

Abstract
Epidermal growth factor receptor (EGFR) is highly expressed in head and neck squamous cell carcinoma (HNSCC) and related to cancer progression. The resistance to anti-EGFR therapy remains a major clinical problem in HNSCC. In this study, we found that TOLL-like receptor 4 (TLR4) was highly expressed in 50% of EGFR overexpressed HNSCC biopsies, which correlated to worse prognosis in patients. In HNSCC cell lines, activation of TLR4 reversed cetuximab-induced the inhibition of proliferation, migration and invasion. LPS induced of TLR4 signaling was potentiated under cetuximab treatment, showing increased activation of downstream NF-κB and MAPK pathways. Accordingly, cetuximab treatment also increased expression of TNF-α, COX2, and other molecules involved in TLR4 induced tumor inflammation. Mechanistically, we found inhibition of EGFR by cetuximab led to decreased phosphorylation of Src and sequentially Src-medicated activation of Cbl-b. This inhibited Cbl-b-mediated degradation of the key TLR4 adaptor protein MyD88 and activated TLR4 signaling. TLR4 or MyD88 overexpressed CAL27 and SCC4 cells grew faster and were more resistant to cetuximab and gefitinib both in vitro and in vivo. Out study delineates a crosstalk between EGFR and TLR4 pathways and identified TLR4 as a potential biomarker as well as a therapeutic target in overcoming the resistance to anti-EGFR therapy of HNSCC.
Keywords: TLR4, HNSCC, EGFR, cetuximab
Introduction
Epidermal growth factor receptor (EGFR) protein as well as mRNA are reported to be overexpressed in 40% to 90% of head and neck squamous cell carcinomas (HNSCC), which is correlated to increased tumor growth and metastasis, poor prognosis, and resistance to chemotherapy and radiotherapy [1,2]. Several EGFR-targeted therapies have been approved for the treatment of HNSCC, including small-molecule tyrosine kinase inhibitors (e.g., gefitinib and erlotinib) and monoclonal antibodies against EGFR (e.g., cetuximab, panitumumab and zalutumumab) [3]. Among these, cetuximab is the most intensively studied and the only FDA approved EGFR inhibitor for use in HNSCC [4]. Cetuximab is a chimeric human-murine immunoglobulin (Ig) G1 monoclonal antibody that can bind to EGFR with similar affinity to its natural ligands (EGF and TGFα) and prevent activation of downstream signaling pathways [5]. It can also induce antibody-dependent cellular cytotoxicity and suppress nuclear EGFR transport and activation of DNA-dependent protein kinase [6]. Currently, for treating HNSCC, cetuximab is approved in combination with radiotherapy in locally-advanced disease [7], as a single agent in platinum-refractory recurrent or metastatic disease [8], and in combination with platinum (carboplatin or cisplatin) and 5-fluorouracil as the first line therapy in recurrent or metastatic disease [9].
However, cetuximab as a monotherapy shows limited efficacy with a low (only 10-13%) initial response rate (known as primary drug resistance), and the majority of patients may develop acquired drug resistance even after a good initial response [10]. This has stimulated a growing interest in understanding the mechanisms of EGFR blockage resistance. Inflammation was reported to play a vital role in cetuximab resistance. Inflammatory cytokines, including IL1A, IL1B and IL8, have been found to be associated with reduced sensitivity and primary resistance to cetuximab in colorectal cancers [11]. Oncogenic transformation of cells may induce inflammatory signaling pathway, resulting in increased secretion of pro-inflammatory cytokines and consequently tumor growth [12]. Therefore, inflammation may be associated with primary resistance to EGFR inhibitors in a wide variety of cancers by supporting the proliferation of malignant cells [13].
Toll-like receptor 4 (TLR4) is an innate immune receptor involved in recognition of inflammation-associated microbial and self-ligands [14]. Activation of TLR4 and similar receptors in immune cells has important defense functions such as increased survival, motility, and invasion of pathogen-fighting cells [15]. TLR4, a natural lipopolysaccharide (LPS) receptor, is also highly upregulated in cancer cells in breast, ovarian, prostate and other tumors [16,17]. Paclitaxel can be regarded as a functional analogue of LPS, and causes dimerization of TLR4, which in turn activates the NF-κB pathway, leading to upregulation of numerous pro-survival proteins and induces resistance to paclitaxel dependent chemotherapy [18,19]. One study reported that phosphorylation of EGFR depends on the activation of TLR4/Src axis, and EGFR is required for TLR4 to activate the proliferation signaling [20]. This indicates that EGFR and TLR4 pathway may crosstalk when regulating cancer cell growth, invasion, and migration, and affect sensitivity to antitumor drugs. However, the relationship and the underlying mechanism between TLR4 and resistance to EGFR-target therapy in HNSCC remain unknown.
In this study, we detected an increase of TLR4 expression in 50% of HNSCC tumor biopsies, which was correlated to the resistance to cetuximab therapy. Mechanistically, EGFR blockade by cetuximab suppressed activation of Src and led to decreased Cbl-b mediated degradation of MyD88. Increased MyD88 activated TLR4 signaling and induced cytokines as well as anti-apoptotic proteins, resulting a negative feedback which caused resistance to EGFR blockade.
Materials and methods
Reagents
LPS and rapamycin were purchased from Sigma (St. Louis, MO, USA). M-PER® mammalian protein extraction reagent, NE-PER nuclear and cytoplasmic extraction reagents, BCATM Protein Assay Kit and SuperSignal West Femto Maximum Sensitivity Substrate were purchased from Pierce (Rockford, IL, USA). Antibodies against Lamin A (sc-56137), GAPDH (sc-47724), p65 (sc-71675), p-p65 (sc-136548), c-jun (sc-166540), p-c-jun (sc-53182), TAK1 (sc-7967), bcl-2 (sc-509), bcl-xl (sc-8392), iNOS (sc-7271), COX-2 (sc-19999), c-Src (sc-130124), p-c-Src (sc-81521), Cbl-b (sc-376409), MyD88 (sc-136970), and TLR4 (sc-293072) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibody against p-TAK1 (ab109404) was purchased from Abcam (Cambridge, MA, USA). ELISA kits for TNF-α, PGE2 and NO were purchased from R&D Systems (Minneapolis, MN, USA).
Cell culture
Human head and neck squamous cell carcinoma SCC4, SCC9 and CAL27 cell lines were purchased from the American Type Culture Collection and cultured as previously reported [21]. Cells were maintained in DMEM medium containing 10% fetal calf serum (FCS; Gibco GrandIsland, NY), 0.29 mg/ml glutamine, 100 units/ml penicillin, and 100 ug/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2.
Clinical specimens, RNA extraction and qRT-PCR
HNSCC tissues and adjacent non-tumor tissues (at least 5 cm away from the tumor site) were obtained from surgical specimens. The protocol was approved by the Ethical Committee of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, and written informed consent was obtained from all participants. The specimens were excised immediately after resection of tumor and preserved in liquid nitrogen. They were classified into two groups based on the relative expression of TLR4/GAPDH as examined by qRT-PCR. The total RNA was extracted from both cell lines and tumor tissues with Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions, and then reverse transcribed to cDNA. The cDNA was subjected to qPCR assays for determination of TLR4 mRNA levels (primer sequences are listed in Table S1).
Response assessment of cetuximab treatment
Modified Response Evaluation Criteria in Solid Tumors (RECIST) were applied to assess the response after cetuximab treatment. The criteria were, Complete Response (CR): all target lesions disappear and pathological lymph nodes reduce in short axis to <10 mm. Partial Response (PR): the sum of diameters of target lesions decrease at least 30%. Progressive Disease (PD): the sum of diameters of target lesions increase at least 20% and the sum also absolutely increase at least 5 mm. Stable Disease (SD): Neither sufficient shrinkage for PR nor sufficient increase for PD [37]. Patients with CR or PR responses were considered as sensitive to cetuximab treatment, patients with SD or PD were defined as resistant.
Construction of transiently and stably transfected cells
The overexpressed plasmids products (Cbl-b and c-Src) were purchased from Gene Pharma (Shanghai, China) and transfected according to the manufacturer’s instructions. The sequences for the overexpressed plasmids of Cbl-b and c-Src were shown in Table S2. Lentiviral vectors overexpressing TLR4 and their negative control were constructed by Gene Pharma (Shanghai, China). SCC4 and CAL27 cells were infected with 50 MOI (multiplicity of infection) of lentiviral vectors according to the manufacturer’s instructions. Stably transfected cells were cultured with 5 μg/mL puromycin for 2 weeks. The efficiency of plasmids and lentiviral infection was evaluated by qPCR and western blotting.
Cell proliferation assays
Cell growth was measured with CCK-8 according to the manufacturer’s instructions [21]. Cells were seeded in 96-well plates, pretreated with 1 μg/mL LPS for 6 h and then incubated in fresh medium with 1 μg/mL cetuximab for 48 h. Then, each well was incubated with 10 µl of CCK-8 solution for 4 h at 37°C. The absorbance was measured at 450 nm using a spectrophotometer.
Wound-healing assays
Transfected cells were seeded in 6-well plates, and wounds were scraped uniformly in monolayer cells using a 10 µL pipette tip. Cells were washed thrice with PBS to remove floating cells. Cell migration across the wound line was observed after 0 h and 24 h of wounding, and the fields of view were randomly selected and photographed under a microscope. All assays were performed independently in triplicate.
Cell invasion assays
In vitro cell invasion assays were performed using matrigel coated 24-well chambers and microfilters (5 μm pored polycarbonate filters), respectively, according to the manufacturers’ instructions (BD Bioscience, Bedford, MA). Briefly, after rehydration of the chambers, CAL27 cells were stimulated with 1 μg/ml LPS for 24 h in the presence or absence of a gradient concentration of rapamycin. Then, they were harvested and seeded in 200 μl of DMEM plus 5% FBS in the upper chamber (1×105 cells per chamber) and in 600 μl of DMEM plus 10% FBS in the lower chamber, respectively. Migrated CAL27 cells were stained with leucocrystal violet. The number of cells in the membrane was counted in 20 randomly selected field views at 100× for 24 h.
Cell apoptosis analysis
Apoptotic rate of HNSCC cells was detected by Annexin-V-FITC apoptosis detection kit (BD, Franklin Lakes, NJ, USA). Then the HNSCC cells stained with FITC-Annexin V and PI were evaluated by measuring the fluorescence of at least 10,000 cells each sample using FACS Calibur flow cytometer (Becton-Dickinson, CA, USA).
Enzyme linked immunosorbent assays (ELISA)
SCC4 and CAL27 cells were seeded in a T25 culture bottle at a density of 3×106/bottle, and supernatants were collected 24 h later. The TNF-α, PGE2 and NO levels were detected using a human TNF-α, PGE2 and NO enzyme-linked immune detection kit according to the manufacturer’s instructions (R&D Systems).
Extraction of cytoplasmic/nuclear proteins, co-immunoprecipitation (Co-IP) and Western blotting
Cells were cultured and transfected with expression vectors. SCC4 and CAL27 cells were harvested and lysed for CoIP or immunoblotting assays after transfection for 48 h. The total, cytoplasmic and nuclear proteins were extracted using a Nuclear/Cytosol Fractionation Kit (BioVision, San Francisco, USA) according to the manufacturer’s instructions. For CoIP assays, cells were lysed in 500 μl of lysis buffer (20 mM Tris with pH 7.4, 50 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.5% SDS, 0.5% deoxycholate, and protease inhibitors). To efficiently solubilize keratins, cells were treated with 2% Empigen BB (Sigma). Then, 500 μg of lysate aliquots (1 μg/μl) were precleared with 50 μl of protein A-Sepharose beads (Upstate Biotechnology, Lake Placid, NY, USA) for 2 h at 4°C. An appropriate amount of rabbit anti-LRP16, rabbit anti-Flag (Sigma), rabbit anti-ERα (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or rabbit nonspecific IgG (Clontech) was then added and incubated overnight at 4°C. Then, 100 μl of pre-blocked agarose beads were added to the mixture and incubated for another 2 h at 4°C. Bound proteins were washed three times, eluted in SDS sample buffer, resolved by SDS-PAGE, and analyzed by immunoblotting using mouse anti-c-Src (Abgent, San Diego, CA, USA), mouse monoclonal anti-Flag (Sigma), rabbit anti-MyD88, mouse anti-Cbl-b and rabbit anti-β-actin (Santa Cruz Biotechnology). Western blotting was briefly described as the cells were lysed in lysis buffer. Cellular proteins were loaded and separated on SDS-PAGE. After that, they were transferred to a nitrocellulose membrane (Amersham Biosciences Piscataway, NJ) by the standard electric transfer protocol. The membrane was blocked and probed with primary antibodies and then with HRP-labeled second antibodies (DAKO, Carpinteria, CA). Peroxidase color was visualized with TMB membrane Peroxidase Substrate (KPL, Gaithersburg, MD). The results of western blot were normalized using GAPDH in total and in cytoplasm with Lamin A in nucleus, quantitated by software Image J.
Xenograft nude mouse model
For in vivo tumor growth assays, SCC4 and CAL27 cells transfected with lentiviral vectors overexpressing TLR4 were subcutaneously implanted to the left upper flank region of male mice (1×106 cells per mouse). Cetuximab and TLR4 inhibitor were peritoneally injected and tumor volume was measured every 3 days using calipers as follows: tumor volume (mm3) = (L×W2)/2, where L was the length and W was the width. About 6 weeks later, mice were euthanized and tumors were excised for standard histological analysis. Cetuximab-resistent xenograft was selected and transplanted subcutaneously into mice as previously described. TLR4 inhibitor (TAK242) was peritoneally injected and tumor volume was measured every 3 days as previously described. All animal experiments were approved by the Animal Experimental Ethical Committee of the Shanghai Jiao Tong University and performed according to the guidelines on the care and use of animals for scientific use.
Immunohistochemistry
For histological analysis, the tumors of patients and nude mice were fixed in 10% formalin for at least 24 h and embedded with paraffin. The sections were stained with hematoxylin and eosin (H&E) for morphological analysis or with specific primary antibody of TLR4 (Santa Cruz, USA) using a 2-Solution DAB Kit (Invitrogen, USA) according to the manufacturer’s instructions. The staining levels were classified as negative (no or weak) or positive (moderate or strong) staining.
Fifty pairs of HNSCC tissues were examined by IHC. Two pathologists blinded to patient data were invited to independently examine the cellular location of TLR4 and compare the staining between tumor and normal tissues for each case. Immunohistochemistry stain score = positive cell score × staining intensity score [38]. The percentage of positive cells was graded as follows: <10% (grade 0), 10-25% (grade 1), 25-50% (grade 2), 50-75% (grade 3), and >75% (grade 4). Immunohistochemical staining intensity was graded as follows: 0 (no staining), 1 (bright yellow), 2 (orange), and 3 (brown). The total score of ≤1, 2-4, 5-8 and ≥9 was defined as negative, low, moderate and high expressed, respectively.
Statistical analysis
Data were represented as mean ± standard deviation (SD) of at least three independent experiments. Statistical analysis was performed using Student’s t-test. P values less than 0.05 were considered to be significant.
Results
Correlation of TLR4 expression with EGFR expression and cetuximab response in HNSCC patients
TLR4 was previously reported to be implicated in drug resistance and also highly expressed in HNSCC biopsies and promote tumor progression [18-22]. However, its physiological and pathological roles and relationship with EGFR expression remain unknown. Here, we collected data from forty-eight HNSCC patients who received cetuximab therapy (see Table 1 for clinicopathological parameters) and found that mRNA expression of both EGFR and TLR4 were more highly expressed in HNSCC tissues than those found in adjacent normal tissues (Figure 1A). Furthermore, the expression level of TLR4 was significantly correlated to EGFR expression with a Pearson coefficient of 0.895 (Figure 1B). When enrolled patients were divided into sensitive (CR/PR, n=27) and resistant (SD/PD, n=21) groups, we observed by immunohistochemistry a significantly higher protein expression of TLR4 on the surface of tumor cells in the resistant group as compared to the sensitive group (Figure 1C). Consistently, in Kaplan Meier analysis, patients bearing tumors with a higher expression of TLR4 which were more resistant to cetuximab therapy displayed a poorer overall survival (OS) rate as well as disease free survival (DFS) rate (Figure 1D, 1E). These results indicated that TLR4 expression levels in HNSCC cells were significantly correlated to EGFR expression and cetuximab therapy response.
Table 1.
Clinicopathologic characteristics and COX regression analysis of prognostic factors in forty eight HNSCC patients who received cetuximab therapy
| Clinicopathologic parameters | Number of cases | COX regression analysis | |
|---|---|---|---|
|
| |||
| Chi-Square scores | P value | ||
| Tobacco | |||
| Yes | 29 | 1.7732 | 0.1830 |
| No | 16 | ||
| Alcohol | |||
| Yes | 21 | 0.0737 | 0.7861 |
| No | 24 | ||
| Sex | |||
| Male | 33 | 1.7732 | 0.1830 |
| Female | 12 | ||
| Age | |||
| ≥60 | 27 | 2.3838 | 0.1226 |
| <60 | 18 | ||
| Tumor site | |||
| Tongue | 27 | 0.2058 | 0.6501 |
| Gingiva | 4 | ||
| Buccal | 1 | ||
| Palate | 3 | ||
| Oropharynx | 1 | ||
| Retromolar region | 2 | ||
| Month floor | 7 | ||
| Tumor stage | |||
| T1 | 0 | 0.1699 | 0.6802 |
| T2 | 5 | ||
| T3 | 8 | ||
| T4 | 32 | ||
| Nodal stasus | |||
| N0 | 13 | 1.8247 | 0.1768 |
| N1 | 16 | ||
| N2 | 16 | ||
| N3 | 0 | ||
| Pathological differentiation grade | |||
| I | 13 | 1.2210 | 0.2692 |
| I-II | 14 | ||
| II | 15 | ||
| III | 1 | ||
Figure 1.

High TLR4 expression was related to resistance to anti-EGFR therapy. A. Relative mRNA expression of TLR4 and EGFR (n=25). B. Correlation between TLR4 and EGFR expression. C. Representative image of different TLR4 staining intensity (negative, low, moderate and high) in HNSCC patients before the cetuximab therapy who were resistant (n=21) or sensitive (n=27) to cetuximab. D. Five-year overall survival (Kaplan-Meier method and log-rank test) in HNSCC patients who received cetuximab therapy (n=48). E. Disease free survival (DFS) in HNSCC patients who received cetuximab therapy (n=48). Data were represented as mean ± SD of at least three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Activation of TLR4 promoted resistance to cetuximab therapy
In order to investigate the role of TLR4 in anti-EGFR therapy, we first determined the concentration of cetuximab in tissue culture experiments as 100 μg/mL according to its IC50s in HNSCC cell line SCC4, SCC9 and CAL27 (Figure 2A-C), which are cetuximab sensitive cell lines. Cells were then pretreated with LPS for 6 hours to activate TLR4 and subsequently treated with cetuximab for 48 hours. We found that cetuximab inhibited HNSCC cell proliferation, while activation of TLR4 enhanced cell proliferation and reversed the inhibitory effect of cetuximab in SCC4, SCC9 and CAL27 cells (Figure 2D). LPS treatment also reversed cetuximab-induced the inhibition of HNSCC cell migration and invasion (Figure 2E, 2F) and reversed cetuximab-induced the apoptosis in CAL27 cells (Figure 2I). To confirm that the LPS effect was mainly mediated by TLR4, we also overexpressed or knocked down TLR4 expression in CAL27 cells. Furthermore, cells were transfected by siRNA or plasmid to inhibit or activate TLR4 expression and subsequently treated with cetuximab for 48 hours. As expected, overexpression of TLR4 increased CAL27 cell migration and invasion and showed more resistant to the cetuximab treatment, whereas knocking down TLR4 in CAL27 led to reduced migration and invasion and showed more sensitive to the cetuximab treatment (Figure 2G, 2H). On the molecular level, LPS treatment or TLR4 overexpression led to activation of NF-κB and MAPK pathways (Figure 2J, 2K), both of which were critical for cell survival under EGFR blockage. Accordingly, the expression of anti-apoptotic proteins, such as Bcl-2 and Bcl-xl, was upregulated by LPS priming or TLR4 overexpression (Figure 2J, 2K). Thus, activation of TLR4 could lead to primary resistance to cetuximab therapy.
Figure 2.

TLR4-ligation led to resistance to growth inhibition of EGFR blocking. (A-C) IC50 of cetuximab in SCC4, SCC9 and CAL27 cells were measured; (D) SCC4, SCC9 and CAL27 cells were seeded in 96-well plates and cultured in media containing serum, pretreated with 1 μg/ml LPS or PBS for 6 h. EGFR was inhibited with 100 μg/mL cetuximab for 48 h, cell proliferation was measured using CCK8 assay; CAL27 cells were seeded in 6-well plates, pretreated with 1 μg/ml LPS or PBS for 6 h and cultured with 100 μg/mL cetuximab for 48 h, cell invasion (E) was measured using transwell assay and cell migration (F) was measured by the wound healing assay; CAL27 cells were transfected by TLR4 siRNA or TLR4 overexpressed plasmid, then cultured with 100 μg/mL cetuximab for 48 h, cell invasion (G) was measured using transwell assay and cell migration (H) was measured by the wound healing assay respectively; (I) The cell apoptosis rate was examined by Annexin-V-FITC staining using flow cytometry; (J, K) Cell extracts for MAPK kinase (c-jun, p-c-jun), NF-κB kinase (TAK1, p-TAK1, IKΚα/ß, p-IKΚα/ß, p65, p-p65) and anti-apoptotic proteins (Bcl-2, Bcl-xl) were subjected to Western blotting. Data were represented as mean ± SD of at least three independent experiments; *P<0.05, **P<0.01, ***P<0.001.
Inhibition of EGFR increased signaling events downstream of TLR4
In order to further investigate whether TLR4 activation was associated with acquired resistance to anti-EGFR therapy, we examined TLR4 activation induced signaling and cytokines under EGFR inhibition. CAL-27 cells were pretreated with cetuximab and then were transfected by siRNA or plasmid to inhibit or activate TLR4 expression. It revealed that overexpression of TLR4 after cetuximab treatment aggravated cell migration and invasion compared with cells disposed with cetuximab alone, whereas knocking down TLR4 led to reduced migration and invasion compared with cells disposed with cetuximab alone (Figure 2A, 2B). SCC4 cells were pretreated with cetuximab and then stimulated with LPS. Cetuximab induced a time-dependent activation of phosphorylated p65/RelA and phosphorylated c-jun, which were key transcription factors of NF-κB and MAPK pathways, respectively (Figure 3C, 3D). Similar results were obtained when SCC4 cells were pretreated with gefitinib and stimulated with LPS (Figure 3C). We also fractionated cell lysates and observed that p65 and c-jun were increased in the nucleus while p-p65 and p-c-jun were increased in the cytoplasm (Figure 3E, 3F). All these results indicated that inhibition of EGFR by cetuximab and gefitinib led to increased signaling events downstream of TLR4.
Figure 3.

Inhibition of EGFR increased TLR4 signaling. CAL-27 cells were pretreated with 100 μg/mL cetuximab for 2 h, and then were transfected by TLR4 siRNA or TLR4 overexpression plasmid, cell invasion (A) was measured using transwell assay and cell migration (B) was measured by the wound healing assay respectively; (C) SCC4 cells were pretreated with 100 μg/mL cetuximab for 2 h, and then stimulated with 1 μg/mL LPS for 0, 15, 30, 60 min, the expression of p-p65 and p-c-jun were detected by western blot; (D) The relative expression of p-p65 and p-c-jun/GAPDH were measured by Image J; (E) SCC4 cells were pretreated with 100 μg/mL cetuximab for 2 h, and then stimulated with 1 μg/mL LPS for 24 h, then the expression of p-p65 and p-c-jun were detected; (E) The nucleus proteins were extracted and the expression of p65, p-p65, c-jun and p-c-jun in nuclear were detected by Western blotting respectively; the relative expression of p65, p-p65, c-jun and p-c-jun/Lamin A in nucleus were measured by Image J; (F) The cytoplasmic proteins were extracted and the expression of p65, p-p65, c-jun and p-c-jun in cytoplasm were detected by Western blott; the relative expression of p65, p-p65, c-jun and p-c-jun/GAPDH in cytoplasm were measured by Image J; Data were represented as mean ± SD of at least three independent experiments; *P<0.05, **P<0.01, ***P<0.001.
EGFR inhibition increased TLR4 induced tumor-associated inflammation
TLR4 activation is known to upregulate TNF-α, PGE2 and NO which are critical for tumor immune microenvironment. As inhibition of EGFR increased signaling events downstream of TLR4, we further investigated whether it might have an effect on TLR4 induced tumor-associated inflammation. Indeed, treatment with cetuximab or gefitinib resulted in a substantial increase in TNF-α (Figure 4A), PGE2 (Figure 4B) and NO (Figure 4C) in CAL27 cells. There was also an increase in the expression of COX2 (PGE2 synthetase) and iNOS (NO synthetase) (Figure 4D). These results suggest TLR4 induced tumor-associated inflammation was increased after EGFR inhibition.
Figure 4.
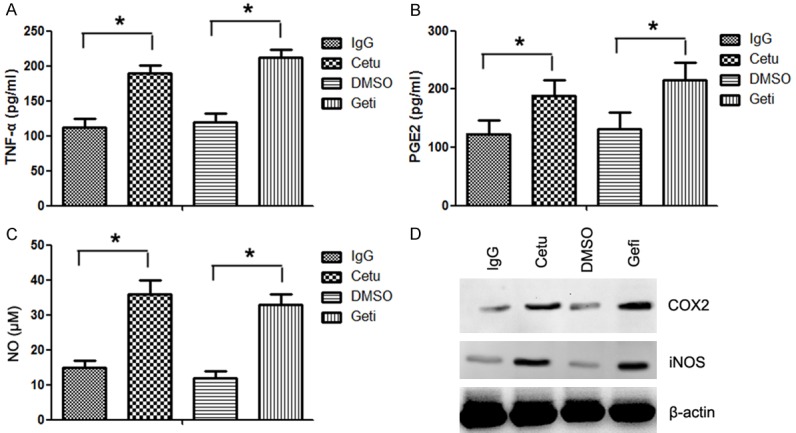
Inhibition of EGFR increased TLR4 induced tumor-associated inflammation. CAL27 cells were cultured in 12-well plates, pretreated with 100 μg/mL cetuximab or gefitinib for 2 h, and then stimulated with 1 μg/mL LPS for 24 h. Supernatants were collected and analyzed for cytokine TNF-α (A), PGE2 (B) and NO (C) production using ELISAs. (D) The expression of PGE2 and NO synthetase was detected by western blotting using anti-COX2 and anti-iNOS antibodies. Data were represented as mean ± SD of at least three independent experiments; *P<0.05, **P<0.01; ***P<0.001.
EGFR inhibition stabilized TLR4 adaptor MyD88
Degradation of MyD88, a TLR adaptor protein, was reported to be an important negative regulation mechanism in TLR4 signaling [32]. As TLR4 downstream signals (NF-κB and MAPK) were increased by EGFR inhibition, we investigated whether cetuximab and gefitinib pretreatment may alter TLR4 adaptor proteins. Western blot analysis demonstrated that TLR4 ligation induced gradual down-regulation of MyD88 protein over time (Figure 5A). In contrast, cetuximab or gefitinib pretreatment led to increased level of MyD88 proteins (Figure 5A). Previous research found that Src may promote Cbl-b medicated degradation of MyD88. The activation of Src after cetuximab or gefitinib pretreatment was decreased irrespective of TLR4 activation status, as assessed from a phospho-Src antibody (Figure 5A). We next performed immune-precipitation experiments to confirm the interactions between c-Src, Cbl-b, and MyD88 proteins. As shown in Figure 5B, 5C, cetuximab or gefitinib treatment led to reduction of c-Src and Cbl-b proteins and increased level of MyD88. Thus, EGFR inhibition may decrease the activation of Src and Cbl-b-mediated degradation of MyD88 and potentiate TLR4 signaling.
Figure 5.
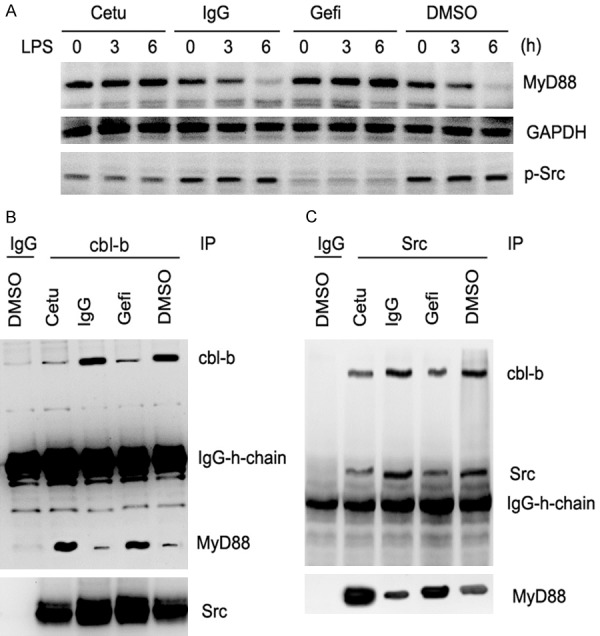
EGFR inhibition led to decreased Cbl-b-mediated degradation of MyD88 by attenuating Src activation. (A) CAL27 cells were pretreated with 100 μg/mL cetuximab, gefitinib, IgG or DMSO for 2 h and then stimulated with 1 μg/mL LPS for 24 h. Western blotting was performed using anti-p-Src and anti-MyD88 antibodies at 0, 3, and 6 h, respectively. Co-IP and western blotting showed the interaction of Cbl-b with MyD88 (B) and that of Src with Cbl-b and MyD88 (C). Data were represented as mean ± SD of at least three independent experiments; *P<0.05, **P<0.01; ***P<0.001.
TLR4 overexpressed HNSCC cells grew faster and were more resistant to cetuximab therapy in vivo and in vitro
The above results showed that TLR4-induced inflammatory response played a critical role in anti-EGFR therapy in vitro, we further explored the role of TLR4 signaling in vivo. Compared with control animals, TLR4 overexpressed CAL27 cells grew faster and were more resistant to cetuximab therapy (Figure 6A). The protein expression of iNOS was assayed in order to further confirm pro-tumor inflammation in nude mice upon anti-EGFR therapy (Figure 6B). Similarly, there was an increase in the protein expression of iNOS, indicting high pro-tumor inflammation for TLR4 overexpressed CAL27 cells. Cetuximab therapy led to more severe pro-tumor inflammation in TLR4 overexpressed CAL27 cells than in control CAL27 cells. Meanwhile, cetuximab therapy upregulated the expression of TLR4 and MyD88, but had no effect on TRIF expression (Figure 6C). To investigate the therapeutic effect of TLR4, cetuximab and TLR4 inhibitor (TAK242) were peritoneally injected and tumor volume was measured every 3 days. The results showed that cetuximab in combination with TAK242 revealed the best response in all groups (Figure 6D, 6E). Furthermore, we established cetuximab-resistant in vivo model to examine the response to the combined therapy. It showed that TAK242 combined therapy could overcome the acquired cetuximab resistance (Figure 6F). All these results indicated that enhanced TLR4 signaling by overexpressing TLR4 promoted tumor growth and resistance to cetuximab therapy through increasing pro-tumor inflammation.
Figure 6.

TLR4 overexpressed CAL-27 cells grew faster and were more resistant to cetuximab in vivo and in vitro. (A) CAL-27 cells transfected with overexpression vectors of TLR4 or control vectors were injected into nude mice. Cetuximab was peritoneally injected 7 days later and tumor volume was measured every 3 days. (B) IHC analyses of iNOS expression in tumor tissues (200×). (C) The expression of TLR4, MyD88 and TRIF in xenograft was measured by RT-PCR. CAL-27 cells transfected with overexpression vectors of TLR4 (E) or control vectors (D) were injected into nude mice. Cetuximab combined with TAK242 was peritoneally injected 7 days later and tumor volume was measured every 3 days. (F) Xenograft resistant to cetuximab was implanted subcutaneously. Cetuximab and TAK242 were peritoneally injected 7 days later and tumor volume was measured every 3 days. Data were represented as mean ± SD of at least three independent experiments; *P<0.05, **P<0.01; ***P<0.001.
MyD88 overexpressed HNSCC cells grew faster and were more resistant to cetuximab therapy in vivo
We then investigated the in vivo function of TLR4 signal in tumor progression and anti-EGFR therapy. MyD88 overexpressed animal models were established, and then cetuximab was peritoneally injected and tumor volume was measured every 3 days (Figure 7A). MyD88 overexpressed SCC4 cells grew faster and were almost resistant to cetuximab therapy. In order to further confirm pro-tumor inflammation in tumor nude mice upon anti-EGFR therapy, the mRNA expression of iNOS (Figure 7B), COX2 (Figure 7C) and TNF-α (Figure 7D) was assayed. Again, there was an increase in mRNA expression of TNF-α, iNOS and COX2, indicating higher pro-tumor inflammation for MyD88 overexpressed SCC4 cells. Cetuximab therapy also led to more severe pro-tumor inflammation in MyD88 overexpressed SCC4 cells than in control SCC4 cells. Also, cetuximab and TLR4 inhibitor (TAK242) were peritoneally injected in MyD88 overexpressed animal models. The combined therapy revealed the best response in all groups (Figure 7E, 7F). These results indicated that enhanced TLR4 signaling by overexpressing MyD88 promoted tumor growth and resistance to cetuximab therapy through increasing pro-tumor inflammation.
Figure 7.

MyD88 overexpressed SCC4 cells grew faster and were more resistant to cetuximab in vivo. (A) SCC4 cells transfected with overexpression vectors of MyD88 or control vectors were injected into nude mice. Cetuximab was peritoneally injected 7 days later and tumor volume was measured every 3 days. The relative mRNA expression of iNOS (B), COX2 (C) and TNF-α (D) extracted from tumor tissues was measured. (E) The expression of TLR4, MyD88 and TRIF in xenograft was measured by RT-PCR. SCC4 cells transfected with overexpression vectors of MyD88 (F) or control vectors (E) were injected into nude mice. Cetuximab and TAK242 were peritoneally injected 7 days later and tumor volume was measured every 3 days. Data were represented as mean ± SD of at least three independent experiments; *P<0.05, **P<0.01; ***P<0.001.
Discussion
The clinical efficacy of EGFR-targeted therapies remains limited in HNSCC due to primary and acquired drug resistance [23]. This study showed that TLR4 was significantly correlated to primary and acquired resistance to cetuximab in HNSCC, and a cross talk between TLR4 and EGFR signaling was observed through a MyD88-Cbl-b-Src pathway. Thus, targeting TLR4 signaling might improve clinical efficacy of EGFR-targeted therapies in HNSCC by reversing cetuximab resistance.
TLRs are crucial components of the innate immune response to bacterial and viral pathogens [24]. However, they may also contribute to tumor growth and progression [25]. Our previous study has shown that TLR4 was expressed in HNSCC cells and played a vital role in inducing tumor proliferation by activation of NF-κB and secretion of pro-inflammatory cytokines (IL-6, VEGF, TGFβ and PGE2) [26]. This study further investigated the dysregulation of TLR4 in clinic. We found that activation of TLR4 promoted cell proliferation, migration and invasion in HNSCC by NF-κB and MAPK pathway. A high expression of NF-κB in tumor cells was associated with tumor progression and chronic inflammation due to its anti-apoptotic properties [27]. In this study, anti-apoptotic proteins and the secretion of TNF-α, PGE2 and NO were upregulated. TNF-α, PGE2 and NO were major mediators of inflammation-induced cancers that were upregulated in various solid tumors [28,29]. TNF-α was involved in primary and acquired resistance to anti-EGFR therapy in non-small cell lung cancer [30]. COX-2/PGE2 could regulate HIF2α activity to reduce the sensitivity of sorafenib (EGFR tyrosine kinase inhibitor) [31], indicating that overexpression of TLR4 might induce continuous activation of EGFR signaling and primary resistance to cetuximab therapy.
Another important finding of our study was that EGFR signaling could feedback via TLR4 signaling to induce acquired resistance to cetuximab (Figure 8). Cetuximab could lead to decreased Cbl-b-mediated degradation of MyD88 by attenuating Src activation, which stabilized the structure of TLR4 and ensured continuous activation of TLR4 signaling. A previous study found that phosphorylation of Syk could promote degradation of MyD88 and TRIF via Cbl-b and subsequently inhibit TLR4 signaling in macrophages [32]. Cbl-b was an E3 ubiquitin-protein ligase that could degrade MyD88 and TRIF via polyubiquitination. The phosphorylation of TLR4 would activate the signaling pathway of its two downstream molecules MyD88 and TRIF [33]. We found that c-Src could form a functional protein complex with Cbl-b and MyD88. Src was the direct downstream molecular of EGFR, and cetuximab would cause the blockade of Src phosphorylation. TLR4 exerts its functions through MyD88 dependent and independent pathways [34]. In MyD88-independent pathway, TRIF phosphorylates TRAF3 to activate IRF3, recruit IKKε/TBK1 and promote the secretion of β-interferon [35]. Our results showed that EGFR inhibitors could continuously activate TLR4 signaling rather than TRIF signaling by Src-Cbl-b-MyD88 pathway, indicating that EGFR regulated TLR4 signaling by MyD88 ubiquitination. The sensitization of TLR4 in turn promoted tumor growth, and the cross talk between EGFR and TLR4 signaling ultimately resulted in acquired resistance to EGFR targeted agents.
Figure 8.
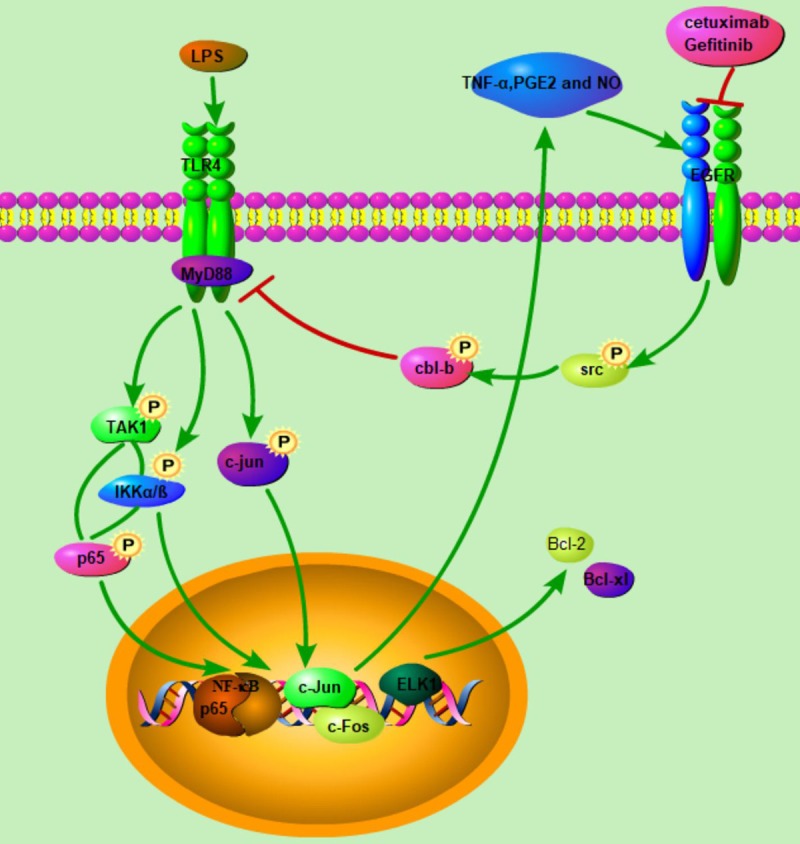
The crosstalk between EGRF inhibition and TLR4 pathways in HNSCC. The increased TLR4 signaling resulted from decreased Cbl-b-mediated degradation of key adaptor protein MyD88, and EGFR inhibition led to decreased activation of Src and sequentially Src-medicated phosphorylation of MyD88. TLR4 activation induced activation of NF-κB and MAPK signal pathway, which further induced the release of proinflammatory cytokines and anti-apoptosis protein feedback led to the resistance to EGFR inhibition.
In our in vivo study, we examined the effect of TLR4 and EGFR inhibition in orthotopic and cetuximab-resistant xenograft models. The combination of cetuximab with TAK242 was highly effective in inhibiting tumor growth in both models, while EGFR inhibition or TAK242 alone was not so effective. TAK242 was a TLR4 inhibitor that could selectively inhibit TLR4-mediated production of cytokines and NO [36]. We further established a MyD88 overexpressed xenograft model to verify the role of MyD88 in the cross talk between EGFR and TLR4 signaling. As expected, we found a significant reduction in tumor growth by combined inhibition of TLR4 and EGFR compared with that by EGFR inhibition alone. Importantly, the secretion of pro-inflammatory cytokines (TNF-α, PGE2 and NO) was also reduced upon the combined therapy, suggesting that this approach is therapeutically useful in HNSCC.
In summary, our results demonstrate for the first time that TLR4 signaling is involved in primary and acquired resistance to cetuximab in HNSCC, and the combination of EGFR and TLR4 is more effective than EGFR alone. The underlying mechanism may be attributed to the cross talk between TLR4 and EGFR signaling through a MyD88-Cbl-b-Src pathway. Our results may provide some insights into further clinical investigation of concomitant EGFR/TLR4 inhibition as a novel strategy for the treatment of cetuximab-resistant HNSCC, especially when tumors are progressed after cetuximab therapy.
Acknowledgements
This study was supported by grants from the National Program on Key Research Project of China (2016YFC0902700), National Natural Science Foundation of China (No. 31140007, 81872185 and 81902748) and sponsored by Shanghai Sailing Program (19YF1427100).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Sacco AG, Cohen EE. Current treatment options for recurrent or metastatic head and neck squamous cell carcinoma. J. Clin. Oncol. 2015;33:3305–3313. doi: 10.1200/JCO.2015.62.0963. [DOI] [PubMed] [Google Scholar]
- 2.Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J. Clin. Oncol. 2006;24:2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 3.Sundvall M, Karrila A, Nordberg J, Grenman R, Elenius K. EGFR targeting drugs in the treatment of head and neck squamous cell carcinoma. Expert Opin Emerg Drugs. 2010;15:185–201. doi: 10.1517/14728211003716442. [DOI] [PubMed] [Google Scholar]
- 4.Azoury SC, Gilmore RC, Shukla V. Molecularly targeted agents and immunotherapy for the treatment of head and neck squamous cell cancer (HNSCC) Discov Med. 2016;21:507–516. [PubMed] [Google Scholar]
- 5.Bauman JE, Ferris RL. Integrating novel therapeutic monoclonal antibodies into the management of head and neck cancer. Cancer. 2014;120:624–632. doi: 10.1002/cncr.28380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu BS, Xia HW, Zhou S, Liu Q, Tang QL, Bi NX, Zhou JT, Gong QY, Nie YZ, Bi F. Inhibition of YAP reverses primary resistance to EGFR inhibitors in colorectal cancer cells. Oncol Rep. 2018;40:2171–2182. doi: 10.3892/or.2018.6630. [DOI] [PubMed] [Google Scholar]
- 7.Gyawali B, Shimokata T, Honda K, Ando Y. Chemotherapy in locally advanced head and neck squamous cell carcinoma. Cancer Treat Rev. 2016;44:10–16. doi: 10.1016/j.ctrv.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Lala M, Chirovsky D, Cheng JD, Mayawala K. Clinical outcomes with therapies for previously treated recurrent/metastatic head-and-neck squamous cell carcinoma (R/M HNSCC): a systematic literature review. Oral Oncol. 2018;84:108–120. doi: 10.1016/j.oraloncology.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Argiris A, Harrington KJ, Tahara M, Schulten J, Chomette P, Ferreira Castro A, Licitra L. Evidence-based treatment options in recurrent and/or metastatic squamous cell carcinoma of the head and neck. Front Oncol. 2017;7:72. doi: 10.3389/fonc.2017.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caponigro F, Milano A, Basile M, Ionna F, Iaffaioli RV. Recent advances in head and neck cancer therapy: the role of new cytotoxic and molecular-targeted agents. Curr Opin Oncol. 2006;18:247–252. doi: 10.1097/01.cco.0000219253.53091.fb. [DOI] [PubMed] [Google Scholar]
- 11.Gelfo V, Rodia MT, Pucci M, Dall’Ora M, Santi S, Solmi R, Roth L, Lindzen M, Bonafè M, Bertotti A, Caramelli E, Lollini PL, Trusolino L, Yarden Y, D’Uva G, Lauriola M. A module of inflammatory cytokines defines resistance of colorectal cancer to EGFR inhibitors. Oncotarget. 2016;7:72167–72183. doi: 10.18632/oncotarget.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Wang S, Zhu R, Li H, Han Q, Zhao RC. Lung tumor exosomes induce a pro-inflammatory phenotype in mesenchymal stem cells via NFkappaB-TLR signaling pathway. J Hematol Oncol. 2016;9:42. doi: 10.1186/s13045-016-0269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, Srinivasan S, Nowak J, Izard T, Rangarajan ES, Carlson KE, Katzenellenbogen JA, Yao XQ, Grant BJ, Leong HS, Lin CY, Frasor J, Nettles KW, Glass CK. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol Cell. 2017;65:1122–1135. e1125. doi: 10.1016/j.molcel.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirsch I, Janovec V, Stranska R, Bendriss-Vermare N. Cross talk between inhibitory immunoreceptor tyrosine-based activation motif-signaling and toll-like receptor pathways in macrophages and dendritic cells. Front Immunol. 2017;8:394. doi: 10.3389/fimmu.2017.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, Ahuja AK, Lopes M, Hausmann A, Hardt WD, Gomariz Á, Nombela-Arrieta C, Manz MG. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. 2017;21:225–240. e225. doi: 10.1016/j.stem.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Haricharan S, Brown P. TLR4 has a TP53-dependent dual role in regulating breast cancer cell growth. Proc Natl Acad Sci U S A. 2015;112:E3216–3225. doi: 10.1073/pnas.1420811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain S, Suklabaidya S, Das B, Raghav SK, Batra SK, Senapati S. TLR4 activation by lipopolysaccharide confers survival advantage to growth factor deprived prostate cancer cells. Prostate. 2015;75:1020–1033. doi: 10.1002/pros.22983. [DOI] [PubMed] [Google Scholar]
- 18.Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, Pereira JA, Silva CM, Silva CR, Silva RL, Speck-Hernandez CA, Mota JM, Alves-Filho JC, Lima-Junior RC, Cunha TM, Cunha FQ. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018;78:5891–5900. doi: 10.1158/0008-5472.CAN-17-3480. [DOI] [PubMed] [Google Scholar]
- 19.Zimmer SM, Liu J, Clayton JL, Stephens DS, Snyder JP. Paclitaxel binding to human and murine MD-2. J Biol Chem. 2008;283:27916–27926. doi: 10.1074/jbc.M802826200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong WJ, Liu JY, Yin JY, Cui JJ, Xiao D, Zhuo W, Luo C, Liu RJ, Li X, Zhang W, Zhou HH, Liu ZQ. Resistin facilitates metastasis of lung adenocarcinoma through the TLR4/Src/EGFR/PI3K/NF-kappaB pathway. Cancer Sci. 2018;109:2391–2400. doi: 10.1111/cas.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin S, Zhang R, An X, Li Z, Fang C, Pan B, Chen W, Xu G, Han W. LncRNA HOXA-AS3 confers cisplatin resistance by interacting with HOXA3 in non-small-cell lung carcinoma cells. Oncogenesis. 2019;8:60. doi: 10.1038/s41389-019-0170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huaitong X, Yuanyong F, Yueqin T, Peng Z, Wei S, Kai S. Microvesicles releasing by oral cancer cells enhance endothelial cell angiogenesis via Shh/RhoA signaling pathway. Cancer Biol Ther. 2017;18:783–791. doi: 10.1080/15384047.2017.1373213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, Meder L, Lovly CM, Persigehl T, Merkelbach-Bruse S, Bos M, Michels S, Fischer R, Albus K, König K, Schildhaus HU, Fassunke J, Ihle MA, Pasternack H, Heydt C, Becker C, Altmüller J, Ji H, Müller C, Florin A, Heuckmann JM, Nuernberg P, Ansén S, Heukamp LC, Berg J, Pao W, Peifer M, Buettner R, Wolf J, Thomas RK, Sos ML. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. 2016;22:4837–4847. doi: 10.1158/1078-0432.CCR-15-1915. [DOI] [PubMed] [Google Scholar]
- 24.van Vliet SJ, Garcia-Vallejo JJ, van Kooyk Y. Dendritic cells and C-type lectin receptors: coupling innate to adaptive immune responses. Immunol Cell Biol. 2008;86:580–587. doi: 10.1038/icb.2008.55. [DOI] [PubMed] [Google Scholar]
- 25.Braunstein MJ, Kucharczyk J, Adams S. Targeting toll-like receptors for cancer therapy. Target Oncol. 2018;13:583–598. doi: 10.1007/s11523-018-0589-7. [DOI] [PubMed] [Google Scholar]
- 26.Ren G, Hu J, Wang R, Han W, Zhao M, Zhou G, Zhang C, Zhang Z. Rapamycin inhibits Toll-like receptor 4-induced pro-oncogenic function in head and neck squamous cell carcinoma. Oncol Rep. 2014;31:2804–2810. doi: 10.3892/or.2014.3134. [DOI] [PubMed] [Google Scholar]
- 27.Shanmugam MK, Nguyen AH, Kumar AP, Tan BK, Sethi G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: potential role in prevention and therapy of cancer. Cancer Lett. 2012;320:158–170. doi: 10.1016/j.canlet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 28.Basudhar D, Glynn SA, Greer M, Somasundaram V, No JH, Scheiblin DA, Garrido P, Heinz WF, Ryan AE, Weiss JM, Cheng RYS, Ridnour LA, Lockett SJ, McVicar DW, Ambs S, Wink DA. Coexpression of NOS2 and COX2 accelerates tumor growth and reduces survival in estrogen receptor-negative breast cancer. Proc Natl Acad Sci U S A. 2017;114:13030–13035. doi: 10.1073/pnas.1709119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong JL, Obermajer N, Odunsi K, Edwards RP, Kalinski P. Synergistic COX2 induction by IFNgamma and TNFalpha self-limits type-1 immunity in the human tumor microenvironment. Cancer Immunol Res. 2016;4:303–311. doi: 10.1158/2326-6066.CIR-15-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong K, Guo G, Gerber DE, Gao B, Peyton M, Huang C, Minna JD, Hatanpaa KJ, Kernstine K, Cai L, Xie Y, Zhu H, Fattah FJ, Zhang S, Takahashi M, Mukherjee B, Burma S, Dowell J, Dao K, Papadimitrakopoulou VA, Olivas V, Bivona TG, Zhao D, Habib AA. TNF-driven adaptive response mediates resistance to EGFR inhibition in lung cancer. J Clin Invest. 2018;128:2500–2518. doi: 10.1172/JCI96148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong XF, Liu TQ, Zhi XT, Zou J, Zhong JT, Li T, Mo XL, Zhou W, Guo WW, Liu X, Chen YY, Li MY, Zhong XG, Han YM, Wang ZH, Dong ZR. COX-2/PGE2 axis regulates HIF2alpha activity to promote hepatocellular carcinoma hypoxic response and reduce the sensitivity of sorafenib treatment. Clin Cancer Res. 2018;24:3204–3216. doi: 10.1158/1078-0432.CCR-17-2725. [DOI] [PubMed] [Google Scholar]
- 32.Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11:734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 34.Srivastava RM, Trivedi S, Concha-Benavente F, Hyun-Bae J, Wang L, Seethala RR, Branstetter BF 4th, Ferrone S, Ferris RL. STAT1-induced HLA class I upregulation enhances immunogenicity and clinical response to anti-EGFR mAb cetuximab therapy in HNC patients. Cancer Immunol Res. 2015;3:936–945. doi: 10.1158/2326-6066.CIR-15-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tzieply N, Kuhn AM, Morbitzer D, Namgaladze D, Heeg A, Schaefer L, von Knethen A, Jensen LE, Brüne B. OxLDL inhibits LPS-induced IFNbeta expression by Pellino3- and IRAK1/4-dependent modification of TANK. Cell Signal. 2012;24:1141–1149. doi: 10.1016/j.cellsig.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 36.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 37.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 38.Scheel AH, Dietel M, Heukamp LC, Jöhrens K, Kirchner T, Reu S, Rüschoff J, Schildhaus HU, Schirmacher P, Tiemann M, Warth A, Weichert W, Fischer RN, Wolf J, Buettner R. Harmonized PD-L1 immunohistochemistry for pulmonary squamous-cell and adenocarcinomas. Mod Pathol. 2016;29:1165–72. doi: 10.1038/modpathol.2016.117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.