table 2). While in all duplex DNA dG residues are a better target, in single stranded DNA (oligonucleotides 27, 28, 29), dA alkylation frequency is similar (28) or higher (27 and 29) to dG alkylation frequency (Figure 8).
Figure 9.

HPLC chromatograms of enzymatic digests of DMC-oligonucleotide complexes formed under bifunctional conditions with duplex oligonucleotides 22 (a), 23 (b) and 24 (c) which have no end-chain dAs. Adducts 5a and 5b could not be detected.
Reactivity of terminal dA Residues Versus Internal Ones in Short Oligonucleotides (Table S1 and 2, Figures 9 and 10).
Figure 10.

dA alkylation frequency of single stranded 26, 27 which have no end-chain dA residue and 25, 28 and 29 which have end-chain dA residues.
Alkylation Frequency is not affected by the organization of the bases located internally of duplex oligonucleotides. This was deduced by comparing the alkylation frequency of oligonucleotides 17 to 21 that contain dA with different flanking residues (17 contains 5’-TAT and 5’ TAG; 18 contains 5’-TAT and 5’-TAC; 19 contains 5’-TAT, 5’-AAT, 5’-TAA, 5’-TAG and 5’-CAA; 20 contains 5’-TAT, 5’-AAT, 5’-TAA, 5’-AAC and 5’-GAT; 21 contains 5’-TAT, 5’-AAT, 5’-TAA and 5’-GAT; Table 1). In addition, dA alkylation does not occur if no dA is present at the 5’ or 3’end position of the substrates (oligonucleotides 22 and 23 and 24, Figure 9 and Table S1, supporting information). This suggests that chain terminal dA residues are more reactive than internal ones in duplex DNA. In single stranded DNA, we observed a much milder end-chain effect. Oligonucleotides 26 and 27 did generate adducts 5a and 5b despite having no end-chain dA. However, the dA alkylation frequency of 26 and 27 (single stranded, no end-chain dA residue) is lower than oligonucleotides 25, 28 and 29 which all have end-chain dA residues (Figure 10).
Diastereoselectivity of dA Alkylation and End-chain Dependent Diastereodivergence (Table S1, supporting information and Figures 11 and 12).
Figure 11.

log10 (5a/5b) for oligonucleotides 11: d(GAAAAAAAAAAA).(TTTTTTTTTTTC); 12 d(GAAAAAAAAAGA).(TCTTTTTTTTTC); 13: d(GAAAAAAAAACA).(CTTTTTTTTTGT); 10: d(TATATGCATATA).(TATATGCATATA).
Figure 12.

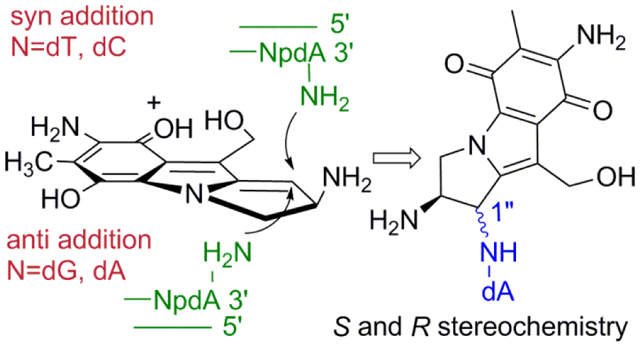
Syn or anti additions of dA to reduced DMC depend on the nature of the adjacent 3’ base. Anti addition is favored at 3’-AG and 3’-AA. At 3’-AT and 3’-AC syn addition is favored.
In duplex DNA, our results demonstrate that the stereochemical outcome of the reaction between DMC and end-chain dA depends on the end-chain composition of the oligonucleotides studied. Nucleophilic addition on the α-face of reduced DMC is favored by dA residues located at the 5’-end to generate 5a as the major adduct (Oligonucleotides 14 (5’AA); 15 (5’-AG); 16 (5’-AC); 17 (5’-AT); Table S1, supporting information). At the 3’ end, the composition of the dinucleotide 3’-ApN determines the stereochemical outcome (Figure 11). Oligonucleotides 11 (3’-AA) and 12 (3’-AG) favor the formation of 5a whereas oligonucleotides 13 (3’-AC) 10 (3’-AT) and 8 (3’-AT) favor the formation of 5b. The presence of a pyrimidine next to a dA residue in the 3’ direction correlates with an orientation of reduced DMC which favors nucleophilic addition on the β-face of the drug’s tetrahydropyrrole ring to generate 5b as the major adduct (Figure 12). In all single stranded oligonucleotides studied (25 to 30), 5a was always the major adduct formed. However, the stereochemical outcome of the reaction between dA residues in single stranded DNA and reduced DMC was not systematically assessed in all possible sequences in this work.
Alkylation of dA residues is Temperature Dependent in Duplex DNA (Table 2).
In calf thymus DNA, M. Luteus DNA and oligonucleotides 31 to 34, the frequency of dA alkylation varies according to the temperature. At low temperature, dA alkylation occurs in all cases. At higher temperature (37°C), dA alkylation is not detected in any duplex DNA substrate studied. This strongly suggests that dA adducts are formed when reactions are run at low temperature in duplex DNA. This may explain why they have not been detected in cells treated with MC and DMC at 37°C in previous studies yet. Reactions between single stranded oligonucleotides and reduced DMC at 37°C yielded complex chromatograms, hampering in most cases the detection of dA adducts. However, with single stranded oligonucleotide 30, we were able to establish that dA adducts are formed at 37 °C in single stranded oligonucleotides, with yields of 0.076 % for 5a and 0,031 % for 5b respectively (Table S1, supporting information).
DMC-dA Monoadducts do not Form ICLs.
No crosslinking activity was observed by DMC-dA adducts 5a and 5b in any substrate studied and at any temperature. This was also observed with the MC-dA adduct previously detected.23
Discussion
Mitomycin C and Decarbamoylmitomycin C are bioreductive drugs: They are unreactive towards DNA, or other nucleophiles, in their original structure but reductive activation of the quinone moiety triggers DNA alkylation (Scheme 1). Until now, DMC-DNA adducts have only been observed at dG sites. DMC forms Interstrand Crosslinks (ICLs) between exocyclic amino groups of opposing deoxyguanosine residues and dG N2-monoadducts.15, 29 Reduction of MC and DMC triggers several consecutive transformation steps leading to a bifunctional alkylating species (36a or 36b) and/or a monofunctional species (35a or 35b) (Scheme 1).30 The bifunctional pathway results in the activation of both C1 and C10 to generate Interstrand Crosslinks (ICLs) as well as monoadducts whereas the monofunctional pathway consists of an autocatalytic process and produces solely monoadducts at C1 (Scheme 1).31
Scheme 1.

MC and DMC Reductive Activation.
In order to understand the molecular basis of DMC-DNA alkylation, we have reacted short oligonucleotides, calf thymus and M. Luteus DNA with DMC using bio-mimetic conditions. These investigations revealed that the mode of reduction has a crucial influence on the stereoisomeric outcome of dG alkylation by DMC.20 Intermediate 36a, generated under bifunctional reductive activation, can undergoe nucleophilic addition of dG both on its α- and β-faces to generate trans (α) and cis (β) dG adducts (2a or 2b and 3a or 3b, Figure 1). On the other hand, the product of the auto-catalytic pathway, aziridinomitosene 35a, only undergoes nucleophilic addition on its α-face to yield trans adduct 2a.20
Analysis of the enzymatic digests of oligonucleotides alkylated by DMC led to the discovery of dA adducts 5a and 5b, analogous to a MC-dA adduct previously characterized. This MC N6-dA adduct was formed at a 100-fold lower frequency than dG N2 adducts in calf thymus DNA and has never been detected in cells.23 Our results show similarities between MC and DMC in the alkylation of dA residues in duplex DNA: 1) the two new dA-DMC adducts identified in this work are formed at similar low frequency in all substrates studied and 2) the 5’-phosphodiester linkage of the DMC dA adducts is also resistant to SVD.
A main difference between MC and DMC in the formation of dA adducts is that DMC produced 2 isomeric adducts (5a and 5b) whereas only the trans (α) adduct was detected with MC. We propose that the difference in the stereochemical outcome of the reaction is due to the competition amongst the monofunctional and bifunctional activation pathways (Scheme 1). In the present study, we used optimized bifunctional reductive conditions to activate DMC and to better simulate cellular DNA alkylation.26 This is in contrast to previous experiments with MC, which were performed using sequential additions of sub-stoichiometric amounts of dithionite or slow enzymatic reductions.15, 23, 31 As a result, these previous reactions favored the monofunctional pathway (Scheme 1). Therefore, the alkylation of dA in DNA was performed by aziridinomitosene 35b and resulted in the production of α-adducts only.23 Our reaction conditions (Materials and Methods) promote the bifunctional activation pathway and this implies that dA alkylation by DMC is effected by intermediate 36a.20 The results presented here show that nucleophilic addition of dA N6 can occur on both the α- and β-faces of 36a to yield adducts 5a and 5b. This is in agreement with the reactivity observed in the alkylation of dG residues by DMC: decarbamoylaziridinomitosene 35a only produces α-dG adducts whereas intermediate 36a yields both α- and β-dG adducts.20
The results of this work also highlight main differences between the reactivity of dA residues in duplex and single stranded DNA. In duplex DNA internal dAs are not reactive, and only chain-terminal dAs can undergo alkylation by DMC (Figure 9). In single stranded DNA, both internal and end-chain dAs can be alkylated. Figure 10 suggests, however, that end-chain dAs in single stranded oligonucleotides still display a higher reactivity toward activated DMC than internal ones.
Why is the alkylation of oligonucleotides selective for terminal dAs? A potential explanation could be found in the kinetics of the reaction. In duplex DNA, terminal dAs may react faster because there are more accessible than internal ones due to the frayed structure of the ends of oligonucleotides. This may explain as well why internal dA residues in single stranded oligonucleotides are able to react with reduced DMC (albeit at a lower frequency than terminal ones) since they are more accessible than internal dA residues in duplex DNA. Another reason for the increased reactivity of terminal dAs in duplex DNA could be that alkylated terminal dAs induce duplex stabilization through hydrogen bonds or π-π interactions between the reduced DMC moiety and adjacent bases or bases located on the opposite strand. Such stabilization by aromatic hydrocarbon groups tethered to 5’-dAs of duplex oligonucleotides has been previously observed.32
Interestingly, although 5a is the major adduct formed in most oligonucleotides studied, some duplex oligonucleotides produced 5b as the major adduct when reacted with reduced DMC (Figures 3b and 11). This was observed when a pyrimidine was adjacent to a dA residue in the 3’direction. In that case, the orientation of reduced DMC favored nucleophilic addition on the β-face of the drug’s tetrahydropyrrole ring to generate 5b as the major adduct (Figure 12). In contrast, at 3’-ApA, GpA and all 5’-NpA, anti-addition of the dA residue to reduced DMC was favored to generate 5a as the major adduct (Figure 12). The reason for this divergence in diastereoselectivity is not clear, but it is likely to be caused by pre-covalent interactions between reduced DMC and terminal bases in duplex DNA, which determine the orientation of reduced DMC prior to nucleophilic addition by terminal dAs.
Results also show that DMC only forms dA adducts in duplex DNA when reactions are run at cold temperature (Table 2). At 37°C, no dA adduct is produced either with calf thymus DNA, M. Luteus DNA or short duplexes. However, in single-stranded oligonucleotides, the formation of dA adducts seems to be favored at 37 °C (oligonucleotide 30 and reduced DMC at 37 °C, Table S1, supporting information). The generality of this reactivity could not be confirmed with other single-stranded oligonucleotides because the adduct profiles from these reaction mixtures were too complex to provide a clear picture. If this temperature-dependent reactivity translates to live cells, then the formation of dA adducts in duplex DNA is unlikely to be of biological significance, because the reaction will take place at temperatures that do not result in the formation of adducts. The nature of in vitro reactions, however, means that they are not a fully accurate representation of in vivo reactivity. Therefore, although we did not detect dA adducts formation in duplex DNA with our model systems, it cannot be ruled out that such adducts are formed in vivo with duplex DNA. Detection of theses adducts in cells would require the use of modern analytical techniques such as a full scan mass spectrum experiment to identify potential ions, followed by a full scan MS/MS experiment on chosen parent ions. Such experiments have not been performed yet but will be the object of future endeavors. Furthermore, our results indicate that unfolded single-stranded structures of nucleic acids could be targets for dA alkylation by DMC in cells, as the alkylation of dA in single stranded oligonucleotides occurs equally efficiently, or better, at higher temperatures. Biologically relevant single-stranded targets for DMC alkylation could be telomere repeats (exemplified by oligonucleotide 27 in this work) or unfolded RNA motifs. Such substrates would then join the growing list of cellular components targeted by Mitomycins such as rRNA and Thioredoxin Reductase.33, 34
Finally, Table 2 and Table S1, supporting information, also demonstrate that the frequency of dA alkylation is highest in M. Luteus DNA compared to other DNA fragments. This was unexpected, as M. Luteus DNA contains the lowest proportion of AT bases of all duplex DNA substrates studied in this work. A plausible explanation may be that as the CG content increases in DNA, the binding affinity of DMC for DNA also increases and this leads to a higher dA alkylation frequency overall.
Conclusion
In the present work, we have successfully identified two novel stereoisomeric deoxyadenosine adducts of decarbamoylmitomycin C and provided a rationale for their formation. A crucial discovery was to establish that temperature governs the regioselectivity of duplex DNA alkylation by DMC: At 0°C, both dG and dA adducts are produced, but at 37 °C, DMC only targets dG nucleobases in our model systems. Another interesting finding is that the nucleophilic addition of dA can occur on the β- or α-face of reduced DMC, as is the case for dG alkylation.20 Reduction conditions (bifunctional versus monofunctional) and reaction temperature are therefore essential factors that determine the regio- and stereoselectivity of the alkylation of dA residues by DMC. As a final remark, it should be noted that single stranded DNA seems more prone to dA alkylation by DMC than duplex DNA, in contrast to what happens with dG alkylation. The prevailing concept is that DMC cytotoxicity is due to its ability to form ICLs in duplex DNA. The results presented here show that DMC is also able to react with single stranded nucleic acids and this may have biological relevance. For instance, DMC may bind to unfolded RNAs in vivo or telomere overhangs, as evidenced by the alkylation observed in the TTAGG human telomere repeat sequence, and this phenomenon may contribute to its cytotoxicity.
Supplementary Material
Figure S1: HPLC chromatogram of pure 5a and 5b; Figure S2. HPLC chromatograms of (a) digest of DMC-oligonucleotide complex formed with oligonucleotide 9 (b) co-injection with adduct 5a (c) co-injection with adduct 5b; Figure S3. HPLC chromatograms of (a) digest of DMC-oligonucleotide complex formed with duplex 6 (b) co-injection with 5a (c) co-injection with 5b; Figure S4. HRMS of adduct 5a; Figure S5. HRMS of adduct 5b; Figure S6: HPLC of (a) digest of duplex 6 treated with DMC without nuclease P1 (b) same sample with co-injection of standard 5a (c) same sample with co-injection of standard 5b (d) same sample but digest of duplex 6 includes nuclease P1; Figure S7: UV spectra of compounds X and Y; Table S1: Frequencies of DNA adducts detected in short oligonucleotides after alkylation by DMC under bifunctional conditions; Figure S8: 1H NMR spectrum of compound 5a; Figure S9: 2-D COSY spectrum of compound 5a; Figure S10: 1H NMR spectrum of compound 5b; Figure S11: 2-D COSY spectrum of compound 5b.
ACKNOWLEDGMENT
The authors would like to thank the PRISM (Program for Research Initiatives in Science and Math) program for support to O. Zacharias.
Funding Sources
This research was supported by a grant from the National Institute of Health (5SC3GM105460-04) to E.C.
ABBREVIATIONS
- dG
deoxyguanosine
- dA
deoxyadenosine
- dT
deoxythymine
- dI
deoxyinosine
- dC
deoxycytosine
- MC
mitomycin C
- DMC
decarbamoylmitomycin C
- SVD
Snake Venom Diesterase
- AP
Alkaline Phosphatase
REFERENCES
- (1).Beland FA, and Poirier MC (1994) DNA adducts and their consequences In Methods to assess DNA damage and repair: Interspecies comparisons (Tardiff RG, Lohman PHM, and Wogan GN, Eds.) pp 29–55, John Wiley and Sons Ltd, Chichester. [Google Scholar]
- (2).Zang H, and Gates KS (2003) Sequence specificity of DNA alkylation by the antitumor natural product leinamycin. Chem. Res. Toxicol 16, 1539–1546. [DOI] [PubMed] [Google Scholar]
- (3).Sugiyama H, Lian C, Isomura M, Saito I, and Wang AH-J (1996) Distamycin A modulates the sequence specificity of DNA alkylation by duocarmycin A. Proc. Natl. Acad. Sci. USA 93, 14405–14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tse WC, and Boger DL (2004) Sequence-selective DNA recognition: Natural products and nature’s lessons. Chem. Biol 11, 1607–1617. [DOI] [PubMed] [Google Scholar]
- (5).Bass PD, Gubler DA, Judd TC, and Williams RM (2013) Mitomycinoid alkaloids: mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev 113, 6816–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hata T, Hoshi T, Kanamori K, Matsumae A, Sano Y, Shima T, and Sugawara R (1956) Mitomycin, a new antibiotic from Streptomyces. I. J. Antibiot 9, 141–146. [PubMed] [Google Scholar]
- (7).Wakaki S, Marumo H, Tomioka K, Shimizu G, Kato E, Kamada H, Kudo S, and Fujimoto Y (1958) Isolation of new fractions of antitumor mitomycins. Antibiot. Chemother 8, 228–240. [PubMed] [Google Scholar]
- (8).Tomasz M (1995) Mitomycin C: small, fast and deadly (but very selective). Chem. Biol. 2, 575–579. [DOI] [PubMed] [Google Scholar]
- (9).Bradner WT (2001) Mitomycin C: a clinical update. Cancer Treat. Rev 27, 35–50. [DOI] [PubMed] [Google Scholar]
- (10).Cincik H, Güngör A, Cekin E, Saglam O, Yildirim S, Poyrazoglu E, and Candan H (2005) Effects of topical application of mitomycin-C and 5-fluorouracil on myringotomy in rats. Otol. Neurotol 26, 351–354. [DOI] [PubMed] [Google Scholar]
- (11).Kim TI, Choi SI, Lee HK, Cho YJ, and Kim EK (2008) Mitomycin C induces apoptosis in cultured corneal fibroblasts derived from type II granular corneal dystrophy corneas. Mol. Vis 14, 1222–1228. [PMC free article] [PubMed] [Google Scholar]
- (12).Lee JY, Stenzel W, Impekoven P, Theisohn M, Stützer H, Löhr M, Reithmeier T, Ernestus RI, Ebel H, and Klug N (2006) The effect of mitomycin C in reducing epidural fibrosis after lumbar laminectomy in rats. J. Neurosurg. Spine 5, 53–60. [DOI] [PubMed] [Google Scholar]
- (13).Wang S, Li X, Yan L, Chen H, Wang J and Sun Y (2017) Upregulation of P27Kip1 by mitomycin C induces fibroblast apoptosis and reduces epidural fibrosis Int. J. Clin. Exp. Pathol. 10, 11779–11788. [PMC free article] [PubMed] [Google Scholar]
- (14).Tomasz M, Chowdary D, Lipman R, Shimotakahara S, Veiro D, Walker V, and Verdine GL (1986) Reaction of DNA with chemically or enzymatically activated mitomycin C: Isolation and structure of the major covalent adduct. Proc. Natl. Acad. Sci. U. S. A 83, 6702–6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tomasz M, Lipman R, Chowdary D, Pawlak J, Verdine GL, and Nakanishi K (1987) Isolation and structure of a covalent cross-link adduct between mitomycin C and DNA. Science 235, 1204–1208. [DOI] [PubMed] [Google Scholar]
- (16).Paz MM, Ladwa S, Champeil E, Liu Y, Rockwell S, Boamah EK, Bargonetti J, Callahan J, Roach J, and Tomasz M (2008) Mapping DNA adducts of mitomycin C and decarbamoyl mitomycin C in cell lines using liquid chromatography/electrospray tandem mass spectrometry. Chem. Res. Toxicol 21, 2370–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rockwell S, and Kim SY (1995) Cytotoxic potential of monoalkylation products between mitomycins and DNA: Studies of decarbamoyl mitomycin C in wild-type and repair-deficient cell lines. Oncol. Res. 7, 39–47. [PubMed] [Google Scholar]
- (18).Kinoshita S, Uzu K, Nakano K, and Takahashi T (1971) Mitomycin derivatives. 2. Derivatives of decarbamoylmitosane and decarbamoylmitosene. J. Med. Chem 14, 109–112. [DOI] [PubMed] [Google Scholar]
- (19).Palom Y, Suresh Kumar G, Tang LQ, Paz MM, Musser SM, Rockwell S, and Tomasz M (2002) Relative toxicities of DNA cross-links and monoadducts: New insights from studies of decarbamoyl mitomycin C and mitomycin C. Chem. Res. Toxicol 15, 1398–1406. [DOI] [PubMed] [Google Scholar]
- (20)(a).Aguilar W, Paz MM, Vargas A, Cheng S-Y, Clement C, and Champeil E (2018) Sequence-dependent diastereospecific and diastereodivergent crosslinking of DNA by decarbamoylmitomycin C. Chem. Eur. J. 24, 6030–6035. . [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) : Aguilar W, Paz MM, Vargas A, Zheng M, Cheng S-Y, and Champeil E (2018) Interdependent sequence-selectivity and diastereoselectivity in the alkylation of DNA by decarbamoylmitomycin C. Chem. Eur. J, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiao G, Kue P, Bhosle RC, and Bargonetti J (2015) Decarbamoyl mitomycin C (DMC) activates p53-independent ataxia telangiectasia and rad3 related protein (ATR) chromatin eviction. Cell Cycle 14, 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cheng S-Y, Seo J, Huang BT, Napolitano T, and Champeil E (2016) Mitomycin C and decarbamoyl mitomycin C induce p53-independent p21 activation. Int. J. Oncol 49, 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Palom Y, Lipman R, Musser SM, and Tomasz M (1998) A mitomycin-N6-deoxyadenosine adduct isolated from DNA. Chem. Res. Toxicol 11, 203–210. [DOI] [PubMed] [Google Scholar]
- (24).NIH Website, https://pubchem.ncbi.nlm.nih.gov/compound/mitomycin_C#, last accessed 4-17-2018. [Google Scholar]
- (25).Kumar S, Lipman R, and Tomasz M (1992) Recognition of specific DNA sequences by mitomycin C for alkylation. Biochemistry 31, 1399–1407. [DOI] [PubMed] [Google Scholar]
- (26).Suresh Kumar G, Lipman R, Cummings J, and Tomasz M (1997) Mitomycin C-DNA adducts generated by DT-diaphorase. Revised mechanism of the enzymatic reductive activation of mitomycin C. Biochemistry 36, 14128–14136. [DOI] [PubMed] [Google Scholar]
- (27).Champeil E, Cheng S-Y, Huang BT, Concheiro-Guisan M, Martinez T, Paz MM and Sapse AM (2016) Synthesis of Mitomycin C and Decarbamoylmitomycin C N2 deoxyguanosine-adducts. Bioorg. Chem 65, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tomasz M, Jung M, Verdine GL, and Nakanishi K (1984) Circular dichroism spectroscopy as a probe for the stereochemistry of aziridine cleavage reactions of Mitomycin C. Applications to adducts of mitomycin with DNA constituents. J. Am. Chem. Soc 106, 7367–7370. [Google Scholar]
- (29).Paz MM and Pritsos CA (2012) The molecular toxicology of mitomycin C In Adv. Mol. Toxicol. Vol. 6 (Fishbein JC, Eds.) pp. 244–286, Elsevier, Amsterdam. [Google Scholar]
- (30).Tomasz M, Chawla AK, and Lipman R (1988) Mechanism of monofunctional and bifunctional alkylation of DNA by mitomycin C. Biochemistry 27, 3182–3187. [DOI] [PubMed] [Google Scholar]
- (31).Li V-S, and Kohn H (1991) Studies on the bonding specificity for mitomycin C-DNA monoalkylation processes. J. Am. Chem. Soc 113, 275–283. [Google Scholar]
- (32).Nakano S-I, Uotani Y, Nakashima S, Anno Y, Fujii M, and Sugimoto N (2003) Large stabilization of a DNA duplex by the deoxyadenosine derivatives tethering an aromatic hydrocarbon group. J. Am. Chem. Soc 125, 8086–8087. [DOI] [PubMed] [Google Scholar]
- (33).Snodgrass RG, Collier AC, Coon AE, and Pritsos CA (2010) Mitomycin C inhibits ribosomal RNA: A novel cytotoxic mechanism for bioreductive drugs. J. Biol. Chem 285, 19068–19075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Paz MM, Zhang X, Lu J, and Holmgren A (2012) A new mechanism of action for the anticancer drug Mitomycin C: Mechanism-based Inhibition of thioredoxin reductase. Chem. Res. Toxicol 25, 1502–1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: HPLC chromatogram of pure 5a and 5b; Figure S2. HPLC chromatograms of (a) digest of DMC-oligonucleotide complex formed with oligonucleotide 9 (b) co-injection with adduct 5a (c) co-injection with adduct 5b; Figure S3. HPLC chromatograms of (a) digest of DMC-oligonucleotide complex formed with duplex 6 (b) co-injection with 5a (c) co-injection with 5b; Figure S4. HRMS of adduct 5a; Figure S5. HRMS of adduct 5b; Figure S6: HPLC of (a) digest of duplex 6 treated with DMC without nuclease P1 (b) same sample with co-injection of standard 5a (c) same sample with co-injection of standard 5b (d) same sample but digest of duplex 6 includes nuclease P1; Figure S7: UV spectra of compounds X and Y; Table S1: Frequencies of DNA adducts detected in short oligonucleotides after alkylation by DMC under bifunctional conditions; Figure S8: 1H NMR spectrum of compound 5a; Figure S9: 2-D COSY spectrum of compound 5a; Figure S10: 1H NMR spectrum of compound 5b; Figure S11: 2-D COSY spectrum of compound 5b.