

Abstract
Snyder–Robinson syndrome (SRS) is an X-linked intellectual disability syndrome caused by a loss-of-function mutation in the spermine synthase (SMS) gene. Primarily affecting males, the main manifestations of SRS include osteoporosis, hypotonic stature, seizures, cognitive impairment, and developmental delay. Because there is no cure for SRS, treatment plans focus on alleviating symptoms rather than targeting the underlying causes. Biochemically, the cells of individuals with SRS accumulate excess spermidine, whereas spermine levels are reduced. We recently demonstrated that SRS patient-derived lymphoblastoid cells are capable of transporting exogenous spermine and its analogs into the cell and, in response, decreasing excess spermidine pools to normal levels. However, dietary supplementation of spermine does not appear to benefit SRS patients or mouse models. Here, we investigated the potential use of a metabolically stable spermine mimetic, (R,R)-1,12-dimethylspermine (Me2SPM), to reduce the intracellular spermidine pools of SRS patient-derived cells. Me2SPM can functionally substitute for the native polyamines in supporting cell growth while stimulating polyamine homeostatic control mechanisms. We found that both lymphoblasts and fibroblasts from SRS patients can accumulate Me2SPM, resulting in significantly decreased spermidine levels with no adverse effects on growth. Me2SPM administration to mice revealed that Me2SPM significantly decreases spermidine levels in multiple tissues. Importantly, Me2SPM was detectable in brain tissue, the organ most affected in SRS, and was associated with changes in polyamine metabolic enzymes. These findings indicate that the (R,R)-diastereomer of 1,12-Me2SPM represents a promising lead compound in developing a treatment aimed at targeting the molecular mechanisms underlying SRS pathology.
Keywords: polyamine; spermidine; neurodevelopment; neurological disease; osteoporosis; (R,R)-1,12-dimethylspermine (Me2SPM); alpha-methylated polyamine; polyamine mimetic; Snyder–Robinson Syndrome; spermine synthase
Introduction
Spermine synthase (SMS)2 (EC 2.5.1.22) is an integral member of the polyamine biosynthetic pathway that catalyzes the production of spermine (SPM) from spermidine (SPD), using decarboxylated S-adenosylmethionine (dcAdoMet) as the aminopropyl donor (Fig. 1A) (1). Inactivating or partial loss-of-function mutations of the SMS gene result in Snyder–Robinson syndrome (SRS), a recessive X-linked intellectual disability syndrome (2–4). Males affected with SRS present with clinical manifestations that most commonly include osteoporosis, hypotonic stature, seizures, cognitive impairment, and developmental delay (5, 6). Because there is no cure for SRS, treatment plans emphasize symptom management, and to date there have been no studies focused on pharmacologically targeting the underlying cause of the disease.
Figure 1.
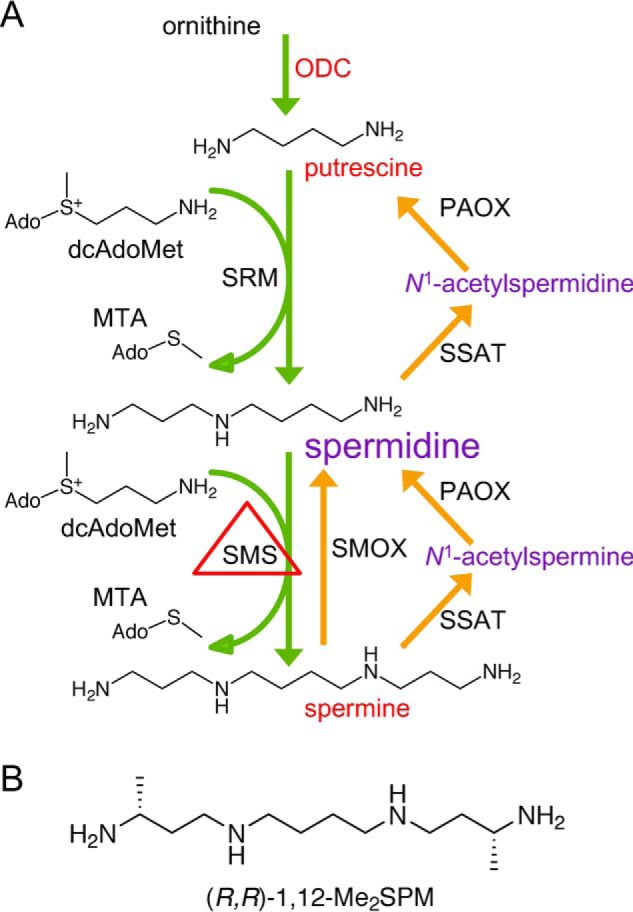
A, the mammalian polyamine metabolic pathway. The diamine PUT is derived from ornithine via ODC, the first rate-limiting step in polyamine biosynthesis. Sequential aminopropyl group additions by spermidine synthase (SRM) and SMS, using decarboxylated S-adenosylmethionine (dcAdoMet) as a donor, create SPD and SPM, respectively. Catabolism of SPM and SPD occurs via acetylation by SSAT followed by oxidation by PAOX. SPM can also be directly oxidized to SPD by SMOX. In SRS, SMS function is decreased or absent, spermidine accumulates, and spermine levels are severely reduced. B, structure of (R,R)-1,12-Me2SPM mimetic.
Biochemically, SMS is the sole source of SPM biosynthesis (Fig. 1A) (7). Although SPM is also a dietary component and can be produced by intestinal microbiota, a defining characteristic of cells from SRS patients is an elevated SPD:SPM ratio (4, 5, 8–12). The extent of this elevated ratio in individual patient cells depends on the nature of the mutation and appears to correlate with enzyme activity level and the resulting pathologic severity, although existing data are insufficient to conclude a genotype–phenotype correlation. A compilation of the mutations, their effects on SMS protein and activity, and clinical features of the SRS patients was recently reported (12). The mammalian polyamines, which include SPM, SPD, and their precursor putrescine (PUT), are absolutely required for many critical cellular functions, and their levels are under strict homeostatic control. However, the skewed ratio in SRS patients clearly has greater pathological consequences for certain tissues, including brain, bone, and muscle.
Initially, it was thought that dietary SPM supplementation would be a simple treatment strategy for SRS. Based on data from an early mouse model of SRS (13, 14) as well as patient experiences,3 this approach appears to be ineffective because of complexities not yet understood. Because polyamine transport is generally down-regulated in the presence of high intracellular polyamine concentrations (15), it was proposed that cells of SRS patients might lack the ability to transport extracellular SPM. However, our recent study using SRS patient-derived lymphoblastoid cells clearly demonstrated that polyamine transport activity in these cells is similar to that of WT SMS cells (16). Not only can SRS lymphoblastoid cells efficiently transport SPM, they also respond to its accumulation by decreasing the excessive SPD pool to within normal levels. However, it is conceivable that cells of the lymphoblast lineage, which do not appear to be major contributors to the SRS pathology, differ in their polyamine transport ability from those of other lineages. Thus, the current study used a panel of fibroblast cell lines of SRS patient origin to investigate the transport of both native SPM and the SPM mimetic (R,R)-1,12-dimethylspermine (Me2SPM) (Fig. 1B). This metabolically stable SPM analog was used with the intention of circumventing the limitations known to exist for the use of native SPM as a treatment in vivo, including acute toxicities when administered to animals by intravenous, intraperitoneal, or oral routes (17–19), as well as the low availability that is apparent with oral SPM consumption in SMS-deficient animals (13, 14).
α-Methylation of SPM provides protection against degradation by mono- and diamine oxidases inside the cell as well as in the presence of serum amine oxidases found in human and bovine serums (20). More metabolically stable than spermine, Me2SPM is not acetylated by the catabolic enzyme spermidine/spermine N1-acetyltransferase (SSAT) and has been shown to support cell growth following polyamine depletion (20–22). It is capable of functionally substituting for native SPM in functions independent of hypusination, a SPD-dependent, post-translational modification of eukaryotic translation initiation factor 5A (23). Like native SPM, Me2SPM has been shown to increase polyamine catabolism through increasing SSAT biosynthesis and activity, thus reducing intracellular SPD pools (24).
Using lymphoblastoid and fibroblast cell lines from SRS patients, the current study investigated the effects of Me2SPM treatment on proliferation, intracellular polyamine concentrations, and polyamine metabolic gene expression and enzyme activity. We also performed preliminary feasibility and toxicity testing in C57Bl/6J male mice that included determinations of Me2SPM accumulation, native polyamine concentrations, and activity assays of the rate-limiting polyamine biosynthetic enzyme ornithine decarboxylase (ODC) and catabolic enzyme SSAT in brain, skeletal muscle, heart, liver, and kidney tissue from treated animals. These studies identified the (R,R)-diastereomer of 1,12-Me2SPM as a promising lead compound capable of reducing SPD levels in SRS cells while sustaining growth. Moreover, our in vivo study revealed that Me2SPM is distributed to and can modulate polyamine homeostasis and SPD levels in multiple tissues, including the brain, the primary organ affected in SRS. Our results form the basis for future preclinical studies aimed at optimization of in vivo Me2SPM dosing schedules to minimize toxic effects while correcting the imbalanced polyamine pools characteristic of Snyder–Robinson syndrome.
Results
The polyamine transport system is functional in SRS patient–derived fibroblasts
We previously demonstrated that lymphoblastoid lines originating from SRS patients were able to transport exogenous SPM and restore a more normal polyamine distribution profile (16). However, because the shift in polyamine distribution in these cells has not been associated with an overt phenotype in the patients, the possibility that cells of other lineages might not have the same transport capabilities was investigated. Fibroblast cell lines derived from the skin of five individual SRS patients, as well as two WT male donors, were used to ascertain whether polyamine transport was dysregulated in SRS fibroblast cells. The cells were treated for 24 h with 5 μm SPM, in the presence of aminoguanidine (a bovine serum amine oxidase inhibitor), followed by preparation of acid-extracted lysates for HPLC analysis of intracellular polyamines. As with SRS lymphoblastoid lines (16), the untreated SRS fibroblast lines demonstrate significantly elevated SPD:SPM ratios (ranging from ∼3 to 13) as compared with cells with fully active spermine synthase (∼0.62) (Fig. 2A). Of note, the line SRS2 was derived from a patient harboring one of the most severe mutations with regard to SMS activity and SPD:SPM ratio (here measured as ∼13.23 versus WT ratios of ∼0.62) (9, 25). Importantly, treating each of these SRS fibroblast lines with SPM not only increased the intracellular concentration of SPM; it also drastically reduced the accumulation of SPD in each, resulting in SPD:SPM ratios substantially closer to the range observed in untreated WT cells. Additionally, the level of total polyamines in line SRS2 was more than double that of WT cells (39.2 versus 14.2 nmol/mg protein), and treatment with SPM reduced the total polyamine concentration to ∼16.5 nmol/mg protein. These results provide support that SRS cells of different lineages maintain their ability to transport SPM and consequently control the homeostatic distribution of the individual polyamines.
Figure 2.
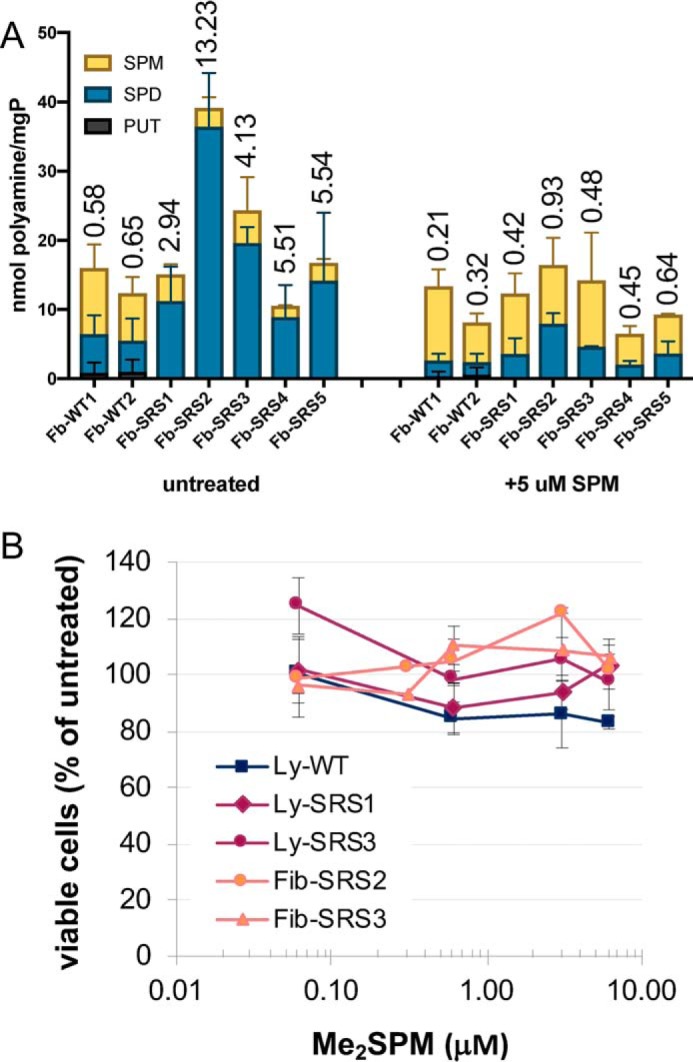
A, polyamine distribution profiles in SRS fibroblasts and response to SPM supplementation. Fibroblast (Fb) cell lines derived from SRS patients or WT donors were treated for 24 h with 5 μm SPM in the presence of 1 mm aminoguanidine and analyzed for polyamine content by HPLC. The columns indicate the means with SE of each polyamine, stacked to indicate the total concentration of all polyamines in the cell, normalized to total protein. The values above the columns indicate the average SPD:SPM ratios. All values were calculated from duplicate determinations of three biological replicates. B, effects of Me2SPM on proliferation in SRS patient-derived cell lines. SRS or WT lymphoblastoid (Ly) or fibroblast (Fib) cell lines were grown for 96 h in the presence of increasing doses of Me2SPM. The cells were collected and counted for viability using Trypan blue exclusion. The data points indicate the means with SE of duplicate determinations of at least two biological replicates.
Me2SPM reduces SPD accumulation without affecting growth of SRS cells
SRS lymphoblastoid and fibroblast cell lines (two each) were incubated for 96 h in the presence of increasing concentrations of Me2SPM. Cell lines chosen for these studies represented severely affected and more mildly affected patients for each cell type. In each of the cell lines, treatment had no adverse effect on proliferation (Fig. 2B), whereas the intracellular concentration of SPD was efficiently reduced as the analog accumulated (Table 1). The resulting SPD:SPM ratios, calculated by incorporating the intracellular Me2SPM concentration as a portion of the SPM pool (SPD/SPM + Me2SPM), rapidly decreased with increasing mimetic uptake.
Table 1.
Effects of Me2SPM on polyamine distribution in SRS patient-derived cell lines
The Me2SPM-treated SRS or WT lymphoblastoid or fibroblast cells collected for viability analysis in Fig. 2B were subsequently used for HPLC analysis of polyamine concentrations. The data represent the means ± S.E. of at least two separate 96-h treatments, with each sample analyzed in duplicate. Intracellular Me2SPM concentration was included with the native SPM concentration in the determination of SPD:SPM ratios.
| Cell line | Me2SPM | PUT | SPD | SPM | Me2SPM | SPD/SPM + Me2SPM |
|---|---|---|---|---|---|---|
| μm | nmol/mg protein | |||||
| WT lymphoblastoid | 0 | 6.63 ± 1.21 | 15.44 ± 1.40 | 9.12 ± 0.63 | 0 | 1.69 |
| SRS1 lymphoblastoid | 0 | 2.66 ± 0.22 | 35.17 ± 6.40 | 4.51 ± 0.96 | 0 | 7.80 |
| 0.1 | 2.73 ± 0.14 | 28.71 ± 3.36 | 3.42 ± 0.34 | 0.41 ± 0.06 | 7.50 | |
| 0.6 | 2.94 ± 0.27 | 24.30 ± 1.51 | 2.72 ± 0.12 | 4.36 ± 0.18 | 3.43 | |
| 3.1 | 2.73 ± 0.22 | 21.59 ± 5.29 | 2.65 ± 0.62 | 24.64 ± 7.00 | 0.79 | |
| 6.1 | 3.08 ± 0.23 | 14.17 ± 2.03 | 1.91 ± 0.29 | 20.26 ± 4.66 | 0.64 | |
| SRS3 lymphoblastoid | 0 | 3.73 ± 0.71 | 39.43 ± 4.52 | 2.85 ± 0.21 | 0 | 13.84 |
| 0.1 | 3.24 ± 0.38 | 34.45 ± 3.90 | 2.25 ± 0.13 | 0.83 ± 0.07 | 11.19 | |
| 0.6 | 5.60 ± 1.40 | 39.27 ± 10.18 | 1.61 ± 0.32 | 10.42 ± 1.58 | 3.26 | |
| 3.1 | 4.71 ± 0.72 | 12.82 ± 1.33 | 0.75 ± 0.03 | 29.04 ± 3.85 | 0.43 | |
| 6.1 | 4.80 ± 0.35 | 12.71 ± 1.01 | 0.79 ± 0.03 | 32.61 ± 3.03 | 0.38 | |
| WT fibroblast | 0 | 0 | 3.09 ± 0.89 | 11.11 ± 3.24 | 0 | 0.28 |
| SRS3 fibroblast | 0 | 0.85 ± 0.34 | 11.62 ± 3.53 | 3.00 ± 0.85 | 0 | 3.87 |
| 0.1 | 0.98 ± 0.36 | 13.77 ± 4.45 | 2.04 ± 0.60 | 6.08 ± 1.32 | 1.70 | |
| 0.6 | 0 | 1.83 ± 0.27 | 1.42 ± 0.42 | 33.15 ± 8.99 | 0.05 | |
| 3.1 | 0 | 1.81 ± 0.42 | 1.23 ± 0.38 | 27.22 ± 7.45 | 0.06 | |
| 6.1 | 0 | 1.15 ± 0.14 | 0.82 ± 0.19 | 20.03 ± 4.15 | 0.06 | |
Our previous report found that SPM treatment of SRS lymphoblasts further decreased their already lowered levels of putrescine (16). The importance of this decreased PUT in SRS patients is unknown but could be of consequence particularly in the brain, because PUT can serve as a precursor to GABA (26–28). Unlike native SPM, Me2SPM treatment did not further reduce the intracellular PUT levels observed in SRS patient lymphoblasts. Together, these results indicate the ability of Me2SPM to functionally substitute for SPM in supporting growth and proliferation of SRS cells while also reducing the intrinsic accumulation of SPD that is characteristic of a lack of spermine synthase activity.
Me2SPM reduces SPD levels without significantly altering polyamine metabolic enzyme activities of SRS cells
Me2SPM was previously shown to induce SSAT activity in a lung cancer cell line capable of extremely high SSAT induction (24). Although treatment with Me2SPM is clearly capable of reducing SPD levels in SRS cells after 96 h, treatment of SRS lymphoblastoid cells for 24 h resulted in no significant change in SAT1 mRNA expression or SSAT activity (Fig. 3A). A related polyamine analog, bis(ethyl)spermine, was used as a positive control and induced SSAT activity by ∼20-fold in line Ly-SRS3. Additional analysis of the treated lymphoblastoid cells revealed no induction of N1-acetylpolyamine oxidase (PAOX) mRNA and a modest increase in activity (Fig. 3B). Spermine oxidase (SMOX) activity, which has the potential to produce toxic reactive oxygen species and aldehydes, was below the limits of detection in both untreated and treated cells. ODC activity was decreased in both WT and SRS lymphoblasts after 24 h and appeared to be post-transcriptional in nature (Fig. 3C). Although the observed changes in enzyme activities were modest, treatment did yield a significant decrease in intracellular SPD in the SRS cells, suggesting the potential for product inhibition of other biosynthetic enzymes in the pathway (Fig. 3D). Assays were also performed in line Ly-SRS1 with similar nonsignificant results.
Figure 3.
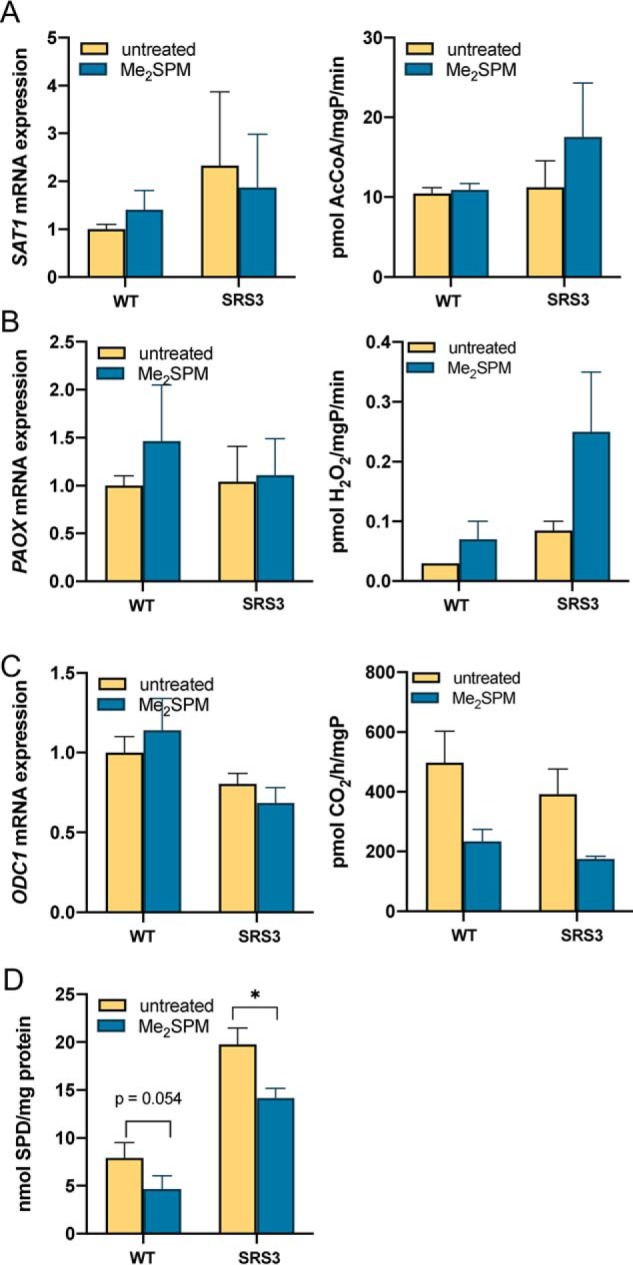
Effects of Me2SPM on polyamine metabolic enzymes in SRS lymphoblastoid cell lines. The cells were treated for 24 h with 6 μm Me2SPM. Lysates were analyzed for SSAT (A), PAOX (B), and ODC (C) mRNA expression levels (left panels) and activities (right panels). Intracellular SPD concentrations were determined from the same lysates used for SSAT assay (D). The results indicate the means of at least two biological replicates, each measured in triplicate. Error bars indicate S.E.
Safety of Me2SPM in vivo
WT, male C57Bl/6J mice received intraperitoneal injections of the (R,R)-diastereomer of 1,12-Me2SPM to determine the tolerable dose range, as well as its tissue distribution and ability to alter tissue polyamine concentrations. Daily injections of 0, 5, and 10 mg/kg Me2SPM for 4 days had no adverse effect on mouse body mass (Fig. 4A), and no overt change in behavior was observed. Because mice treated with 50 and 100 mg/kg doses began losing weight after the first injection, the second injection was delayed until day 2. On day 3, one mouse each receiving 50 or 100 mg/kg Me2SPM had died overnight; the one other mouse in the 100 mg/kg group exhibited tremors, weakness, and anorexia. The majority of remaining mice in the 50 mg/kg group appeared weak, with body mass losses approaching 20%. Per protocol, mice in the 50 and 100 mg/kg groups were sacrificed for necropsy on day 3.
Figure 4.
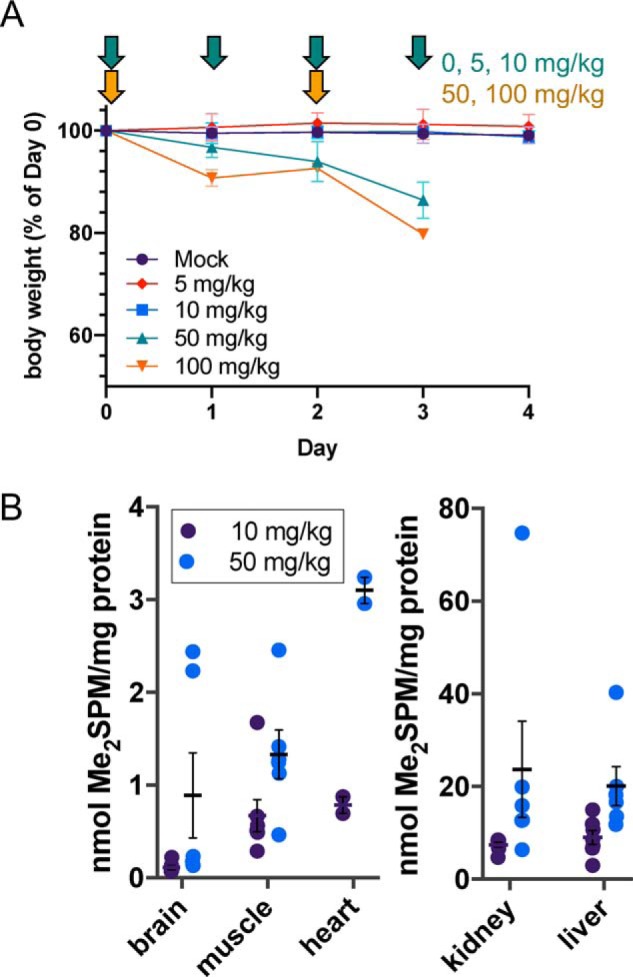
Dosing schedule, safety, and tissue distribution of Me2SPM in vivo. C57Bl/6J mice (n = 24) were treated for 4 days with increasing doses of Me2SPM (0 mg/kg, n = 7; 5 mg/kg, n = 2; 10 mg/kg, n = 7; 50 mg/kg, n = 6; 100 mg/kg, n = 2). Changes in body mass and timing of intraperitoneal injections are indicated by arrows in Fig. 4A. Tissues were collected for enzyme activity assays and HPLC analysis of polyamine and Me2SPM concentrations on day 4 (0, 5, and 10 mg/kg groups) or day 3 (50 and 100 mg/kg groups). Tissue concentrations of Me2SPM relative to total protein in the mock, 10, and 50 mg/kg groups are shown in B, with individual mice plotted as circles and the means and S.E. of each group indicated with horizontal lines and error bars.
Me2SPM is distributed to SRS-affected tissues
At set time points, mice were necropsied and tissues were collected and divided for biochemical analyses and/or histopathology. Brain, liver, kidney, heart, and hindlimb striated muscle were sampled from all mice. These tissues are either known to be involved in the SRS phenotype or provide information on potential dose-limiting toxicity. At the subtoxic dose of 10 mg/kg, intracellular Me2SPM was detected in all organs examined, with the kidney and liver concentrations exceeding those of striated muscle and heart by ∼10-fold. A very small amount of Me2SPM was detected in the brain tissue of 10 mg/kg treated animals, likely because of passive diffusion across the blood–brain barrier (Fig. 4B). In animals receiving 50 mg/kg Me2SPM, there was greater accumulation of Me2SPM in all organs, with average brain tissue levels similar to those of muscle. Notably, a bimodal distribution was observed in Me2SPM concentration in the brains of mice in the 50 mg/kg treatment group, which segregated the two mice in the pilot study (7-week-old mice, top two data points) from the remaining four mice that were treated at 20 weeks of age.
Tissue-specific modulation of polyamine metabolism by Me2SPM
SSAT activity tended to increase in the brains of mice receiving 10 mg/kg Me2SPM, and a significant increase was detected in those in the 50 mg/kg group (Fig. 5A). Half of the mice in the high-dose group demonstrated increases in ODC activity as well, some of which were extremely high (Fig. 5B). This elevation of both SSAT and ODC likely contributed to the significantly increased PUT levels detected in the brain as well as the absence of a decrease in SPD at the 50 mg/kg dose (Fig. 5C).
Figure 5.

Changes in polyamine levels and metabolic enzyme activities in Me2SPM-treated mouse brain, muscle, and heart. Brain tissue from treated mice was assayed for SSAT (A) and ODC (B) activities and intracellular PAs (C). Skeletal (D) and heart muscles (E) were analyzed for native polyamine distributions. The data points in A and B indicate the average of three determinations per mouse, relative to total protein, with horizontal lines indicating the means with S.E. The data in C–E represent duplicate determinations of each tissue (brain and muscle, mock, n = 7; 10 mg/kg, n = 7; 50 mg/kg, n = 6; heart, n = 2 each). For box-and-whisker plots, the horizontal lines indicate medians, + indicates the means, the boxes span the interquartile range, and the whiskers indicate the extremes.
Hypotonia is a common phenotype in SRS males. Analyses of muscle tissue taken from the hind limb also indicated a low level of Me2SPM accumulation at the nontoxic doses (Fig. 4B), with decreased abundance of SPM rather than SPD with increasing dose (Fig. 5D). It is worth noting the organ-specific differences in SPD and SPM distribution in the absence of treatment, as the more abundant polyamine may be reduced first in WT mice. However, because SPD concentration presumably far exceeds SPM concentration in all tissues of SRS patients, we anticipate that Me2SPM will be capable of reducing the accumulation of SPD in all tissues. These studies await the availability of a phenotyped mouse model, which is currently being established (The Jackson Laboratory). Hearts from two mice of each group were used for polyamine analysis. The 10 mg/kg dose of Me2SPM had no apparent effect on polyamine levels in the heart muscle, an organ that is not often affected in SRS patients, despite the detection of Me2SPM. However, dosing with 50 mg/kg resulted in higher analog accumulation (Fig. 4B) with reductions in both SPD and SPM pools (Fig. 5E).
Renal abnormalities have been reported in several SRS patients, particularly those with more severe phenotypes (9, 12). Liver and kidney tissues were examined for their response to Me2SPM and as potential indicators of toxicity. High polyamine levels are associated with both hepatic and nephrotoxicity, and both of these organs respond to injury and/or stress by inducing polyamine catabolism, particularly through SSAT (29–31). At nontoxic doses, Me2SPM was detected in both organs, with the greatest levels in the kidneys (Fig. 4B). SPD levels in the kidney decreased significantly in both the 10 and 50 mg/kg treatment groups without an overall significant induction of SSAT activity (Fig. 6A); however, three of the six mice at the high dose displayed elevated renal SSAT activity (Fig. 6B). Compared with other tissues, baseline renal ODC activity was very high and varied among the mice receiving mock or 10 mg/kg Me2SPM. However, ODC activity in the 50 mg/kg group was depleted, with only two of the six mice having detectable levels of activity (Fig. 6C). In liver tissues, SPD levels also decreased, and a significant increase in hepatic SSAT activity was detected in the 50 mg/kg group (Fig. 6, D and E). Hepatic ODC activity tended to increase with increasing dose (Fig. 6F).
Figure 6.
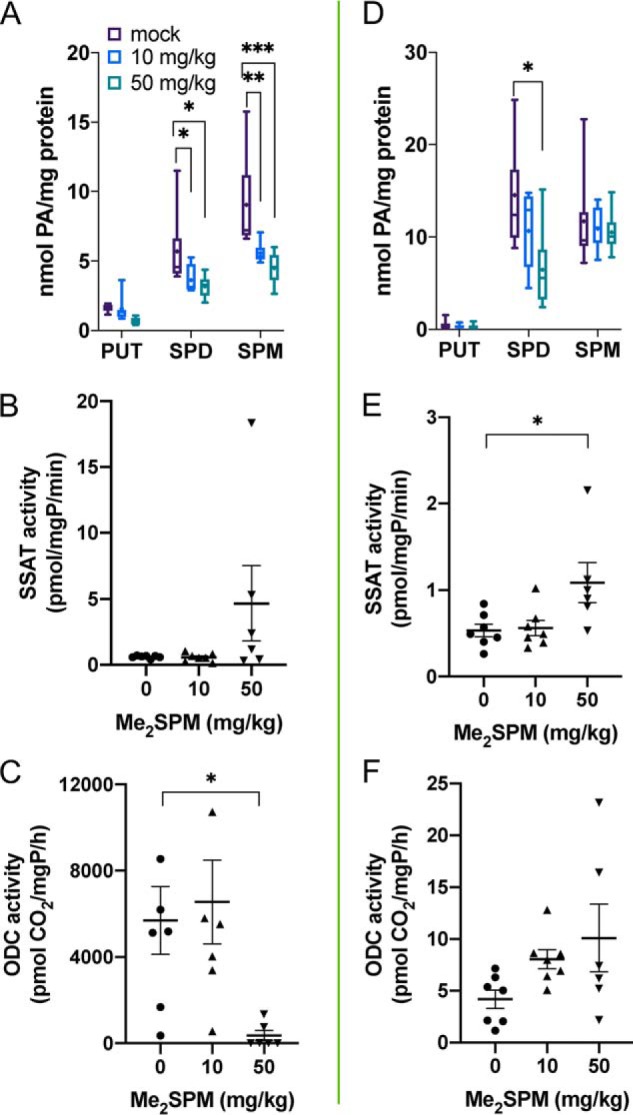
Changes in polyamine levels and key metabolic enzyme activities in Me2SPM-treated mouse kidney (A–C) and liver (D–F). Tissues from treated mice were assayed for intracellular polyamine concentrations (A and D), SSAT (B and E), and ODC activities (C and F). The data points in B, C, E, and F indicate the averages of three determinations per mouse, relative to total protein, with horizontal lines indicating the means with S.E. The data in A and D represent duplicate determinations of each tissue (mock, n = 7; 10 mg/kg, n = 7; 50 mg/kg, n = 6). The lines indicate medians, + indicates means, the boxes span the interquartile range, and the whiskers indicate the extremes.
Histopathological responses to Me2SPM
Sections of brain, striated hindlimb muscle, heart, liver, and kidney were sampled from mice in the mock, 10 mg/kg, or 50 mg/kg treatment groups to observe for histopathological changes indicative of toxicity. Representative images of each organ in each treatment group following standard hematoxylin and eosin staining are provided in the supporting information (Figs. S1–S3). There were no remarkable differences in brain, kidney, or muscle (striated or heart) histology among the three treatment groups. One of the four mice in the 50 mg/kg high-dose group displayed microvesicular fatty changes throughout its hepatocytes. Glycogen accumulation varied among the treatment groups but was likely unrelated to treatment.
Discussion
SRS males are hypotonic at birth and begin to exhibit an asthenic habitus, facial asymmetry, and often a prominent lower lip within the first years of life. As they age, but prior to puberty, muscle hypoplasia, osteoporosis, kyphoscoliosis, and long great toes are observed along with other clinical findings that are not consistent across the patient population. Because hypotonia is observed at birth, screening newborn males would be possible. Testing would consist of determining the spermidine/spermine ratio as well as the SMS activity in white blood cells. Because carriers of SMS mutations have ratio and activity values in the normal range, it would not be productive to screen the mothers biochemically, and targeted sequencing of the SMS gene would be of limited value based on the low prevalence of SRS in the population. Thus, a targeted treatment strategy, such as Me2SPM, that is intended to rebalance polyamine pools in the SRS male would likely be best implemented upon diagnosis. Considering the emergence of symptoms over time as the patient reaches adulthood, it is reasonable to predict that individuals receiving treatment earlier in life might receive the greatest benefits.
The selection of Me2SPM for these studies was based on several characteristics that differentiate it from the widely studied bis(ethyl) polyamine analogs, which have been analyzed for their antiproliferative/chemotherapeutic potential in cancer (32). Most importantly for the treatment of SRS, the methylation of SPM on its α carbons (versus alkylation on the primary amines) allows Me2SPM to substitute for the natural polyamines in their growth-sustaining functions. α-Methylation of SPM prevents its acetylation and subsequent oxidation via the SSAT/PAOX catabolic pathway and protects it against degradation by cellular monoamine and diamine oxidases as well as serum amine oxidases (20). The (R,R)-diastereomer of 1,12-Me2SPM in particular was chosen because it has been shown to be also resistant to oxidation by SMOX (23), the expression of which is high in certain organs, including the brain and liver. Although Me2SPM can serve as a poor substrate for PAOX (33), we detected no degradation to α-methylspermidine (α-MeSPD, the resulting metabolite) in any of the mouse tissues examined. In addition to degrading the native polyamines or their alkylated analogs, activity of these amine oxidases produces toxic by-products including aldehydes and hydrogen peroxide (34). Me2SPM is a therefore a metabolically stable SPM mimetic that sustains cell growth while avoiding oxidative degradation and the production of toxic by-products.
Overall, Me2SPM did demonstrate the ability to decrease SPD levels in several tissues when administered to WT mice. Unfortunately, at the dosing schedule used, the toxicity associated with the required doses of 1,12-Me2SPM may have limited its ability to decrease SPD levels in certain tissues, particularly the brain, which is severely affected in SRS. It is important to note that these in vivo studies were designed primarily to detect acute toxicity, which occurs following the administration of native polyamines (18, 35). The short duration of these experiments (4 days) may have been insufficient to elicit the desired decrease in SPD throughout the body with the nontoxic dose of Me2SPM. Because patient therapy would likely require long-term treatment, future studies will focus on optimizing the dosing schedule for prolonged treatment in the absence of toxicity.
PUT levels in the brain increased in the animals receiving the higher doses, and these appeared to feed forward to prevent a reduction in SPD. Although the high dose of Me2SPM was associated with increased ODC activity in certain tissues, including the brain, the mice in which this occurred were not the same mice that accumulated higher levels of the mimetic. The mouse with extremely high brain ODC levels (top dot in Fig. 5B) was the same mouse in which lipid-containing microvesicles were observed in hepatocytes (Fig. S1). Whether these observations have a common cause is unknown, but the accumulation of Me2SPM in both of these tissues was not remarkably different from others in the same dosage group. This ODC induction could rather be due to an induction of the stress response by the high dose of Me2SPM, because both ODC and SSAT are known stress-response genes. Additionally, induction of SSAT could further contribute to the toxicity through producing acetylated SPD for oxidation by PAOX, which produces reactive oxygen species and toxic aldehydes as by-products.
In the 50 mg/kg group, high levels of Me2SPM accumulated in the liver and kidney and were associated with increases in SSAT activity, which could result from toxicity-associated damage in these organs. However, histopathological analyses of these organs revealed no evidence indicative of acute renal or hepatic toxicity, suggesting this induction is due to either accumulation of the analog itself or induction of the stress response. Although we did not detect evidence of oxidative degradation of Me2SPM into α-MeSPD in our mouse study, a metabolite consistent with α-MeSPD was apparent in our SRS lymphoblastoid cell lines, which express high levels of PAOX. However, this oxidation had no adverse effect on proliferation, likely because of the localization of PAOX in the peroxisome (36). The fate of this metabolite in vivo might have eluded detection because of excretion or other compartmentalization. Thus, the possibility that oxidation of Me2SPM might occur to some extent in organs with high PAOX expression remains and could be a source of toxicity.
The ability of Me2SPM to efficiently reduce the high accumulation of SPD in the SRS patient cell lines raises the question as to the mechanism of this depletion. With the exception of the extremely sensitive lung cancer cell line previously described (24), other studies have reported results similar to ours, where polyamine pools are altered by Me2SPM exposure in a manner consistent with increased SSAT activity but with limited induction of the protein (37). Me2SPM may displace native polyamines from their intracellular binding sites, thereby increasing the pools of free SPM and SPD available for acetylation by basal levels of SSAT. Furthermore, because Me2SPM is a competitive inhibitor of SSAT (22), the timing of the treatment prior to assay may need to be adjusted to avoid high analog accumulation. SPD biosynthesis might be limited by reduced dcAdoMet levels subsequent to Me2SPM-mediated repression of S-adenosylmethionine decarboxylase (SAMDC) (37). The diluting effect of cell division would further decrease SPD concentrations, and Me2SPM could further affect spermidine biosynthesis through product inhibition. However, the SRS fibroblast lines have a greater response to low doses of the mimetic even though they proliferate much more slowly than the lymphoblastoid lines.
In addition to the elevated SPD:SPM ratio, SRS patient-derived cells tend to have reduced or nondetectable PUT levels compared with WT cells (Fig. 2A) (16). This could have important implications, particularly in the brain where PUT can serve as a precursor for GABA. Treatment with exogenous SPM reduced these levels even further in SRS lymphoblasts (16), whereas treatment with Me2SPM sustained the PUT level, with higher treatment doses tending to increase PUT content (Table 1). In brain tissue from WT mice receiving subtoxic doses, Me2SPM treatment successfully maintained the baseline PUT concentration (Fig. 5C).
The results of these studies indicate that Me2SPM is capable of targeting the underlying molecular mechanisms responsible for Snyder–Robinson syndrome, specifically the imbalance of SPD:SPM ratios. Our studies with this compound were limited by the lack of an appropriate mouse model for this disease. Although recently made available (The Jackson Laboratory), these mice have not yet been fully phenotyped. Additionally, the physiological relevance of the originally described SRS mouse model, known as the Gyro mouse (13, 14), is questionable. This Gyro mouse completely lacks the Sms gene, whereas the majority of SRS patients have partial loss-of-function mutations. Furthermore, the phenotype is confounded by the partial deletion of an adjacent gene that results in defective phosphate metabolism.
Our results with 1,12-Me2SPM are promising but do indicate the potential for toxicity, the source of which remains to be established. However, daily treatment at 10 mg/kg gave no indication of toxicity, and delivery of Me2SPM to tissues and the resulting effects on polyamine distribution were evident, although at low levels. Consequently, these studies form the basis for future studies, the goal of which will be to optimize the dosing schedule of 1,12-Me2SPM for further investigation in a mouse model of Snyder–Robinson syndrome. Additionally, 1,12-Me2SPM may serve as a lead compound for the design of related molecules with improved safety profiles. In particular, recent analyses of structurally similar compounds have indicated that positioning of the methyl groups of these compounds alters their biochemical properties with regard to polyamine metabolic enzymes (38). Thus, the results reported in this study provide proof of principle that dimethylated SPM mimetics are effective in decreasing SPD levels in SRS-affected cells as well as in multiple tissues in vivo, paving the way for further discovery and development in the understanding and treatment of SRS.
Experimental procedures
Cell lines, culture conditions, and Me2SPM synthesis
The lymphoblastoid cell lines derived from male SRS patients or healthy donors were previously described (4, 8, 10, 16) and maintained in RPMI 1640 containing 15% fetal bovine serum (Gemini Bio-Products, Sacramento, CA), 2 mm glutamine, sodium pyruvate, nonessential amino acids, and penicillin/streptomycin at 5% CO2 and 37 °C. Fibroblast cell lines established from skin biopsies of SRS males or healthy donor males were maintained in Dulbecco's modified Eagle's medium containing 15% fetal bovine serum, l-glutamine, sodium pyruvate, nonessential amino acids, and antibiotics at 5% CO2 and 37 °C. In experiments cultured with exogenous SPM, 1 mm aminoguanidine (Sigma) was added to prevent extracellular oxidation of SPM by bovine serum amine oxidase. (R,R)-1,12-Me2SPM was synthesized as previously described (33, 39).
Polyamine concentration determinations and enzyme activity assays
Intracellular polyamine concentrations of cell or tissue lysates were determined by HPLC following acid extraction and dansylation of the supernatant, as originally described by Kabra et al. (40). Standards prepared for HPLC included diaminoheptane (internal standard), PUT, SPD, SPM, and acetylated SPD and SPM, all of which were purchased from Sigma. Enzyme activity assays were performed for SSAT and ODC using radiolabeled ornithine and acetyl-CoA, respectively, as previously described (41, 42). SMOX and PAOX activities were measured using luminol-based detection of H2O2 in the presence of either SPM (for SMOX) or N1-acetylated spermine (for PAOX) as substrates (43). All enzyme activities and intracellular polyamine concentrations are presented relative to total cellular protein, as determined using Bio-Rad protein dye with interpolation on a BSA standard curve.
RNA isolation and quantitative RT-PCR
Total cellular RNA was extracted from treated cells using TRIzol reagent (Invitrogen). One μg of each sample was used to generate cDNA using qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD), followed by SYBR-green–mediated, quantitative real-time PCR (Bio-Rad iQ2 detection system) with primers targeting polyamine metabolism–associated genes. Custom primers specific for human ODC1, SAT1, PAOX, and GAPDH were synthesized by Integrated DNA Technologies (Coralville, IA) and optimized using annealing temperature gradients followed by analyses of melt curves and agarose gel electrophoresis. The threshold cycle for each gene in each sample (of which there were at least two biological replicates) was determined in triplicate and normalized to GAPDH expression. Fold change was determined using the 2−ΔΔCt algorithm.
Pilot in vivo safety study
Male C57Bl/6J mice (The Jackson Laboratory) at 7 weeks of age were randomly divided into five treatment groups for initial toxicity testing (two mice/dose). (R,R)-1,12-Me2SPM was prepared in PBS, pH 7.4, and filter-sterilized. The mice were weighed daily and injected intraperitoneally with ≤0.1 ml/injection of 0 (PBS only), 5, 10, 50, or 100 mg Me2SPM/kg body weight. The mice in the 0, 5, or 10 mg/kg groups were injected daily for a total of four doses and were sacrificed for necropsy 24 h following the final dose. The mice in the 50 and 100 mg/kg groups were injected every 48 h, thereby receiving a total of two doses. According to protocol, the animals were sacrificed upon body weight loss nearing 20%. All animals were humanely euthanized using CO2, and necropsies were performed immediately for the collection of brain, kidney, liver, skeletal muscle, and heart tissue. Tissues were quick frozen and stored at −80 °C for assay of polyamine metabolic enzymes and polyamine concentrations as well as tissue distribution of Me2SPM. All animal studies were conducted according to protocol following Johns Hopkins University Institutional Animal Care and Use Committee approval.
Expansion cohort
An expansion cohort of 14 WT male mice at 20 weeks of age was used for further testing using a narrowed dose range of Me2SPM as determined in the pilot safety study. Five mice each were treated at 0 or 10 mg/kg daily, and four mice were treated at 50 mg/kg every other day, as depicted in Fig. 4A and described above. The mice were again weighed daily, and tissues were collected for enzyme analyses and polyamine determinations, as well as for histological observation.
Histopathological analyses
Brain, striated muscle, heart, liver, and kidney tissues were collected from 14 mice (5 each from mock and 10 mg/kg groups and 4 from 50 mg/kg), fixed in formalin, and processed for standard hematoxylin and eosin tissue staining. The slides were analyzed and interpreted by a pathologist (K. G.) blinded to the study parameters.
Statistical analyses
Statistically significant differences between untreated and treated samples were determined using multiple t tests (GraphPad Prism software, La Jolla, CA). *, p values ≤ 0.05 were considered statistically significant; **, p ≤ 0.005; ***, p ≤ 0.0005.
Author contributions
T. M. S. and J. R. F. data curation; T. M. S. validation; T. M. S., J. R. F., X. G., C. E. H., and T. T. D. investigation; T. M. S. and K. G. visualization; T. M. S., M. K., C. E. S., A. K., and R. A. C. methodology; T. M. S. writing-original draft; T. M. S. and R. A. C. project administration; T. M. S., M. K., J. R. F., C. E. S., K. G., A. K., and R. A. C. writing-review and editing; M. K., C. E. S., K. G., A. K., and R. A. C. resources; J. R. F., X. G., and K. G. formal analysis; C. E. S., K. G., A. K., and R. A. C. funding acquisition; R. A. C. conceptualization; R. A. C. supervision.
Supplementary Material
This work was supported by NCI, National Institutes of Health Grants R01CA204345 and R01CA235863 (to R. A. C.), P30CA006973 (to S. K. C. C. C.) and R21CA229582 (to K. G.) and NINDS, National Institutes of Health Grant R01NS073854 (to C. E. S.). This work was also supported by funding from the Million Dollar Bike Ride, Orphan Disease Center at the University of Pennsylvania Grants MDBR-18-127-SR (to R. A. C.) and MDBR-20-135-SRS (to R. A. C. and T. M. S.) and a grant from the South Carolina Department of Disabilities and Special Needs (to C. E. S.). Synthesis of 1,12-Me2SPM was supported by Russian Science Foundation Grant 17-74-20049 (to A. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
C. E. Schwartz, private communication.
- SMS
- spermine synthase
- Me2SPM
- (R,R)-1,12-dimethylspermine
- SRS
- Snyder–Robinson syndrome
- SPD
- spermidine
- SPM
- spermine
- PUT
- putrescine
- SSAT
- spermidine/spermine N1-acetyltransferase
- ODC
- ornithine decarboxylase
- PAOX
- N1-acetylpolyamine oxidase
- SMOX
- spermine oxidase
- α-MeSPD
- α-methylspermidine.
References
- 1. Pegg A. E., and Michael A. J. (2010) Spermine synthase. Cell Mol. Life Sci. 67, 113–121 10.1007/s00018-009-0165-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Snyder R. D., and Robinson A. (1969) Recessive sex-linked mental retardation in the absence of other recognizable abnormalities: report of a family. Clin. Pediatr. (Phila.) 8, 669–674 10.1177/000992286900801114 [DOI] [PubMed] [Google Scholar]
- 3. Arena J. F., Schwartz C., Ouzts L., Stevenson R., Miller M., Garza J., Nance M., and Lubs H. (1996) X-linked mental retardation with thin habitus, osteoporosis, and kyphoscoliosis: linkage to Xp21.3–p22.12. Am. J Med. Genet. 64, 50–58 [DOI] [PubMed] [Google Scholar]
- 4. Cason A. L., Ikeguchi Y., Skinner C., Wood T. C., Holden K. R., Lubs H. A., Martinez F., Simensen R. J., Stevenson R. E., Pegg A. E., and Schwartz C. E. (2003) X-linked spermine synthase gene (SMS) defect: the first polyamine deficiency syndrome. Eur. J. Hum. Genet. 11, 937–944 10.1038/sj.ejhg.5201072 [DOI] [PubMed] [Google Scholar]
- 5. Schwartz C. E., Wang X., Stevenson R. E., and Pegg A. E. (2011) Spermine synthase deficiency resulting in X-linked intellectual disability (Snyder–Robinson syndrome). Methods Mol. Biol. 720, 437–445 10.1007/978-1-61779-034-8_28 [DOI] [PubMed] [Google Scholar]
- 6. Starks R., Kirby P., Ciliberto M., and Hefti M. (2018) Snyder–Robinson syndrome. Autops. Case Rep. 8, e2018031 10.4322/acr.2018.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pegg A. E. (2009) Mammalian polyamine metabolism and function. IUBMB Life 61, 880–894 10.1002/iub.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Alencastro G., McCloskey D. E., Kliemann S. E., Maranduba C. M., Pegg A. E., Wang X., Bertola D. R., Schwartz C. E., Passos-Bueno M. R., and Sertié A. L. (2008) New SMS mutation leads to a striking reduction in spermine synthase protein function and a severe form of Snyder–Robinson X-linked recessive mental retardation syndrome. J. Med. Genet. 45, 539–543 10.1136/jmg.2007.056713 [DOI] [PubMed] [Google Scholar]
- 9. Albert J. S., Bhattacharyya N., Wolfe L. A., Bone W. P., Maduro V., Accardi J., Adams D. R., Schwartz C. E., Norris J., Wood T., Gafni R. I., Collins M. T., Tosi L. L., Markello T. C., Gahl W. A., et al. (2015) Impaired osteoblast and osteoclast function characterize the osteoporosis of Snyder–Robinson syndrome. Orphanet. J. Rare Dis. 10, 27 10.1186/s13023-015-0235-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becerra-Solano L. E., Butler J., Castaneda-Cisneros G., McCloskey D. E., Wang X., Pegg A. E., Schwartz C. E., Sanchez-Corona J., and Garcia-Ortiz J. E. (2009) A missense mutation, p.V132G, in the X-linked spermine synthase gene (SMS) causes Snyder–Robinson syndrome. Am. J. Med. Genet. A 149A, 328–335 10.1002/ajmg.a.32641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peron A., Spaccini L., Norris J., Bova S. M., Selicorni A., Weber G., Wood T., Schwartz C. E., and Mastrangelo M. (2013) Snyder–Robinson syndrome: a novel nonsense mutation in spermine synthase and expansion of the phenotype. Am. J. Med. Genet. A 161A, 2316–2320 10.1002/ajmg.a.36116 [DOI] [PubMed] [Google Scholar]
- 12. Larcher L., Norris J. W., Lejeune E., Buratti J., Mignot C., Garel C., Keren B., Schwartz C. E., and Whalen S. (2019) The complete loss of function of the SMS gene results in a severe form of Snyder–Robinson syndrome. Eur. J. Med. Genet. 103777 10.1016/j.ejmg.2019.103777 [DOI] [PubMed] [Google Scholar]
- 13. Mackintosh C. A., and Pegg A. E. (2000) Effect of spermine synthase deficiency on polyamine biosynthesis and content in mice and embryonic fibroblasts, and the sensitivity of fibroblasts to 1,3-bis-(2-chloroethyl)-N-nitrosourea. Biochem. J. 351, 439–447 10.1042/0264-6021:3510439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X., Levic S., Gratton M. A., Doyle K. J., Yamoah E. N., and Pegg A. E. (2009) Spermine synthase deficiency leads to deafness and a profound sensitivity to α-difluoromethylornithine. J. Biol. Chem. 284, 930–937 10.1074/jbc.M807758200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seiler N., Delcros J. G., and Moulinoux J. P. (1996) Polyamine transport in mammalian cells: an update. Int. J. Biochem. Cell Biol. 28, 843–861 10.1016/1357-2725(96)00021-0 [DOI] [PubMed] [Google Scholar]
- 16. Murray-Stewart T., Dunworth M., Foley J. R., Schwartz C. E., and Casero R. A. Jr. (2018) Polyamine homeostasis in Snyder–Robinson syndrome. Med. Sci. 6, E112 10.3390/medsci6040112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pegg A. E. (2013) Toxicity of polyamines and their metabolic products. Chem. Res. Toxicol. 26, 1782–1800 10.1021/tx400316s [DOI] [PubMed] [Google Scholar]
- 18. Tabor C. W., and Rosenthal S. M. (1956) Pharmacology of spermine and spermidine: some effects on animals and bacteria. J. Pharmacol. Exp. Ther. 116, 139–155 [PubMed] [Google Scholar]
- 19. Til H. P., Falke H. E., Prinsen M. K., and Willems M. I. (1997) Acute and subacute toxicity of tyramine, spermidine, spermine, putrescine and cadaverine in rats. Food Chem. Toxicol. 35, 337–348 10.1016/S0278-6915(97)00121-X [DOI] [PubMed] [Google Scholar]
- 20. Lakanen J. R., Coward J. K., and Pegg A. E. (1992) α-Methyl polyamines: metabolically stable spermidine and spermine mimics capable of supporting growth in cells depleted of polyamines. J. Med. Chem. 35, 724–734 10.1021/jm00082a013 [DOI] [PubMed] [Google Scholar]
- 21. Byers T. L., Lakanen J. R., Coward J. K., and Pegg A. E. (1994) The role of hypusine depletion in cytostasis induced by S-adenosyl-l-methionine decarboxylase inhibition: new evidence provided by 1-methylspermidine and 1,12-dimethylspermine. Biochem. J. 303, 363–368 10.1042/bj3030363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Järvinen A., Grigorenko N., Khomutov A. R., Hyvönen M. T., Uimari A., Vepsäläinen J., Sinervirta R., Keinänen T. A., Vujcic S., Alhonen L., Porter C. W., and Jänne J. (2005) Metabolic stability of α-methylated polyamine derivatives and their use as substitutes for the natural polyamines. J. Biol. Chem. 280, 6595–6601 10.1074/jbc.M412788200 [DOI] [PubMed] [Google Scholar]
- 23. Hyvönen M. T., Keinänen T. A., Cerrada-Gimenez M., Sinervirta R., Grigorenko N., Khomutov A. R., Vepsäläinen J., Alhonen L., and Jänne J. (2007) Role of hypusinated eukaryotic translation initiation factor 5A in polyamine depletion-induced cytostasis. J. Biol. Chem. 282, 34700–34706 10.1074/jbc.M704282200 [DOI] [PubMed] [Google Scholar]
- 24. Yang J., Xiao L., Berkey K. A., Tamez P. A., Coward J. K., and Casero R. A. Jr. (1995) Significant induction of spermidine/spermine N1-acetyltransferase without cytotoxicity by the growth-supporting polyamine analogue 1,12-dimethylspermine. J. Cell. Physiol. 165, 71–76 10.1002/jcp.1041650109 [DOI] [PubMed] [Google Scholar]
- 25. Li C., Brazill J. M., Liu S., Bello C., Zhu Y., Morimoto M., Cascio L., Pauly R., Diaz-Perez Z., Malicdan M. C. V., Wang H., Boccuto L., Schwartz C. E., Gahl W. A., Boerkoel C. F., et al. (2017) Spermine synthase deficiency causes lysosomal dysfunction and oxidative stress in models of Snyder–Robinson syndrome. Nat. Commun. 8, 1257 10.1038/s41467-017-01289-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sequerra E. B., Gardino P., Hedin-Pereira C., and de Mello F. G. (2007) Putrescine as an important source of GABA in the postnatal rat subventricular zone. Neuroscience 146, 489–493 10.1016/j.neuroscience.2007.01.062 [DOI] [PubMed] [Google Scholar]
- 27. Seiler N., and Eichentopf B. (1975) 4-Aminobutyrate in mammalian putrescine catabolism. Biochem. J. 152, 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim J. I., Ganesan S., Luo S. X., Wu Y. W., Park E., Huang E. J., Chen L., and Ding J. B. (2015) Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 350, 102–106 10.1126/science.aac4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zahedi K., Barone S., Kramer D. L., Amlal H., Alhonen L., Jänne J., Porter C. W., and Soleimani M. (2010) The role of spermidine/spermine N1-acetyltransferase in endotoxin-induced acute kidney injury. Am. J. Physiol. Cell Physiol. 299, C164–C174 10.1152/ajpcell.00512.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zahedi K., Barone S. L., Xu J., Steinbergs N., Schuster R., Lentsch A. B., Amlal H., Wang J., Casero R. A. Jr, and Soleimani M. (2012) Hepatocyte-specific ablation of spermine/spermidine-N1-acetyltransferase gene reduces the severity of CCl4-induced acute liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G546–G560 10.1152/ajpgi.00431.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zahedi K., Lentsch A. B., Okaya T., Barone S., Sakai N., Witte D. P., Arend L. J., Alhonen L., Jell J., Jänne J., Porter C. W., and Soleimani M. (2009) Spermidine/spermine-N1-acetyltransferase ablation protects against liver and kidney ischemia–reperfusion injury in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G899–G909 10.1152/ajpgi.90507.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Casero R. A. Jr., and Woster P. M. (2001) Terminally alkylated polyamine analogues as chemotherapeutic agents. J. Med. Chem. 44, 1–26 10.1021/jm000084m [DOI] [PubMed] [Google Scholar]
- 33. Järvinen A. J., Cerrada-Gimenez M., Grigorenko N. A., Khomutov A. R., Vepsäläinen J. J., Sinervirta R. M., Keinänen T. A., Alhonen L. I., and Jänne J. E. (2006) α-Methyl polyamines: efficient synthesis and tolerance studies in vivo and in vitro: first evidence for dormant stereospecificity of polyamine oxidase. J. Med. Chem. 49, 399–406 10.1021/jm050872h [DOI] [PubMed] [Google Scholar]
- 34. Murray Stewart T., Dunston T. T., Woster P. M., and Casero R. A. Jr. (2018) Polyamine catabolism and oxidative damage. J. Biol. Chem. 293, 18736–18745 10.1074/jbc.TM118.003337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seiler N. (1981) Polyamine metabolism and function in brain. Neurochem. Int. 3, 95–110 10.1016/0197-0186(81)90027-9 [DOI] [PubMed] [Google Scholar]
- 36. Hölttä E. (1977) Oxidation of spermidine and spermine in rat liver: purification and properties of polyamine oxidase. Biochemistry 16, 91–100 10.1021/bi00620a015 [DOI] [PubMed] [Google Scholar]
- 37. Hyvönen M. T., Howard M. T., Anderson C. B., Grigorenko N., Khomutov A. R., Vepsäläinen J., Alhonen L., Jänne J., and Keinänen T. A. (2009) Divergent regulation of the key enzymes of polyamine metabolism by chiral α-methylated polyamine analogues. Biochem. J. 422, 321–328 10.1042/BJ20090737 [DOI] [PubMed] [Google Scholar]
- 38. Khomutov M., Hyvönen M. T., Simonian A., Formanovsky A. A., Mikhura I. V., Chizhov A. O., Kochetkov S. N., Alhonen L., Vepsäläinen J., Keinänen T. A., and Khomutov A. R. (2019) Unforeseen possibilities to investigate the regulation of polyamine metabolism revealed by novel C-methylated spermine derivatives. J. Med. Chem. 62, 11335–11347 10.1021/acs.jmedchem.9b01666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grigorenko N. A., Khomutov A. R., Keinänen T. A., Järvinen A., Alhonen L., Jänne J., and Vepsäläinen J. (2007) Synthesis of novel optical isomers of α-methylpolyamines. Tetrahedron 63, 2257–2262 10.1016/j.tet.2006.12.065 [DOI] [Google Scholar]
- 40. Kabra P. M., Lee H. K., Lubich W. P., and Marton L. J. (1986) Solid-phase extraction and determination of dansyl derivatives of unconjugated and acetylated polyamines by reversed-phase liquid chromatography: improved separation systems for polyamines in cerebrospinal fluid, urine and tissue. J. Chromatogr. 380, 19–32 10.1016/S0378-4347(00)83621-X [DOI] [PubMed] [Google Scholar]
- 41. Casero R. A. Jr, Celano P., Ervin S. J., Porter C. W., Bergeron R. J., and Libby P. R. (1989) Differential induction of spermidine/spermine N1-acetyltransferase in human lung cancer cells by the bis(ethyl)polyamine analogues. Cancer Res. 49, 3829–3833 [PubMed] [Google Scholar]
- 42. Seely J. E., and Pegg A. E. (1983) Ornithine decarboxylase (mouse kidney). Methods Enzymol. 94, 158–161 10.1016/S0076-6879(83)94025-9 [DOI] [PubMed] [Google Scholar]
- 43. Goodwin A. C., Murray-Stewart T. R., and Casero R. A. Jr. (2011) A simple assay for mammalian spermine oxidase: a polyamine catabolic enzyme implicated in drug response and disease. Methods Mol. Biol. 720, 173–181 10.1007/978-1-61779-034-8_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.