

Abstract
To investigate whether p16 deletion can prevent osteoporosis caused by estrogen deficiency, we first confirmed that p16 protein expression levels were significantly up-regulated in bony tissue of ovariectomized (OVX) wild-type mice. Eight-week-old wild-type and p16-/- mice were then sham-operated or bilateral OVX. After 12 weeks, the bone phenotypes of all models were analyzed by radiography, micro-computed tomography, histology, immunohistochemistry, and molecular biology. The results showed that p16 deficiency could rescue OVX-induced osteoporosis by significantly increased bone mineral density, trabecular bone volume, total collagen positive area, osteoblast number, type I collagen positive area, fibroblast colony-forming unit (CFU-f) and alkaline phosphatase-positive CFU-f with up-regulation of the mRNA expression levels of Alp, Runx2, type I collagen and osteocalcin, and significantly reduced osteoclast surface and the ratio of RANKL/OPG mRNA expression level. Furthermore, we also demonstrated that p16 deletion inhibited OVX-induced oxidative stress and bone cell senescence, such as a significant decrease in reactive oxygen species levels, up-regulation of superoxide dismutase 1 and 2 protein expression levels, and reduction of the percentage of β-galactosidase-positive osteocytes and p21 protein expression levels in bony tissue. Our results indicate that p16 deletion can prevent estrogen deficiency-induced osteoporosis by inhibiting oxidative stress, osteocyte senescence and osteoclastic bone resorption, stimulating osteogenesis and osteoblastic bone formation. Therefore, this study provides new insights into the potential of p16 as a novel therapeutic target for estrogen deficiency-induced osteoporosis.
Keywords: Estrogen deficiency, osteoporosis, p16, oxidative stress, osteocyte senescence
Introduction
Osteoporosis is characterized by low bone mass, microarchitectural disruption, and skeletal fragility, resulting in decreased bone strength and an increased risk of fracture [1]. Osteoporosis has become a global health problem and research hotspot of many countries all over the world in the past decade. The bone loss triggered by declining serum sex steroids in postmenopausal humans has been extensively investigated [2]. As the most common type and with the highest incidence rate, postmenopausal osteoporosis leading bone loss resulted mainly from stimulation of bone resorption via increased osteoclast formation or increased osteoclast lifespan coupled with insufficient osteoblastic bone formation [3]. Currently, clinical treatment of postmenopausal osteoporosis mainly includes hormone replacement therapy, calcium, vitamin D, bisphosphonate, selective estrogen receptor modulator, and parathyroid hormone [4]. However, the therapeutic effects are not ideal. Exploring the mechanism of postmenopausal osteoporosis is a primary task for scientific researchers so that it can develop drugs that prevent and treat postmenopausal osteoporosis.
Accumulating studies have shown that the oxidative stress induced by reactive oxygen species (ROS) is increased with aging, which is implicated in the pathophysiology of postmenopausal osteoporosis [5-10]. Studies have shown that estrogen protects bone by acting as an antioxidant and resists oxidative stress [11]. Besides, studies have suggested that the transition from “estrogen-centered” pathogenesis to oxidative stress is also involved in the mechanism of osteoporosis [2]. Oxidative stress is an important cause of cellular senescence. Cellular senescence is the process by which a cell enters a permanent cell cycle block, and senescent cells display a senescence-associated secretory phenotype (SASP) [12]. Our recent study has demonstrated that estrogen deficiency-induced bone loss was not only associated with increased oxidative stress and osteoclastic bone resorption but also associated with increased osteocyte senescence, as shown in increased the percentages of p16 positive osteocytes and p16 protein expression levels in bony tissue [13].
P16 is one of the products of the INK4/ARF locus and was molecularly cloned by virtue of its interaction with cyclin-dependent kinase 4 (CDK4). It was soon realized that p16 controls the G1 phase of the cell cycle, is a key regulator of cell senescence, and is frequently inactivated in cancer and also is regarded as related to many age-related pathologies, including myocardial infarction, type 2 diabetes, and osteoporosis [14]. The cell cycle-dependent kinase inhibitor p16 is not only a recognized indicator of cellular senescence, but it also acts as a critical effector of cellular senescence [15]. During the development of physiological aging and aging-related diseases, the expression level of p16 is gradually increased [16]. Recent studies have shown that p16-positive cells in different tissues contribute to the development and progression of aging-related lesions, resulting in a shortened healthy life span, while the elimination of p16-positive senescent cells can delay the development and progression of senescence-related lesions in different tissues. Deletion of p16-positive senescent cells not only prolongs the lifespan of premature aging mice but also extends the lifespan of natural aging mice [17,18]. However, it remains unknown whether p16 deletion can rescue estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence, suppressing osteoclastic bone resorption, stimulating osteogenesis of bone marrow mesenchymal stem cells (BM-MSCs) and osteoblastic bone formation.
In this study, we confirmed that p16 protein expression level was upregulated significantly in wild-type ovariectomized (WT-OVX) mice compared with WT-sham mice. Then 8-week-old WT and p16-/- mice were sham-operated or bilaterally ovariectomized (OVX). After 12 weeks, bone phenotypes of all models were analyzed by radiography, micro-computed tomography (micro-CT), histology immunohistochemistry, and molecular biology to determine whether deletion of p16 could rescue estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence.
Materials and methods
Mice and genotyping
Male and female p16+/- mice of the FVB N2 background were mated to produce offspring heterozygous at both loci, which were then mated to generate p16-/- and wild-type (WT) pups. Their genotypes were analyzed, as described previously [19]. All animal experiments were carried out in compliance with, and approval by approved by the Institutional Animal Care and Use Committee of Nanjing Medical University.
Ovariectomy
Eight-week-old wild-type and p16-/- mice were sham-operated or bilaterally ovariectomized (OVX) according to previously established methods [20]. After 12 weeks, the success of ovariectomy was confirmed by X-ray images of the coccygeal vertebra that were taken with a Faxitron and shown significantly lower bone mineral density (data not shown).
Radiographs and micro-computed tomography
The vertebral bodies were removed, and all soft tissues were dissected for contact radiography or micro-computed tomography (micro-CT) as described [21]. The measurements of bone mineral density (BMD), trabecular volume relative to tissue volume were measured as previously described [22].
Histology
All animals were sacrificed by cervical dislocation after inhaled anesthesia with ether. Vertebrae were removed and dissected free of soft tissue, fixed with 4% paraformaldehyde, decalcified with EDTA glycerol solution, and embedded in paraffin. Sections were stained with H&E or histochemically for total collagen or tartrate-resistant acid phosphatase (TRAP) as previously described [21].
Immunohistochemical staining
Immunohistochemical staining for type I collagen and β-galactosidase (β-gal) was performed using the avidin-biotin-peroxidase complex technique with affinity-purified rabbit anti-mouse β-gal (Santa Cruz, CA, USA) and p21 (Santa 192 Cruz, CA, USA) as described previously [13].
Fibroblast colony-forming unit assay
Bone marrow cells were flushed out from femurs and tibias. For fibroblast colony-forming unit (CFU-f) and alkaline phosphatase (ALP)-expressing CFU-f assays, total bone marrow cells were cultured in six-well-plates at 1×106 cells/well in 2 mL of a modified essential medium (α-MEM) containing 10% fetal calf serum (FBS) (Hyclone Laboratories, Logan, UT, USA), 50 mg/mL ascorbic acid and 10 mM β-glycerophosphate for 10 days. At the end of the culture period, cells were washed with PBS, fixed with PLP fixative, and stained with methyl blue or cytochemical for ALP. Following staining, the quantitation of total colony (CFU-f) area and of ALP-positive (CFU-fap) area was determined using computer-assisted image analysis.
Quantitative real-time RT-PCR
Total RNA was isolated from mouse bone tissue or bone marrow mesenchymal stem cells (BM-MSCs) with Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Real-time RT-PCR was performed as described previously [13].
Western blot analysis
For examination of protein expression level, proteins were extracted from the vertebral bodies, quantitated with a protein assay kit (Bio-Rad, Mississauga, Ontario, Canada). Protein samples (15 µg) were fractionated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blotted with primary antibodies against SOD1 (Abcam), SOD2 (Novus Biological), p16 (Santa Cruz, MA), p21 (Santa Cruz, MA) and β-actin (Bioworld Technology, St. Louis Park, MN, USA) were used as a loading control. Immunoblotting was carried out, as described previously [23].
Reactive oxygen species (ROS) examination
Bone marrow cells were filtered and incubated with 5 mM diacetyldichlorofluorescein (DCFDA) (Invitrogen) and 10% FBS for 30 mins at 37°C. Then cells were analyzed with a FACS Calibur flow cytometer (Becton Dickinson, Heidelberg, Germany) for the mean fluorescent intensity (MFI) and relative fluorescent intensity to MFI of shame-WT mice.
Statistical analysis
All the described data represent at least three separate experiments (n>5/group) and are expressed as the Mean ± SEM. Comparisons between the two groups were analyzed using a two-tailed unpaired Student’s t-test. Comparisons between more than two groups were performed using one-way ANOVA followed by Bonferroni post-hoc multiple comparisons. The significant level was P<0.05.
Results
Effect of p16 deletion on OVX-induced bone loss
Our recent study showed that OVX induced the upregulation of p16 expression levels in wild-type (WT) mice. In this study, we confirmed that the protein expression levels were upregulated dramatically in WT-OVX mice relative to WT-sham mice (Figure 1A and 1B). P16-/- mice and their WT littermates were surgically OVX or sham-operated to determine whether p16 deletion could rescue OVX-induced bone loss. After 12 weeks, bone phenotypes of all models were analyzed by radiography, micro-CT, and total collagen staining. The results showed that bone mineral density, trabecular bone volume, and total collagen positive area were decreased significantly in WT-OVX mice and increased substantially in p16-/--sham (KO-sham) mice. However, they were normalized in p16-/--OVX (KO-OVX) mice compared with WT-sham mice (Figure 1C-H). These parameters were markedly increased in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 1C-H). These results demonstrated that p16 deletion could rescue OVX-induced bone loss.
Figure 1.
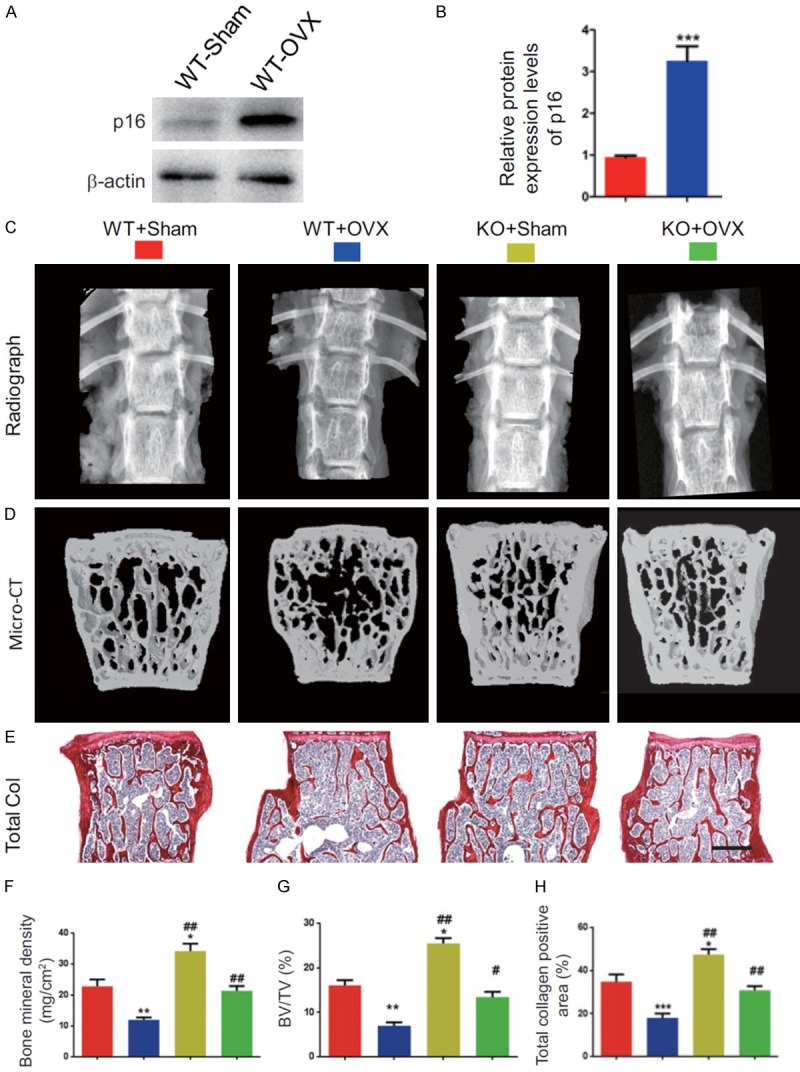
p16 deletion rescues OVX-induced bone loss. (A) The protein expression levels of p16 were detected in bony tissue from WT-Sham and WT-OVX mice by Western blotting. β-actin was used as the loading control. (B) p16 protein levels were assessed by densitometric analysis calculated as a ratio relative to β-actin protein levels and expressed relative to levels of WT-Sham mice. (C) The representative radiographs and (D) micro-CT images of three-dimensional reconstructed images of the vertebral bodies from WT-Sham, WT-OVX, p16 knockout sham (KO-Sham) and p16 knockout OVX (KO-OVX) mice. (E) Representative micrographs of paraffin-embedded sections of vertebral bodies stained histochemically for total collagen. Scale bars represent 400 μm. (F) Bone mineral density. (G) Trabecular bone volume/total volume (BV/TV). (H) Total collagen positive areas. Data represented as mean ± SEM of determinations in 5 mice of each group. *: P<0.05; **: P<0.01; ***: P<0.001, compared with WT-Sham mice. #: P<0.05 and ##: P<0.01, compared with the same operational WT mice.
Effect of p16 deletion on osteoblastic bone formation in OVX mice
To determine whether rescued OVX-induced bone loss by p16 deletion is associated with increased osteoblastic bone formation, osteoblast number and collagen deposition in the bone matrix were examined by histology, immunohistochemical staining for type I collagen and computer-assisted image analyses. The results showed that osteoblast number and type I collagen positive area were decreased significantly in WT-OVX mice and increased considerably in KO-sham mice, however, they were normalized in KO-OVX mice compared with WT-sham mice (Figure 2A-D). These parameters were increased significantly in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 2A-D). These results implied that p16 deletion could rescue OVX-induced bone loss through increasing osteoblastic bone formation.
Figure 2.
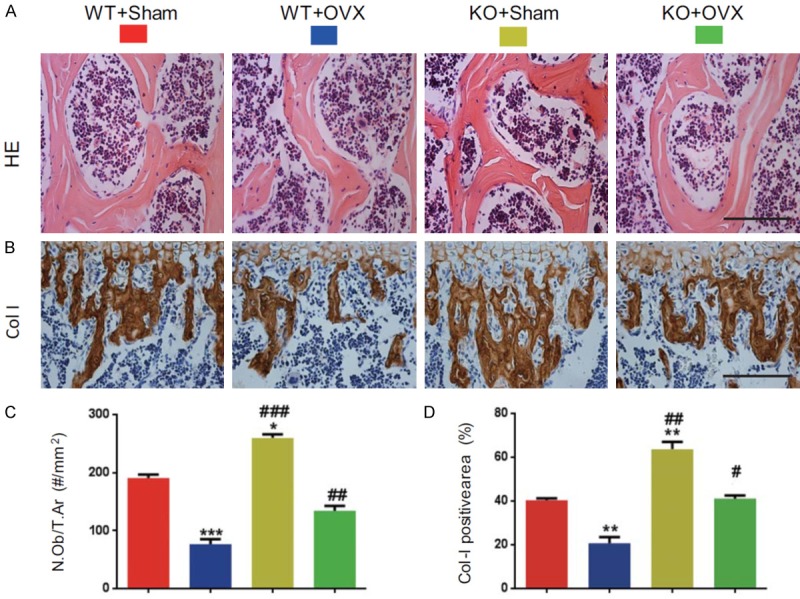
p16 deletion increased osteoblastic bone formation in OVX mice. (A) Representative micrographs of paraffin-embedded sections of vertebral bodies stained with H&E and (B) immunostaining for type I. Scale bars represent 50 μm in (A and B). (C) Osteoblast number relative to tissue area (N.Ob/T. Ar, #/mm2). (D) Type I collagen (Col-I) positive areas were measured by computer-assisted image analysis. Data represented as mean ± SEM of determinations in 5 mice of each group. **: P<0.01; P<0.05; ***: P<0.001, compared with WT-Sham mice. ##: P<0.01 and ###: P<0.001, compared with the same operational WT mice.
Effect of p16 deletion on osteogenesis of bone marrow mesenchymal stem cells in OVX mice
To determine whether rescued OVX-induced bone loss by p16 deletion is associated with enhanced osteogenesis of bone marrow mesenchymal stem cells (BM-MSCs), CFU-f assays were performed by ex vivo bone marrow cell cultures. Results revealed that CFU-f and CFU-fap areas were decreased significantly in WT-OVX mice and increased significantly in KO-sham mice; however, they were normalized in KO-OVX mice compared with WT-sham mice (Figure 3A-D). These parameters were increased considerably in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 3A-D). We also examined the effect of p16 deletion on osteogenic gene expression using real-time RT-PCR. Results showed that mRNA expression levels of Alp, Runx2, type I collagen, and osteocalcin were down-regulated significantly in WT-OVX mice and upregulated significantly in KO-sham mice. However, they were normalized in KO-OVX mice compared with WT-sham mice (Figure 3E-H). They were markedly upregulated in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 3E-H). These results suggest that p16 deletion could rescue OVX-induced osteogenic defect through stimulating osteogenesis of BM-MSCs.
Figure 3.
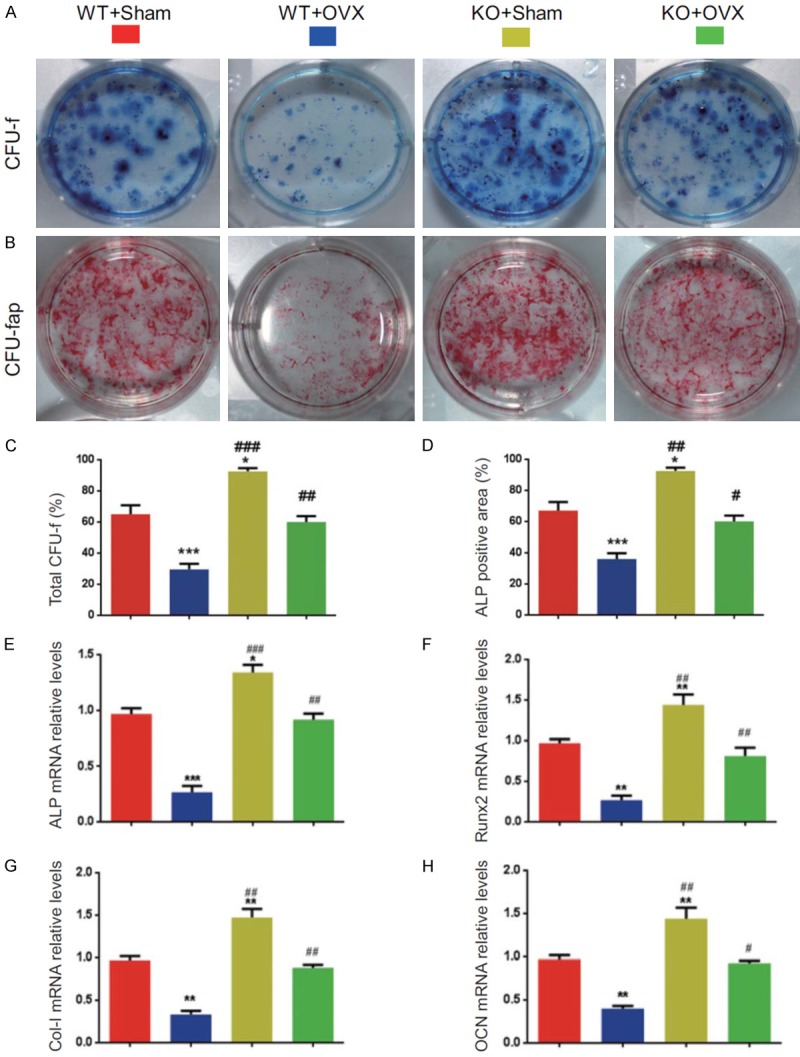
p16 deletion stimulated osteogenesis of BM-MSCs in OVX mice. Primary bone marrow cells from 6-month-old WT-Sham, WT-OVX, KO-Sham, and KO-OVX mice were cultured ex vivo in osteogenic differentiation medium for 18 days, and resulting cultures were stained with (A) methylene blue for the total number of colonies (CFU-F) and (B) cytochemically for ALP to show CFU-Fap. (C) Total CFU-f-positive areas and (D) ALP-positive areas relative to the culture dish area. Real-time RT-PCR analysis of bone marrow MSC extracts for the expression of (E) Alp, (F) Runx2, (G) Col-I, and (H) osteocalcin (OCN). Messenger RNA expression assessed by real-time RT-PCR is expressed as a ratio to Gapdh expression level and expressed relative to WT-Sham mice. Data represented as mean ± SEM of determinations in 5 mice of each group. *: P<0.05; **: P<0.01; ***: P<0.001, compared with WT-Sham mice. #: P<0.05; ##: P<0.01 and ###: P<0.001, compared with the same operational WT mice.
Effect of p16 deletion on osteoclastic bone resorption in OVX mice
To determine whether rescued OVX-induced bone loss by p16 deletion is associated with decreased osteoclastic bone resorption, osteoclast surface and the ratio of RANKL/OPG mRNA expression levels were examined using histochemical staining for TRAP and real-time RT-PCR. Results showed that osteoclast surface and the ratio of RANKL/OPG mRNA expression levels were increased dramatically in WT-OVX mice and reduced significantly in KO-sham mice, however, they were normalized in KO-OVX mice compared with WT-sham mice (Figure 4A-C). These parameters were decreased significantly in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 4A-C). These results implied that p16 deletion could rescue OVX-induced bone loss through inhibiting osteoclastic bone resorption.
Figure 4.
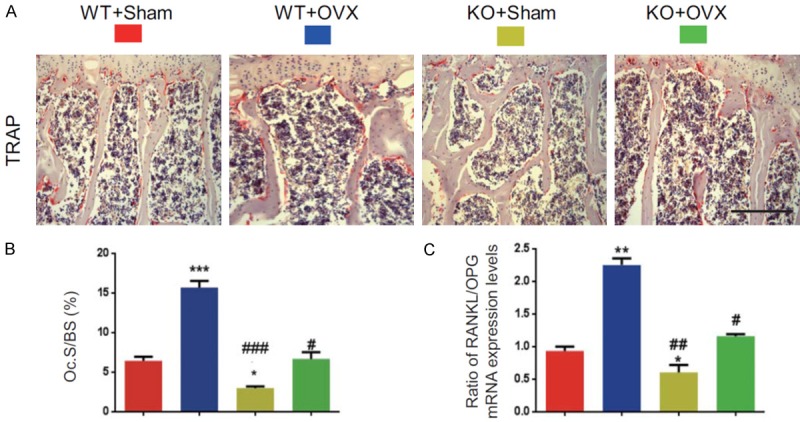
p16 deletion inhibited osteoclastic bone resorption in OVX mice. (A) Representative micrographs of paraffin-embedded sections of vertebral bodies stained histochemically for TRAP. Scale bars represent 50 μm in (A). (B) Osteoclast surface/bone surface (Oc.S/BS, %). (C) Real-time RT-PCR analysis of vertebral extracts for the expression of NANKL and OPG and expressed the ratio of NANKL/OPG mRNA expression levels. Data represented as mean ± SEM of determinations in 5 mice of each group. *: P<0.05; **: P<0.01; ***: P<0.001, compared with WT-Sham mice. #: P<0.05; ##: P<0.01 and ###: P<0.001, compared with the same operational WT mice.
Effect of p16 deletion on redox balance in OVX mice
To determine whether the estrogen deficiency-induced osteoporosis rescued by p16 deletion is associated with reduced oxidative stress induced by OVX, we examined the alterations of ROS levels and expression levels of antioxidant enzymes in bony tissue. Results showed that ROS levels increased dramatically in WT-OVX mice and reduced significantly in KO-sham mice, however, they were normalized in KO-OVX mice compared with WT-sham mice and were decreased significantly in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 5A, 5B). The protein expression levels of superoxide dismutase (SOD) 1 and 2 were down-regulated significantly in WT-OVX mice and upregulated clearly in KO-sham mice, however, they were normalized in KO-OVX mice compared with WT-sham mice (Figure 5C-E). The protein expression levels of SOD1 and SOD2 were upregulated significantly in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 5C-E). These results demonstrated that p16 deletion could inhibit oxidative stress in OVX mice by inhibiting the expression levels of antioxidant enzymes.
Figure 5.
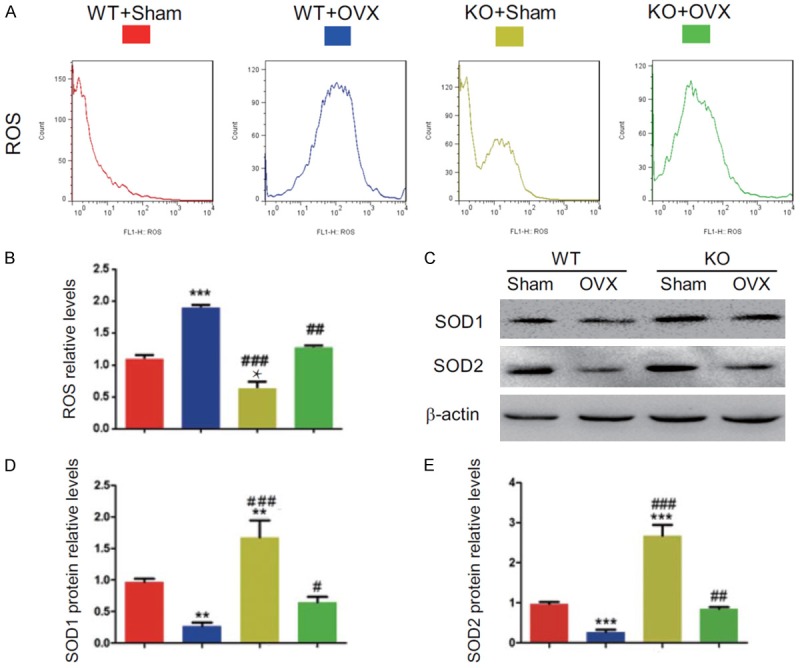
p16 deletion reduced oxidative stress in OVX mice. (A) Representative graphs of flow cytometry analysis for ROS levels in the bone marrow and (B) ROS relative levels. (C) The protein expression levels of SOD1 and SOD2 were detected by Western blotting. β-actin was used as the loading control. (D) SOD1 and (E) SOD2 protein levels were assessed by densitometric analysis calculated as a ratio relative to β-actin protein levels and expressed relative to levels of WT-Sham mice. Data represented as mean ± SEM of determinations in 5 mice of each group. *: P<0.05; **: P<0.01; ***: P<0.001, compared with WT-Sham mice. ##: P<0.01 and ###: P<0.001, compared with the same operational WT mice.
Effect of p16 deletion on osteocyte senescence in OVX mice
To determine whether the estrogen deficiency-induced osteoporosis rescued by p16 deletion is associated with reduced osteocyte senescence induced by OVX, we examined the alterations of osteocyte senescence by immunostaining for β-galactosidase (β-gal) and Western blot for p21 expression. Results showed that the percentages of β-gal positive osteocytes and p21 protein expression levels in bony tissue were increased dramatically in WT-OVX mice and reduced significantly in KO-sham mice, however, they were normalized in KO-OVX mice compared with WT-sham mice. They were decreased significantly in KO-sham or KO-OVX mice compared with WT-sham or WT-OVX mice (Figure 6A-D). These results demonstrated that p16 deletion could inhibit osteocyte senescence induced by OVX.
Figure 6.
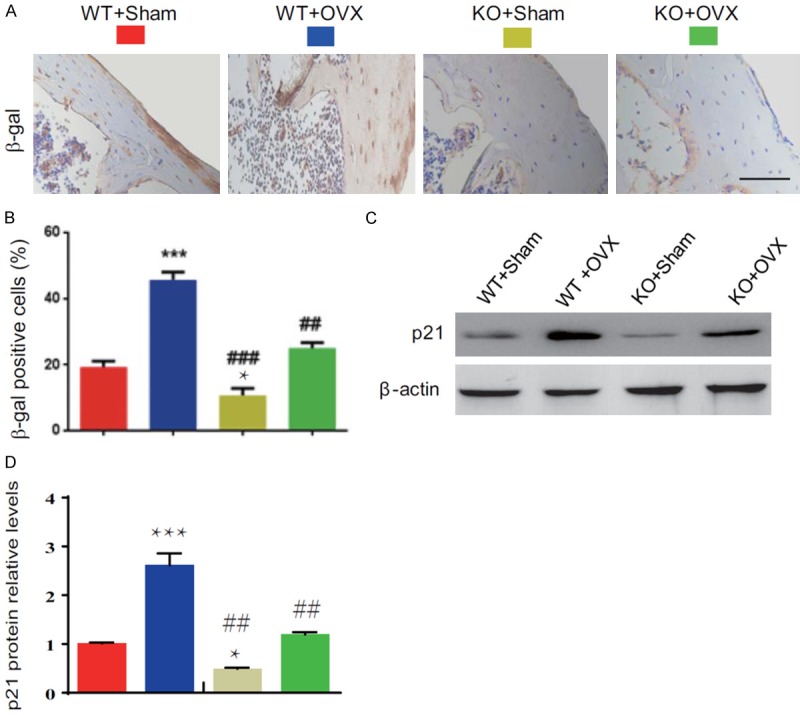
p16 deletion inhibited osteocyte senescence in OVX mice. (A) Representative micrographs of paraffin-embedded sections of vertebral bodies immunostained for β-gal. Scale bars represent 50 μm in (A). (B) The percentage of β-gal positive cells. (C) The protein expression levels of p21 were detected by Western blotting. β-actin was used as the loading control. (D) p21 protein levels were assessed by densitometric analysis calculated as a ratio relative to β-actin protein levels and expressed relative to levels of WT-Sham mice. Data represented as mean ± SEM of determinations in 5 mice of each group. *: P<0.05; ***: P<0.001, compared with WT-Sham mice. ##: P<0.01 and ###: P<0.001, compared with the same operational WT mice.
Discussion
In the present study, we employed OVX mouse model, which is a well-established and widely used animal model in the study of postmenopausal osteoporosis [24-26] to assess the effect of p16 deletion on the estrogen deficiency-induced osteoporosis. We first confirmed that OVX resulted in bone loss with reduced BMD, trabecular bone volume, and total collagen positive areas. Simultaneously, we demonstrated that p16 deletion rescued bone loss induced by OVX.
Although the significant consequence of estrogen deficiency is an increase in bone resorption, estrogen is also essential for maintaining bone formation at the cellular level [27]. Thus, we evaluated the effects of OVX and p16 deletion on bone turnover parameters. Our current study demonstrated that OVX induced not only osteoclastic bone resorption, as shown by increased TRAP-positive osteoclast surface and RANKL/OPG ratio but also reduced osteoblastic bone formation and osteogenesis of BM-MSCs as shown decreased osteoblast number, type I collagen positive areas, CFU-f and ALP positive CFU-f formation. In contrast, all parameters for osteoclastic bone resorption and osteoblastic bone formation and osteogenesis were normalized by p16 deletion. Our results indicate that p16 deletion can prevent estrogen deficiency-induced osteoporosis by inhibiting osteoclastic bone resorption, stimulating osteoblastic bone formation, and osteogenesis of BM-MSCs.
Next, we asked whether the estrogen deficiency-induced osteoporosis prevented by p16 deletion is associated with reduced oxidative stress induced by OVX. Recent studies have powerfully shown that estrogen deficiency accelerates bone aging as it dramatically diminishes the defense mechanisms against oxidative stress [2,28]. Research has demonstrated that BMI-1 works as a direct downstream target of estrogen signaling. In that study, 17β-estradiol treatment upregulated BMI-1 expression levels in ERα-positive MCF-7 cells but not in ERα-negative MDA-MB-231 cells, and ERα upregulated BMI-1 expression at a transcriptional level by directly binding to BMI-1 core promoter [29]. Our recent studies have shown that estrogen deficiency down-regulates Bmi1, increases ROS, T cell activation, and RANKL production in T cells, thereby enhancing osteoclastogenesis and accelerating bone loss. Our previous studies have also shown that estrogen deficiency causes oxidative stress in mouse bone tissue with reduced antioxidant enzyme levels and activity, and impairs osteogenesis and osteoblastic bone formation, while the use of antioxidant N-acetyl-l-cysteine can significantly improve osteogenesis and osteoblastic bone formation in OVX mice [20]. The previous study has shown that oxidative stress increases p16 expression, which in turn, accelerates senescence in human dental pulp stem cells cultured under ambient oxygen tension [30]. Previous studies suggest that the relationship between p16 levels and oxidative stress may be highly dependent on the circumstances and cell system utilized. For example, it was reported that over-expression of p16 increases cellular ROS levels in a human diploid fibroblast line (TIG-3), and p16 knockdown decreases ROS in a conditionally immortalized human fibroblast line (SVts8) [31]. In contrast, another study has presented evidence that p16 deficiency leads to dysregulation of intracellular ROS in multiple cell types [32]. Our current study confirmed that estrogen deficiency could induce oxidative stress, including increased ROS levels and reduced antioxidase levels including down-regulating SOD1 and SOD2 protein expression levels in bony tissue, whereas p16 deletion could reduce oxidative stress by increasing antioxidase levels in OVX mice. Results from previous and this study, therefore, support that estrogen plays a role in stimulating osteoblastic bone formation and inhibiting osteoclastic bone resorption in vivo, at least partially through defense against oxidative stress and inactivation of p16 signaling.
We recently reported that p16 deletion could partly prevent aging resulting from 1,25(OH)2D3 deficiency by enhancing cell proliferative ability and reducing cell senescence [23]. In the current study, we found that OVX could activate p16 signaling, whereas p16 deletion rescues OVX-induced osteoporosis by stimulating osteoblastic bone formation, inhibiting osteoclastic bone resorption, and osteocyte senescence. Studies have used the “suicide” transgenic INK-ATTAC to eliminate relatively small senescent cells, which permits inducible elimination of senescent cells expressing p16, prolongs health span, and prevents the development of multiple age-related morbidities in both progeroid and normal chronologically-aged mice [17,18]. In old mice with established bone loss, activation of the INK-ATTAC caspase 8 in senescent cells or treatment with senolytics or the JAKi for 2-4 months resulted in higher bone mass and strength and better bone microarchitecture compared to vehicle-treated mice [33], suggesting that targeting cellular senescence could prevent age-related bone loss in mice. Our findings indicate that estrogen could prevent bone loss by targeting the p16 cellular senescence pathway.
In conclusion, this study demonstrated that p16 deletion rescued estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence, suppressing osteoclastic bone resorption, stimulating osteogenesis of BM-MSCs, and osteoblastic bone formation. Our results from this study, therefore, provide new insight indicating that p16 possesses excellent potential as a novel class of therapeutic target for osteoporosis induced by estrogen deficiency.
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2018YFA0800802 to DM) and the National Natural Science Foundation of China (81730066 to DM).
Disclosure of conflict of interest
None.
References
- 1.Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, Lindsay R National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int. 2014;25:2359–2381. doi: 10.1007/s00198-014-2794-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black DM, Rosen CJ. Clinical practice. Postmenopausal osteoporosis. N Engl J Med. 2016;374:254–262. doi: 10.1056/NEJMcp1513724. [DOI] [PubMed] [Google Scholar]
- 4.Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D. Pharmacological management of osteoporosis in postmenopausal women: an endocrine society* clinical practice guideline. J Clin Endocrinol Metab. 2019;104:1595–1622. doi: 10.1210/jc.2019-00221. [DOI] [PubMed] [Google Scholar]
- 5.Barrett-Connor E, Grady D, Stefanick ML. The rise and fall of menopausal hormone therapy. Annu Rev Public Health. 2005;26:115–140. doi: 10.1146/annurev.publhealth.26.021304.144637. [DOI] [PubMed] [Google Scholar]
- 6.Kawai M, Modder UI, Khosla S, Rosen CJ. Emerging therapeutic opportunities for skeletal restoration. Nat Rev Drug Discov. 2011;10:141–156. doi: 10.1038/nrd3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khosla S, Bellido TM, Drezner MK, Gordon CM, Harris TB, Kiel DP, Kream BE, LeBoff MS, Lian JB, Peterson CA, Rosen CJ, Williams JP, Winer KK, Sherman SS. Forum on aging and skeletal health: summary of the proceedings of an ASBMR workshop. J Bone Miner Res. 2011;26:2565–2578. doi: 10.1002/jbmr.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377:1276–1287. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid IR. Short-term and long-term effects of osteoporosis therapies. Nat Rev Endocrinol. 2015;11:418–428. doi: 10.1038/nrendo.2015.71. [DOI] [PubMed] [Google Scholar]
- 10.Seeman E, Delmas PD. Bone quality--the material and structural basis of bone strength and fragility. N Engl J Med. 2006;354:2250–2261. doi: 10.1056/NEJMra053077. [DOI] [PubMed] [Google Scholar]
- 11.Cervellati C, Bonaccorsi G, Cremonini E, Bergamini CM, Patella A, Castaldini C, Ferrazzini S, Capatti A, Picarelli V, Pansini FS, Massari L. Bone mass density selectively correlates with serum markers of oxidative damage in post-menopausal women. Clin Chem Lab Med. 2013;51:333–338. doi: 10.1515/cclm-2012-0095. [DOI] [PubMed] [Google Scholar]
- 12.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geng Q, Gao H, Yang R, Guo K, Miao D. Pyrroloquinoline quinone prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Int J Biol Sci. 2019;15:58–68. doi: 10.7150/ijbs.25783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riggs BL, Khosla S, Melton LJ 3rd. Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. 2002;23:279–302. doi: 10.1210/edrv.23.3.0465. [DOI] [PubMed] [Google Scholar]
- 15.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16 (Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin J, Tao J, Gu X, Yu Z, Wang R, Zuo G, Li Q, Lv X, Miao D. P16INK4a deletion ameliorated renal tubulointerstitial injury in a stress-induced premature senescence model of Bmi-1 deficiency. Sci Rep. 2017;7:7502. doi: 10.1038/s41598-017-06868-8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Shi C, Wu J, Yan Q, Wang R, Miao D. Bone marrow ablation demonstrates that estrogen plays an important role in osteogenesis and bone turnover via an antioxidative mechanism. Bone. 2015;79:94–104. doi: 10.1016/j.bone.2015.05.034. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Li J, Zhang H, Wang H, Yin G, Miao D. Pyrroloquinoline quinone prevents testosterone deficiency-induced osteoporosis by stimulating osteoblastic bone formation and inhibiting osteoclastic bone resorption. Am J Transl Res. 2017;9:1230–1242. [PMC free article] [PubMed] [Google Scholar]
- 22.Sun W, Zhang H, Wang H, Chiu YG, Wang M, Ritchlin CT, Kiernan A, Boyce BF, Xing L. Targeting notch-activated M1 macrophages attenuates joint tissue damage in a mouse model of inflammatory arthritis. J Bone Miner Res. 2017;32:1469–1480. doi: 10.1002/jbmr.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L, Yang R, Qiao W, Zhang W, Chen J, Mao L, Goltzman D, Miao D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell. 2019;18:e12951. doi: 10.1111/acel.12951. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Bouxsein ML, Myers KS, Shultz KL, Donahue LR, Rosen CJ, Beamer WG. Ovariectomy-induced bone loss varies among inbred strains of mice. J Bone Miner Res. 2005;20:1085–1092. doi: 10.1359/JBMR.050307. [DOI] [PubMed] [Google Scholar]
- 25.Khosla S, Melton LJ 3rd, Riggs BL. The unitary model for estrogen deficiency and the pathogenesis of osteoporosis: is a revision needed? J Bone Miner Res. 2011;26:441–451. doi: 10.1002/jbmr.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Syed FA, Melim T. Rodent models of aging bone: an update. Curr Osteoporos Rep. 2011;9:219–228. doi: 10.1007/s11914-011-0074-z. [DOI] [PubMed] [Google Scholar]
- 27.Khosla S, Oursler MJ, Monroe DG. Estrogen and the skeleton. Trends Endocrinol Metab. 2012;23:576–581. doi: 10.1016/j.tem.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manolagas SC, Parfitt AM. What old means to bone? Trends Endocrinol Metab. 2010;21:369–374. doi: 10.1016/j.tem.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Liu H, Li X, Zhao J, Zhang H, Mao J, Zou Y, Zhang H, Zhang S, Hou W, Hou L, McNutt MA, Zhang B. Estrogen receptor alpha-coupled Bmi1 regulation pathway in breast cancer and its clinical implications. BMC Cancer. 2014;14:122. doi: 10.1186/1471-2407-14-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mas-Bargues C, Vina-Almunia J, Ingles M, Sanz-Ros J, Gambini J, Ibanez-Cabellos JS, Garcia-Gimenez JL, Vina J, Borras C. Role of p16 (INK4a) and BMI-1 in oxidative stress-induced premature senescence in human dental pulp stem cells. Redox Biol. 2017;12:690–698. doi: 10.1016/j.redox.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 32.Jenkins NC, Liu T, Cassidy P, Leachman SA, Boucher KM, Goodson AG, Samadashwily G, Grossman D. The p16 (INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene. 2011;30:265–274. doi: 10.1038/onc.2010.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017;23:1072–1079. doi: 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]