

Summary
The power of single-cell RNA sequencing (scRNA-seq) stems from its ability to uncover cell type-dependent phenotypes, which rests on the accuracy of cell type identification. However, resolving cell types within and, thus, comparison of scRNA-seq data across conditions is challenging owing to technical factors such as sparsity, low number of cells, and batch effect. To address these challenges, we developed scID (Single Cell IDentification), which uses the Fisher's Linear Discriminant Analysis-like framework to identify transcriptionally related cell types between scRNA-seq datasets. We demonstrate the accuracy and performance of scID relative to existing methods on several published datasets. By increasing power to identify transcriptionally similar cell types across datasets with batch effect, scID enhances investigator's ability to integrate and uncover development-, disease-, and perturbation-associated changes in scRNA-seq data.
Subject Areas: Biological Sciences, Bioinformatics, Mathematical Biosciences, Omics, Transcriptomics
Graphical Abstract
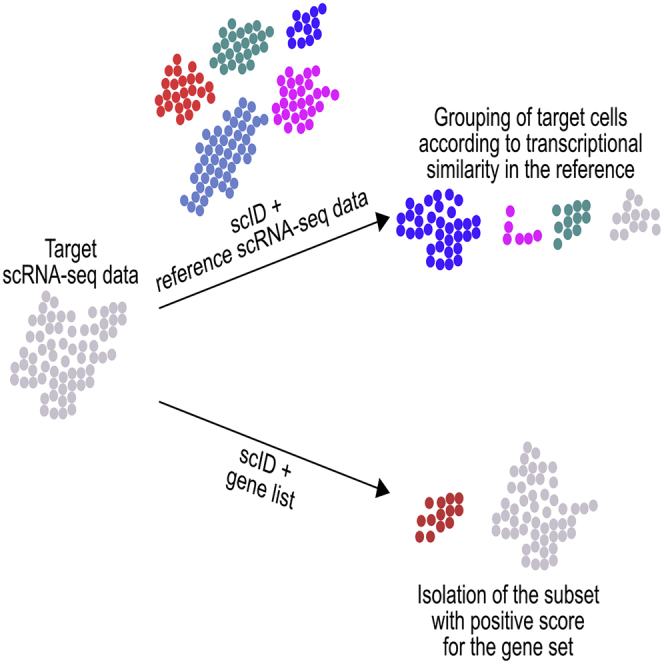
Highlights
-
•
scID identifies transcriptionally equivalent cell populations across datasets
-
•
scID's accuracy relies on the goodness of cluster enriched genes in the reference
-
•
scID can also be used to score data against a user-provided gene list
-
•
R package and use cases are at https://github.com/BatadaLab/scID
Biological Sciences; Bioinformatics; Mathematical Biosciences; Omics; Transcriptomics
Introduction
Single-cell RNA-sequencing (scRNA-seq) (Hashimshony et al., 2012, Jaitin et al., 2014, Macosko et al., 2015, Picelli et al., 2014, Ramskold et al., 2012, Zheng et al., 2017) is now being routinely used to characterize cell-type-specific changes in development, disease, and perturbations (Regev et al., 2017), which require analysis of cross-data comparison (Butler et al., 2018, Haghverdi et al., 2018, Kiselev et al., 2018). However, there are technical and biological variations between the different datasets that may hamper the joint analysis of these data (Butler et al., 2018, Haghverdi et al., 2018, Kiselev et al., 2019).
Batch correction approaches such as canonical correlation analysis (CCA) (Butler et al., 2018), mutual nearest neighbours (MNN) (Haghverdi et al., 2018), and single cell variational inference (scVI) (Lopez et al., 2018) have been designed to integrate scRNA-seq datasets. The advantage of these methods is that batch correction allows merging of datasets that provide more power to resolve cell types (Heimberg et al., 2016), particularly those that are rare. In both CCA and MNN, cells that have similar local correlation or neighborhood structure in the datasets are paired. However, when the data are significantly imbalanced in the number of cells and library size per cell, both of which influence accuracy of clustering, it is unclear how these methods perform. Moreover, depending on the extent of technical bias or extent of overlap in cell types between the data, untangling technical from biological variation between data may not be possible and runs the risk of over-correcting, i.e., the biological variance between the data may be lost.
As deeply characterized, extensively validated, and annotated tissue-, organ-, and organism-level atlases are increasingly being generated (Regev et al., 2017), it is worthwhile to reuse such high-quality information to identify known populations of cells in the target data with lower quality. In addition to the propagation of cell labels and metadata across datasets, reference data can also be used to group cells in the target data in which unsupervised clustering does not resolve consistently given the partial knowledge of the cell types in the literature or with the clusters found in the superior quality reference data. In such cases, a biomarker-based approach for identifying transcriptionally equivalent cells across datasets involves determining whether the top marker genes derived from the reference clusters are present in the target cells. However, this biomarker-based approach does not work effectively owing to the dropout problem with scRNA-seq data, i.e., for a large proportion of genes, the expression values are not sampled (Bacher and Kendziorski, 2016, Stegle et al., 2015, Vallejos et al., 2017). To map cells from target scRNA-seq data to a reference data, scmap (Kiselev et al., 2018) extracts features from each reference cluster and uses a distance- and correlation-based metric to quantify the closeness between each reference cluster's centroid and the cell in the target data to assign target cells to reference clusters. However, distance metrics in high-dimensional space may not work as expected (Aggarwal et al., 2001). CaSTLe uses genes with high expression in both the target and reference data or those that have high mutual information to clusters identities in the reference datasets to identify equivalent populations (Lieberman et al., 2018). However, CaSTLe's accuracy has not been systematically assessed against newer developed methods. Additional mapping methods have also been developed (de Kanter et al., 2019).
Here we present a novel method called scID for identifying transcriptionally related groups of cells across datasets by exploiting information from both the reference and the target datasets without making any assumptions regarding the nature of technical and biological variation in these datasets. More specifically, scID extracts a list of cluster-specific genes (markers) from the reference dataset and weighs their relevance in the target dataset by learning a classifier with respect to a putative population of cells either expressing or not expressing those genes. Through extensive analysis of published datasets with batch effect and strong asymmetry in cell number and library sizes per cell in which the independent clustering of the target data via unsupervised method is not obviously similar to that in the reference, we show that scID is more accurate than the above-mentioned methods. Thus, scID helps uncover hidden biological variation present between scRNA-seq datasets that can vary broadly in batch effect and quality (i.e., different number of cells, dropout levels, and dynamic range of gene expression).
Results
Identification of Transcriptionally Similar Cells across scRNA-Seq Datasets
Identifying target cells that match a reference cluster (C) can be seen as a binary classification problem fC(gj) by treating the reference cluster of interest as one class (C) and the rest of the reference clusters as another class (C−), i.e., . Assuming that cell types can be linearly separable, one can, for example, use Fisher's linear discriminant analysis to learn a direction, , from the reference data that maximizes the separation between C and C− population where is the number of signature genes or features for cluster C. Thus, to each target cell we can assign an ‘scID score”:
| (Equation 1) |
where is the normalized gene expression of feature i in cell j and is the weight that reflects the discriminatory power of the i-th gene, such that in the projected space, the C and the C− populations are maximally separated. To mitigate the influence of batch effect, it may be more suitable to estimate weights from the target data instead of the reference. Given putative and c− populations selected from the target data, the weights are estimated as
| (Equation 2) |
where represents the mean expression of the putative target cluster c (c−) and () represents standard deviation of gene expression within the putative target cluster c (c−). A target cell can then be assigned to the reference cluster C by choosing a suitable threshold.
scID maps cells in the target data that are equivalent to clusters in the reference data in three key stages (Figure 1A).
Figure 1.

Overview and Assessment of scID
(A) The three main stages involved in mapping cells across scRNA-seq data with scID are as follows: In stage 1, gene signatures are extracted from the reference data (shown as clustered groups on a reduced dimension). In stage 2, discriminative weights are estimated from the target data for each reference cluster-specific gene signature. In stage 3, every target cell is scored for each feature and is assigned to the corresponding reference cluster.
(B) Quantification of accuracy of DPR classification (stage 2 of scID). Boxplot shows interquartile range for TPR (black) or FPR (white) for all the cell types in each published dataset listed in the x axis. See also Figure S1.
(C) Quantification of TPR and FPR of stage 2 (black) and stage 3 (white) of scID. Significance was computed using two-sided paired Kruskal-Wallis test for difference in TPR or FPR between stage 2 and stage 3.
(D) Assessment of accuracy of scID via self-mapping of published datasets. The indicated published data (x axis labels) were self-mapped, i.e., used as both reference and target, by scID and the assigned labels were compared with the published cell labels.
(E) Assessment of classification accuracy of scRNA-seq data integration. Human pancreas Smart-seq2 data (Segerstolpe et al.) were used as reference and CEL-seq1 as target (white; Grun et al., 2016) or CEL-seq2 as target (black; Muraro et al., 2016). See also Figures S2 and S3.
In the first stage, genes that are differentially expressed in each cluster (herein referred to as gene signatures) are extracted from each cluster of the reference data. In the second stage, for each reference cluster C, the weights of the genes in the respective gene signature (which reflect their discriminability) are estimated either from the reference or from the target data, depending on the nature of the data, by identifying putative and c− cells using an approach we term differential precision-recall (DPR). In the last stage, the extracted features (i.e., the combination of gene signatures and their weights) are used to identify target cells that are most concordant with extracted features. A finite mixture of Gaussians is used to model the scores of the target cells. Cells with high likelihood of falling under the distribution with the highest average score are assigned to the reference cluster from which the gene signatures were extracted. Ideally a target cell should be assigned to only one reference cluster but when closely related cells are present in the reference data, it is possible that the features extracted are not able to highly discriminate between them leading to assignment of a cell to multiple clusters in the reference. Conflicts in assignments are resolved using a relative normalized score strategy. Further details are provided below and in the Methods section.
As the accuracy of weights depends strongly on selection of true cells, we assessed the accuracy with which the DPR approach identified true cells in several published reference data. We observed that the median true-positive rate of the DPR approach was between 45% and 80% and the median false-positive rate was between 0% and 5% (Figure 1B). Moreover, the estimated weights after the DPR approach were consistent with weights from the reference data using published label (Figure S1). Thus, although the DPR approach is stringent and misses higher-than-desired levels of true cells, its role in the overall scID methodology is the recovery of true cells with very low false-positive rate so that the estimated gene weights are accurate. To assess the accuracy of the scoring approach (stage 3 of scID) and to quantify its contribution to the improvement in classification accuracy, we used the mouse retinal bipolar DropSeq data (Shekhar et al., 2016) and compared the true-positive and false-positive rates of cells classified by the DPR approach to the subsequent weight-based classification. We find that, relative to the accuracy of the DPR approach (Figure 1B), the accuracy of the weight-based classification of cells has a significantly higher true-positive rate without increase in the false-positive rate (Figure 1C). To assess the stability and generalizability of scID across scRNA-seq data across a range of quality and cell type composition, we performed self-mapping (i.e., using the same dataset as reference and target) of four published datasets spanning different tissues and technologies. The accuracy of self-mapping, as assessed by Adjusted Rand Index, was relatively high (Figure 1D).
We next tested our assumption that different target data with different batch effect lead to different gene weights and quantified its impact on classification accuracy. We selected two reference-target pairs from similar tissue so that the differences are largely due to technical factors rather than biological factors. To this end, we sought to detect equivalent cells across multiple human pancreas datasets from different technologies (Smart-Seq2 [Segerstolpe et al., 2016], CEL-Seq1 [Baron et al., 2016], and CEL-Seq2 [Muraro et al., 2016]) in which the published cell identity labels for each dataset is available. We used the Smart-Seq2 as reference data and CEL-Seq or CEL-Seq2 as target. We confirmed that the two reference/target data pairs (i.e., Smart-Seq2/CEL-Seq1 and Smart-Seq2/CEL-Seq2) had different extent of batch effect (Figure S2). As expected, the scID estimated weights of the same cell-type-specific genes calculated from the two target datasets are significantly different, yet the overall accuracy of cell type classification for the two target datasets by scID were similar (Figure S3). scID was one of the two most accurately and consistently well-performing methods (Figure 1E).
However, these datasets were relatively balanced in terms of cell number and quality and therefore with mild batch effect. We next assessed these methods in dataset pairs with significant asymmetry in cell number and cell coverage between reference and target datasets in cell number and coverage and have stronger batch effect (Buttner et al., 2019) (Figure S2).
Reference-Based Identification of Equivalent Cells by scID across the Mouse Retinal Bipolar Neurons scRNA-Seq Datasets
In general, a large number of cells despite shallow coverage per cell provides more power to resolve highly similar cells than low number of cells with deep coverage per cell (Heimberg et al., 2016). We sought to identify equivalent cell types in scRNA-seq data with strong imbalance in the number of cells or read coverage per cell. Shekhar et al (Shekhar et al. 2016) generated scRNA-seq data from mouse retinal bipolar neurons using two different technologies: Drop-seq (Macosko et al., 2015) (with ∼26,800 cells and ∼700 genes per cell) and the plate-based Smart-seq2 (with 288 cells and ∼5,000 genes per cell). Unsupervised clustering of the full Drop-seq data with Seurat resulted in ∼18 discrete clusters, whereas unsupervised clustering of the Smart-seq2 data with Seurat and several other clustering methods (data not shown) identified only 4 clusters (Figure 2A) possibly due to low cell numbers.
Figure 2.

Reference-Based Identification of Equivalent Cells across the Mouse Retinal Bipolar Neurons scRNA-Seq Datasets
(A) t-SNE plot showing clusters in Drop-seq (reference) and Smart-seq2 (target) data of mouse retinal bipolar cells from Shekhar et al. Cluster membership of reference cells (∼26,800 cells) were taken from the publication. Smart-seq2 data (288 cells) were clustered using Seurat, and cluster names were assigned arbitrarily.
(B) Heatmap showing Z score normalized average expression of gene signatures (row) in the clusters (column) of the reference Drop-seq data (left) and in the target Smart-seq2 data (right). Red (khakhi) indicates enrichment and blue (turquois) indicates depletion of the reference gene signature levels relative to average expression of gene signatures across all clusters of reference (target) data.
(C) Identification of target (Smart-seq2) cells that are equivalent to reference (Drop-seq) clusters using marker-based approach. The top two differentially enriched (or marker) genes in each reference (Drop-seq) cluster were used to identify equivalent cells in the target (Smart-seq2) data using a thresholding approach. Bars represent percentage of classified and unassigned cells using various thresholds for normalized gene expression of the marker genes as indicated on the x axis. Gray represents the percentage of cells that express markers of multiple clusters, yellow represents the percentage of cells that can be unambiguously classified to a single cluster, and blue represents the percentage of cells that do not express markers of any of the clusters. These cells are referred to as orphans. X axis represents different thresholds of normalized gene expression (see Methods).
(D) Assessment of accuracy of various methods methods for classifying target cells using Adjusted Rand Index.
(E) Assessment of accuracy of various methods methods for classifying target cells using Variation of Information.
To determine how the transcriptional signatures of the reference clusters are distributed in the target data, we computed the average gene signature per cluster (Figure 2B; see Methods). The dominant diagonal pattern in the gene signature matrix for the reference data indicates the specificity of the extracted gene signatures. All the subtypes of bipolar cells (BCs) that were well separated in the reference Drop-seq data co-clustered in the target Smart-seq data (Figure 2B). Surprisingly, despite over seven times greater number of genes per cell in the target Smart-seq2 data compared with the reference Drop-seq data, the biomarker approach of using top markers of clusters from the reference Drop-seq data resulted in unambiguous assignment of only a small proportion of cells (Figure 2C).
Thus, we assessed the ability of scID and competing methods to assign cluster identity to cells in the target Smart-seq2 data using Drop-seq data as reference. scID and CaSTLe assigned 100% (n = 288) of the Smart-seq2 cells into 15 Drop-seq clusters, whereas scmap assigned only 63.2% (n = 182) of the Smart-seq2 cells into 10 clusters. Using target cells that specifically expressed cell-type-specific markers (yellow, Figure 2C) as ground truth, two independent metrics for accuracy suggested that scID was one of the two most accurately performing methods on these datasets (Figures 2D and 2E).
Reference-Based Identification of Equivalent Cells in scRNA-Seq and Ultra-Sparse Single Nuclei RNA-Seq from Mouse Brain
In many clinical settings where only postmortem tissues are available, single nuclei-RNA seq (snRNA-seq) (Habib et al., 2017, Lake et al., 2016) may be possible. However, the transcript abundance in the nuclei is significantly lower than in the cytoplasm and as a consequence the complexity of scRNA-seq libraries from nuclei-scRNA-seq from the same tissue is significantly lower than in whole-cell scRNA-seq (Habib et al., 2017) thus making it challenging to cluster despite large number of cells. To assess how well scID performs compared with alternatives in this setting, we obtained publicly available Chromium 10X (Zheng et al., 2017) -based scRNA-seq data on mouse brain cells and snRNA-seq on mouse brain nuclei (see Methods).
The brain whole-cell scRNA-seq data had ∼9K cells, and the brain nuclei snRNA-seq data had ∼1K cells. Unsupervised clustering of the data separately identified different number of clusters, which did not have an obvious one-to-one correspondence (Figure 3A). As the whole-cell scRNA-seq data have more cells than the nuclei-seq, we used the former as reference and obtained gene sets that are differentially expressed in each cluster compared with the rest of the cells (Figure 3B). Efforts to identify cluster membership of nuclei-seq data based on top markers in each of the clusters from the whole-cell scRNA-seq data allowed unambiguous classification of only a small number of cells possibly due to shallow coverage that leads to low dynamic range in gene expression. Not surprisingly, there was significant number of orphan cells (i.e., those in which transcripts of cell markers were not captured) (Figure 3C).
Figure 3.

Reference-Based Identification of Equivalent Cells in Single Cell and Nuclei RNA-seq from Mouse Brain
(A) t-SNE plot showing clusters in the mouse brain scRNA-seq data with ∼9,000 cells (left) and tSNE of the mouse brain single nuclei RNA-seq (snRNA-seq) data with ∼1,000 cells (right). Data were clustered with Seurat (v3).
(B) Heatmap showing Z score normalized average expression of gene signatures (rows) in the reference (left) and in the target (right) clusters (columns). Red (khakhi) indicates enrichment and blue (turquois) indicates depletion of the gene signature levels relative to average expression of that gene signature across all clusters of reference (target) data.
(C) Marker-based identification of cell types in target data. Data were binarized using different thresholds (see Methods) that represent expression value of each marker gene relative to the maximum. Two most differentially expressed markers from each reference cluster were used. Cells that express markers of multiple clusters (gray) are labeled as Ambiguous. Cells that only express markers of a single cluster (yellow) are labeled as Classified. Cells that do not express markers of any clusters (blue) are labeled as Orphans.
(D) Assessment of accuracy of various methods methods for classifying target cells using Adjusted Rand Index.
(E) Assessment of accuracy of various methods methods for classifying target cells using Variation of Information.
Thus, we assessed the ability of scID and competing methods to assign cluster identity to cells in the target (snRNA-seq) data using scRNA-seq data as reference. scID assigned 99.5% (n = 949) of the snRNA-seq cells into 14 scRNA-seq clusters, whereas scmap assigned 60.2% (n = 574) of the snRNA-seq cells into 11 clusters and CaSTLe assigned 100% (n = 954) of the snRNA-seq cells into 15 clusters. Using target cells that specifically expressed cell-type-specific markers (yellow, Figure 2C) as ground truth, two independent metrics for accuracy suggested that scID had the highest accuracy (Figures 3D and 3E).
Discussion
Comparison of multiple scRNA-seq datasets across different tissues, individuals, and perturbations is necessary in order to reveal potential biological mechanisms underlying phenotypic diversity. However, comparison of scRNA-seq datasets is challenging because even scRNA-seq data from the same tissue but with different technology or donor can have significant batch effect confounding the technical with the biological variability. scRNA-seq datasets generated from different scRNA-seq methods and quality (i.e., similar number of cells and library depth per cell) are in general much easier to correct than data generated from different methods and quality. Particularly, in cases of strong asymmetry in quality between datasets to be compared, mapping target cells across data at the individual cell level rather than performing batch correction can be a more appropriate route for identifying known transcriptionally related cells.
We have developed scID that uses the framework of Fisher’s linear discriminant analysis to map cells from a target data to a reference dataset (Figure 1A). scID adapts to characteristics of the target data in order to learn the discriminative power of cluster-specific genes extracted from the reference in the target data. scID down-weighs the genes within the signature that are not discriminatory in the target data. Through extensive characterization of multiple published scRNA-seq datasets, we assessed scID’s accuracy relative to alternative approaches in situations where the reference and target datasets have strong asymmetry in quality and have shown that scID outperforms existing methods for recovering transcriptionally related cell populations from the target data, which does not cluster concordantly with the reference.
As the scale of single-cell RNA-seq data increases and the numbers of clusters obtained are so large that manual annotation is cumbersome, scID can enable automatic propagation of annotations and metadata across clusters in these datasets. In addition, scID can be used for ordering single transcriptomes based on an arbitrary user-specified gene list without the need for providing information on gene expression levels.
Limitations of the Study
scID can estimate gene signature weights from either the reference or the target data. When the reference and the target data have strong batch effect, estimation of weights from the target appears to be more accurate. However, in cases where there are very similar cell types in the datasets to be compared, estimation of weights from the reference may be more suitable. Moreover, if the target data contain a cell type that is only weakly related to but not exactly the cell type in the reference, scID is susceptible to falsely classifying such cells.
Owing to reliance on gene signatures to discriminate between clusters, a limitation of scID is that it works only for reference clusters that have distinct gene signatures. Additionally, the accuracy of scID depends on the correctness of the labels of the reference data. For a dataset to serve as a reference, we expect well-defined clusters by cell type such that cluster-specific gene signatures exist and they are mutually exclusive as seen in Figures 2B and 3B. If neither of the two data is well clustered and contains good gene signatures, scID is not a recommended approach for identification of equivalent cell types. Imputation of gene expression (Zhang et al., 2019) particularly when the target scRNA-seq is highly shallow may increase the power for identification of equivalent cells and is worth exploring in the future.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
N.N.B. was funded by the Chancellor's Fellowship from the University of Edinburgh.
Author Contributions
K.B. and N.N.B. conceived the study. K.B, S.S., and N.N.B. developed the computational framework. K.B. implemented the method and performed the analysis. K.B., S.S., and N.N.B. interpreted the results. N.N.B. wrote the manuscript with input from K.B and S.S. N.N.B. supervised the study. All authors read and approved the manuscript.
Declaration of Interests
The authors declare that they have no competing interest.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100914.
Data and Code Availability
No datasets were generated. The R implementation for scID is available on Github (https://batadalab.github.io/scID/), and all the scripts to generate the figures shown in this manuscript are available at https://github.com/BatadaLab/scID_manuscript_figures.
Supplemental Information
References
- Aggarwal C.C., Hinneburg A., Keim D.A. On the Surprising Behavior of Distance Metrics in High Dimensional Space. In: Van den Bussche J., Vianu V., editors. Database Theory — ICDT 2001. ICDT 2001. Lecture Notes in Computer Science, vol 1973. Springer, Berlin; Heidelberg: 2001. https://link.springer.com/chapter/10.1007/3-540-44503-X_27 [Google Scholar]
- Bacher R., Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol. 2016;17:63. doi: 10.1186/s13059-016-0927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M., Veres A., Wolock S.L., Faust A.L., Gaujoux R., Vetere A., Ryu J.H., Wagner B.K., Shen-Orr S.S., Klein A.M. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3:346–360.e4. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner M., Miao Z., Wolf F.A., Teichmann S.A., Theis F.J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods. 2019;16:43–49. doi: 10.1038/s41592-018-0254-1. [DOI] [PubMed] [Google Scholar]
- de Kanter J.K., Lijnzaad P., Candelli T., Margaritis T., Holstege F.C.P. CHETAH: a selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 2019;47:e95. doi: 10.1093/nar/gkz543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grun D., Muraro M.J., Boisset J.C., Wiebrands K., Lyubimova A., Dharmadhikari G., van den Born M., van Es J., Jansen E., Clevers H. De novo prediction of stem cell identity using single-cell transcriptome data. Cell Stem Cell. 2016;19:266–277. doi: 10.1016/j.stem.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N., Avraham-Davidi I., Basu A., Burks T., Shekhar K., Hofree M., Choudhury S.R., Aguet F., Gelfand E., Ardlie K. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods. 2017;14:955–958. doi: 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T., Wagner F., Sher N., Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Heimberg G., Bhatnagar R., El-Samad H., Thomson M. Low dimensionality in gene expression data enables the accurate extraction of transcriptional programs from shallow sequencing. Cell Syst. 2016;2:239–250. doi: 10.1016/j.cels.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin D.A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev V.Y., Andrews T.S., Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019;20:273–282. doi: 10.1038/s41576-018-0088-9. [DOI] [PubMed] [Google Scholar]
- Kiselev V.Y., Yiu A., Hemberg M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods. 2018;15:359–362. doi: 10.1038/nmeth.4644. [DOI] [PubMed] [Google Scholar]
- Lake B.B., Ai R., Kaeser G.E., Salathia N.S., Yung Y.C., Liu R., Wildberg A., Gao D., Fung H.L., Chen S. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman Y., Rokach L., Shay T. CaSTLe - classification of single cells by transfer learning: harnessing the power of publicly available single cell RNA sequencing experiments to annotate new experiments. PLoS One. 2018;13:e0205499. doi: 10.1371/journal.pone.0205499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R., Regier J., Cole M., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro M.J., Dharmadhikari G., Grun D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M.A., Carlotti F., de Koning E.J. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 2016;3:385–394.e3. doi: 10.1016/j.cels.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Bjorklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Ramskold D., Luo S., Wang Y.C., Li R., Deng Q., Faridani O.R., Daniels G.A., Khrebtukova I., Loring J.F., Laurent L.C. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M. The human cell atlas. Elife. 2017;6:e27041. doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerstolpe A., Palasantza A., Eliasson P., Andersson E.M., Andreasson A.C., Sun X., Picelli S., Sabirsh A., Clausen M., Bjursell M.K. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 2016;24:593–607. doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar K., Lapan S.W., Whitney I.E., Tran N.M., Macosko E.Z., Kowalczyk M., Adiconis X., Levin J.Z., Nemesh J., Goldman M. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016;166:1308–1323.e30. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegle O., Teichmann S.A., Marioni J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015;16:133–145. doi: 10.1038/nrg3833. [DOI] [PubMed] [Google Scholar]
- Vallejos C.A., Risso D., Scialdone A., Dudoit S., Marioni J.C. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat. Methods. 2017;14:565–571. doi: 10.1038/nmeth.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.W., O’Flanagan C., Chavez E.A., Lim J.L.P., Ceglia N., McPherson A., Wiens M., Walters P., Chan T., Hewitson B. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods. 2019;16:1007–1015. doi: 10.1038/s41592-019-0529-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated. The R implementation for scID is available on Github (https://batadalab.github.io/scID/), and all the scripts to generate the figures shown in this manuscript are available at https://github.com/BatadaLab/scID_manuscript_figures.