

Abstract
We investigated the role of insufficiency of the active form of vitamin D, 1,25-dihydroxyvitamin D [1,25(OH)2D] in age-related bone loss. We employed mice with heterozygous deletion of Cyp27b1, the gene encoding the enzyme that synthesizes 1,25(OH)2D, as a model for 1,25(OH)2D insufficiency and compared the phenotype of lumber vertebrae from 3-, 9- and 18-month-old Cyp27b1+/- mice and their wild-type littermates. We found that in wild-type mice, bone mineral density, bone volume, and Cyp27b1 protein expression levels decreased progressively with age, accompanied by declining osteoblastic bone formation and increasing osteoclastic bone resorption, however these age-related skeletal alterations were more severe in Cyp27b1+/- mice which had significantly lower serum 1,25(OH)2D levels. We then assessed the effect of 1,25(OH)2D haploinsufficiency on oxidative stress and DNA damage, cell senescence and senescence-associated secretory phenotype (SASP) in 9-month-old wild-type and Cyp27b1+/- mice. Our results demonstrated that, in Cyp27b1+/- mice compared with their wild-type littermates, the parameters of oxidative stress and DNA damage were significantly increased, whereas the expression levels of antioxidant enzymes were significantly down-regulated; the percentage of senescent osteocytes and bone marrow mesenchymal stem cells, and the expression levels of SASP molecules and p16, p19 and p53 proteins were all significantly increased in bone tissues. Taken together, the results of this study indicate that 1,25(OH)2D insufficiency accelerates age-related bone loss by increasing oxidative stress and DNA damage, inducing bone cell senescence and SASP, and subsequently inhibiting osteoblastic bone formation while stimulating osteoclastic bone resorption.
Keywords: 1,25(OH)2D; Cyp27b1; osteoporosis; oxidative stress; cell senescence
Introduction
Osteoporosis is a chronic progressive disease characterized by decreases in bone mineral density and bone mass, as well as microarchitectural deterioration of bone, all of which lead to increased susceptibility to fractures [1]. Both genetic and environmental factors contribute to the risk of developing osteoporosis. Many non-hereditary factors have been implicated as risk factors, including tobacco smoking, aging, estrogen deficiency and increased oxidative stress [2,3]. Vitamin D deficiency has been assumed to be an osteoporotic risk factor, and the vitamin D level of 25-hydroxyvitamin D [25(OH)D] has been reported to be negatively correlated with the occurrence of osteoporosis in Western populations [4,5]. Therefore, vitamin D deficiency may play an important role in the development of osteoporosis.
Vitamin D, synthesized in skin tissue or absorbed via the intestine, is first metabolized by the action of liver 25-hydroxylase, to 25(OH)D, which is then converted to the active form, 1,25(OH)2D, by the action of a 25-hydroxyvitamin D 1α-hydroxylase (1α-OHase), which is encoded by the Cyp27b1 gene [6,7]. The active 1,25(OH)2D moiety then exerts its action by binding to the vitamin D receptor (VDR). Previous studies have demonstrated that both 1,25(OH)2D and VDR play vital roles in maintaining the balance of calcium and phosphorus, and bone homeostasis [6,7]. Our previous studies, using mouse models of combined genetic deletion of Cyp27b1 and other genes related to calcium and phosphorus metabolism, including the calcium sensing receptor (Casr) and parathyroid hormone (Pth) [8-12] have demonstrated that both endogenous and exogenous 1,25(OH)2D could stimulate bone formation. Other studies, using mouse models with homozygous deletion of Cyp27b1 or Vdr have revealed many new mechanisms of action of the 1,25 (OH)2D/VDR system in maintaining calcium and phosphorus balance and protecting bone and extraskeletal health [13,14]. However, complete 1,25(OH)2D deficiency is very rare in humans, whereas 1,25 (OH)2D insufficiency may be more common. We therefore employed an animal model of 1,25 (OH)2D insufficiency for our studies, to explore its role in skeletal aging.
Although bone loss is a common feature of human aging, the molecular mechanisms that mediate this effect remain unclear. Oxidative stress increases in the skeleton with age [15], and pharmacological and genetic studies in mice have shown that oxidative stress has a detrimental effect on bone, whereas antioxidants can correct osteoporosis caused by male and female gonadectomy [16-18]. The premature aging mouse model caused by oxidative damage exhibits an osteoporotic phenotype [19,20]. Antioxidant SOD1 knockout mice exhibit increased oxidative stress and decreased bone mass, while antioxidant supplementation can correct bone loss caused by SOD1 deficiency [21]. Overall, therefore, the results of these animal models suggest that oxidative stress is a key cause of bone loss. However, it is unclear whether 1,25 (OH)2D insufficiency can accelerate age-related bone loss by increasing oxidative stress.
Cellular senescence is a process in which a cell enters permanent cell cycle arrest, and senescent cells acquire a senescence-associated secretory phenotype (SASP) [22]. SASP includes pro-inflammatory cytokines, growth factors, chemokines, and matrix remodeling enzymes [23]. Senescent cells cause or aggravate the development of aging-related diseases through their growth arrest phenotype and SASP factors. It is unclear whether 1,25(OH)2D insufficiency accelerates age-related bone loss by inducing bone cell senescence and SASP.
In order to investigate whether 1,25(OH)2D haploinsufficiency accelerates age-related bone loss and whether this occurs by increasing oxidative stress and bone cell senescence, the phenotype of lumber vertebrae from 3-, 9- and 18-month-old Cyp27b1 +/- aging mice and their wild-type littermates were compared. We additionally analyzed alterations of serum calcium, phosphorus, PTH, 25(OH)D and 1,25(OH)2D levels, and parameters of oxidative stress, DNA damage and bone cell senescence in 9-month-old Cyp27b1 +/- mice and their wild-type littermates.
Materials and methods
Animals
The Cyp27b1 heterozygous (Cyp27b1 +/-) mice were generated as previously described by Panda et al. [8] and maintained in an SPF laboratory Animal Center in Nanjing Medical University (Nanjing, China). All experimental procedures involved in this study were pre-approved by the Committee on the Ethics of Animal Experiments of Nanjing Medical University (Permit Number: IACUC-1802007) and carried out strictly in compliance with guidelines of the Institute for Laboratory Animal Research of Nanjing Medical University.
Serum isolation
Blood was collected from 9-month-old wild-type (WT) and Cyp27b1 +/- mice, separated by centrifugation (3000 rpm, 15 min, 4°C) and stored at -80°C. The serum calcium and phosphorus levels were determined in Nanjing Medical University Medical Laboratory Animal Center (Nanjing, China).
Enzyme-linked immunosorbent assay
The serum levels of 25(OH)D (Nanjing Jiancheng Bioengineering Institute), 1,25(OH)2D (Nanjing Jiancheng Bioengineering Institute) and PTH (Nanjing Jiancheng Bioengineering Institute) of 9-month-old WT and Cyp27b1 +/- mice were measured. The serum calcium and phosphorus levels were determined in Nanjing Medical University Medical Laboratory Animal Center (Nanjing, China).
Radiological studies
WT and Cyp27b1 +/- mice were euthanized to obtain the lumber vertebral samples at the age of 3-, 9- or 18-months. Vertebrae were analyzed by radiography, bone densitometry and micro-computed tomography (Μicro-CT) as we described previously [24].
Histology, histochemistry and immunohistochemistry
The vertebral samples were obtained and histologically processed as described previously [24]. Briefly, the vertebral samples were embedded with paraffin and cut into 5-μm sections with a rotary microtome (Leica Biosystems Nussloch GmbH, Nussloch, Germany). The staining of total collagen (T-col), hematoxylin and eosin (H&E), alkaline phosphatase (ALP) activity, tartrate resistant acid phosphatase (TRAP) activity or immunohistochemistry of p16 (Abcam) and β-gal (Abcam) was carried out as previously described [25].
Cell cultures
Primary bone marrow mesenchymal stem cells (BM-MSCs) were isolated from 9-month-old WT and Cyp27b1 +/- mouse and cultured in six-well dish as we previous described [26]. The cellular senescence β-galactosidase staining was performed, when cells grew to 40-50% confluence with a cellular senescence β-galactosidase staining kit (# C0602, Beyotime Biotechnology, Wuhan, China) according to the manufacturer’s protocol.
Western blot analysis
Total protein was extracted from the vertebral tissue of each group of mice and protein concentration was measured using the BCA kit (Beyotime Biotechnology, Wuhan, China). Western blotting was performed as described previously [27], by incubating overnight at 4°C with primary antibodies against Sirt1 (ab110304, Abcam), Nrf2 (L593, Bioworld), γ-H2AX (Ser139, #80312S, Cell Signaling Technology), p19 (sc-1665, Santa Cruz Biotechnology, USA), p16 (ab211542, Abcam), p53 (2524, Cell Signaling Technology) and β-actin (ab616, Abcam). Membranes were then incubated with HRP-conjugated antibodies for 2 hours at room temperature, visualized by ECL solution (Nanjing Institute of Bioengineering, China) and quantified by Scion Image Beta 4.02 (Scion Corporation, NIH).
Intracellular reactive oxygen species (ROS) analysis
To analyze the oxidative stress levels, BM-MSCs of WT, and Cyp27b1 +/- mouse were prepared as single cell suspensions and labeled with diacetyldichlorofluorescein (DCFDA) for 30 minutes in the dark at room temperature. Fluorescence intensity was analyzed by flow cytometry (Becton Dickinson, Heidelberg, Germany).
Quantitative real-time RT-PCR
To detect gene expression, total RNA was extracted from vertebral tissue using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Reverse-transcription reactions were performed using PrimeScript RT Master Mix (Perfect Real Time, Takara Bio Inc., Shiga, Japan). Quantitative real-time RT-PCR amplifications were performed as described previously [28], and the primer sequences are shown in Table 1.
Table 1.
Primers used in this study for real time RT-PCR
| Name | S/AS | Sequence (5’ to 3’) | Tm (°C) | bp |
|---|---|---|---|---|
| TNFα | S | CCTGTAGCCCACGTCGTAG | 62 | 148 |
| AS | GGGAGTAGACAAGGTACAACCC | |||
| IL-6 | S | TAGTCCTTCCTACCCCAATTTCC | 61 | 76 |
| AS | TTGGTCCTTAGCCACTCCTTC | |||
| IL-1α | S | CGAAGACTACAGTTCTGCCATT | 60 | 126 |
| AS | GACGTTTCAGAGGTTCTCAGAG | |||
| IL-1β | S | GCAACTGTTCCTGAACTCAACT | 61 | 89 |
| AS | ATCTTTTGGGGTCCGTCAACT | |||
| Mmp3 | S | ACATGGAGACTTTGTCCCTTTTG | 61 | 192 |
| AS | TTGGCTGAGTGGTAGAGTCCC | |||
| Mmp13 | S | CTTCTTCTTGTTGAGCTGGACTC | 62 | 173 |
| AS | CTGTGGAGGTCACTGTAGACT | |||
| ATG-7 | S | GTTCGCCCCCTTTAATAGTGC | 62 | 161 |
| AS | TGAACTCCAACGTCAAGCGG | |||
| PAI-1 | S | TTCAGCCCTTGCTTGCCTC | 62 | 116 |
| AS | ACACTTTTACTCCGAAGTCGGT | |||
| IGFbp2 | S | CAGACGCTACGCTGCTATCC | 63 | 140 |
| AS | CCCTCAGAGTGGTCGTCATCA | |||
| Nqo1 | S | AGGATGGGAGGTACTCGAATC | 59 | 144 |
| AS | AGGCGTCCTTCCTTATATGCTA | |||
| Txnrd1 | S | CCCACTTGCCCCAACTGTT | 60 | 134 |
| AS | GGGAGTGTCTTGGAGGGAC | |||
| Nrf2 | S | ACCAAGGGGCACCATATAAAAG | 60 | 114 |
| AS | CTTCGCCGAGTTGCACTCA | |||
| Hmox1 | S | AAGCCGAGAATGCTGAGTTCA | 60 | 100 |
| AS | GCCGTGTAGATATGGTACAAGGA | |||
| GSR | S | GACACCTCTTCCTTCGACTACC | 60 | 116 |
| AS | CCCAGCTTGTGACTCTCCAC | |||
| CAT | S | GCAGATACCTGTGAACTGTCCCT | 60 | 472 |
| AS | TTACAGGTTAGCTTTTCCCTTCG | |||
| SOD1 | S | ATTACAGGATTAACTGAAGG | 50 | 238 |
| AS | CAATGATGGAATGCTCTC | |||
| GAPDH | S | TGGATTTGGACGCATTGGTC | 55 | 211 |
| AS | TTTGCACTGGTACGTGTTGAT |
Statistical analysis
All the statistical results were showed as mean ± SEM. Differences between two groups were analyzed using 2-tailed unpaired Student’s t-test and differences among more than 2 groups were analyzed using one-way ANOVA by GraphPad Prism software (Version 6.07; GraphPad Software Inc., San Diego, CA, USA) [29]. Each experiment was repeated at least three times (n > 6/group). P values <0.05 were considered statistically significant.
Results
1,25(OH)2D insufficiency accelerates age-related bone loss
In order to investigate the effect of 1,25(OH)2D insufficiency on age-related bone loss, we compared bone mineral density (BMD), bone microarchitecture (by Μicro-CT), and total collagen in lumbar vertebrae from 3-, 9- and 18-month-old Cyp27b1 +/- mice and their wild-type littermates. The BMD, trabecular bone volume and trabecular number decreased, whereas the trabecular separation gradually increased with age in both wild-type and Cyp27b1 +/- mice, however, these alterations were more dramatic in Cyp27b1 +/- mice compared with their wild-type littermates (Figure 1A-H). These results demonstrated that 1,25(OH)2D insufficiency reduces bone density and bone volume and disrupts trabecular microarchitecture, thus accelerating age-related bone loss.
Figure 1.
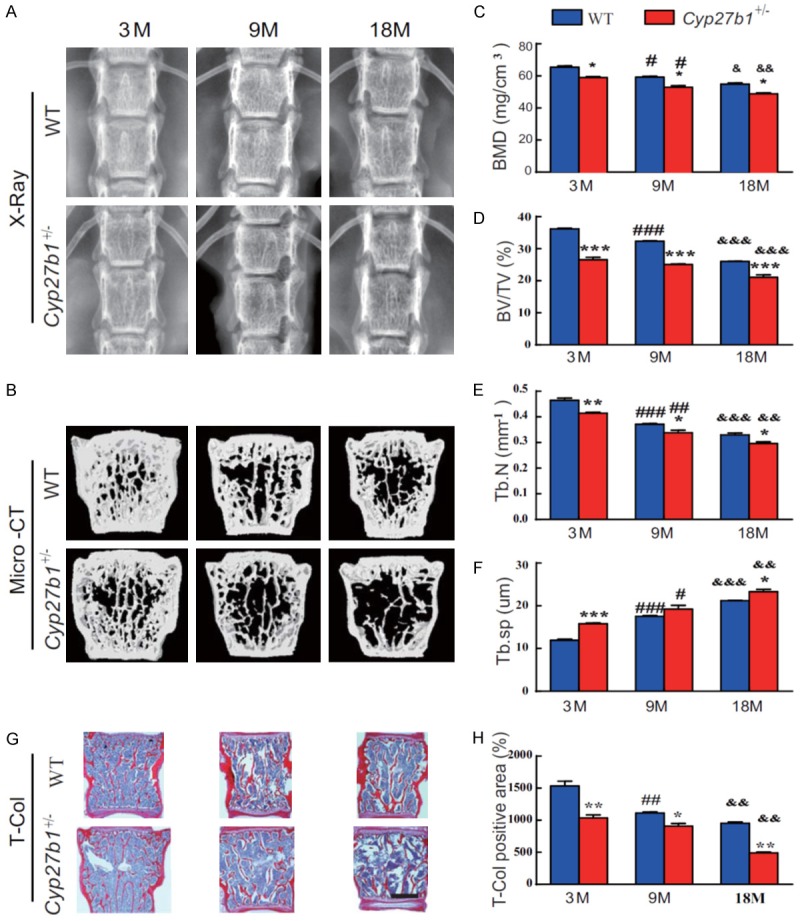
1,25(OH)2D insufficiency accelerates age-related bone loss. (A) Representative radiographs and (B) three-dimensional reconstruction of Micro-CT scan images of vertebrae from 3-, 9- and 18-month-old Cyp27b1 +/- mice and wild-type (WT) mice. Quantitative analysis of (C) bone mineral density (BMD), (D) the bone mass parameter, bone volume relative to tissue volume (BV/TV), (E) the bone microstructural parameter, trabecular number (Tb.N) and (F) the bone microstructural parameter, trabecular separation (Tb.Sp). (G) Representative micrographs of first lumbar vertebral sections stained histochemically for total collagen (T-Col). (H) The percentage of T-Col-positive area relative to total bone tissue. Values are means ± SEM of 6 determinations per group. ***P<0.001, **P<0.01, *P<0.05, compared with age-matched WT mice age. ###P<0.001, ##P<0.01, #P<0.05, Compared with genotype-matched 3-month-old mice. &&&P<0.001, &&P<0.01, &P<0.05, compared with genotype-matched 9-month-old mice.
1,25(OH)2D insufficiency increases the reduction in age-related osteoblastic bone formation
We then compared osteoblastic bone formation parameters in aging 3-, 9- and 18-month-old Cyp27b1 +/- mice and their wild-type littermates. We found that osteoblast numbers, and type I collagen-positive and alkaline phosphatase (ALP)-positive areas were gradually reduced with age in both wild-type and Cyp27b1 +/- mice, however, these parameters were reduced more dramatically in Cyp27b1 +/- mice compared with their wild-type littermates (Figure 2A-F). These results demonstrated that 1,25(OH)2D insufficiency increased the severity of reductions in age-related osteoblastic bone formation.
Figure 2.
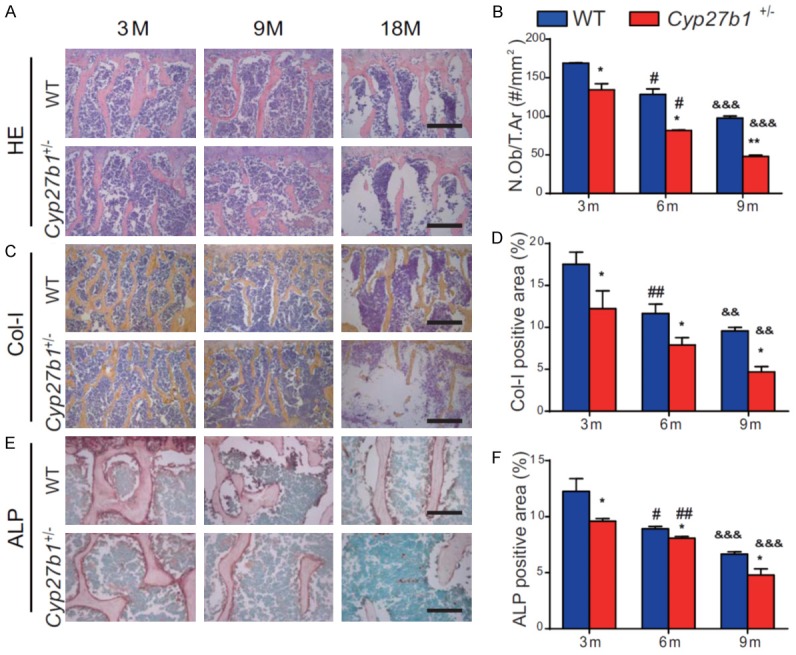
1,25(OH)2D insufficiency increases the severity of reduced, age-related, osteoblastic bone formation. Representative micrographs of the first lumbar vertebral sections from 3-, 9- and 18-month-old Cyp27b1 +/- mice and wild-type (WT) mice stained (A) with H&E, (C) immunohistochemically for type I collagen (Col-I), an osteoblastic differentiation marker, and (E) histochemically for ALP, another osteoblastic differentiation marker. (B) The number of osteoblasts relative to tissue area (N.Ob/T.Ar). The percentages of (D) Col-I-positive and (F) ALP-positive areas. Values are means ± SEM of 6 determinations per group. **P<0.01, *P<0.05, compared with age-matched WT mice. ##P<0.01, #P<0.05, compared with genotype-matched 3-month-old mice. &&&P<0.001, &&P<0.01, compared with genotype-matched 9-month-old mice.
1,25(OH)2D insufficiency enhances the severity of increased age-related osteoclastic bone resorption
We then compared osteoclastic bone resorption parameters in aging 3-, 9- and 18-month-old Cyp27b1 +/- mice and their wild-type littermates. We found that surfaces staining for the osteoclast marker, tartrate-resistant acid phosphatase (TRAP), and the osteoclast surface relative to bone surface gradually increased with age in both wild-type and Cyp27b1 +/- mice, however, the increases were more dramatic in Cyp27b1 +/- mice compared with their wild-type littermates (Figure 3A and 3B). These results demonstrated that 1,25(OH)2D insufficiency increased the of age-related augmentation of osteoclastic bone resorption.
Figure 3.
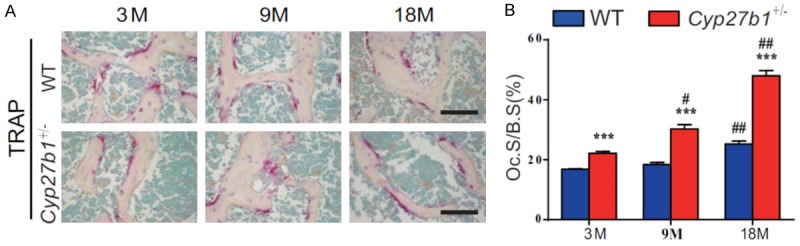
1,25(OH)2D insufficiency increases the augmented, age-related, osteoclastic bone resorption. A. Representative micrographs of sections of the first lumbar vertebral from 3-, 9- and 18-month-old Cyp27b1 +/- mice and wild-type (WT) mice stained histochemically for TRAP, a specific osteoclastic marker. B. The surface of osteoclasts relative to bone surface (Oc.S/B.S). Values are means ± SEM of 6 determinations per group. ***P<0.001, compared with age-matched WT mice. ##P<0.01, #P<0.05, compared with genotype-matched 3-month-old mice. &&P<0.01, &P<0.05, Compared with genotype-matched 9-month-old mice.
Down-regulation of cyp27b1 expression with age and effects of cyp27b1 heterozygous deletion on circulating analytes
To assess whether Cyp27b1 expression levels were down-regulated with age, we examined the protein expression levels of Cyp27b1 in kidney, intestine and bone of 3-, 9- and 18-month-old wild-type mice using Western blots. The results showed that the protein expression levels of Cyp27b1 were gradually down-regulated with age (Figure 4A, Figure S1A). We next assessed whether heterozygous deletion of Cyp27b1 resulted in alterations of serum levels of hormones regulating calcium and phosphate and/or in changes of serum minerals. We found that serum calcium, phosphorus, PTH and 25(OH)D levels were not altered significantly, despite the fact that serum 1,25(OH)2D levels were reduced significantly in Cyp27b1 +/- mice compared with their WT littermates (Figure 4B-F). We also confirmed that mRNA expression levels of Cyp27b1 in bone tissues were reduced significantly in Cyp27b1 +/- mice compared with their wild-type littermates (Figure 4G).
Figure 4.
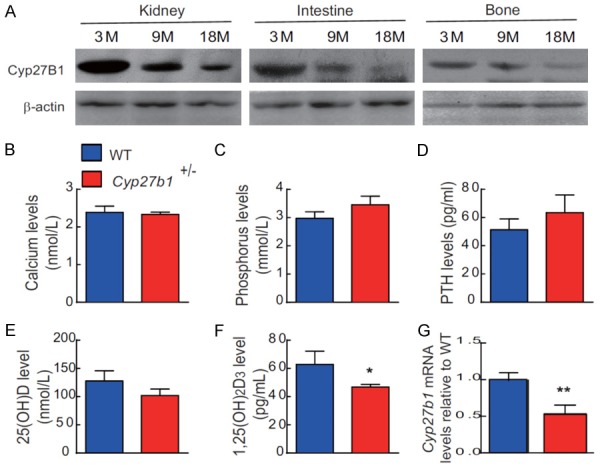
Down-regulation of Cyp27b1 expression with age and Cyp27b1 heterozygous deletion results in 1,25(OH)2D insufficiency. (A) Western blots of kidney, and intestine and bone extracts for the expression of Cyp27b1 in 3-, 9- and 18-month-old WT mice. ß-actin was used as loading control for Western blots. (B) Serum calcium, (C) phosphorus, (D) PTH, (E) 25(OH)D and (F) 1,25(OH)2D levels in 9-month-old Cyp27b1 +/- mice and WT mice. (G) Cyp27b1 mRNA levels in vertebrae of 9-month-old Cyp27b1+/- mice and WT mice were determined by real-time RT-PCR, and calculated as a ratio relative to Gapdh mRNA. Results are shown relative to WT. Values are means ± SEM of 6 determinations per group. *P<0.05, **P<0.01, compared with WT mice.
1,25(OH)2D insufficiency increases oxidative stress and DNA damage in bone by down-regulation of antioxidant enzyme expression
We then determined whether the acceleration of age-related bone loss induced by 1,25(OH)2D insufficiency was associated with increased oxidative stress and DNA damage. The ROS levels in BM-MSCs (Figure 5A), serum MDA levels (Figure 5B), and the protein expression levels in bone of γ-H2AX (Figure 5C and 5D), a biomarker for DNA double-strand breaks, were all significantly increased. In contrast, the protein expression levels of Sirt1, which induces FOXO3a (Forkhead box O3a) deacetylation and inhibits oxidative stress and Nrf2, a regulator of the expression of genes encoding antioxidant proteins involved in cellular redox homeostasis, and the mRNA expression levels of Nrf2 and of the antioxidant enzymes superoxide dismutase1 (SOD1), NAD(P)H dehydrogenase [quinone]1 (Nqo1), glutathione-disulfide reductase (GSR), heme oxygenase (decycling) 1 (Hmox1), thioredoxin reductase 1, cytoplasmic (Txnrd1) and catalase (CAT) were all significantly down-regulated in bone of Cyp27b1 +/- mice compared with their wild-type littermates (Figure 5C-E, Figure S1B). These results demonstrated that 1,25(OH)2D insufficiency increases osseous oxidative stress and DNA damage by down-regulation of antioxidant enzyme expression.
Figure 5.
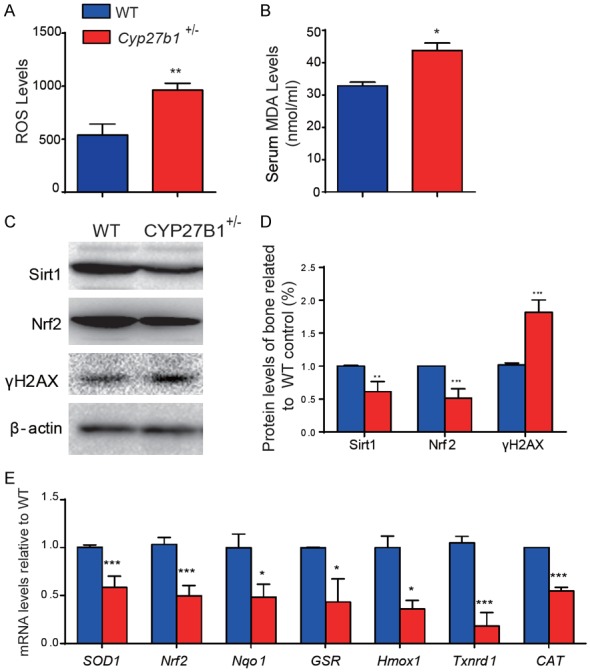
1,25(OH)2D insufficiency increases oxidative stress and DNA damage in bone by down-regulation of antioxidant enzyme expression. Parameters of oxidative stress were detected in 9-month-old Cyp27b1 +/- and WT mice including (A) ROS levels in BM-MSCs and (B) serum levels of malondialdehyde (MDA). (C) Western blots of bone extracts for the expression of Sirt1, a deacetylase and inhibitor of oxidative stress, Nrf2, a regulator of the expression of genes encoding antioxidant proteins involved in cellular redox homeostasis, and γ-H2AX, a DNA damage marker, in 9-month-old Cyp27b1 +/- and WT mice. ß-actin was used as loading control for Western blots. (D) Quantification of protein relative expression levels of Sirt1, Nrf2 and γ-H2AX assessed by densitometric analysis. (E) mRNA levels of Nrf2, and of the antioxidant enzymes superoxide dismutase1 (SOD1), NAD(P)H dehydrogenase [quinone]1 (Nqo1), glutathione-disulfide reductase (GSR), heme oxygenase (decycling) 1 (Hmox1), thioredoxin reductase 1, cytoplasmic (Txnrd1) and catalase (CAT) in vertebrae were determined by real-time RT-PCR, calculated as ratio relative to Gapdh mRNA and expressed relative to WT. Values are means ± SEM of 6 determinations per group. ***P<0.001, **P<0.01, *P<0.05 compared to littermate WT mice.
1,25(OH)2D insufficiency increases cell senescence and SASP in bone tissues by up-regulating p16, p19 and p53
To determine whether the acceleration of age-related bone loss induced by 1,25(OH)2D insufficiency was associated with altered cell senescence and SASP, we examined these processes in osteocytes and bone marrow mesenchymal stem cell (BM-MSC). We found that, in 9-month-old Cyp27b1 +/- mice compared with their wild-type littermates, the percentage of β-galactosidase (β-gal)-positive and p16-positive osteocytes and senescence associated β-gal (SA-β-gal)-positive BM-MSCs were significantly increased, and the mRNA expression levels of SASP molecules, including tumor necrosis factor α (TNFα), interleukin-6 (IL-6), IL-1α, IL-1β, matrix metalloproteinase3 (mmp3), mmp13, autophagy related-7 (ATG-7), plasminogen activator inhibitor-1 (PAI-1) and insulin-like growth factor-binding protein 2 (IGFbp2) were also significantly increased (Figure 6A-G, Figure S1C). We also found, in bone tissues from 9-month-old Cyp27b1 +/- mice compared with their wild-type littermates, that the protein expression levels of the senescence -associated markers p16 (cyclin-dependent kinase inhibitor 2A, multiple tumor suppressor 1), p19 (RNA silencing suppressor p19) and the tumor suppressor p53 were all up-regulated significantly (Figure 6H). These results demonstrated that 1,25(OH)2D insufficiency increases cell senescence and SASP in bone tissues with up-regulation of p16, p19 and p53.
Figure 6.

1,25(OH)2D insufficiency increases cell senescence and SASP in bone tissues by up-regulating p16, p19 and p53. (A) Representative micrographs of vertebral sections from 9-month-old Cyp27b1 +/- mice and WT mice stained immuhistochemically for (A) cellular senescence markers, β-gal and (C) p16. The percentages of (B) β-gal-positive and (D) p16-positive osteocytes. (E) Representative micrographs of BM-MSCs from 9-month-old Cyp27b1 +/- mice and WT mice stained cytochemically for SA-β-gal and (F) the percentage of SA-β-gal-positive BM-MSCs. (G) mRNA expression levels of SASP molecules, including TNFα, IL-6, IL-1α, IL-1β, mmp3, mmp13, ATG-7, PAI-1 and IGFbp2 in vertebrae were determined by real-time RT-PCR, calculated as a ratio relative to Gapdh mRNA and expressed relative to WT mice. (H) Western blots of bone extracts for the expression of p16, p19 and p53 in 9-month-old Cyp27b1 +/- and WT mice. ß-actin was used as loading control for Western blots. Values are means ± SEM of 6 determinations per group. ***P<0.001, **P<0.01, compared with WT mice.
Discussion
In this study, we first investigated whether 1,25(OH)2D insufficiency accelerates age-related bone loss. By analyzing the phenotype of lumber vertebrae from 3-, 9- and 18-month-old mice, we found that bone mass decreased progressively with age in wild-type mice accompanied by reduced osteoblastic bone formation and increased osteoclastic bone resorption. These changes were accelerated in Cyp27b1 +/- mice, which manifest 1,25(OH)2D insufficiency, and age-related bone loss was thus augmented.
Previous studies have shown that there is an age-related decline in expression of Cyp27b1 in human mesenchymal stem cells, associated with decreased osteoblastogenesis [30]. As well, in human studies, as a result of declining renal function with age, a decrease in 1,25(OH)2D production by approximately 50% has been reported [31]. These findings suggest that the down-regulation of Cyp27b1 expression levels with age may result in age-related bone loss by decreasing 1,25(OH)2D production, inhibiting osteogenesis by mesenchymal stem cells, and thus reducing osteoblastic bone formation. To address whether bone loss was associated with down-regulation of Cyp27b1 expression levels with age, we examined the protein expression levels of Cyp27b1 in kidney, intestine and bone of 3-, 9- and 18-month-old wild-type mice using Western blots and found that they were indeed progressively down-regulated.
In our previous studies we found that in mutant homozygous mouse models with genetic deletion of Cyp27b1 (Cyp27b1-/-) or VDR (Vdr-/-), osteoblast numbers, mineral apposition rate and bone volume were suppressed below levels seen in wild-type mice, even when hypocalcemia and secondary hyperparathyroidism were prevented by feeding the animals a high calcium, high phosphorus, lactose-containing rescue diet [10]. This suggested that 1,25(OH)2D has a direct bone anabolic action besides its role in maintaining calcium and phosphorus balance. However, complete deletion of 1,25(OH)2D is very rare in humans, whereas its insufficiency may be more common. Therefore we asked whether Cyp27b1 heterozygous null mice could be used as a model for 1,25(OH)2D insufficiency. We examined serum calcium, phosphorus and PTH, 25(OH)D and 1,25(OH)2D levels in 9-month-old Cyp27b1 +/- mice and their wild-type littermates and found that the serum 1,25(OH)2D levels were significantly reduced in Cyp27b1 heterozygous mice, while serum calcium, phosphorus, PTH and 25(OH)D levels were not obviously altered compared to wild-type. These results suggest that Cyp27b1 +/- mice can be used as an animal model to investigate the mechanism of 1,25(OH)2D insufficiency-induced age-related diseases.
We recently demonstrated that 1,25(OH)2D exerts an antioxidative stress role by activation of Nrf2-antioxidant signaling [32]. In the current study, we examined parameters of oxidative stress and DNA damage in bone of 9-month-old wild-type and Cyp27b1 +/- mice and found that 1,25(OH)2D insufficiency increased oxidative stress and DNA damage, but reduced antioxidant capacity by down-regulating expression levels of antioxidant enzymes. Previous studies in animal models suggest that oxidative stress is a key cause of bone loss [16-18]. Our results indicate that 1,25(OH)2D insufficiency is associated with increased oxidative stress and DNA damage in bone by down-regulation of antioxidant enzyme expression, and this mechanism may therefore contribute to the acceleration of age-related bone loss.
We recently demonstrated that the aging induced by 1,25(OH)2D deficiency was accompanied by increased cellular senescence and SASP in multiple organs, whereas exogenous 1,25(OH)2D supplementation could largely rescue the aging phenotypes through reduced cellular senescence and SASP [32]. Previous reports have found that older mice have a higher proportion of senescent osteocytes and a higher expression level of multiple SASP markers than younger mice [33], suggesting that senescent osteocytes and SASP may contribute to age-related bone loss. In our current study, we found that 1,25(OH)2D insufficiency increased osteocyte and BM-MSC senescence and SASP in bone tissues by up-regulating p16, p19 and p53. Our results therefore indicate that the acceleration of age-related bone loss induced by 1,25(OH)2D insufficiency is associated with increases bone cell senescence and SASP.
In summary, results from this study indicate that the down-regulation of Cyp27b1 expression levels with age may result in age-related bone loss by reducing 1,25(OH)2D production, and thus inhibiting osteogenesis of mesenchymal stem cells, and osteoblastic bone formation. Cyp27b1 +/- mice can be used as an animal model of 1,25(OH)2D insufficiency. We therefore employed this mouse model and demonstrated that 1,25(OH)2D insufficiency accelerated age-related bone loss by increasing oxidative stress and DNA damage, inducing bone cell senescence and SASP, and thus inhibiting osteoblastic bone formation while stimulating osteoclastic bone resorption.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81730066 to DM) and from National Key R&D Program of China (2018YFA0800800 to DM), from General topic of Nanjing Medical Science Development Project (YKK15239 to XW) and from the Canadian Institutes of Health Research (CIHR) to DG.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kanis JA, Mccloskey EV, Johansson H, Oden A, Khaltaev N. A reference standard for the description of osteoporosis. Bone. 2008;42:467–475. doi: 10.1016/j.bone.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Hendrickx G, Boudin E, Van Hul W. A look behind the scenes: the risk and pathogenesis of primary osteoporosis. Nat Rev Rheumatol. 2015;11:462–474. doi: 10.1038/nrrheum.2015.48. [DOI] [PubMed] [Google Scholar]
- 3.Tilg H, Moschen AR, Kaser A, Pines A, Dotan I. Gut, inflammation and osteoporosis: basic and clinical concepts. Gut. 2008;57:684–694. doi: 10.1136/gut.2006.117382. [DOI] [PubMed] [Google Scholar]
- 4.Jagelaviciene E, Vaitkeviciene I, Silingaite D, Sinkunaite E, Daugelaite G. The relationship between vitamin D and periodontal pathology. Medicina (Kaunas) 2018;54 doi: 10.3390/medicina54030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Schoor NM, Visser M, Pluijm SM, Kuchuk N, Smit JH, Lips P. Vitamin D deficiency as a risk factor for osteoporotic fractures. Bone. 2008;42:260–266. doi: 10.1016/j.bone.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Feldman D, J Malloy P. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. Bonekey Rep. 2014;3:510. doi: 10.1038/bonekey.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glorieux FH, St-Arnaud R. Molecular cloning of (25-OH D)-1 alpha-hydroxylase: an approach to the understanding of vitamin D pseudo-deficiency. Recent Prog Horm Res. 1998;53:341–349. discussion 350. [PubMed] [Google Scholar]
- 8.Panda DK, Miao D, Tremblay ML, Sirois J, Farookhi R, Hendy GN, Goltzman D. Targeted ablation of the 25-hydroxyvitamin D 1alpha -hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98:7498–7503. doi: 10.1073/pnas.131029498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Lv F, Sun W, Tao C, Ding G, Karaplis A, Brown E, Goltzman D, Miao D. The abnormal phenotypes of cartilage and bone in calcium-sensing receptor deficient mice are dependent on the actions of calcium, phosphorus, and PTH. PLoS Genet. 2011;7:e1002294. doi: 10.1371/journal.pgen.1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN, Goltzman D. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279:16754–16766. doi: 10.1074/jbc.M310271200. [DOI] [PubMed] [Google Scholar]
- 11.Shu L, Ji J, Zhu Q, Cao G, Karaplis A, Pollak MR, Brown E, Goltzman D, Miao D. The calcium-sensing receptor mediates bone turnover induced by dietary calcium and parathyroid hormone in neonates. J Bone Miner Res. 2011;26:1057–1071. doi: 10.1002/jbmr.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue Y, Karaplis AC, Hendy GN, Goltzman D, Miao D. Exogenous 1,25-dihydroxyvitamin D3 exerts a skeletal anabolic effect and improves mineral ion homeostasis in mice that are homozygous for both the 1alpha-hydroxylase and parathyroid hormone null alleles. Endocrinology. 2006;147:4801–4810. doi: 10.1210/en.2006-0403. [DOI] [PubMed] [Google Scholar]
- 13.Christakos S, Seth T, Hirsch J, Porta A, Moulas A, Dhawan P. Vitamin D biology revealed through the study of knockout and transgenic mouse models. Annu Rev Nutr. 2013;33:71–85. doi: 10.1146/annurev-nutr-071812-161249. [DOI] [PubMed] [Google Scholar]
- 14.Goltzman D. Vitamin D action: lessons learned from genetic mouse models. Ann N Y Acad Sci. 2010;1192:145–152. doi: 10.1111/j.1749-6632.2009.05226.x. [DOI] [PubMed] [Google Scholar]
- 15.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 16.Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, Kousteni S, O’Brien CA, Bellido T, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282:27285–27297. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, Urry ZL, Chambers TJ. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112:915–923. doi: 10.1172/JCI18859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi C, Wu J, Yan Q, Wang R, Miao D. Bone marrow ablation demonstrates that estrogen plays an important role in osteogenesis and bone turnover via an antioxidative mechanism. Bone. 2015;79:94–104. doi: 10.1016/j.bone.2015.05.034. [DOI] [PubMed] [Google Scholar]
- 19.de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, Meradji M, Hoeijmakers JH. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- 20.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Park SH, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 21.Nojiri H, Saita Y, Morikawa D, Kobayashi K, Tsuda C, Miyazaki T, Saito M, Marumo K, Yonezawa I, Kaneko K, Shirasawa T, Shimizu T. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J Bone Miner Res. 2011;26:2682–2694. doi: 10.1002/jbmr.489. [DOI] [PubMed] [Google Scholar]
- 22.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ovadya Y, Krizhanovsky V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology. 2014;15:627–642. doi: 10.1007/s10522-014-9529-9. [DOI] [PubMed] [Google Scholar]
- 24.Wu X, Li J, Zhang H, Wang H, Yin G, Miao D. Pyrroloquinoline quinone prevents testosterone deficiency-induced osteoporosis by stimulating osteoblastic bone formation and inhibiting osteoclastic bone resorption. Am J Transl Res. 2017;9:1230–1242. [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang HW, Ding J, Jin JL, Guo J, Liu JN, Karaplis A, Goltzman D, Miao D. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res. 2010;25:640–652. doi: 10.1359/jbmr.090812. [DOI] [PubMed] [Google Scholar]
- 26.Zhou X, Dai X, Wu X, Ji J, Karaplis A, Goltzman D, Yang X, Miao D. Overexpression of Bmi1 in lymphocytes stimulates skeletogenesis by improving the osteogenic microenvironment. Sci Rep. 2016;6:29171. doi: 10.1038/srep29171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai X, Zhang Q, Yu Z, Sun W, Wang R, Miao D. Bmi1 deficient mice exhibit male infertility. Int J Biol Sci. 2018;14:358–368. doi: 10.7150/ijbs.23325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geng Q, Gao H, Yang R, Guo K, Miao D. Pyrroloquinoline quinone prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Int J Biol Sci. 2019;15:58–68. doi: 10.7150/ijbs.25783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, Tan J, Chothani SP, Ye L, Rackham OJL, Ko NSJ, Sahib NE, Pua CJ, Zhen NTG, Xie C, Wang M, Maatz H, Lim S, Saar K, Blachut S, Petretto E, Schmidt S, Putoczki T, Guimaraes-Camboa N, Wakimoto H, van Heesch S, Sigmundsson K, Lim SL, Soon JL, Chao VTT, Chua YL, Tan TE, Evans SM, Loh YJ, Jamal MH, Ong KK, Chua KC, Ong BH, Chakaramakkil MJ, Seidman JG, Seidman CE, Hubner N, Sin KYK, Cook SA. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–115. doi: 10.1038/nature24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geng S, Zhou S, Glowacki J. Age-related decline in osteoblastogenesis and 1alpha-hydroxylase/CYP27B1 in human mesenchymal stem cells: stimulation by parathyroid hormone. Aging Cell. 2011;10:962–971. doi: 10.1111/j.1474-9726.2011.00735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallagher JC. Vitamin D and aging. Endocrinol Metab Clin North Am. 2013;42:319–332. doi: 10.1016/j.ecl.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Yang R, Qiao W, Zhang W, Chen J, Mao L, Goltzman D, Miao D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell. 2019;18:e12951. doi: 10.1111/acel.12951. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Sims NA. Senescent osteocytes: do they cause damage and can they be targeted to presrve the skeleton? J Bone Miner Res. 2016;31:1917–1919. doi: 10.1002/jbmr.2994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.