

Abstract
The cyclic nucleotide-gated (CNG) channel - composed of CNGA3 and CNGB3 subunits - mediates the influx of cations in cone photoreceptors after light stimulation and thus is a key element in cone phototransduction. Mutations in CNGA3 and CNGB3 are associated with achromatopsia, a rare autosomal recessive retinal disorder. Here, we demonstrate that the presence of an early nonsense mutation in CNGA3 induces the usage of a downstream alternative translation initiation site giving rise to a short CNGA3 isoform. The expression of this short isoform was verified by Western blot analysis and DAB staining of HEK293 cells and cone photoreceptor-like 661W cells expressing CNGA3-GST fusion constructs. Functionality of the short isoform was confirmed by a cellular calcium influx assay. Furthermore, patients carrying an early nonsense mutation were analyzed for residual cone photoreceptor function in order to identify a potential role of the short isoform to modify the clinical outcome in achromatopsia patients. Yet the results suggest that the short isoform is not able to compensate for the loss of the long isoform leaving the biological role of this variant unclear.
Keywords: Achromatopsia, Cone photoreceptor, CNGA3, Alternative translation initiation site
1. Introduction
The cone photoreceptor cyclic nucleotide-gated (CNG) channel represents a non-selective cation channel which is located in the cone photoreceptor outer segments (Biel and Michalakis, 2009; Kaupp and Seifert, 2002; Matulef and Zagotta, 2003). Recent investigations suggest a channel structure comprising three CNGA3 and one modulatory CNGB3 subunits (Shuart et al., 2011). Both subunits are structurally homologous proteins consisting of six transmembrane domains (S1–S6) with a pore forming region between S5 and S6 and a cyclic nucleotide binding domain which is connected to S6 by a C-linker. Under physiological conditions, the heteromeric CNG channel is activated by intracellular binding of cGMP allowing the influx of sodium and calcium ions into the cone photoreceptor outer segment.
This CNG channel plays a crucial role in the phototransduction cascade in cones and mutations in the genes for both channel subunits are the main cause for autosomal recessive achromatopsia (ACHM; OMIM 216900; OMIM 262300), a rare retinal disorder with a prevalence of approximately 1:30,000 (Wissinger et al., 2001; Sharpe et al., 1999). Two clinical subtypes of achromatopsia have been described, a complete and an incomplete form. In complete achromatopsia, cone photoreceptor function is completely absent causing a total lack of color discrimination, a best-corrected visual acuity of 20/200 or less, severe photophobia and a nystagmus (Kohl et al., 1998, 2000). The incomplete form is characterized by less severe symptoms: color discrimination may vary from sustained to severely impaired, visual acuity may reach values of 20/80 compared to 20/200 in complete achromatopsia and photophobia may be completely absent (Tränkner et al., 2004; Kohl et al., 2004). More than 100 different disease-causing mutations have been reported for CNGA3 [see HGMD (Wissinger et al., 2001; Kohl et al., 1998; Ahuja et al., 2008; Azam et al., 2010; Fahim et al., 2013; Genead et al., 2011; Goto-Omoto et al., 2006; Nishiguchi et al., 2005; Koeppen et al., 2008, 2010; Johnson et al., 2004; Li et al., 2014; Liang et al., 2015; Reuter et al., 2008; Thomas et al., 2012; Saqib et al., 2011, 2015; Shaikh et al., 2015; Sundaram et al., 2014; Thiadens et al., 2010; Tränkner et al., 2004; Vincent et al., 2011; Wang et al., 2011; Wiszniewski et al., 2007; Zelinger et al., 2015);], comprising mainly missense mutations and a smaller fraction of nonsense mutations, splice site mutations, insertions and/or deletions.
Here we report the identification of an alternative translation initiation site (TIS) downstream of a nonsense mutation in CNGA3 causing the expression of a short isoform of CNGA3 with residual activity.
2. Material and methods
2.1. Subjects and molecular genetic analysis
This study was performed according to the tenets of the Declaration of Helsinki, and the participants gave written consent, approved by the respective local research and ethical review boards. Achromatopsia patients were examined at an ophthalmologic centre and biosamples referred to the Molecular Genetics Laboratory at the Centre for Ophthalmic Research, University of Tübingen for genetic testing. The clinical assessment of the family CHRO 590 was based on visual acuity testing using a Snellen vision chart and a saturated Farnsworth D-15 color vision test. Venous blood was taken and total genomic DNA was extracted according to standard procedures. All coding exons and flanking intronic regions of CNGA3 (OMIM 216900) were sequenced as reported previously (Kohl et al., 1998).
2.2. Heterologous expression of human CNGA3
The generation of the wild type human CNGA3 expression construct (RefSeq NM_001298.2) was described previously (Tränkner et al., 2004). In vitro mutagenesis PCR was used to generate the mutant CNGA3 expression constructs CNGA3R23X, CNGA3R23X,ΔM52, CNGA3W171X and CNGA3E228K for functional characterization using the bioassay described below. CNGA3E228K is a well studied mutation causing incomplete achromatopsia and was used as a control with residual functional activity whereas CNGA3W171X does not produce a functional protein serving as negative control.
A shortened CNGA3-glutathione S-transferase (GST)-fusion construct was generated in pcDNA3.1/MycHis A(+) (Life Technologies Corporation, Carlsbad, CA, USA) by overlap extension PCR comprising a 306 bp fragment of the human CNGA3 gene (c.−117C to c.189C) and the GST sequence from pETM-30 (EMBL Heidelberg, Heidelberg, Germany). Mutant CNGA3-GST fusion constructs CNGA3R23X-GST and CNGA3R23X,ΔM52-GST were generated by in vitro mutagenesis. The plasmid pCAEQ encoding for apoaequorin was kindly provided by Perkin Elmer (Waltham, MA) and was modified by excision of the mitochondrial targeting sequence.
Human embryonic kidney 293 (HEK293) cells and the murine cone photoreceptor derived 661W cells (kindly provided by Muayyad R. Al-Ubaidi; University of Oklahoma Health Sciences, Oklahoma City, OK) were maintained in Dulbecco’s Modified Eagle Medium (Life Technologies Corporation, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS; Life Technologies Corporation, Carlsbad, CA, USA), 1% fungizone (PAA, Linz, Austria) and 1% penicillin/streptomycine (Sigma Aldrich GmbH, Munich, Germany) at 37 °C and 5% CO2. Transfections were carried out using lipofectamine 2000 (Life Technologies Corporation, Carlsbad, CA, USA) following manufacturer’s instruction. HEK293 cells were seeded at a density of approximately 2 × 105 cells/cm2 and transfected with plasmids encoding for apoaequorin and CNGA3 at a ratio of 1.0:2.1 for bioassay experiments or 7.5 μg of plasmid encoding for CNGA3-GST fusion proteins for Western blot. For immunocytochemical staining, 661W cells were seeded at a density of 5 × 104 cells/cm2 and transfected with 3 μg of plasmid encoding for CNGA3-GST fusion constructs.
2.3. Aequorin-based bioassay
Six hours post-transfection, HEK293 cells were washed with Dulbecco’s phosphate buffered saline (DPBS; Life Technologies Corporation, Carlsbad, CA, USA), detached by Trypsin-EDTA (Life Technologies Corporation, Carlsbad, CA, USA) and seeded in opaque 96-well plates at a density of 7.5 × 105 cells per well. After overnight incubation, cells were treated with 3 mM sodium butyrate (Sigma Aldrich GmbH, Munich, Germany) for 24 h. Subsequently, cells were incubated with 8 μM coelenterazine (Biomol GmbH, Hamburg, Germany) in calcium imaging solution (5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES pH 7.4, 30 mM Glucose, 150 mM NaCl) to reconstitute the aequorin. Luminescence measurements were carried out using an Orion luminometer equipped with an injection unit (Titertek-Berthold Detection Systems, Pforzheim, Germany). Cells were kept in calcium imaging solution containing 10 mM CaCl2 and luminescence was recorded 6 s before and 94 s after automatic application of the cGMP analogue 8-bromoguanosine-3′,5′-cyclic monophosphate (8-Br-cGMP; BIOLOG Life Science Institute, Bremen, Germany) at a final concentration of 10 mM. For data analysis, the base line before addition of the ligand 8-Br-cGMP was subtracted from all values of a single measurement and the area under the curve was calculated for a time interval of t = 14–20 s after begin of the measurement. For comparison of wild type and mutant luminescence responses, the AUC data were normalized to the wild type values to obtain the fold change (FC). Statistical analysis was done using Kruskal-Wallis one-way analysis of variance in Mystat (Systat Software Inc. London, UK). Data are presented as mean ± SE, significance levels are p > 0.05: not significant (n.s.); p < 0.01: **; p < 0.001: ***.
2.4. SDS-PAGE and Western blot
Six hours post-transfection, HEK293 cells were seeded in 6-cm dishes and following an overnight incubation treated with 3 mM sodium butyrate for 30 h. Cells were trypsinized, washed gently with DPBS and transferred into ice cold lysis buffer (1 mM DTT, 1 mM EDTA, 50 mM Tris and 150 mM NaCl, pH 7.4) containing 1% proteinase inhibitor cocktail (Merck Millipore, Billerica, MA). Cell lysis was performed by four freeze/thaw cycles using liquid nitrogen. After cell debris removal by centrifugation at 500×g for 20 min at 4 °C, membrane fractions were enriched by centrifugation at 30 000×g for 45 min at 4 °C. The pellet was resuspended in 50 μl lysis buffer containing 1% NP-40 and 0.25% sodium deoxycholate. Protein samples were incubated with Laemmli buffer for 1 h at 4 °C, separated on a 10% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane. Blots were blocked overnight with 5% milk powder (Bio-Rad Laboratories GmbH, Munich, Germany) in TBST and probed with mouse anti-c-myc antibody (1:700; Enzo Life Sciences, Lörrach, Germany) or mouse anti-β-actin antibody (1:4000; Merck Millipore, Billerica, MA) as loading control. For detection, a HRP-conjugated (horse radish peroxidase) goat anti-mouse (1:10 000; Merck Millipore, Billerica, MA) secondary antibody was used.
2.5. Immunocytochemical staining
Six hours post-transfection, 661W cells were trypsinized and seeded on poly-L-lysine (Sigma Aldrich GmbH, Munich, Germany) coated glass cover slips. After overnight incubation, cells were treated with 3 mM sodium butyrate for 24 h, washed with DPBS and then fixed with 4% paraformaldehyde for 15 min. Cells were blocked in PBS with 10% FCS and 0.1% Triton X for 30 min, followed by an incubation of primary antibody against c-myc (1:700, Enzo Life Sciences, Lörrach, Germany) diluted in PBS containing 10% FCS and 0.1% Triton X for 1 h. Secondary antibody staining was performed using HRP-conjugated goat-anti-mouse secondary antibody (1:10 000, Merck Millipore, Billerica, MA) diluted in PBS containing 10% FCS and incubation for 1 h. For visualization, a diaminobenzidine (DAB) staining was performed applying 0.005% DAB and 0.12% H2O2 in PBS. After 2 min incubation, the DAB solution was replaced by PBS. All incubation steps were performed at RT and between each step, cells were washed with PBS. Images from stained cells were acquired using the Axio Imager Z1 (Carl Zeiss, Oberkochen, Germany).
3. Results
3.1. Residual channel function with CNGA3 harbouring the p.R23Xmutation
To analyze the functionality of CNGA3R23X mutant channels, a cellular assay using the calcium-sensitive photoprotein aequorin, which allows the quantification of CNG channel mediated calcium influx, was used. For comparison we assayed in parallel CNGA3 wild type channels as well as the variants CNGA3E228K, a well studied mutant CNGA3 channel with residual activity causing incomplete achromatopsia (Reuter et al., 2008), and CNGA3W171X with no residual activity associated with complete achromatopsia. In HEK293 cells expressing CNGA3WT, the application of 8-Br-cGMP resulted in a transient luminescence signal with a maximum increase of 2.38 ± 0.13 × 105 RLU (Fig. 1 A), which is not observed in untransfected control cells and thus represents CNGA3 channel-mediated calcium influx. Surprisingly, the mutant CNGA3R23X showed significant albeit reduced channel function (FC = 0.25 ± 0.01) comparable to that of the hypomorphic mutant CNGA3E228K (FC = 0.23 ± 0.02) whereas CNGA3W171X channels yield barely detectable signals (FC = 0.01 ± 0.00; p < 0.01; Fig. 1 B). This finding suggests that CNGA3R23X can form functional homomeric channels in our heterologous expression system.
Fig. 1. Relative quantification of the ligand-induced calcium influx through mutant and wild type human CNGA3 channels.
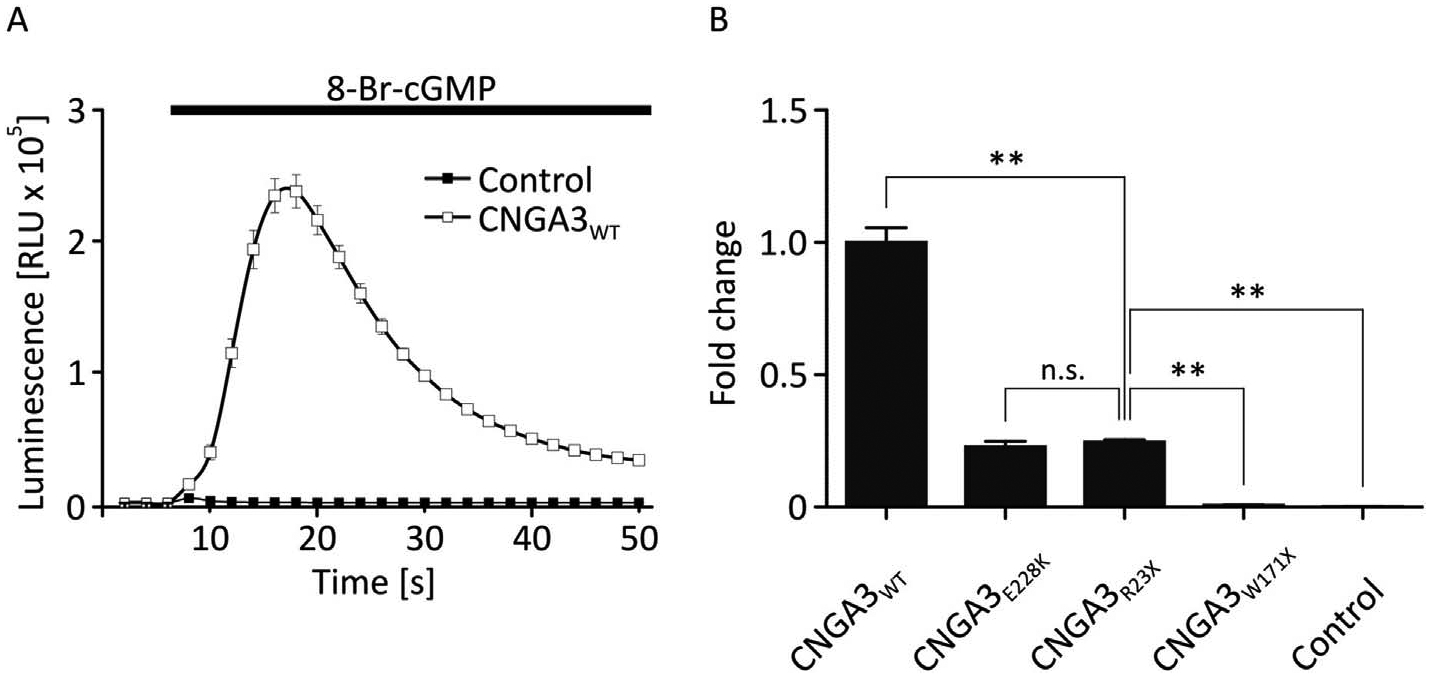
(A) A bioassay was used for the relative quantification of the calcium influx through CNG channels in a heterologous expression system using the calcium sensitive photoprotein aequorin. For all measurements the ligand 8-Br-cGMP was applied at the same time after begin of the measurement. HEK293 cells expressing wild type CNG channels and aequorin showed a transient luminescence peak after application of the ligand 8-Br-cGMP (empty boxes) which was absent in cells expressing only the photoprotein (filled boxes). A high signal-to-noise ratio was obtained indicating that the assay was suitable for analyzing the calcium influx through CNGA3. (B) The area under the curve (AUC) of the luminescence signal was calculated for a time interval of t =14–20 s after begin of the measurement corresponding to the peak values of the signal in wild type CNGA3. Two CNGA3 mutants with stop mutations were analyzed with the bioassay. No calcium influx was measured for CNGA3W171X, whereas CNGA3R23X yielded a calcium signal which was about 4-fold reduced compared to the wild type protein (n = 6) but not significantly different compared to the hypomorphic mutant channel CNGA3E228K, which served as a control for a mutant with residual channel function. Cells expressing only aequorin served as a control
3.2. Identification of a second translation initiation site in CNGA3
Since the mutation p.R23X is located close to the N-terminus of CNGA3, we asked whether a second in-frame TIS is present in CNGA3 mRNA that facilitates the expression of a shorter protein variant and thereby may explain the channel function observed in the bioassay. The open reading frame of the human CNGA3 gene bears another AUG codon at p.M52. In order to evaluate the use of this potential alternative TIS, we deleted p.M52 in the construct encoding for CNGA3R23X thus generating CNGA3R23X, ΔM52. In the bioassay, the deletion of p.M52 resulted in a significant reduction of the luminescence signal in comparison to CNGA3R23X (CNGA3R23X, ΔM52: FC = 0.04 ± 0.01; CNGA3R23X: FC = 0.26 ± 0.01) suggesting that the expression of CNGA3 polypeptide is virtually abolished by the deletion of the alternative TIS (Fig. 2).
Fig. 2. Calcium influx through mutant human CNGA3 channels confirms the p.R23X induced activation of an alternative TIS in CNGA3.
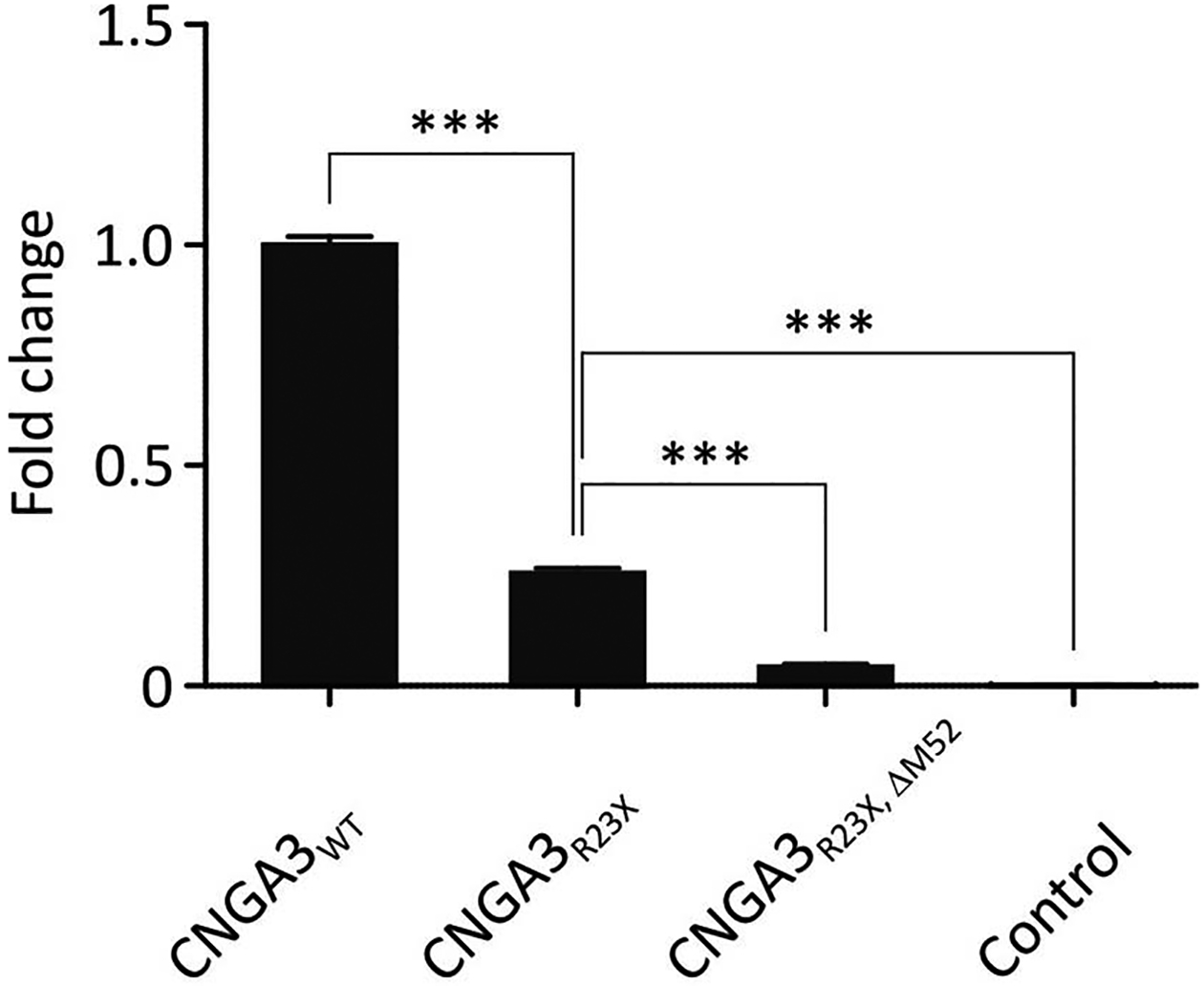
The bar diagram shows AUC calculations of the luminescence signal normalized to the wildtype CNGA3 value. The ligand-induced calcium influx observed in HEK293 cells expressing CNGA3R23X was virtually abolished after deletion of p.M52 (n ≥ 6). Wild type CNGA3 expressing cells and cells expressing no CNG channel served as a positive and negative control, respectively.
3.3. Verification of expression of the amino-terminal truncated CNGA3 protein
For verification of protein expression initiated at p.M52, we performed Western blot experiments. In order to detect differences in protein sizes the first 63 codons of the CNGA3 open reading frame were fused to c-myc tagged GST. HEK293 cells expressing CNGA3WT-GST showed two bands on the Western blot, a longer isoform of about 35 kDa in size corresponding to a polypeptide with p.M1 as amino-terminus and a shorter isoform of about 30 kDa corresponding to a polypeptide with p.M52 as amino-terminus (Fig. 3 A). The short isoform was about 5-fold less abundant compared to the long transcript. Upon transfection of the CNGA3R23X-GST construct, only the short isoform was observed but with a higher abundance (about 3-fold) than in CNGA3WT-GST. No protein bands were observed in lysates of HEK293 cells transfected with the CNGA3R23X, ΔM52-GST construct.
Fig. 3. Expression of the short CNGA3 isoform in HEK293 and 661W cells verified by Western blot experiments and by DAB staining.
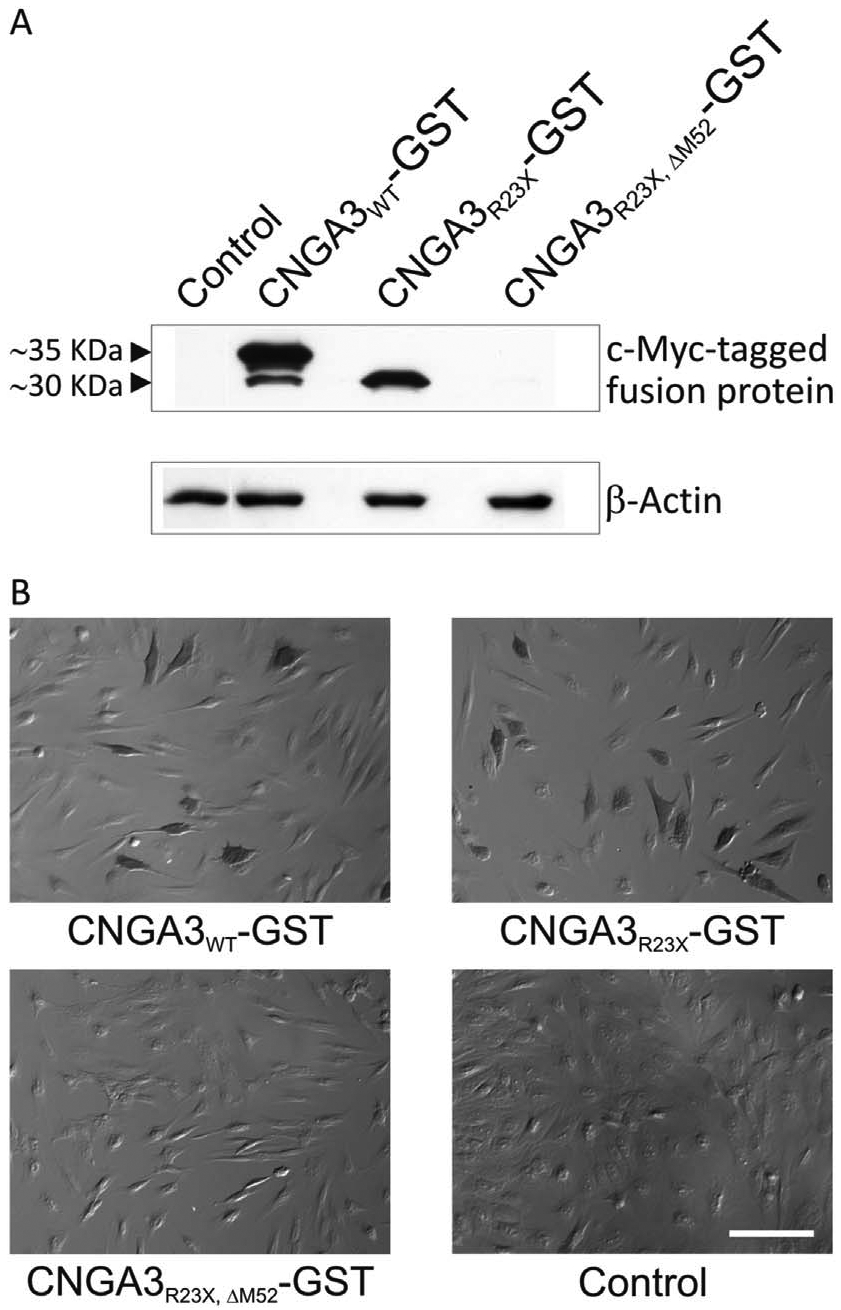
(A) Shortened C-terminal myc-tagged CNGA3-GST fusion constructs were used to detect the small differences in the size of the short and long CNGA3 isoform by Western blot. Wild type and mutant CNGA3-GST fusion proteins in lysates of transfected HEK293 cells were visualized using a c-myc antibody and β-actin served as loading control. Lysates of HEK293 cells transfected with the CNGA3WT-GST construct showed two bands of about 35 kDa and 30 kDa, which were absent in the untransfected control. The mutant CNGA3R23X-GST construct only yielded the 30 kDa band, which was absent after deletion of the alternative TIS in the CNGA3R23X,ΔM52-GST construct. (B) DAB staining of murine cone-photoreceptor-like 661W cells transfected with different CNGA3-GST fusion constructs in comparison with untransfected cells (control). DAB staining was observed in cells transfected with the CNGA3WT-GST and the CNGA3R23X-GST constructs but absent in cells transfected with the double mutant CNGA3R23X,ΔM52-GST construct. Scale bar represents 100 μm.
To verify that the expression of the short isoform is not an artefact observed in HEK293 cells, we performed a DAB staining using the cone-like cell line 661W after heterologous expression of the GST fusion constructs. After expression of CNGA3WT-GST and CNGA3R23X-GST, DAB-positive cells were observed indicating that both isoforms are expressed in these cone photoreceptor-like cells (Fig. 3 B). No DAB-positive cells were found upon transfection of 661W cells with the CNGA3R23X, ΔM52-GST construct.
3.4. Clinical examination of achromatopsia patients with early nonsense mutations in CNGA3
In our ongoing genetic screening of the CNGA3 gene, we identified two siblings with a clinical diagnosis of ACHM at young age carrying compound heterozygous nonsense mutations in CNGA3. Both siblings carry the previously described early nonsense mutation c.67C > T; p.R23X (Johnson et al., 2004) and the known nonsense mutation c.1030G > T; p.E344X (Nishiguchi et al., 2005). Both siblings of family CHRO590 presented with strongly reduced visual acuity of 20/400 and the saturated Farnsworth D-15 test showed severely perturbed color vision in one patient. Taken together, the clinical data of both subjects suggest that both patients have complete achromatopsia.
4. Discussion
Here, we report the identification of a novel isoform of human CNGA3 resulting from an in-frame alternative TIS 154 bp downstream of the first TIS. Translation starting from the alternative TIS gives rise to a protein lacking the N-terminal 51 amino acids of the long isoform of CNGA3. This short isoform was identified by expression of mutant CNGA3R23X channels carrying an early stop codon and performing functional studies by measuring calcium influx in a heterologous expression system. Deleting the second start codon (p.M52) in CNGA3R23X completely abolished the calcium signal.
The use of the alternative TIS was verified by Western blot experiment and immunocytochemical staining. Those experiments showed a reduced but well detectable expression of the shortened CNGA3. Interestingly, this isoform was also present in CNGA3WT-expressing cells, although much less abundant. This suggests that the short and the long isoform are both expressed in wild type HEK cells. The immunocytochemical staining using 661W cells showed that cone photoreceptor-like cells are in principle capable of expressing the short isoform of CNGA3. Since we have used the human CNGA3 including upstream sequences in all experiments, we reason that the short protein isoform is also expressed in human cone photoreceptors.
The higher amount of the long isoform of CNGA3 after expression of CNGA3WT compared to the short isoform indicates that p.M1 is the preferred TIS. In CNGA3R23X-expressing cells only the short isoform of CNGA3 was detected with a higher abundance than in CNGA3WT. This may suggest that the expression level of the short isoform is elevated if an early nonsense mutation abolishes the expression of the long isoform. Compared with HEK293 cells transfected with the wild type CNGA3 construct, ligand-induced calcium influx is reduced in cells expressing CNGA3R23X, most likely due to the lower total CNGA3 level. Yet calcium influx in cells expressing CNGA3R23X is similar to that in cells expressing CNGA3E228K. Since CNGA3E228K allows patients carrying this mutation to maintain residual cone photoreceptor function (Reuter et al., 2008), we speculated that patients carrying early nonsense mutations would also show less severe symptoms typical for incomplete achromatopsia. However, the clinical data available for the two siblings of family CHRO590 both being compound heterozygous for p.R23X and a further downstream stop mutation rather support a complete lack of cone photoreceptor function. Published clinical data from other achromatopsia patients carrying early nonsense mutations confirm a complete achromatopsia phenotype with no evidence for residual cone function (Johnson et al., 2004; Li et al., 2014; Liang et al., 2015; Zelinger et al., 2015). Among these are three patients homozygous for the mutation p.R23X. One patient showed at young age a decreased visual acuity of 3/60 and a complete lack of cone ERG response (Johnson et al., 2004). In the other two patients, the visual acuity could not be assessed but the 30 Hz cone flicker ERG was not detectable (Zelinger et al., 2015), and thus in all three patients no cone photoreceptor function was observed.
Therefore, the clinical findings suggest that the short isoform of CNGA3 is not able to compensate the loss of the long isoform. The reason for the lack of residual cone photoreceptor function is yet unknown, but an impaired assembly of the short CNGA3 isoform with the CNGB3 subunit or a hampered transport to the cone photoreceptor outer segments may be involved. Effects such as a loss of known ion channel modulator interaction sites can be excluded, since those sites are located further downstream of the alternative TIS of CNGA3 (Dai et al., 2013; Dai and Varnum, 2013; Grunwald et al., 1999).
Alternative translation initiation is an evolutionary conserved mechanism increasing the diversity of the proteome of a cell and has already been shown for multiple genes, including other ion channels such as the chloride channels CFTR and TMEM16A as well as the K2P channels TREK-1 and TREK-2 (Ramalho et al., 2009; Thomas et al., 2008; Kisselbach et al., 2014; Sondo et al., 2014). Three different mechanisms have been proposed facilitating expression from alternative TIS (Kozak, 1999). The scanning for the start codon of the cap-mediated canonical translation initiation may be “leaky” and allow translation initiation at a downstream start site (Chenik et al., 1995). Secondly, after translation termination of an upstream open reading frame, the ribosome may continue scanning for a second TIS when the translation termination signal is followed shortly after the first TIS (Meyers, 2003). Alternatively, internal ribosome entry sites (IRES) may allow the binding of ribosomes close to the alternative TIS, similar to the IRES-mediated translation initiation in viruses (Allam and Ali, 2010; Chan et al., 2013). We suggest that the short isoform is the result of a “leaky” ribosomal scanning, because (I) the short isoform is also present in cells expressing wild type CNGA3 (see Fig. 3A) and (II) in CNGA3R23X-expressing cells, the short isoform is present with higher abundance compared to CNGA3WT suggesting that the ribosome continues scanning more frequently after initiating the translation at the first start side and reaching the stop codon.
Acknowledgments
This work was supported by the German Ministry for Education and Research (BMBF, program on rare diseases, IonNeurONet: 01GM1105A).
Footnotes
Conflicts of interest
None.
References
- Ahuja Y, Kohl S, Traboulsi EI, 2008. CNGA3 mutations in two United Arab Emirates families with achromatopsia. Mol. Vis 14, 1293–1297 [PMC free article] [PubMed] [Google Scholar]
- Allam H, Ali N, 2010. Initiation factor eIF2-independent mode of c-Src mRNA translation occurs via an internal ribosome entry site. J. Biol. Chem 285 (8), 5713–5725. 10.1074/jbc.M109.029462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M, Collin RW, Shah ST, Shah AA, Khan MI, Hussain A, et al. , 2010. Novel CNGA3 and CNGB3 mutations in two Pakistani families with achromatopsia. Mol. Vis 16, 774–781. [PMC free article] [PubMed] [Google Scholar]
- Biel M, Michalakis S, 2009. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol 191, 111–136. 10.1007/978-3-540-68964-5_7. [DOI] [PubMed] [Google Scholar]
- Chan CP, Kok KH, Tang HM, Wong CM, Jin DY, 2013. Internal ribosome entry site-mediated translational regulation of ATF4 splice variant in mammalian unfolded protein response. Biochim. Biophys. Acta 1833 (10), 2165–2175. 10.1016/j.bbamcr.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Chenik M, Chebli K, Blondel D, 1995. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA is mediated by a ribosomal leaky scanning mechanism. J. Virol 69 (2), 707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai G, Varnum MD, 2013. CNGA3 achromatopsia-associated mutation potentiates the phosphoinositide sensitivity of cone photoreceptor CNG channels by altering inter-subunit interactions. Am. J. Physiol. Cell Physiol 305 (2), C147–C159. 10.1152/ajpcell.00037.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai G, Peng C, Liu C, Varnum MD, 2013. Two structural components in CNGA3 support regulation of cone CNG channels by phosphoinositides. J. Gen. Physiol 141 (4), 413–430. 10.1085/jgp.201210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahim AT, Khan NW, Zahid S, Schachar IH, Branham K, Kohl S, et al. , 2013. Diagnostic fundus autofluorescence patterns in achromatopsia. e2 Am. J. Ophthalmol 156 (6), 1211–1219. 10.1016/j.ajo.2013.06.033. [DOI] [PubMed] [Google Scholar]
- Genead MA, Fishman GA, Rha J, Dubis AM, Bonci DM, Dubra A, et al. , 2011. Photoreceptor structure and function in patients with congenital achromatopsia. Invest. Ophthalmol. Vis. Sci 52 (10), 7298–7308. 10.1167/iovs.11-7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Omoto S, Hayashi T, Gekka T, Kubo A, Takeuchi T, Kitahara K, 2006. Compound heterozygous CNGA3 mutations (R436W, L633P) in a Japanese patient with congenital achromatopsia. Vis. Neurosci 23 (3–4), 395–402. 10.1017/s095252380623308x. [DOI] [PubMed] [Google Scholar]
- Grunwald ME, Zhong H, Lai J, Yau KW, 1999. Molecular determinants of the modulation of cyclic nucleotide-activated channels by calmodulin. Proc. Natl. Acad. Sci. U. S. A 96 (23), 13444–13449. 10.1073/pnas.962313444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Michaelides M, Aligianis IA, Ainsworth JR, Mollon JD, Maher ER, et al. , 2004. Achromatopsia caused by novel mutations in both CNGA3 and CNGB3. J. Med. Genet 41 (2) e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R, 2002. Cyclic nucleotide-gated ion channels. Physiol. Rev 82 (3), 769–824. 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- Kisselbach J, Seyler C, Schweizer PA, Gerstberger R, Becker R, Katus HA, Thomas D, 2014. Modulation of K2P 2.1 and K2P 10.1 K(+) channel sensitivity to carvedilol by alternative mRNA translation initiation. Br. J. Pharmacol 171 (23), 5182–5194. 10.1111/bph.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen K, Reuter P, Kohl S, Baumann B, Ladewig T, Wissinger B, 2008. Functional analysis of human CNGA3 mutations associated with colour blindness suggests impaired surface expression of channel mutants A3(R427C) and A3(R563C). Eur. J. Neurosci 27 (9), 2391–2401. 10.1111/j.1460-9568.2008.06195.x. [DOI] [PubMed] [Google Scholar]
- Koeppen K, Reuter P, Ladewig T, Kohl S, Baumann B, Jacobson SG, et al. , 2010. Dissecting the pathogenic mechanisms of mutations in the pore region of the human cone photoreceptor cyclic nucleotide-gated channel. Hum. Mutat 31 (7), 830–839. 10.1002/humu.21283. [DOI] [PubMed] [Google Scholar]
- Kohl S, Marx T, Giddings I, Jagle H, Jacobson SG, Apfelstedt-Sylla E, et al. , 1998. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat. Genet 19 (3), 257–259. 10.1038/935. [DOI] [PubMed] [Google Scholar]
- Kohl S, Baumann B, Broghammer M, Jagle H, Sieving P, Kellner U, et al. , 2000. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum. Mol. Genet 9 (14), 2107–2116. [DOI] [PubMed] [Google Scholar]
- Kohl S, Jagle H, Wissinger B, 2004. Achromatopsia In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K (Eds.), GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle, pp. 1993–2015. [PubMed] [Google Scholar]
- Kozak M, 1999. Initiation of translation in prokaryotes and eukaryotes. Gene 234 (2), 187–208. [DOI] [PubMed] [Google Scholar]
- Li S, Huang L, Xiao X, Jia X, Guo X, Zhang Q, 2014. Identification of CNGA3 mutations in 46 families: common cause of achromatopsia and cone-rod dystrophies in Chinese patients. JAMA Ophthalmol 132 (9), 1076–1083. 10.1001/jamaophthalmol.2014.1032. [DOI] [PubMed] [Google Scholar]
- Liang X, Dong F, Li H, Li H, Yang L, Sui R, 2015. Novel CNGA3 mutations in Chinese patients with achromatopsia. Br. J. Ophthalmol 99 (4), 571–576. 10.1136/bjophthalmol-2014-305432. [DOI] [PubMed] [Google Scholar]
- Matulef K, Zagotta WN, 2003. Cyclic nucleotide-gated ion channels. Annu. Rev. Cell Dev. Biol 19, 23–44. 10.1146/annurev.cellbio.19.110701.154854. [DOI] [PubMed] [Google Scholar]
- Meyers G, 2003. Translation of the minor capsid protein of a calicivirus is initiated by a novel termination-dependent reinitiation mechanism. J. Biol. Chem 278 (36), 34051–34060. 10.1074/jbc.M304874200. [DOI] [PubMed] [Google Scholar]
- Nishiguchi KM, Sandberg MA, Gorji N, Berson EL, Dryja TP, 2005. Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum. Mutat 25 (3), 248–258. 10.1002/humu.20142. [DOI] [PubMed] [Google Scholar]
- Ramalho AS, Lewandowska MA, Farinha CM, Mendes F, Goncalves J, Barreto C, Harris A, Amaral MD, 2009. Deletion of CFTR translation start site reveals functional isoforms of the protein in CF patients. Cell. Physiol. Biochem 24 (5–6), 335–346. 10.1159/000257426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter P, Koeppen K, Ladewig T, Kohl S, Baumann B, Wissinger B, 2008. Mutations in CNGA3 impair trafficking or function of cone cyclic nucleotide-gated channels, resulting in achromatopsia. Hum. Mutat 29 (10), 1228–1236. 10.1002/humu.20790. [DOI] [PubMed] [Google Scholar]
- Saqib MA, Awan BM, Sarfraz M, Khan MN, Rashid S, Ansar M, 2011. Genetic analysis of four Pakistani families with achromatopsia and a novel S4 motif mutation of CNGA3. Jpn. J. Ophthalmol 55 (6), 676–680. 10.1007/s10384-011-0070-y. [DOI] [PubMed] [Google Scholar]
- Saqib MA, Nikopoulos K, Ullah E, Sher Khan F, Iqbal J, Bibi R, et al. , 2015. Homozygosity mapping reveals novel and known mutations in Pakistani families with inherited retinal dystrophies. Sci. Rep 5, 9965 10.1038/srep09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh RS, Reuter P, Sisk RA, Kausar T, Shahzad M, Maqsood MI, et al. , 2015. Homozygous missense variant in the human CNGA3 channel causes cone-rod dystrophy. Eur. J. Hum. Genet 23 (4), 473–480. 10.1038/ejhg.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe LT, Stockman A, Jagle H, Nathans J, 1999. Opsin genes, cone photopigments, color vision, and color blindness In: Gegenfurtner K, Sharpe LT (Eds.), Color Vision: from Genes to Perception. Cambridge University Press, Cambridge, UK, pp. 3–52. [Google Scholar]
- Shuart NG, Haitin Y, Camp SS, Black KD, Zagotta WN, 2011. Molecular mechanism for 3:1 subunit stoichiometry of rod cyclic nucleotide-gated ion channels. Nat. Commun 2, 457 10.1038/ncomms1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondo E, Scudieri P, Tomati V, Caci E, Mazzone A, Farrugia G, Ravazzolo R, Galietta LJ, 2014. Non-canonical translation start sites in the TMEM16A chloride channel. Biochem Biophys Acta 1838 (1 Pt B), 89–97. 10.1016/j.bbamem.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram V, Wilde C, Aboshiha J, Cowing J, Han C, Langlo CS, et al. , 2014. Retinal structure and function in achromatopsia: implications for gene therapy. Ophthalmology 121 (1), 234–245. 10.1016/j.ophtha.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiadens AA, Roosing S, Collin RW, van Moll-Ramirez N, van Lith-Verhoeven JJ, van Schooneveld MJ, et al. , 2010. Comprehensive analysis of the achromatopsia genes CNGA3 and CNGB3 in progressive cone dystrophy. e1. Ophthalmology 117 (4), 825–830. 10.1016/j.ophtha.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Thomas D, Plant LD, Wilkens CM, McCrossan ZA, Goldstein SA, 2008. Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 58 (6), 859–870. 10.1016/j.neuron.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MG, McLean RJ, Kohl S, Sheth V, Gottlob I, 2012. Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. Br. J. Ophthalmol 96 (9), 1232–1236. 10.1136/bjophthalmol-2012-301737.PubMed. [DOI] [PubMed] [Google Scholar]
- Tränkner D, Jägle H, Kohl S, Apfelstedt-Sylla E, Sharpe LT, Kaupp UB, et al. , 2004. Molecular basis of an inherited form of incomplete achromatopsia. J. Neurosci 24 (1), 138–147. 10.1523/JNEUROSCI.3883-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A, Wright T, Billingsley G, Westall C, Heon E, 2011. Oligocone trichromacy is part of the spectrum of CNGA3-related cone system disorders. Ophthalmic Genet. 32 (2), 107–113. 10.3109/13816810.2010.544366. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang H, Cao M, Li Z, Chen X, Patenia C, et al. , 2011. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Hum. Mutat 32 (12), 1450–1459. 10.1002/humu.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissinger B, Gamer D, Jagle H, Giorda R, Marx T, Mayer S, et al. , 2001. CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet 69 (4), 722–737. 10.1086/323613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiszniewski W, Lewis RA, Lupski JR, 2007. Achromatopsia: the CNGB3 p.T383fsX mutation results from a founder effect and is responsible for the visual phenotype in the original report of uniparental disomy 14. Hum. Genet 121 (3–4), 433–439. 10.1007/s00439-006-0314-y. [DOI] [PubMed] [Google Scholar]
- Zelinger L, Cideciyan AV, Kohl S, Schwartz SB, Rosenmann A, Eli D, et al. , 2015. Genetics and disease expression in the CNGA3 form of achromatopsia: steps on the path to gene therapy. Ophthalmology 122 (5), 997–1007. 10.1016/j.ophtha.2014.11.025. [DOI] [PubMed] [Google Scholar]