

Abstract
The innate immune response is crucial for defense against virus infections where the complement system, coagulation cascade and natural antibodies play key roles. These immune components are interconnected in an intricate network and are tightly regulated to maintain homeostasis and avoid uncontrolled immune responses. Many viruses in turn have evolved to modulate these interactions through various strategies to evade innate immune activation. This review summarizes the current understanding on viral strategies to inhibit the activation of complement and coagulation cascades, evade natural antibody-mediated clearance and utilize complement regulatory mechanisms to their advantage.
Keywords: Complement, natural antibodies, coagulation, viral
Introduction
The first line of defense against foreign pathogens is the innate immune response, comprised of innate cells, physical barriers, and humoral components, consisting of the complement and coagulation cascades, and natural antibodies (NAb). Vital to maintaining a barrier and clearing pathogens that breach the barrier, the innate immune response also removes debris to maintain homeostasis. While there is a plethora of published data on innate immune cells and physical barriers against viral infection, there is still much to discover about the mechanisms of the innate humoral immune response. The individual proteins of the complement and coagulation systems, and NAb production are tightly regulated to mount an innate immune response for viral defense and homeostasis maintenance. In addition, the humoral components are intricately linked at multiple points in the cascade, providing an amplified immune response and viral clearance. Viruses have evolved over millions of years to evade the immune system including modulating activation and regulation of the complement and coagulation cascades and NAb production. Several virus families have evolved conserved genes encoding for proteins that act as virulence factors and inhibit multiple components of the humoral innate immune response by mimicry, incorporation of host molecules in the virion, or detection escape mechanisms. These strategies are advantageous for the virus to increase pathogenicity through activation, suppression or preventing virus neutralization.
This review provides information detailing the individual components of complement regulation and activation, the coagulation system, and NAb production in response to viral infections. Additionally, the complex roles of the complement system, coagulation cascade, and Nab production are discussed, demonstrating the complexity and crosstalk observed during viral infection.
Viral Inhibition of Complement Activation and Complement Regulators
The complement cascade plays a vital role in initiating and regulating the innate and adaptive immune systems responses against invading pathogens such as viruses. The classical, lectin, and alternative pathways initiate clearance of foreign pathogens, reducing the infectious burden. Viruses can successfully evade host immune responses by modulating complement activation through inhibition of major proteins in the complement cascade such as C1q, mannose binding lectin (MBL), C3/C3b, C4/C4b, and C5b-9. These strategies increase viral pathogenicity by suppressing complement activation or effectively halting virus neutralization.
Inhibition of Complement Initiation
C1q and MBL initiate the classical and lectin pathways, respectively. After binding to a ligand, C1q undergoes a necessary conformational change to initiate the classical pathway. The direct binding of the human astrovirus type I capsid protein disrupts this conformational change and complement activation by dissociating the C1s2–C1r2 tetramer from C1q[1, 2]. Astrovirus capsid protein may also inhibit C1q globular heads from binding IgG [3]. Conversely, Hepatitis C virus (HCV) core protein acts as a C1q mimic on activated T cells by binding the C1q receptor with a similar affinity to C1q, thereby inhibiting T cell proliferation [4–6]. The capsid protein of astroviruses also interacts with MBL causing dual inhibition, although the exact mechanism of binding is still not fully understood [2, 3].
Both the classical and lectin pathways activate C4 and subsequently produce C4b, an essential protein in the C3 convertase, C4b2a. Both HCV and herpesvirus saimiri (HVS) inhibit C4 with distinct mechanisms. The HCV NS5A and core proteins inhibit C4 transcription and subsequent translation, thus inhibiting C3 convertase formation [7, 8]. In contrast, HVS encodes a complement control protein homolog with structural homology to complement regulators which binds with high affinity to C4b, thereby accelerating the decay of C3 convertase [9]. Together, these viruses evolved mechanisms to shut down initiation of the complement cascade preventing formation of anaphylatoxins and the lytic pore.
Viral Inhibition of C3, C3b and the Terminal Membrane Attack Complex
As central proteins in the complement cascade, C3 and C3b are the converging point for all three initiation pathways and are critical for the formation of the alternative C3 convertase, C3bBb, and both C5 convertases. In addition to inhibiting C4, the HCV NS5A protein modulates C3 [7]. Similarly, chronic HCV infection represses the C3 promoter, depleting serum C3, and thus C3 and C5 convertase production [8, 10].
Many viruses evolved to encode virulence proteins that bind to C3b, instead of C3. Poxviruses contain inhibitors of complement enzymes (PICES) which bind C3b [11, 12], with varying binding affinity between specific poxvirus [13–15]. Structurally and functionally similar to two regulators of decay accelerating activity against C3 convertase, Factor H and C4b binding protein (C4BP) [16, 17], PICES-like vaccinia virus complement control protein degrades C3b to iC3b1, preventing C3b binding to activated factor B (Bb) [18, 19]. However, smallpox inhibitor of complement enzymes degrades iC3b1 further into C3f and iC3b2 [11, 18]. Herpes simplex virus (HSV) encodes glycoprotein C (gC) which also binds to C3b [20–24]. The C3b binding domain of HSV-1 gC-1 is homologous to the C3b binding sites of factors H, B, complement receptor 1 (CR1), and CR2 [20]. HSV-1 gC-1 inhibits C3b binding of factor H and properdin, an alternative pathway C3 convertase stabilizer [20, 22]. Competitive binding with properdin suggests gC-1 may decrease the stability of C3 convertase. In conjunction with HSV gC binding of C3b, an additional study showed binding of C3b by a CR1-like C3 receptor found on the HSV membrane [25]. Kaposi’s sarcoma-associated herpesvirus (KSHV) encodes a soluble and cell-associated form of a complement control protein (KCP) which functions as a potent cofactor for classical pathway factor I cleavage of C3b [26, 27]. Other viruses that include KCP homologs that inhibit C3b in the same manner as KSHV are rhesus rhadinovirus (RRV) and murine gammaherpesvirus 68 (γHV68) [28, 29]. Overall, viruses binding to C3b evolved similar functions to various complement cofactors and binding receptors to inhibit formation of C3 and C5 convertases as well as inhibiting formation of the lytic pore in all three complement pathways.
The C5b-9 protein complex is the final step in the complement cascade and leads to formation of the membrane attack complex (MAC) and subsequent cell lysis. Flavivirus non-structural protein 1 (NS1) protein inhibits the complement cascade by binding numerous proteins in the C5b-9 complex [30, 31]. Although inhibition of MAC is a novel mechanism for NS1, the exact mechanism and purpose is not fully understood. NS1 binds tightly to C5, C6 and C9, and binds weakly to C7 to inhibit C9 polymerization and prevents the lytic pore. Although NS1 alone decreases MAC formation, vitronectin, a multifunctional glycoprotein with regulatory functions found in serum, the extracellular matrix, and bone, binds C9 and NS1 simultaneously to further decrease MAC formation [30, 32]. Several flavivirus NS1s inhibit MAC formation but Zika viral NS1 binds stronger and with greater efficiency to C9 than other viruses [30]. In addition to inhibiting C5b-9, soluble dengue virus NS1 activates complement in the fluid phase, releasing soluble C5b-9 into the plasma [33]. This triggers increased vascular leakage in patients with dengue hemorrhagic fever, possibly implicating NS1 in the progression of more deadly forms of the dengue infection [33]. Flaviviruses prevent formation of the MAC at the plasma membrane to increase viral replication as well as increase vascular leakage, intensifying lethal infection.
Viruses incorporate host complement regulators
CD55, CD46 and CD59 are frequently found incorporated on the surface of many virions depending on their expression levels in the host cells. Incorporation of CD46 and CD55 promote factor I-mediated cleavage of C4b and C3b and decay accelerating activity against C3 convertase while CD59 suppresses complement mediated cytolysis. Viral acquisition of these regulatory proteins enhances complement resistance and is also speculated to play a role in tropism [34].
Viruses may incorporate only one type of complement regulator, such as Nipah virus [35] with CD46 and influenza A [36] and Infectious bronchitis virus [37] with CD59 on virions. Several other viruses including HCV, human T cell leukemia type I and human cytomegalovirus (HCMV) incorporate both CD55 and CD59 on the virion surfaces [38–41] and upregulate cellular CD55 expression [38].
Viruses in the same family may integrate complement regulators differently. In Paramyxoviridae, Mumps virus (MuV) and Vesicular Stomatitis virus (VSV) incorporate both CD46 and CD55 [42]. However, New Castle disease virus [43, 44] and Parainfluenza virus-5 [45] incorporate CD46, CD55 and CD59 on the surface of the virion and upregulate cellular CD46, CD55 and CD59 expression [44–46]. HIV, Simian immunodeficiency virus [47–49] and extracellular enveloped virions of vaccinia virus [34] are other viruses that also integrate CD46, CD55 and CD59 into the virions.
The potency of virion associated complement regulators appears to be variable. Virion associated CD59 is very potent in HIV, HCV and HCMV [41, 50] while CD55 plays a prominent role in complement evasion in MuV and VSV [42, 44, 45]. Incorporation of other complement regulators has only been described in HIV virions that acquire factor H [49]. Together, multiple types of viruses integrate host derived complement inhibitors into the virions and may also increase complement regulators on the cell surface to protect intracellular viral processes.
Viruses modulate or mimic host complement regulators
Viruses may encode proteins that directly bind to complement regulatory molecules or modulate their expression. Hepatitis B virus X protein binds to the CD59 promoter to upregulates CD59 expression [51]. Flavivirus NS1 discussed above, also recruits C4BP, a co-factor for factor I, triggering C4b cleavage and inhibiting classical and lectin pathway activation [31, 52]. Furthermore, West Nile virus NS1 binds factor H to degrade C3b which decreases alternative pathway activation [30, 53].
Viral proteins that mimic host complement regulatory molecules may show homology to host complement regulators, but their function may differ [54]. Both HSV and KSHV encode proteins with functions homologous to CD55 and/or CD46 as discussed above [22, 26, 27, 55]. Poxvirus PICES not only bind to C3b/C4b but also expresses CD46 cofactor activity and CD55-like activity to inhibit complement dependent cytolysis via both classical and alternative pathways [11–13]. Other viruses, such as T-lymphotropic HVS encode a structural homolog of CD59 [56] while Nipah virus encodes a functional ‘factor I-like’ protease which functions along with factor H to cleave C3b into iC3b [35]. These instances clearly indicate that mimicking host complement regulators enable these viruses to replicate in the cells.
Viruses utilize host complement regulators for attachment
Multiple viruses use complement regulators for cellular adhesion and entry. CD46 serves as the receptor for measles virus [57], human herpes virus 6 [58], different serotypes of adenoviruses (reviewed in [59]) and bovine diarrhea virus [60]. Enterovirus 70 [61] and Cardiovirulent coxsackie virus [62] use CD55 as the cellular receptor. Poxviruses PICES (discussed above) appear to play important roles in virus attachment to the host cell [11, 12].
In summary, viruses encompass multiple strategies, including modulation or acquisition of host complement regulators and mimicry, to evade complement mediated virus neutralization. This may also lead to increased virulence and pathogenicity, and ultimately define the disease outcome. Virus-derived regulatory molecules are also appealing therapeutic agents in treating complement disorders as they may possess higher affinity and inhibitory potential than host regulatory molecules [63–65]. Despite the importance, only a minority of viruses or virus-derived complement regulators have been identified or characterized in this regard so far. Thus, further research is crucial to expand our understanding of their potential application as therapeutic agents against virus infections, inflammatory diseases and autoimmune diseases.
The role of Natural Antibodies in Viral Clearance
As a first line of defense, the immune system generates NAbs that are germline encoded and exist prior to encountering a cognate antigen. IgM isotypes typically respond to infected sites first [66] and provide the majority of NAb protection; however, natural IgG and IgA are also important NAbs that predominately exist in the serum and mucosal membranes, respectively. NAbs are produced by B-1 cells, marginal zone B-cells, and other B-cell types in the absence of external antigen stimulation [67–69], although the exact sources of NAbs are still debated. NAbs are non-specific [70] and have low affinity [71] due to fewer non-templated nucleotide additions and the lack of or minimal somatic hypermutation [72, 73]. The non-specificity permits recognition of more than one viral infection. Natural IgM maintains homeostasis by binding to apoptotic cells for enhanced phagocytosis [74, 75], regulating B cells [76], and recognizing self, thus playing a role in autoimmunity [66, 77]. Nabs recognize oxidized lipids, phospholipids, glycolipids, and glycoproteins, and cross-react with similar epitopes on microbes [78], such as phosphorylcholines, leading to pathogen clearance.
NAbs are critical in clearing virions during infection through 1) direct pathogen neutralization, 2) antigen recruitment to secondary lymphoid organs for subsequent neutralization, and 3) activation of the complement system. However, due to the vast differences of NAb characteristics as well as variability in viruses, the functional role of NAbs is debated. Recently, two new requirements were proposed for an antibody to be considered a NAb including the ability to exert a protective and regulatory function and an immediate response to those functions [79]. The broader definition demonstrates the complexity of NAbs. To evade NAb detection and clearance, viruses employ a variety of escape mechanisms to survive.
Natural Antibodies Aid in Viral Clearance
NAbs neutralize pathogens partially by their high avidity (rather than affinity), allowing the adaptive immune system time to tailor the immune response [80]. Viral neutralization results from antibody interference with proteins on the virion surface, aggregation of virus particles, or blocking virion cell uptake. Initial influenza studies in SCID mice revealed IgM and IgA prophylactically protected the host but were ineffective therapeutically against influenza virus, possibly due to insufficient access to all tissues where the virus is produced [81]. Another study found influenza neutralization depended on natural IgM and complement working in concert to aggregate the virion and coat the viral hemagglutinin receptor [82]. Additionally, the location of infection plays a role in virus neutralization. This is observed with Poliovirus, where the primary infection site is in the gastrointestinal tract. Mucosal IgA is the main antibody to block infection, but it also elicits IgM and IgG to prevent spreading to the central nervous system [83]. While multiple studies reveal NAb production plays an important role in combating viral infections, most require other mechanisms of the immune system to fully neutralize the virus, such as complement activation.
To enhance the immune response, NAbs distribute viruses to secondary lymphoid organs, as seen in VSV, lymphocytic choriomeningitis virus (LCMV), and vaccinia virus (vacc-WR strain) infection [84]. Using antibody-deficient mice, infection with these viruses resulted in 10 to 100 times lower viral titers in secondary lymphoid organs compared to antibody-competent mice [84]. Corroborating these results, NAbs reduced viral organ titers in the kidney, liver, and brain, but increased virus titers in the spleen, thereby preventing vital organs from viral infection [85]. NAbs activate the complement system via the classical pathway. Binding of the antibody Fc portion to C1q activates the complement cascade. Antibody binding to multiple epitopes on the surface of an antigen aggregates the antibody, enabling several C1q heads to bind with improved affinity [86]. Generally, NAbs are more effective for cytopathogenic viruses rather than non-cytopathogenic viruses. Research demonstrated purified human C1, C2, C3, and C4 required the presence of IgM to fully neutralize cytopathic VSV to the same extent as normal human serum [87]. In contrast, other research failed to demonstrate that NAbs participate in VSV-induced antibody responses in wild-type mice [88], suggesting that mouse and human complement requirements differ. Other studies demonstrated a role of complement receptors in viral protection. IgM response to VSV as well as poliomyelitis virus and recombinant vaccinia virus in mice was dependent on CR3 and CR4-expressing macrophages [89]. Neutralizing IgM and IgG responses were independent of CR2-mediated B-cell stimulation with live VSV in mice; however, CR2 was important for B-cell IgG class switching in mice immunized with nonreplicating antigens [89]. On the contrary, NAbs are typically insufficient in responding against poor or non-cytopathic viruses, such as LCMV, because somatic hypermutation is required to rid the host of these viruses [90]. These data demonstrate the complexity and variability of NAbs in response to viral infections.
Natural Antibody Viral Recognition and Viral Escape
Despite not being as effective as humoral antibody responses, NAbs play an important role in clearing pathogens. Viral targets for NAb neutralization are less clear; however, researchers are making headway exploring this area. Antibodies potentially recognize virions that incorporate cell membrane components during the budding process. One study showed natural IgM potentially targets the respiratory syncytial virus (RSV) envelope proteins, including the glycosylated fusion and attachment proteins [91]. This was based on increased newborn RSV-IgM antigen presenting cells and plasma RSV-IgM titers, demonstrating the presence of NAbs since IgM does not cross the placental barrier [91]. These results were contradicted in another study that showed the antibodies that recognize an RSV epitope have little to no poly-reactivity, thus suggesting they are distinct from IgM NAbs [92]. However, it is difficult to test every possible reactivity. Other studies demonstrated LCMV glycoprotein pseudotyped VSV complement lysis was dependent on NAbs recognizing xenoantigens such as galactose-α-(1,3)-galactose or N-glycolylneuraminic acid expressed on nonhuman cell lines [93]. These studies demonstrate the complexity and limited information available on the mechanisms of viral recognition by NAbs and exploring this further would greatly enhance vaccine development.
Viruses such as HIV have high mutation rates, resulting in increased adaptability and improved immune evasion. NAbs may induce long-lasting internalization of the main HIV co-receptor from cell membranes thereby possibly inhibiting HIV infection [94]. Viruses also avoid antibody detection by latent infection. The herpes virus remains hidden and expresses a small of number of genes to become non-immunogenic [95], possibly resulting in a decreased NAb response. Finally, viruses such as Hepatitis B virus (HBV) remain persistent in patients by up-regulating multiple inhibitory receptors and down-regulating antigen presentation genes in B-cells, causing a lack of antibody production towards HBV [96]. Perhaps this viral mechanism targets NAb producing B-1 cells and would be interesting to explore further.
As discussed, NAbs play a crucial role in viral clearance through neutralization, antigen recruitment to secondary lymphoid organs, and complement activation. Viral recognition by NAbs is less clear but research is making headway in this field. Even though NAbs have limited capabilities in clearing viral infections solely on their own, they play an important and intricate role in linking the innate and adaptive immune system as well as complement activation. It is not completely known how viruses inhibit NAbs and further research is needed to explore these multi-functional antibodies of the innate immune system.
The coagulation pathway
The proteolytic coagulation cascade maintains homeostasis in response to blood vessel injury. Rupture of blood vessels activates coagulation that together with platelet mediated hemostasis stops bleeding by forming a blood-obstructing platelet plug at the site of endothelial injury. The coagulation cascade is described as “waterfall sequence for intrinsic blood clotting” [97] because upon activation of the pathway, various proteins interact with their substrates and are converted to enzymatic active forms in a sequential manner. There are two accepted converging coagulation pathways; the extrinsic (tissue factor pathway) and intrinsic (contact activation) pathways, which converge at activation of factor X. Many viruses downregulate the coagulation regulators and/or inhibit fibrinolysis. Viruses also activate coagulation directly by damaging endothelial cells during infection [98]. The cascade may also be indirectly activated through inflammation or viral protein mimics of coagulation proteins. Some of these mechanisms will be discussed in this section.
Some viruses have evolved to induce or mimic host responses. Influenza virus infection stimulates production of large amounts of platelets which increases disease severity and mortality [99]. In contrast, dengue viruses produce proteins mimicking platelets and endothelial proteins to induce cross-reactive autoantibodies capable of inducing coagulation [100, 101]. Coagulation factors such as Xa, IXa, and II are serine proteases just like some dengue virus proteins such as prM, E and NS1. This likely results in the induction of autoantibodies capable of cross-reacting with the above-mentioned coagulation factors [101].
During blood vessel injury, the extrinsic pathway is activated first, and this response is enhanced by activation of the intrinsic pathway. Viruses such as herpes virus [102], dengue virus [103, 104], HIV [105, 106] and Ebola virus [107, 108] activate coagulation via the extrinsic pathway. Other viruses such as HIV [105, 106, 109] and influenza virus [110–112] activate coagulation via both the intrinsic and coagulation pathways.
Viral subversion of the extrinsic and intrinsic pathways
In the extrinsic pathway, damaged endothelial cells release tissue factor (TF) [113], which binds to circulating factor VII (VII) to form a complex TF-VIIa. Herpes virus also causes endothelial cell damage [114, 115] that induces TF in a manner not requiring viral replication [116]. Likewise, HIV and Ebola virus infections induce TF in the bloodstream and within monocytes and macrophages [105, 106, 117–119]. Without directly activating TF, dengue virus upregulates TF receptors to induce vascular cell adhesion molecule 1 expression, leading to endothelial cell activation [120]. Finally, TF increases morbidity and mortality during influenza virus infections, although the mechanism is not clearly understood [121, 122]. The TF-VIIa complex cleaves factor X to activate Xa, but viruses may alter this process as well [123]. For example, herpes viruses activate factor X even before they infect cells using the procoagulant phosphatidylserine with endogenous processes [124–126].
Subclinical levels of viral activation of coagulation increases coagulation factor expression and/or clinical activation resulting in disseminated intravascular coagulopathy (DIC). DIC occurs when numerous micro thrombi form within blood vessels, eventually depleting coagulation factors and resulting in the inability to form clots. This phenomenon is present in viral infections such as in influenza infection [127]. Blood clot formation in DIC is dependent on the extrinsic pathway with TF expressed as a membrane-bound protein on mononuclear cells [128].
The intrinsic pathway forms independently of plasma extraneous components by endothelial surface damage activating factor XII (Hageman factor) to XIIa. Herpes virus infection induces such surface damage and subsequent activation of coagulation [114, 129, 130]. In addition, HIV and influenza infections increase Von Willebrand factor, a measure of endothelial cell damage [109, 110, 131, 132]. Factor XIIa effects the sequential activation of factors XI (PTA) and then IX (Christmas factor) to active forms XIa and IXa respectively [133]. Certain adenoviruses such as adenovirus strain 5 (Adv5) and Adv31 require factors IX or X to efficiently bind during infection [134].
The knob fiber domains of Adv5, Adv18 and Adv31, but not Adv12, interact with complement C4-binding protein as well as factor IX, enabling viral uptake via liver hepatocytes, an understanding that can be exploited when thinking about tissues to target with adenovirus vectors [135]. The Adv5 capsid protein hexon binds to coagulation factor X, thereby enhancing viral entry into hepatocytes [134, 136–139] and activating the innate response during Adv infection [140]. The Adv-factor X interaction is a target for therapeutic agents [135]. When Adv5 is being used as a vector, factor X is essential to ensure viral transduction to the liver as it shields the virus from attack by the classical pathway of the complement system [141]. Adv35 which is an Adv5 containing fibers from Adv B serotype 35, bind with lower affinity to factor X and may thus be better candidates for selective transfer of genes compared to Adv5 alone [142].
Common Pathway
The two coagulation pathways converge into a common pathway upon factor X activation. Factor Xa, through its interaction with cofactor Va on membrane surfaces, cleaves prothrombin, generating thrombin. In a feedback mechanism, thrombin activates factor IX to produce large amounts of thrombin that is sufficient to convert fibrinogen to fibrin. Herpes viruses use fibrin to camouflage their surfaces, thereby reducing their recognition by the immune system [129]. Additionally, thrombin activates protein C and in the presence of protein S leads to the activation of factors V and XIII. Thrombin enhances herpes virus infectivity [143] and is important during Adv hepatic transduction [137]. Additionally, HIV positive individuals are deficient of certain proteins such as protein C, protein S and platelets, denoting a pro-coagulant state [144–146]. However, it is not known if the deficiency is a result of lack of protein production or accelerated consumption during HIV infection. Finally, thrombin cleaves fibrinogen to soluble fibrin which is crosslinked with factor XIIIa to form the fibrin clot. During Hepatitis infection, the liver produces fibrinogen and fibrin resulting in their deposition and leading to clot formation [147]. This is confirmed by an increase of thrombin receptors on hepatic stellate cells [148, 149].
In conclusion, activation of the coagulation system during viral infections can occur as a result of direct endothelial cell damage or blood vessel damage. This ultimately leads to formation of a blood clot at the site of injury or formation of multiple thrombi and eventual depletion of blood clotting factors. Additionally, some viruses activate either the extrinsic pathway, intrinsic pathway or the common pathway. The result of activation of the coagulation cascade may increase or decrease coagulation factors depending on the viral infection.
Figure 1: Complement and virus interactions.
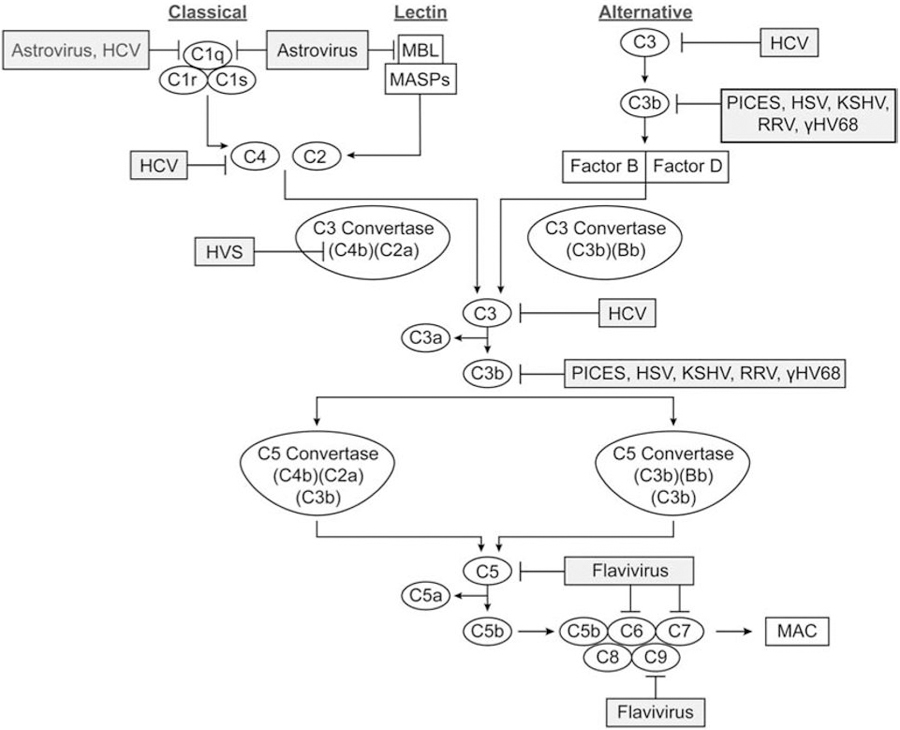
Complement proteins of the three initiation and terminal pathways are indicated in ovals. Virus inhibition of specific complement proteins is indicated by shaded rectangles. MBL indicates Mannose Binding lectin, MASPs indicates MBL associated serine proteases, and MAC indicates membrane attack complex.
Figure 2: Viruses utilize complement regulation to their advantage.
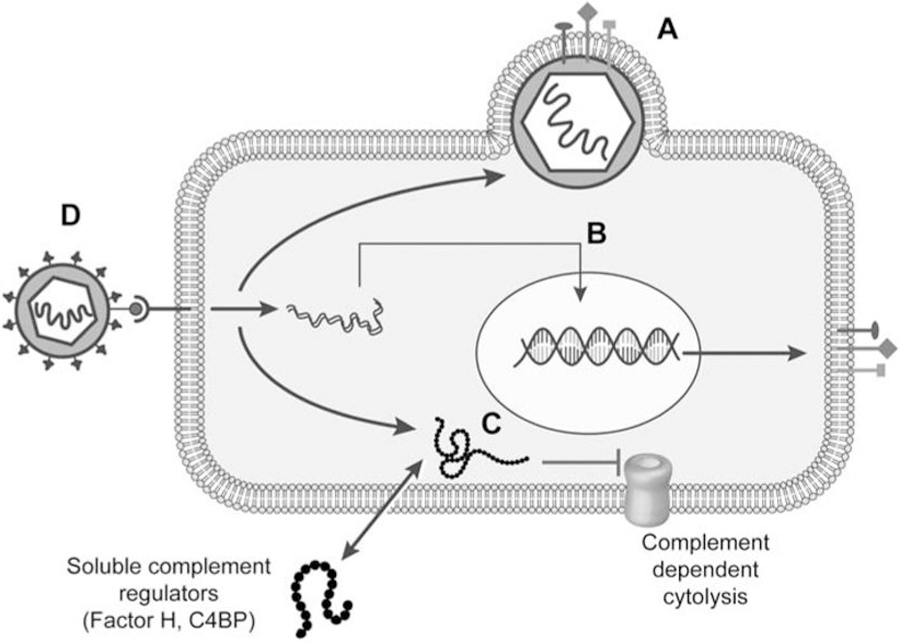
(A)Viruses sequester host complement regulatory molecules to incorporate them on the virion surface. (B) Viruses also upregulate expression of host complement regulatory molecules to avoid complement mediated cell lysis. (C) Viruses encode proteins that bind and modulate complement regulatory molecules or mimic regulator function inhibiting complement dependent cytolysis. (D) Host complement regulatory molecules may serve as cellular receptors for virus attachment.
Figure 3. Roles of NAbs (IgM) during homeostasis and virus infection.
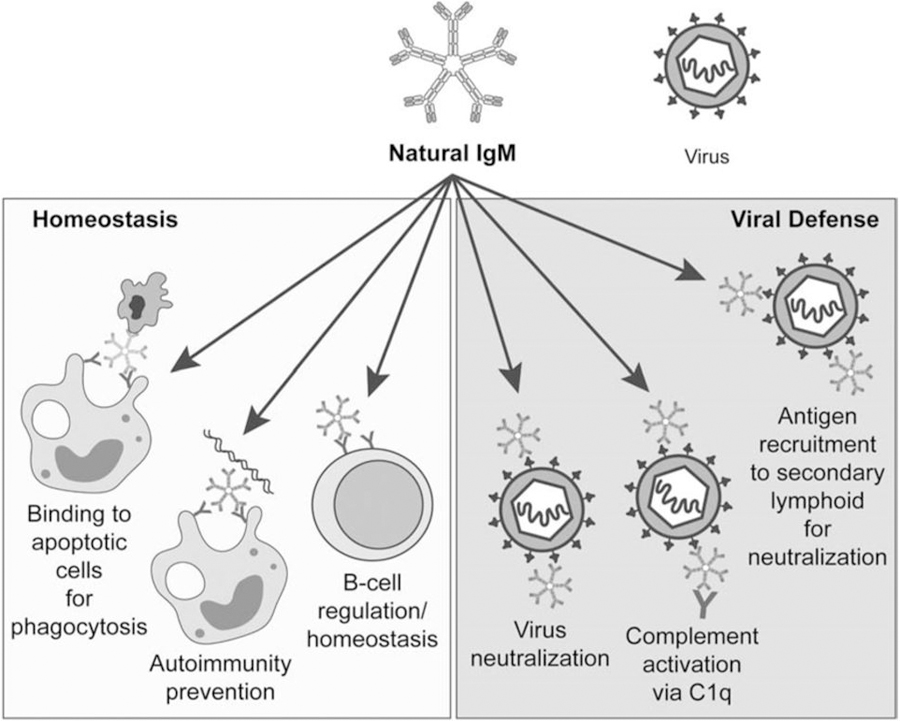
During homeostasis, NAbs bind to apoptotic cells for phagocytosis, prevent autoimmunity through the binding and clearance of damaged proteins such as double stranded DNA, and aid in the regulation of B-cells. During virus infection, NAbs neutralize viruses, activate the complement system, or recruit the virus to secondary lymphoid organs.
Figure 4: Coagulation virus interactions.
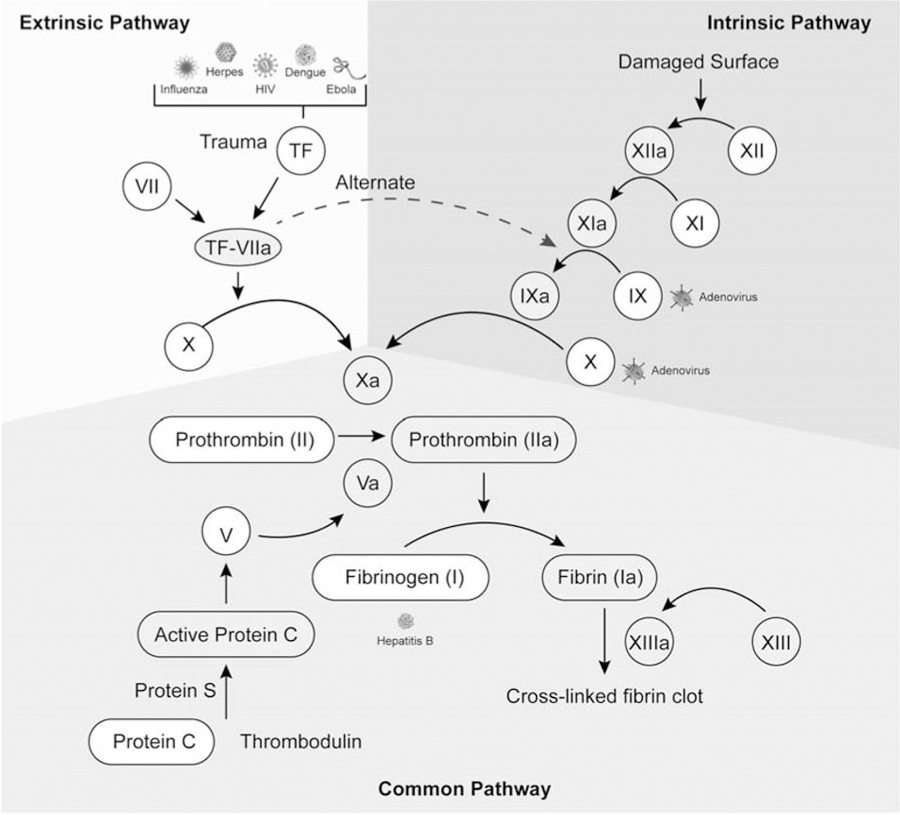
Extrinsic, intrinsic and common pathways are indicated by shaded areas. Specific viruses are located next to the affected coagulation factor. The gray and white circular shapes represent inactive and active coagulation factors respectively.
Highlights.
Viruses inhibit complement activation by protease degradation of initiators.
Viruses produce complement mimics or incorporate regulators into the virion to inhibit complement.
Natural antibodies aid in viral clearance but viruses also inhibit natural antibody recognition
Viruses may evade the coagulation pathway or even utilize it in pathogenesis.
Acknowledgements
We thank Mal Hoover for assistance with the figures. This work was supported by the K-INBRE (NIH P20 GM103418), the H.L Snyder Medical Foundation, the Johnson Cancer Research Center, Kansas State University and the Office of the Assistant Secretary of Defense for Health Affairs, through the Defense Medical Research and Development Program under Award No. W81XWH-18–1-0716. Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense or the National Institutes of Health.
Abbreviations:
- Adv5 (used for strains 5, 18, 31, and 21)
Adenovirus strain 5
- C4BP
C4b binding protein
- CR1 (used for 1, 2, 3 and 4)
Complement receptor 1
- DIC
Disseminated intravascular coagulopathy
- EEV
Extracellular enveloped virons
- gC
Glycoprotein C
- HBV
Hepatitis B virus
- HCV
Hepatitis C virus
- HSV
Herpes simplex virus
- HVS
Herpesvirus saimiri
- HIV
Human immunodeficiency virus
- HCMV
Human cytomegalovirus
- (HTLV-1)
Human T cell leukemia Type I virus
- IBV
Infectious bronchitis virus
- PICES
Poxviral inhibitors of complement enzymes
- IMV
Intracellular mature virons
- (KSHV)
Kaposi’s sarcoma-associated herpesvirus
- KCP
KSHV complement control protein
- LCMV
Lymphocytic choriomeningitis virus
- MBL
Mannose binding lectin
- MAC
Membrane attack complex
- MuV
Mumps virus
- γHV68
Murine gammaherpesvirus 68
- NAb
Natural antibody
- NS1
Non-structural protein 1
- PIV5
Parainfluenza virus-5
- RSV
Respiratory syncytial virus
- RRV
Rhesus rhadinovirus
- TF
Tissue factor
- VSV
Vesicular stomatitis virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bonaparte RS, Hair PS, Banthia D, Marshall DM, Cunnion KM, Krishna NK, Human Astrovirus Coat Protein Inhibits Serum Complement Activation via C1, the First Component of the Classical Pathway, J. Virol 82 (2008) 817–827. 10.1128/JVI.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hair PS, Gronemus JQC, Katrina B, Salvi VP, Cunnion KM, Thielens NM, Arlaud GJ, Rawal N, Krishna NK, Human Astrovirus Coat Protein Binds C1q and MBL and Inhibits the Classical and Lectin Pathways of Complement Activation, Mol. Immunol 47 (2010) 792–798. 10.1016/j.molimm.2009.10.006. [DOI] [PubMed] [Google Scholar]
- [3].Gronemus JQ, Hair PS, Crawford KB, Nyalwidhe JO, Cunnion KM, Krishna NK, Potent inhibition of the classical pathway of complement by a novel C1q-binding peptide derived from the human astrovirus coat protein, Mol. Immunol 48 (2010) 305–313. 10.1016/j.molimm.2010.07.012. [DOI] [PubMed] [Google Scholar]
- [4].Sansonno D, Tucci FA, Ghebrehiwet B, Lauletta G, Peerschke EIB, Conteduca V, Russi S, Gatti P, Sansonno L, Dammacco F, Role of the Receptor for the Globular Domain of C1q Protein in the Pathogenesis of HCV-Related Cryoglobulin Vascular Damage, J. Immunol 183 (2010) 6013–6020. 10.4049/jimmunol.0902038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yao ZQ, Eisen-Vandervelde A, Waggoner SN, Cale EM, Hahn YS, Direct Binding of Hepatitis C Virus Core to gC1qR on CD4+ and CD8+ T Cells Leads to Impaired Activation of Lck and Akt, J. Virol 78 (2004) 6409–6419. 10.1128/JVI.78.12.6409-6419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS, Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation, J. Clin. Invest 106 (2000) 1239–1249. 10.1172/JCI10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R, Transcriptional Repression of C4 Complement by Hepatitis C Virus Proteins, J. Virol 85 (2011) 4157–4166. 10.1128/JVI.02449-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim H, Meyer K, Di Bisceglie AM, Ray R, Inhibition of C3 Convertase Activity by Hepatitis C Virus as an Additional Lesion in the Regulation of Complement Components, PLoS ONE 9 (2014) e101422 10.1371/journal.pone.0101422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Singh AK, Mullick J, Bernet J, Sahu A, Functional Characterization of the Complement Control Protein Homolog of Herpesvirus Saimiri J. Biol. Chem 281 (2006) 23119–23128. 10.1074/jbc.M603085200. [DOI] [PubMed] [Google Scholar]
- [10].Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, Raya R, Hepatitis C Virus Proteins Inhibit C3 Complement Production, J. Virol 86 (2012) 2221–2228. 10.1128/JVI.06577-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liszewski MK, Leung MK, Hauhart R, Buller RML, Bertram P, Wang X, Rosengard AM, Kotwal GJ, Atkinson JP, Structure and Regulatory Profile of the Monkeypox Inhibitor of Complement: Comparison to Homologs in Vaccinia and Variola and Evidence for Dimer Formation, J. Immunol 176 (2006) 3725–3734. 10.4049/jimmunol.176.6.3725. [DOI] [PubMed] [Google Scholar]
- [12].Moulton EA, Bertram P, Chen N, Buller RML, Atkinson JP, Ectromelia Virus Inhibitor of Complement Enzymes Protects Intracellular Mature Virus and Infected Cells from Mouse Complement J. Virol 84 (2010) 9128–9139. 10.1128/JVI.02677-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liszewski MK, Bertram P, Leung MK, Hauhart R, Zhang L, Atkinson JP, Smallpox inhibitor of complement enzymes (SPICE): Regulation of complement activation on cells and mechanism of its cellular attachment, J. Immunol 181 (2008) 4199–4207. 10.4049/jimmunol.181.6.4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rosengard AM, Liu Y, Nie Z, Jimenez R, Variola virus immune evasion design: Expression of a highly efficient inhibitor of human complement, PNAS 99 (2002) 8808–8813. 10.1073/pnas.112220499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yadav VN, Pyaram K, Mullick J, Sahu A, Identification of Hot Spots in the Variola Virus Complement Inhibitor (SPICE) for Human Complement Regulation, J. Virol 82 (2008) 3283–3293. 10.1128/JVI.01935-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mullick J, Bernet J, Panse Y, Hallihosur S, Singh AK, Sahu A, Identification of Complement Regulatory Domains in Vaccinia Virus Complement Control Protein J. Virol 79 (2005) 12382–12393. 10.1128/JVI.79.19.12382-12393.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McKenzie R, Kotwal GJ, Moss B, Hammer CH, Frank MM, Regulation of Complement Activity by Vaccinia Virus Complement-Control Protein, J. Infect. Dis 166 (1992) 1245–1250. 10.1093/infdis/166.6.1245. [DOI] [PubMed] [Google Scholar]
- [18].Sahu A, Isaacs SN, Soulika AM, Lambris JD, Interactions of Vaccinia Virus Complement Control Protein with Human Complement Proteins: Factor I-Mediated Degradation of C3b to iC3b1 Inactivates the Alternative Complement Pathway, J. Immunol 160 (1998) 5596–5604. [PubMed] [Google Scholar]
- [19].Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, Tzekou A, Volokhina E, Granneman JCM, Hauhart R, Bertram P, Liszewski MK, Atkinson JP, Lambris JD, Gros P, Regulators of complement activity mediate inhibitory mechanisms through a common C3b‐ binding mode, EMBO J 35 (2016) 1133–1149. 10.15252/embj.201593673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huemer HP, Wang Y, Garred P, Koistinen V, Oppermann S, Herpes simplex virus glycoprotein C: molecular mimicry of complement regulatory proteins by a viral protein, Immunology 79 (1993) 639–647. [PMC free article] [PubMed] [Google Scholar]
- [21].McNearney TA, Odell C, Holers VM, Spear PG, Atkinson JP, Herpes Simplex Virus Glycoproteins gC-1 and gC-2 Bind to the Third Component of Complement and Provide Protection Against Complement-Mediated Neutralization of Viral Infectivity, J. Exp. Med 166 (1987). 10.1084/jem.166.5.1525Published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kostavasili I, Sahu A, Friedman HM, Eisenberg RJ, Cohen GH, Lambris JD, Mechanism of complement inactivation by glycoprotein C of herpes simplex virus, J. Immunol 158 (1997) 1763–1771. [PubMed] [Google Scholar]
- [23].Friedman HM, Cohen GH, Eisenberg RJ, Seidel CA, Cines DB, Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells, Nature 309 (1984) 633–635. https://doi-org.er.lib.k-state.edu/10.1038/309633a0. [DOI] [PubMed] [Google Scholar]
- [24].Eisenberg RJ, Ponce de Leon M, Friedman HM, Fries LF, Frank MM, Hastings JC, Cohen GH, Complement component C3b binds directly to purified glycoprotein C of herpes simplex virus types 1 and 2, Microb. Pathog 3 (1987) 423–435. [DOI] [PubMed] [Google Scholar]
- [25].Kubota Y, Gaither TA, Cason J, O’Shea JJ, Lawley TJ, Characterization of the C3 receptor induced by herpes simplex virus type 1 infection of human epidermal, endothelial, and A431 cells, J. Immunol 138 (1987) 1137–1142. [PubMed] [Google Scholar]
- [26].Spiller OB, Blackbourn DJ, Mark L, Proctor DG, Blom AM, Functional Activity of the Complement Regulator Encoded by Kaposi’s Sarcoma-associated Herpesvirus, J. Biol. Chem 278 (2003) 9283–9289. 10.1074/jbc.M211579200. [DOI] [PubMed] [Google Scholar]
- [27].Spiller OB, Mark L, Blue CE, Proctor DG, Aitken JA, Blom AM, Blackbourn DJ, Dissecting the Regions of Virion-Associated Kaposi’s Sarcoma-Associated Herpesvirus Complement Control Protein Required for Complement Regulation and Cell Binding, J. Virol 80 (2006) 4068–4078. 10.1128/JVI.80.8.4068-4078.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mark L, Spiller OB, Okroj M, Chanas S, Aitken JA, Wong SW, Damania B, Blom AM, Blackbourn DJ, Molecular Characterization of the Rhesus Rhadinovirus (RRV) ORF4 Gene and the RRV Complement Control Protein It Encodes, J. Virol 81 (2007) 4166–4176. 10.1128/JVI.02069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kapadia SB, Molina H, van Berkel V, Speck SH, Virgin HW, Murine Gammaherpesvirus 68 Encodes a Functional Regulator of Complement Activation, J. Virol 73 (1999) 7658–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Conde JN, da Silva EM, Allonso D, Coelho DR, Andrade I.d.S., de Medeiros LN, Menezes JL, Barbosa AS, Mohana-Borges R, Inhibition of the Membrane Attack Complex by Dengue Virus NS1 through Interaction with Vitronectin and Terminal Complement Proteins, J. Virol 90 (2016) 9570–9581. 10.1128/JVI.00912-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, Atkinson JP, Antagonism of the complement component C4 by flavivirus nonstructural protein NS1, J. Exp. Med 207 (2010). 10.1084/jem.20092545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schvartz I, Seger D, Shaltiel S, Vitronectin, Int. J. Biochem. Cell Biol 31 (1999) 539–544. 10.1016/j.biocel.2010.09.004. [DOI] [PubMed] [Google Scholar]
- [33].Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, Jairungsri A, Kanlaya R, Tangthawornchaikul N, Puttikhunt C, Pattanakitsakul S.-n., Yenchitsomanus P.-t., Mongkolsapaya J, Kasinrerk W, Sittisombut N, Husmann M, Blettner M, Vasanawathana S, Bhakdi S, Malasit P, Vascular Leakage in Severe Dengue Virus Infections: A Potential Role for the Nonstructural Viral Protein NS1 and Complement J. Infect. Dis 193 (2006) 1078–1088. 10.1086/500949. [DOI] [PubMed] [Google Scholar]
- [34].Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, Smith GL, Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope, PNAS 95 (1998) 7544–7549. 10.1073/pnas.95.13.7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Johnson JB, Borisevich V, Rockx B, Parks GD, A Novel Factor I Activity in Nipah Virus Inhibits Human Complement Pathways through Cleavage of C3b, J. Virol 89 (2014) 989–998. 10.1128/JVI.02427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, Palese P, Cellular Proteins in Influenza Virus Particles, PLoS Pathog 4 (2008). 10.1371/journal.ppat.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wei Y, Ji Y, Guo H, Zhi X, Han S, Zhang Y, Gao Y, Chang Y, Yan D, Li K, Liu DX, Sun S, CD59 Association with Infectious Bronchitis Virus Particles Protects against Antibody-Dependent Complement-Mediated Lysis, J. Gen. Virol 98 (2017) 2725–2730. 10.1099/jgv.0.000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, Diamond MS, Atkinson JP, Ray R, Hepatitis C Virus Infection Upregulates CD55 Expression on the Hepatocyte Surface and Promotes Association with Virus Particles, J. Virol 87 (2013) 7902–7910. 10.1128/JVI.00917-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ejaz A, Steinmann E, Bánki Z, Anggakusuma, Khalid S, Lengauer S, Wilhelm C, Zoller H, Schloegl A, Steinmann J, Grabski E, Kleines M, Pietschmann T, Stoiber H, Specific Acquisition of Functional CD59 but Not CD46 or CD55 by Hepatitis C Virus, PLoS ONE 7 (2012) e45770 10.1371/journal.pone.0045770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Amet T, Ghabril M, Chalasani N, Byrd D, Hu N, Grantham A, Liu Z, Qin X, He JJ, Yu Q, CD59 Incorporation Protects Hepatitis C Virus Against Complement-Mediated Destruction, Hepatology 55 (2012) 354–363. 10.1002/hep.24686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Spear GT, Lurain NS, Parker CJ, Ghassemi M, Payne GH, Saifuddin M, Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV), J. Immunol 155 (1995) 4376–4381. [PubMed] [Google Scholar]
- [42].Johnson JB, Lyles DS, Alexander-Miller MA, Parks GD, Virion-Associated Complement Regulator CD55 Is More Potent than CD46 in Mediating Resistance of Mumps Virus and Vesicular Stomatitis Virus to Neutralization, J. Virol 86 (2012) 9929–9940. 10.1128/JVI.01154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Biswas M, Johnson JB, Kumar SRP, Parks GD, Subbiah E, Incorporation of Host Complement Regulatory Proteins into Newcastle Disease Virus Enhances Complement Evasion, J. Virol 86 (2012) 12708–12716. 10.1128/JVI.00886-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rangaswamy US, Cotter CR, Cheng X, Jin H, Chen Z, CD55 is a key complement regulatory protein that counteracts complement-mediated inactivation of Newcastle Disease Virus, J. Gen. Virol 97 (2016) 1765–1770. 10.1099/jgv.0.000498. [DOI] [PubMed] [Google Scholar]
- [45].Li Y, Parks GD, Relative Contribution of Cellular Complement Inhibitors CD59, CD46, and CD55 to Parainfluenza Virus 5 Inhibition of Complement-Mediated Neutralization, Viruses 10 (2018). 10.3390/v10050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li Y, Johnson JB, Parks GD, Parainfluenza Virus 5 Upregulates CD55 Expression to Produce Virions with Enhanced Resistance to Complement-Mediated Neutralization, Virology 497 (2016) 305–313. 10.1016/j.virol.2016.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Montefiori DC, Cornell RJ, Zhou JY, Zhou JT, Hirsch VM, Johnson PR, Complement Control Proteins, CD46, CD55, and CD59, as Common Surface Constituents of Human and Simian Immunodeficiency Viruses and Possible Targets for Vaccine Protection, Virology 205 (1994) 82–92. 10.1006/viro.1994.1622. [DOI] [PubMed] [Google Scholar]
- [48].Marschang P, Sodroski J, Würzner R, Dierich MP, Decay-Accelerating Factor (CD55) Protects Human Immunodeficiency Virus Type 1 from Inactivation by Human Complement, Eur. J. Immunol 25 (1995) 285–290. 10.1002/eji.1830250147. [DOI] [PubMed] [Google Scholar]
- [49].Stoiber H, Pinter C, Siccardi AG, Clivio A, Dierichm MP, Efficient Destruction of Human Immunodeficiency Virus in Human Serum by Inhibiting the Protective Action of Complement Factor H and Decay Accelerating Factor (DAF, CD55), J. Exp. Med 183 (1996) 307–310. 10.1084/jem.183.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Saifuddin M, Hedayati T, Atkinson JP, Holguin MH, Parker CJ, Spear GT, Human Immunodeficiency Virus Type 1 Incorporates Both Glycosyl Phosphatidylinositol-Anchored CD55 and CD59 and Integral Membrane CD46 at Levels That Protect from Complement-Mediated Destruction, J. Gen. Virol 78 (1997) 1907–1911. 10.1099/0022-1317-78-8-1907. [DOI] [PubMed] [Google Scholar]
- [51].Shan C, Zhang S, Cui W, You X, Kong G, Du Y, Qiu L, Ye L, Zhang X, Hepatitis B virus X protein activates CD59 involving DNA binding and let-7i in protection of hepatoma and hepatic cells from complement attack, Carcinogenesis 32 (2011) 1190–1197. 10.1093/carcin/bgr106. [DOI] [PubMed] [Google Scholar]
- [52].Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP, Binding of Flavivirus Non-structural Protein NS1 to C4b Binding Protein Modulates Complement Activation, J. Immunol 187 (2011) 424–433. 10.4049/jimmunol.1100750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, Atkinson JP, Diamond MS, West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H, PNAS 103 (2006) 19111–19116. 10.1073/pnas.0605668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ojha H, Panwar HS, Gorham RD, Morikis D, Sahu A, Viral regulators of complement activation: Structure, function and evolution, Mol. Immunol 61 (2014) 89–99. 10.1016/j.molimm.2014.06.004. [DOI] [PubMed] [Google Scholar]
- [55].Lubinski J, Wang L, Mastellos D, Sahu A, Lambris JD, Friedman HM, In Vivo Role of Complement-interacting Domains of Herpes Simplex Virus Type 1 Glycoprotein gC, J. Immunol 176 (1999) 3725–3734. 10.1084/jem.190.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bramley JC, Davies A, Lachmann PJ, Herpes virus saimiri CD59 - baculovirus expression and characterisation of complement inhibitory activity, Biochm. Soc. Trans 25 (1997) 354S–354S. 10.1042/bst025354s. [DOI] [PubMed] [Google Scholar]
- [57].Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D, Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus, J. Virol 67 (1993) 6025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Campadelli-Fiume G, Virus receptor arrays, CD46 and human herpesvirus 6, Trends Microbiol 8 (2000) 436–438. 10.1016/s0966-842x(00)01804-7. [DOI] [PubMed] [Google Scholar]
- [59].Marttila M, Persson D, Gustafsson D, Liszewski MK, Atkinson JP, Wadell G, Arnberg N, CD46 is a cellular receptor for all species B adenoviruses except types 3 and 7, J. Virol 79 (2005) 14429–14436. 10.1128/JVI.79.22.14429-14436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Maurer K, Krey T, Moennig V, Thiel H-J, Rümenapf T, CD46 is a cellular receptor for bovine viral diarrhea virus, J. Virol 78 (2004) 1792–1799. 10.1128/jvi.78.4.1792-1799.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Karnauchow TM, Tolson DL, Harrison BA, Altman E, Lublin DM, Dimock K, The HeLa Cell Receptor for Enterovirus 70 Is Decay-Accelerating Factor (CD55), J. Virol 70 (1996) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Martino TA, Petric M, Brown M, Aitken K, Gauntt CJ, Richardson CD, Chow LH, Liu PP, Cardiovirulent Coxsackieviruses and the Decay-Accelerating Factor (CD55) Receptor, Virology 244 (1998) 302–314. 10.1006/viro.1998.9122. [DOI] [PubMed] [Google Scholar]
- [63].Abou-El-Hassan H, Zaraket H, Viral-Derived Complement Inhibitors: Current Status and Potential Role in Immunomodulation, Exp. Biol. Med 242 (2017) 397–410. 10.1177/1535370216675772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Reynolds DN, Smith SA, Zhang Y-P, Lahiri DK, Morassutti DJ, Shields CB, Kotwal GJ, Vaccinia Virus Complement Control Protein Modulates Inflammation Following Spinal Cord Injury, Ann. N. Y. Acad. Sci 1010 (2003) 534–539. 10.1196/annals.1299.099. [DOI] [PubMed] [Google Scholar]
- [65].Miller CG, Shchelkunov SN, Kotwal GJ, The Cowpox Virus-Encoded Homolog of the Vaccinia Virus Complement Control Protein Is an Inflammation Modulatory Protein, Virology 229 (1997) 126–133. 10.1006/viro.1996.8396. [DOI] [PubMed] [Google Scholar]
- [66].Boes M, Role of natural and immune IgM antibodies in immune responses, Mol. Immunol 37 (2000) 1141–1149. 10.1016/s0161-5890(01)00025-6. [DOI] [PubMed] [Google Scholar]
- [67].Antin JH, Emerson SG, Martin P, Gadol N, Ault KA, Leu-1+ (CD5+) B cells. A major lymphoid subpopulation in human fetal spleen: phenotypic and functional studies, J. Immunol 136 (1986) 505–510. [PubMed] [Google Scholar]
- [68].Kantor AB, Stall AM, Adams S, Herzenberg LA, Herzenberg LA, Differential development of progenitor activity for three B-cell lineages, Proc. Natl. Acad. Sci. USA 89 (1992) 3320–3324. 10.1073/pnas.89.8.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kasaian MT, Ikematsu H, Casali P, Identification and analysis of a novel human surface CD5- B lymphocyte subset producing natural antibodies, J. Immunol 148 (1992) 2690–2702. [PMC free article] [PubMed] [Google Scholar]
- [70].Ehrenstein MR, Notley CA, The importance of natural IgM: scavenger, protector and regulator, Nat. Rev. Immunol 10 (2010) 778–786. 10.1038/nri2849. [DOI] [PubMed] [Google Scholar]
- [71].Eisen HN, Affinity enhancement of antibodies: how low-affinity antibodies produced early in immune responses are followed by high-affinity antibodies later and in memory B-cell responses, Cancer Immunol. Res 2 (2014) 381–392. 10.1158/2326-6066.CIR-14-0029. [DOI] [PubMed] [Google Scholar]
- [72].Kantor AB, Merrill CE, Herzenberg LA, Hillson JL, An unbiased analysis of V(H)-D-J(H) sequences from B-1a, B-1b, and conventional B cells, J. Immunol 158 (1997) 1175–1186. [PubMed] [Google Scholar]
- [73].Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T, Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme, Cell 102 (2000) 553–563. 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- [74].Chen Y, Park YB, Patel E, Silverman GJ, IgM antibodies to apoptosis-associated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells, J. Immunol 182 (2009) 6031–6043. 10.4049/jimmunol.0804191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chen Y, Khanna S, Goodyear CS, Park YB, Raz E, Thiel S, Gronwall C, Vas J, Boyle DL, Corr M, Kono DH, Silverman GJ, Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis, J. Immunol 183 (2009) 1346–1359. 10.4049/jimmunol.0900948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kaveri SV, Silverman GJ, Bayry J, Natural IgM in immune equilibrium and harnessing their therapeutic potential, J. Immunol 188 (2012) 939–945. 10.4049/jimmunol.1102107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ehrenstein MR, Cook HT, Neuberger MS, Deficiency in serum immunoglobulin (Ig)M predisposes to development of IgG autoantibodies, J. Exp. Med 191 (2000) 1253–1258. 10.1084/jem.191.7.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gronwall C, Vas J, Silverman GJ, Protective Roles of Natural IgM Antibodies, Front. Immunol 3 (2012) 66 10.3389/fimmu.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Holodick NE, Rodriguez-Zhurbenko N, Hernandez AM, Defining Natural Antibodies, Front. Immunol 8 (2017) 872 10.3389/fimmu.2017.00872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Baumgarth N, Tung JW, Herzenberg LA, Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion, Springer Semin. Immunopathol 26 (2005) 347–362. 10.1007/s00281-004-0182-2. [DOI] [PubMed] [Google Scholar]
- [81].Palladino G, Mozdzanowska K, Washko G, Gerhard W, Virus-neutralizing antibodies of immunoglobulin G (IgG) but not of IgM or IgA isotypes can cure influenza virus pneumonia in SCID mice, J. Virol 69 (1995) 2075–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jayasekera JP, Moseman EA, Carroll MC, Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity, J. Virol 81 (2007) 3487–3494. 10.1128/JVI.02128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA, Vaccine-derived polioviruses and the endgame strategy for global polio eradication, Annu. Rev. Microbiol 59 (2005) 587–635. 10.1146/annurev.micro.58.030603.123625. [DOI] [PubMed] [Google Scholar]
- [84].Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, Zinkernagel RM, Control of early viral and bacterial distribution and disease by natural antibodies, Science 286 (1999) 2156–2159. 10.1126/science.286.5447.2156. [DOI] [PubMed] [Google Scholar]
- [85].Matter MS, Ochsenbein AF, Natural antibodies target virus-antibody complexes to organized lymphoid tissue, Autoimmun. Rev 7 (2008) 480–486. 10.1016/j.autrev.2008.03.018. [DOI] [PubMed] [Google Scholar]
- [86].Duncan AR, Winter G, The binding site for C1q on IgG, Nature 332 (1988) 738–740. 10.1038/332738a0. [DOI] [PubMed] [Google Scholar]
- [87].Beebe DP, Cooper NR, Neutralization of vesicular stomatitis virus (VSV) by human complement requires a natural IgM antibody present in human serum, J. Immunol 126 (1981) 1562–1568. [PubMed] [Google Scholar]
- [88].Hangartner L, Senn BM, Ledermann B, Kalinke U, Seiler P, Bucher E, Zellweger RM, Fink K, Odermatt B, Burki K, Zinkernagel RM, Hengartner H, Antiviral immune responses in gene-targeted mice expressing the immunoglobulin heavy chain of virus-neutralizing antibodies, Proc. Natl. Acad. Sci. USA 100 (2003) 12883–12888. 10.1073/pnas.2135542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ochsenbein AF, Pinschewer DD, Odermatt B, Carroll MC, Hengartner H, Zinkernagel RM, Protective T cell-independent antiviral antibody responses are dependent on complement, J. Exp. Med 190 (1999) 1165–1174. 10.1084/jem.190.8.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hangartner L, Zinkernagel RM, Hengartner H, Antiviral antibody responses: the two extremes of a wide spectrum, Nat. Rev. Immunol 6 (2006) 231–243. 10.1038/nri1783. [DOI] [PubMed] [Google Scholar]
- [91].Jans J, Pettengill M, Kim D, van der Made C, de Groot R, Henriet S, de Jonge MI, Ferwerda G, Levy O, Human newborn B cells mount an interferon-α/β receptor-dependent humoral response to respiratory syncytial virus, J. Allergy Clin. Immunol 139 (2017) 1997–2000. 10.1016/j.jaci.2016.10.032. [DOI] [PubMed] [Google Scholar]
- [92].Goodwin E, Gilman MSA, Wrapp D, Chen M, Ngwuta JO, Moin SM, Bai P, Sivasubramanian A, Connor RI, Wright PF, Graham BS, McLellan JS, Walker LM, Infants Infected with Respiratory Syncytial Virus Generate Potent Neutralizing Antibodies that Lack Somatic Hypermutation, Immunity 48 (2018) 339–349. 10.1016/j.immuni.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pipperger L, Koske I, Wild N, Müllauer B, Krenn D, Stoiber H, Wollmann G, Kimpel J, von Laer D, Bánki Z, Xenoantigen-Dependent Complement-Mediated Neutralization of Lymphocytic Choriomeningitis Virus Glycoprotein-Pseudotyped Vesicular Stomatitis Virus in Human Serum, J. Virol 93 (2019) e00567–00519. 10.1128/JVI.00567-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Venuti A, Pastori C, Lopalco L, The Role of Natural Antibodies to CC Chemokine Receptor 5 in HIV Infection, Front. Immunol 8 (2017) 1358 10.3389/fimmu.2017.01358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ, Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency, Proc. Natl. Acad. Sci. USA 105 (2008) 5453–5458. 10.1073/pnas.0711910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Poonia B, Ayithan N, Nandi M, Masur H, Kottilil S, HBV induces inhibitory FcRL receptor on B cells and dysregulates B cell-T follicular helper cell axis, Sci. Rep 8 (2018) 15296 10.1038/s41598-018-33719-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Earl WD, Oscar DR, Waterfall Sequence for Intrinsic Blood Clotting on JSTOR, 1964, pp. 1310–1312. [DOI] [PubMed]
- [98].10.1016/S0008-6363(02)00857-XLevi M, Keller TT, Van Gorp E, Ten Cate H, Infection and inflammation and the coagulation system, 2003, pp. 26–39. [DOI] [PubMed]
- [99].Lê VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin J-L, Adrian I, Errazuriz-Cerda E, Frasquilho S, Antunes L, Lina B, Bordet J-C, Jandrot-Perrus M, Ludwig S, Riteau B, Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis, Am. J. Respir. Crit. Care. Med 191 (2015) 804–819. 10.1164/rccm.201406-1031OC. [DOI] [PubMed] [Google Scholar]
- [100].Anderson R, Wang S, Osiowy C, Issekutz AC, Activation of endothelial cells via antibody-enhanced dengue virus infection of peripheral blood monocytes, J. Virol 71 (1997) 4226–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lin Y-S, Yeh T-M, Lin C-F, Wan S-W, Chuang Y-C, Hsu T-K, Liu H-S, Liu C-C, Anderson R, Lei H-Y, Molecular mimicry between virus and host and its implications for dengue disease pathogenesis, Exp. Biol. Med 236 (2011) 515–523. 10.1258/ebm.2011.010339. [DOI] [PubMed] [Google Scholar]
- [102].Gershom ES, Sutherland MR, Lollar P, Pryzdial ELG, Involvement of the contact phase and intrinsic pathway in herpes simplex virus-initiated plasma coagulation, J. Thromb. Haemost 8 (2010) 1037–1043. 10.1111/j.1538-7836.2010.03789.x. [DOI] [PubMed] [Google Scholar]
- [103].van Gorp EC, Minnema MC, Suharti C, Mairuhu AT, Brandjes DP, ten Cate H, Hack CE, Meijers JC, Activation of coagulation factor XI, without detectable contact activation in dengue haemorrhagic fever, Br. J. Haemat 113 (2001) 94–99. 10.1046/j.1365-2141.2001.02710.x. [DOI] [PubMed] [Google Scholar]
- [104].Lei H-Y, Yeh T-M, Liu H-S, Lin Y-S, Chen S-H, Liu C-C, Immunopathogenesis of dengue virus infection, J. Biomed. Sci 8 (2001) 377–388. 10.1007/BF02255946. [DOI] [PubMed] [Google Scholar]
- [105].Funderburg NT, Zidar DA, Shive C, Lioi A, Mudd J, Musselwhite LW, Simon DI, Costa MA, Rodriguez B, Sieg SF, Lederman MM, Shared monocyte subset phenotypes in HIV-1 infection and in uninfected subjects with acute coronary syndrome, Blood 120 (2012) 4599–4608. 10.1182/blood-2012-05-433946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Taylor FB, Chang A, Ruf W, Morrissey JH, Hinshaw L, Catlett R, Blick K, Edgington TS, Lethal E coli septic shock is prevented by blocking tissue factor with monoclonal antibody, Circ. Shock 33 (1991) 127–134. [PubMed] [Google Scholar]
- [107].Feldmann H, Volchkov VE, Volchkova VA, Klenk HD, The glycoproteins of Marburg and Ebola virus and their potential roles in pathogenesis, Arch. Virol. Suppl (1999) 159–169. 10.1007/978-3-7091-6425-9_11. [DOI] [PubMed]
- [108].10.1016/S0140-6736(10)60667-8Feldmann H, Geisbert TW, Ebola haemorrhagic fever, Lancet Publishing Group, 2011, pp. 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Goeijenbier M, Van Gorp ECM, Van Den Brand JMA, Stittelaar K, Bakhtiari K, Roelofs JJTH, Van Amerongen G, Kuiken T, Martina BEE, Meijers JCM, Osterhaus ADME, Activation of coagulation and tissue fibrin deposition in experimental influenza in ferrets, BMC Microbiol 14 (2014). 10.1186/1471-2180-14-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Akiyama R, Komori I, Hiramoto R, Isonishi A, Matsumoto M, Fujimura Y, H1N1 influenza (swine flu)-associated thrombotic microangiopathy with a markedly high plasma ratio of von Willebrand factor to ADAMTS13, Intern. Med 50 (2011) 643–647. 10.2169/internalmedicine.50.4620. [DOI] [PubMed] [Google Scholar]
- [111].Keller TT, van der Sluijs KF, de Kruif MD, Gerdes VEA, Meijers JCM, Florquin S, van der Poll T, van Gorp ECM, Brandjes DPM, Büller HR, Levi M, Effects on coagulation and fibrinolysis induced by influenza in mice with a reduced capacity to generate activated protein C and a deficiency in plasminogen activator inhibitor type 1, Circ. Res 99 (2006) 1261–1269. 10.1161/01.RES.0000250834.29108.1a. [DOI] [PubMed] [Google Scholar]
- [112].Shibamiya A, Hersemeyer K, Schmidt Wöll T, Sedding D, Daniel J-M, Bauer S, Koyama T, Preissner KT, Kanse SM, A key role for Toll-like receptor-3 in disrupting the hemostasis balance on endothelial cells, Blood 113 (2009) 714–722. 10.1182/blood-2008-02-137901. [DOI] [PubMed] [Google Scholar]
- [113].Mackman N, Role of Tissue Factor in Hemostasis, Thrombosis, and Vascular Development, Arterioscler. Thromb. Vasc. Biol 24 (2004) 1015–1022. 10.1161/01.ATV.0000130465.23430.74. [DOI] [PubMed] [Google Scholar]
- [114].Etingin OR, Silverstein RL, Friedman HM, Hajjar DP, Viral activation of the coagulation cascade: molecular interactions at the surface of infected endothelial cells, Cell 61 (1990) 657–662. 10.1016/0092-8674(90)90477-v. [DOI] [PubMed] [Google Scholar]
- [115].Visser MR, Tracy PB, Vercellotti GM, Goodman JL, White JG, Jacob HS, Enhanced thrombin generation and platelet binding on herpes simplex virus-infected endothelium, Proc. Natl. Acad. Sci. USA 85 (1988) 8227–8230. 10.1073/pnas.85.21.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Key NS, Bach RR, Vercellotti GM, Moldow CF, Herpes simplex virus type I does not require productive infection to induce tissue factor in human umbilical vein endothelial cells, Lab. Invest 68 (1993) 645–651. [PubMed] [Google Scholar]
- [117].Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, Luciano AA, Stevens W, Rodriguez B, Brenchley JM, Douek DC, Lederman MM, Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation, Blood 115 (2010) 161–167. 10.1182/blood-2009-03-210179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Mayne E, Funderburg NT, Sieg SF, Asaad R, Kalinowska M, Rodriguez B, Schmaier AH, Stevens W, Lederman MM, Increased platelet and microparticle activation in HIV infection: upregulation of P-selectin and tissue factor expression, J. Acquir. Immune Defic. Syndr 59 (2012) 340–346. 10.1097/QAI.0b013e3182439355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Geisbert Thomas W.W., Young Howard A.A., Jahrling Peter B.B., Davis Kelly J.J., Kagan E, Hensley Lisa E.E., Mechanisms Underlying Coagulation Abnormalities in Ebola Hemorrhagic Fever: Overexpression of Tissue Factor in Primate Monocytes/Macrophages Is a Key Event, J. Infect. Dis 188 (2003) 1618–1629. 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- [120].Huerta-Zepeda A, Cabello-Gutiérrez C, Cime-Castillo J, Monroy-Martínez V, Manjarrez-Zavala ME, Gutiérrez-Rodríguez M, Izaguirre R, Ruiz-Ordaz BH, Crosstalk between coagulation and inflammation during Dengue virus infection, Thromb. Haemost 99 (2008) 936–943. 10.1160/TH07-08-0438. [DOI] [PubMed] [Google Scholar]
- [121].Antoniak S, Tatsumi K, Hisada Y, Milner JJ, Neidich SD, Shaver CM, Pawlinski R, Beck MA, Bastarache JA, Mackman N, Tissue factor deficiency increases alveolar hemorrhage and death in influenza A virus-infected mice, J. Thromb. Haemost 14 (2016) 1238–1248. 10.1111/jth.13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Visseren FL, Bouwman JJ, Bouter KP, Diepersloot RJ, de Groot PH, Erkelens DW, Procoagulant activity of endothelial cells after infection with respiratory viruses, Thromb. Haemost 84 (2000) 319–324. [PubMed] [Google Scholar]
- [123].Bjarne O, Samuel IR, Activation of Factor IX by the reaction product of tissue factor and Factor VII: Additional pathway for initiating blood coagulation, PNAS 74 (1977) 5260–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Bevers EM, Comfurius P, Zwaal RFA, Changes in membrane phospholipid distribution during platelet activation, Biochem. Biophys. Acta 736 (1983) 57–66. 10.1016/0005-2736(83)90169-4. [DOI] [PubMed] [Google Scholar]
- [125].Pryzdial ELG, Wright JF, Prothrombinase assembly on an enveloped virus: Evidence that the cytomegalovirus surface contains procoagulant phospholipid, Blood 84 (1994) 3749–3757. [PubMed] [Google Scholar]
- [126].Sutherland MR, Raynor CM, Leenknegt H, Wright JF, Pryzdial ELG, Coagulation initiated on herpesviruses, Proc. Natl. Acad. Sci. USA 94 (1997) 13510–13514. 10.1073/pnas.94.25.13510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Yang Y, Tang H, Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia, Cell. Mol. Immunol 13 (2016) 432–442. 10.1038/cmi.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Arai A, Hirano H, Ueta Y, Hamada T, Mita T, Shirahata A, Detection of mononuclear cells as the source of the increased tissue factor mRNA in the liver from lipopolysaccharide-treated rats, Thromb. Res 97 (2000) 153–162. 10.1016/s0049-3848(99)00147-4. [DOI] [PubMed] [Google Scholar]
- [129].Antoniak S, Mackman N, Multiple roles of the coagulation protease cascade during virus infection, Blood 123 (2014) 2605–2613. 10.1182/blood-2013-09-526277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Sutherland M, Friedman H, Pryzdial E, Herpes simplex virus type 1-encoded glycoprotein C enhances coagulation factor VIIa activity on the virus, Thromb. Haemost 92 (2004) 947–955. 10.1160/TH04-04-0242. [DOI] [PubMed] [Google Scholar]
- [131].Aukrust P, Bjørnsen S, Lunden B, Otterdal K, Ng EC, Ameln W, Ueland T, Müller F, Solum NO, Brosstad F, Frøland SS, Persistently elevated levels of von Willebrand factor antigen in HIV infection. Downregulation during highly active antiretroviral therapy, Thromb. Haemost 84 (2000) 183–187. [PubMed] [Google Scholar]
- [132].Jong E, Louw S, Van Gorp ECM, Meijers JCM, Cate HT, Jacobson BF, The effect of initiating combined antiretroviral therapy on endothelial cell activation and coagulation markers in South African HIV-infected individuals, Thromb. Haemost 104 (2010) 1228–1234. 10.1160/TH10-04-0233. [DOI] [PubMed] [Google Scholar]
- [133].Macfarlane RG, An Enzyme Cascade in the Blood Clotting Mechanism, and its Function as a Biochemical Amplifier, Nature 202 (1964) 498–499. 10.1038/202498a0. [DOI] [PubMed] [Google Scholar]
- [134].Jonsson MI, Lenman AE, Frangsmyr L, Nyberg C, Abdullahi M, Arnberg N, Coagulation Factors IX and X Enhance Binding and Infection of Adenovirus Types 5 and 31 in Human Epithelial Cells, J. Virol 83 (2009) 3816–3825. 10.1128/jvi.02562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lenman A, Muller S, Nygren MI, Frangsmyr L, Stehle T, Arnberg N, Coagulation Factor IX Mediates Serotype-Specific Binding of Species A Adenoviruses to Host Cells, J. Virol 85 (2011) 13420–13431. 10.1128/jvi.06088-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL, Shayakhmetov DM, Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo, Proc. Natl. Acad. Sci. USA 105 (2008) 5483–5488. 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Parker AL, Waddington SN, Nicol CG, Shayakhmetov DM, Buckley SM, Denby L, Kemball-Cook G, Ni S, Lieber A, McVey JH, Nicklin SA, Baker AH, Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes, Blood 108 (2006) 2554–2561. 10.1182/blood-2006-04-008532. [DOI] [PubMed] [Google Scholar]
- [138].Parker AL, McVey JH, Doctor JH, Lopez-Franco O, Waddington SN, Havenga MJE, Nicklin SA, Baker AH, Influence of Coagulation Factor Zymogens on the Infectivity of Adenoviruses Pseudotyped with Fibers from Subgroup D, J. Virol 81 (2007) 3627–3631. 10.1128/jvi.02786-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, Pink R, Buckley SMK, Greig JA, Denby L, Custers J, Morita T, Francischetti IMB, Monteiro RQ, Barouch DH, van Rooijen N, Napoli C, Havenga MJE, Nicklin SA, Baker AH, Adenovirus serotype 5 hexon mediates liver gene transfer, Cell 132 (2008) 397–409. 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- [140].Doronin K, Flatt JW, Di Paolo NC, Khare R, Kalyuzhniy O, Acchione M, Sumida JP, Ohto U, Shimizu T, Akashi-Takamura S, Miyake K, MacDonald JW, Bammler TK, Beyer RP, Farin FM, Stewart PL, Shayakhmetov DM, Coagulation factor X activates innate immunity to human species C adenovirus, Science 338 (2012) 795–798. 10.1126/science.1226625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Xu Z, Qiu Q, Tian J, Smith JS, Conenello GM, Morita T, Byrnes AP, Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement, Nat. Med 19 (2013) 452–457. 10.1038/nm.3107. [DOI] [PubMed] [Google Scholar]
- [142].Greig JA, Buckley SMK, Waddington SN, Parker AL, Bhella D, Pink R, Rahim AA, Morita T, Nicklin SA, McVey JH, Baker AH, Influence of coagulation factor X on in vitro and in vivo gene delivery by adenovirus (Ad) 5, Ad35, and chimeric Ad5/Ad35 vectors, Mol.Ther 17 (2009) 1683–1691. 10.1038/mt.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Sutherland MR, Friedman HM, Pryzdial ELG, Thrombin enhances herpes simplex virus infection of cells involving protease-activated receptor 1, J. Thromb. Haemost 5 (2007) 1055–1061. 10.1111/j.1538-7836.2007.02441.x. [DOI] [PubMed] [Google Scholar]
- [144].Erbe M, Rickerts V, Bauersachs RM, Lindhoff-Last E, Acquired protein C and protein S deficiency in HIV-infected patients, Clin. Appl. Thromb. Hemost 9 (2003) 325–331. [DOI] [PubMed] [Google Scholar]
- [145].Feffer SE, Fox RL, Orsen MM, Harjai KJ, Glatt AE, Thrombotic tendencies and correlation with clinical status in patients infected with HIV, South. Med. J 88 (1995) 1126–1130. 10.1097/00007611-199511000-00008. [DOI] [PubMed] [Google Scholar]
- [146].Klein SK, Slim EJ, de Kruif MD, Keller TT, ten Cate H, van Gorp ECM, Brandjes DPM, Is chronic HIV infection associated with venous thrombotic disease? A systematic review, Neth. J. Med 63 (2005) 129–136. [PubMed] [Google Scholar]
- [147].Neubauer K, Knittel T, Armbrust T, Ramadori G, Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury, Gastroenterology 108 (1995) 1124–1135. 10.1016/0016-5085(95)90211-2. [DOI] [PubMed] [Google Scholar]
- [148].Liu M, Chan CW, McGilvray I, Ning Q, Levy GA, Fulminant viral hepatitis: molecular and cellular basis, and clinical implications, Expert Rev. Mol. Med 2001 (2001) 1–19. 10.1017/S1462399401002812. [DOI] [PubMed] [Google Scholar]
- [149].Marra F, DeFranco R, Grappone C, Milani S, Pinzani M, Pellegrini G, Laffi G, Gentilini P, Expression of the thrombin receptor in human liver: up-regulation during acute and chronic injury, Hepatology 27 (1998) 462–471. 10.1002/hep.510270221. [DOI] [PubMed] [Google Scholar]