

Abstract
Introduction:
De novo lipogenesis (DNL) plays a major role in fatty acid metabolism and contributes significantly to triglyceride accumulation within the hepatocytes in patients with nonalcoholic steatohepatitis (NASH). Acetyl-CoA carboxylase (ACC) converts acetyl-CoA to malonyl CoA, and is a rate-controlling step in DNL. Furthermore, malonyl-CoA is an important regulator of hepatic mitochondrial fat oxidation through its ability to inhibit carnitine palmitoyltransferase I. Therefore, inhibiting ACC pharmacologically represents an attractive approach to treating NASH.
Areas Covered:
This article summarizes preclinical and clinical data on the efficacy and safety of the liver targeted ACC inhibitor GS-0976 (Firsocostat) for the treatment of NASH. In a phase 2 trial that included 126 patients with NASH and fibrosis, GS-0976 20 mg daily for 12 weeks showed significant relative reduction in liver fat by 29%; however, treatment was associated with an increase in plasma triglycerides with 16 patients having levels > 500 mg/dL.
Expert Opinion:
Preclinical and preliminary clinical data support the development of GS-0976 as treatment for NASH. ACC-induced hypertriglyceridemia can be mitigated by fish oil or fibrates, but the long-term cardiovascular effects require further investigations.
Drug Summary.
Drug name: GS-0976; Firsocostat; ND-630, NDI 010976
Phase: Currently undergoing phase IIb trial
Indication: Under evaluation for treatment of nonalcoholic steatohepatitis (NASH) with advanced fibrosis
Pharmacology: Acetyl Co-A carboxylase (ACC) inhibitor
Route of administration: Oral
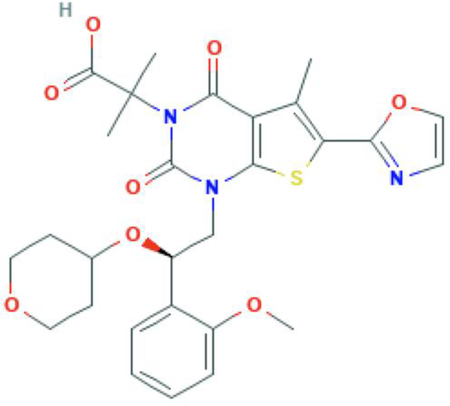
Chemical structure: C28H31N3O8S
Pivotal trial: ATLAS phase 2 trial ( NCT03449446) that will assess the safety and tolerability of GS-0976, selonsertib (Apoptosis signal-regulating kinase1; ASK1 inhibitor), and GS-9674 (Farnesoid X receptor; FXR agonist), administered alone or in combination, in patients with NASH and bridging fibrosis or compensated cirrhosis (F3 or F4 fibrosis).
1. Introduction and Overview of the Market
Nonalcoholic fatty liver disease (NAFLD) is now the most common form of chronic liver disease in the United States and globally1,2. NAFLD includes a histologic spectrum that ranges from simple steatosis or nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH) progressing to advanced liver fibrosis, cirrhosis, and end-stage liver disease requiring liver transplantation3,4. Despite its significant health care burden, there are no FDA-approved medications for NASH. Both vitamin E and pioglitazone have shown some efficacy against NASH in randomized clinical trials5,6. However, both medications have been associated with potentially serious adverse events such as prostate cancer with vitamin E and bladder cancer with pioglitazone limiting their uptake by clinicians7. More recently, the farnesoid X receptor (FXR) agonist obeticholic acid showed consistent efficacy on fibrosis regression in phase 2 and 3 trials in patients with NASH and significant fibrosis paving the way for potential FDA approval in 20208,9. Unfortunately, in the phase 3 REGENERATE trial, obeticholic acid was associated with significant pruritus in 50% of patients and increase in LDL cholesterol raising concerns about long-term cardiovascular morbidity. Moreover, fibrosis improvement by 1 stage occurred in only 23% of patients making it clear that there is need for combination therapy with other drugs to increase efficacy.
Triglyceride accumulation within the hepatocytes is the hallmark of the disease and is considered the first hit that predisposes the liver to subsequent multiple parallel hits that can lead to disease progression10. Consistent with this hypothesis, liver-targeted mitochondrial uncoupling has been shown to decrease hepatic steatosis, eradicate inflammation and reverse liver fibrosis in rodent models of NASH11,12. Further evidence for the role of intrahepatic triglyceride accumulation in disease progression comes from a Mendelian randomization study that showed that increase in liver fat is causally related to inflammation, hepatocyte injury and fibrosis13. Although initial studies suggested that the main source of intrahepatic triglycerides was from adipose tissue lipolysis in the context of insulin resistance14, more recent studies from the same group have found that patients with NAFLD have significantly higher rates of de novo lipogenesis (DNL), even under fasting conditions, compared to obese patients without NAFLD with similar rates of adipose flux of fatty acids (FAs)15. Despite the significant role of DNL, esterification of plasma FAs in a substrate supply-dependent/ insulin-independent manner account for approximately 60% of intrahepatic triglyceride synthesis16.
DNL plays a major role in fatty acid metabolism with the rate-limiting step being the conversion of acetyl-coenzyme A (acetyl-CoA) to malonyl-CoA by the enzyme acetyl-CoA carboxylase (ACC)17. This conversion occurs in two half-reactions, a biotin carboxylase (BC) reaction and a carboxyltransferase (CT) reaction17,18. ACC has two isoforms in mammals19. ACC1 is primarily present in the cytosol of lipogenic tissues such as the liver and adipose tissue where it controls the first committed reaction in DNL. Whereas, ACC2 is primarily present in the mitochondria of oxidative tissues such as the skeletal muscle and catalyzes the formation of malonyl-CoA which functions as a potent allosteric inhibitor of carnitine palmitoyl-transferase 1 (CPT1) which mediates the transfer of FAs into the mitochondria for β-oxidation20,21. Knockdown of ACC1 using antisense oligonucleotides (ASOs) decreases DNL whereas knockdown of ACC2 using the same ASO approach leads to increased mitochondrial fatty acid oxidation22. The net effect of knocking down the expression of both isoforms is a decrease in DNL due to decreased hepatic expression of ACC1 and an increase in mitochondrial fatty acid oxidation due to decreased hepatic expression of ACC2, leading to decreased hepatic steatosis22. ACC activity is regulated through different mechanisms including: (1) phosphorylation (e.g. AMP-activated protein kinase (AMPK) inhibits ACC through phosphorylation), (2) allosteric regulation (e.g. citrate promotes and palmitate inhibits ACC activity), (3) protein-protein interaction (e.g. polymerization mediated by MIG-12 enhances ACC activity)23–25. In support for the role of ACC in NAFLD development and progression and consistent with the ACC1/ACC2 ASO studies, Fullerton et al. demonstrated that ACC double knock-in mice that maintain both ACC1 and ACC2 in a constitutively activated state develop histologic and clinical evidence of NAFLD including increased DNL (elevation in hepatic malonyl-CoA, diacylglycerol, and TGs), leading to PKCε activation, hepatic insulin resistance, and liver fibrosis26.
2. Rationale for ACC inhibition as treatment for NAFLD
Because of the central role of ACC1/ACC2 in stimulating DNL and impairing fatty acid β-oxidation, inhibition of ACC enzymes pharmacologically provides an attractive approach to treating NAFLD/NASH by decreasing hepatic lipid synthesis while simultaneously increasing fatty acid oxidation. Several studies have demonstrated that genetic ablation of ACC isoforms in mice protects from hepatic steatosis and liver injury. Abu-Elheiga and colleagues showed that ACC2 knockout mice fed a high fat diet were protected from weight gain with decreased liver TG and improved hepatic insulin sensitivity27,28. However, Olson et al. were unable to replicate these findings in their ACC2 knockout mice29. Furthermore, Hoehn et al. demonstrated that inhibition of ACC2 pharmacologically by the administration of an AMPK activator or genetically did not alter energy expenditure or adiposity despite an increase in whole-body fatty acid oxidation30. Liver specific ACC1 knockout mice have reduced liver TG accumulation but no effect on glucose homeostasis31. However, long-term reduction in liver fat may have an indirect effect on insulin sensitivity by improving liver fibrosis which may have a causal relationship with insulin resistance based on a Mendelian randomization analysis13.
Based on these results several classes of small molecule inhibitors of ACC have been developed and designed to: (1) compete with acetyl-CoA, (2) inhibit the CT reaction, or (3) inhibit the BC reaction which may also inhibit the dimerization of ACC32.
3. Preclinical experience with GS-0976 in NAFLD
GS-0976 (synonyms: ND-630, NDI 010976, Firsocostat) is a liver targeted, small molecule allosteric inhibitor of both ACC1 and ACC2 that binds to the BC domain, thus preventing dimerization and inhibiting enzyme activity33 (Figure 1). The BC domain is a shallow, hydrophilic pocket with superior physiochemical properties making it ideal for pharmaceutical targeting. GS-0976 is liver specific because it was designed to be a substrate for hepatic organic anion-transporting polypeptide (OATP) transporters, resulting in liver-directed bio-distribution and ensuring NASH focused effects.
Figure 1. Mechanism of Action of GS-0976: A Liver-Directed Allosteric Inhibitor of ACC1 and ACC2.
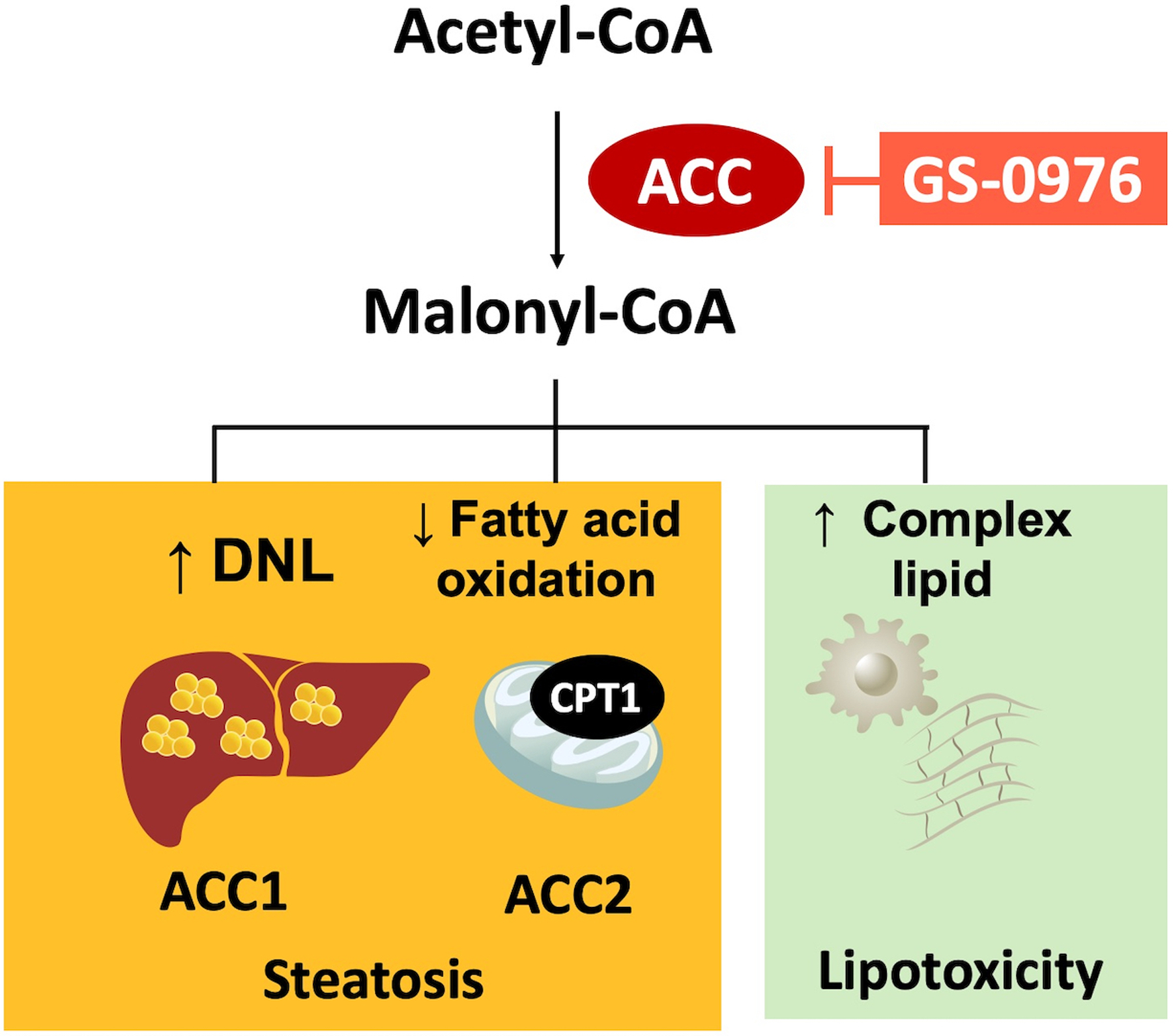
ACC catalyzes the rate-limiting step in hepatic DNL. ACC1 primarily is present in the cytosol and catalyzes the first committed reaction in DNL. ACC2 primarily is present in the mitochondria and catalyzes the formation of malonyl-CoA which functions as a potent allosteric inhibitor of carnitine palmitoyl-transferase 1 (CPT1) thereby inhibiting the transfer of FAs into the mitochondria for β-oxidation. The net effect of activating both isoforms is an increase in hepatic TG and complex lipids leading to lipotoxicity.
Several lines of evidence from pre-clinical data support the development of GS-0976 as a treatment for NASH. Harriman and colleagues demonstrated the ability of GS-0976 (previously called ND-630) to inhibit fatty acid synthesis in cultured human hepatic (HepG2) cells as evidenced by reduction of [14C] acetate incorporation into fatty acids33. Importantly, total cell number, cellular protein concentration, and incorporation of [14C]acetate into cholesterol were not changed, indicating that GS-0976 inhibition of fatty acid synthesis was not due to decreased cell viability or nonspecific metabolic effects. GS-0976 also enhanced fatty acid oxidation in cultured cells as shown by its ability to stimulate [14C]palmitate oxidation and the release of [14C]O2. When administered chronically to rats with diet- or genetically-induced obesity, GS-0976 inhibition decreased hepatic steatosis and improved insulin sensitivity. Our team demonstrated that although long-term treatment (21 days) with an allosteric liver-directed inhibitor of ACC1/ACC2 significantly reduced hepatic steatosis in diet-induced animal models of NAFLD, there was a significant increase in serum TGs (from 30% to 130%)34. The mechanism for hypertriglyceridemia was related to a reduction in the production of polyunsaturated fatty acids (PUFAs) from malonyl-CoA, which decreased the activation of PPARα and induced the expression of multiple LXR/ SREBP1 target genes leading to increased hepatic VLDL secretion as well as a reduction in triglyceride clearance by lipoprotein lipase, which could be attributed in part to increased plasma apolipoprotein C3 (ApoC3) concentrations. These findings are consistent with other reports showing increased expression of LXR/ SREBP1 target genes caused by reduced levels of PUFAs35.
4. Phase I/ proof of concept data with GS-0976 and effects on DNL
A randomized, double-blind, pharmacodynamic study involving overweight/ obese but otherwise healthy adult males evaluated the effects of single doses of GS-0976 at 20 mg, 50 mg, and 200 mg on hepatic DNL36. Hepatic lipogenesis was triggered by oral fructose administration and quantified by infusing a stable isotope tracer and then monitoring its incorporation into palmitate of circulating VLDL via gas chromatography-mass spectrometry analysis. Significant inhibition of DNL by 70%, 85%, and 104% was noted with the 20 mg, 50 mg, and 200 mg doses of GS-0976, respectively, compared to placebo. Indeed, individuals that achieved plasma GS-0976 concentrations > 4 ng/mL had >90% inhibition of fractional DNL.
In an open-label, proof-of-concept study we evaluated GS-0976 given orally 20 mg once daily for 12 weeks in ten patients with suspected NASH diagnosed based on MRI proton density fat fraction (MRI-PDFF) ≥ 10% and liver stiffness by magnetic resonance elastography (MRE) ≥ 2.88 kPa37. GS-0976 reduced liver fat content by 43% with 70% of patients experiencing ≥ 30% reduction in MRI-PDFF. This reduction in hepatic TGs was associated with 22% decrease in hepatic DNL as determined by quantification of newly synthesized palmitate in blood sampled in the fasting state after a 14-day deuterated water labeling period. In addition, noninvasive markers of fibrosis showed significant improvement after 12 weeks of GS-0976 treatment including liver stiffness reduction by 9% on MRE. Overall, GS-0976 was well tolerated but two patients experienced grade 3 laboratory abnormalities including one patient who had significant elevation in ALT (peak value of 355 U/L) and another patient who had significant elevation in TG (peak value of 683 mg/dL).
5. Phase II trial of GS-0976
The phase 2, randomized, placebo-controlled trial by Loomba et al. included 126 patients who were randomized to receive GS-0976 high-dose of 20 mg once daily (n=49), GS-0976 low-dose of 5 mg daily (n=51), or placebo (n=26) for 12 weeks38 (Figure 2). All patients in the study were diagnosed with NASH and liver fibrosis stages F1 through F3 based on biopsy or by MRI-PDFF/ MRE (liver steatosis ≥8% and liver stiffness ≥ 2.5 kPa). The primary endpoint was the proportion of patients who achieved a relative decrease of at least 30% in hepatic steatosis by MRI-PDFF from baseline to week 12 (PDFF response) based on previous data demonstrating that this threshold is associated with a histologic response in NASH39.
Figure 2. Phase 2 Clinical Data on GS-0976 in Patients with NAFLD.
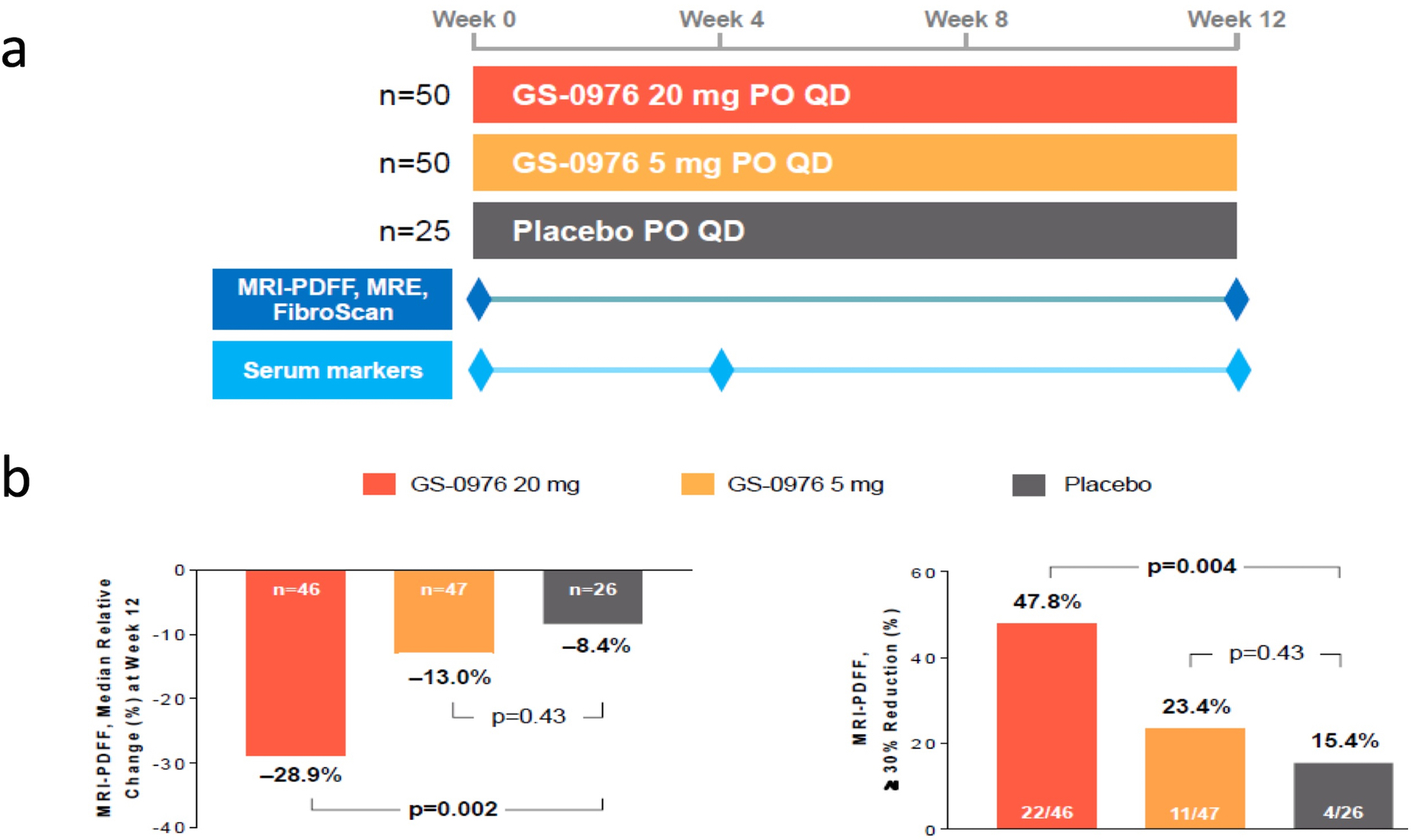
(a) Design of the phase 2 trial with GS-0976. Patients were randomized 2:2:1 into arm 1 (GS-0976 at 20 mg PO daily), arm 2 (GS-0976 at 5 mg PO daily), and arm 3 (placebo) for 12 weeks. Patients underwent MRI-PDFF at baseline and at week 12 and the primary endpoint was the proportion of patients who achieved a relative decrease of at least 30% in hepatic steatosis. (b) Efficacy data showing significant reduction in hepatic steatosis in the high-dose GS-0976 arm compared to placebo as expressed by relative steatosis reduction from baseline (28.9%) and the proportion of patients that had relative decrease of at least 30% (47.8%).
-
Efficacy
Patients receiving the higher dose of GS-0976 (20 mg once daily) demonstrated a significant 29% decrease in liver fat content measured by MRI-PDFF compared to placebo after 12 weeks of treatment (P = 0.002). The percentage of patients achieving the PDFF response of a relative decrease of at least 30% was 48% of patients receiving GS-0976 20 mg daily versus 15% in those receiving placebo (Figure 2). Further, patients treated with GS-0976 20 mg experienced a significant decrease in tissue inhibitor of metalloproteinase 1 (TIMP-1), a serum marker associated with liver fibrosis. No significant differences were noted for other markers of fibrosis such as MRE or the Enhanced Liver Fibrosis (ELF) test. Importantly, differences between GS-0976 low dose of 5 mg daily and placebo were not statistically significant for steatosis or fibrosis surrogate markers.
-
Safety and Tolerability
Nausea, abdominal pain, diarrhea and headache were the most common adverse events. At week 12, a median relative change in TGs from baseline of +11 percent, +13 percent and −4 percent was observed in patients receiving GS-0976 20 mg, GS-0976 5 mg and placebo, respectively (Figure 3). Asymptomatic Grade 3 or 4 TG elevations (>500 mg/dL) were observed in 16 patients receiving GS-0976 20 mg (n=7) or 5 mg (n=9); all of these patients had a baseline TG level >250 mg/dL (p<0.001). The majority of these patients with TG elevations either responded to fibrate or fish oil therapy (n=4) or resolved without additional treatment or cessation of GS-0976 (n=7). Total cholesterol, HDL cholesterol, LDL cholesterol and the number of small dense atherogenic LDL particles did not differ at week 12 compared to baseline. Moreover, GS-0976 did not affect plasma glucose, insulin, HbA1c or body weight. Fatty acids that are generated by DNL through ACC1 are essential in regulating platelet function and activation40; however, no effects on platelet count or the risk of bleeding were noted in the trial.
Figure 3.
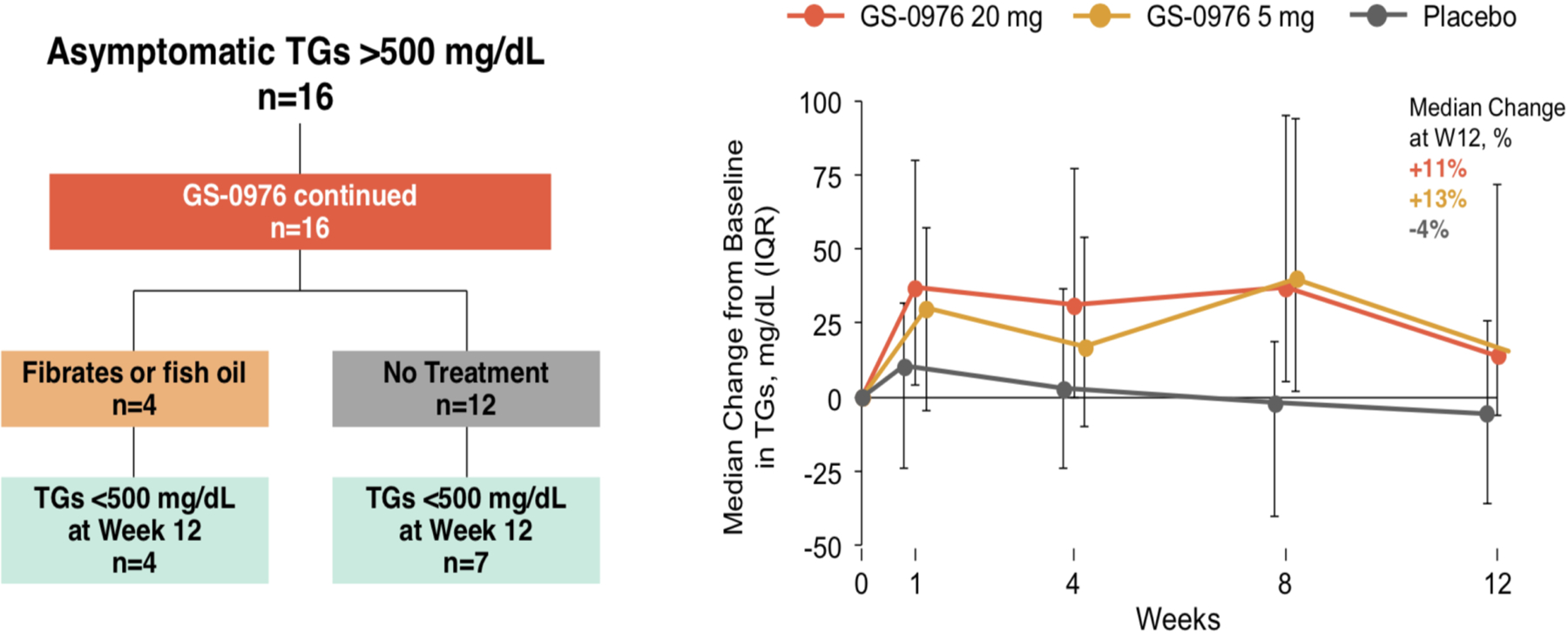
Changes in Serum Triglyceride Levels and the Clinical Course of Grade 3 or 4 Hypertriglyceridemia During the Phase 2 Trial with GS-0976.
6. Future development of GS-0976
Based on the safety and efficacy data presented above, Gilead Sciences advanced GS-0976 at a dose of 20 mg daily to be part of the ATLAS phase 2 trial ( NCT03449446) that will assess the safety and tolerability of GS-0976, selonsertib (Apoptosis signal-regulating kinase1; ASK1 inhibitor), and GS-9674 (Farnesoid X receptor; FXR agonist), administered alone or in combination, in patients with NASH and bridging fibrosis or compensated cirrhosis (F3 or F4 fibrosis). The primary efficacy endpoint is the proportion of participants who achieve a ≥ 1-stage improvement in fibrosis without worsening of NASH at week 48 based on liver histology obtained via biopsy. Other primary endpoints include the proportion of subjects experiencing adverse events or laboratory abnormalities.
The decision to study GS-0976 in combination with other agents stems from the fact that NASH development and progression starts with TG and other lipid accumulation within the liver (considered the first hit) followed by multiple parallel hits (hepatocyte apoptosis, inflammation, oxidative stress, and activation of stellate cells) that lead to fibrosis progression.
Patients with NASH and advanced fibrosis, defined as the presence of bridging fibrosis (F3) or cirrhosis (F4), have the highest rates of liver-related morbidity and mortality41–43 making them the group with the highest unmet need for treatment.
Targeting fibrosis without affecting upstream injury due to lipotoxicity may not be sufficient to induce any benefit. This is supported by the failure of simtuzumab, a monoclonal antibody against lysyl oxidase-like 2 that promotes fibrogenesis by catalyzing cross-linkage of collagen, in showing any significant fibrosis improvement in NASH patients44. In fact, simtuzumab did not significantly decrease fibrosis stage, progression to cirrhosis in patients with bridging fibrosis, or liver-related clinical events in patients with cirrhosis in a large program that included over 450 patients with biopsy-proven NASH.
7. Expert opinion
There is consensus among NASH key-opinion leaders that combination therapy may represent the future of NASH treatment for the following reasons: (1) NASH is a heterogeneous disorder with multiple pathogenic mechanisms leading to disease progression; (2) Lipotoxicity, resulting from excess fatty acids generated from increased hepatic de novo lipogenesis and white adipose tissue lipolysis, is a key driver of NASH and fibrosis development; (3) Showing efficacy in terms of fibrosis improvement, delaying or halting progression to cirrhosis, and eventually improving liver-related outcomes are pre-requisites for any long-term approval of NASH drugs; (4) Improving liver fibrosis is likely to require drugs that target upstream lipotoxicity and inflammation, as well as drugs that target the final steps of hepatic stellate cells activation and collagen production.
In this context, GS-0976 is an attractive therapeutic target to inhibit hepatic de novo lipogensis and decrease the deleterious effects of lipotoxicity. Combining GS-0976 with other NASH drugs that are expected to have anti-fibrotic effects represents a sound strategy that is supported by preclinical data that suggest enhanced effects with combination therapy45.
Recent evidence suggests that increased DNL through diets that are high in fructose may trigger the development of hepatocellular carcinoma (HCC), a devastating complication of NASH that can develop even in the absence of cirrhosis46–48. A recent study demonstrated that DNL inhibition via the ACC inhibitor ND-645 decreased the development of HCC and improved survival of tumor-bearing rats49. In fact, the combination of ND-645 and sorafenib significantly reduced the incidence of HCC by 81%. Whether GS-0976 has a protective effect against the development of HCC in patients with NASH and whether it may have a role as an add-on therapy to existing HCC drugs remains to be determined in future studies. Blocking other enzymes involved in the DNL pathway such as stearoyl-CoA desaturase has been shown to suppress HCC growth lending further support to the concept of targeting lipid metabolism as therapeutic target50.
Despite impressive preliminary data on the efficacy of GS-0976 on inhibiting DNL and reducing hepatic steatosis, the drug has several drawbacks that may make it less competitive in the future NASH landscape including:
-
Hypertriglyceridemia:
As discussed previously, GS-0976 was associated with an increase in plasma TG concentration and the development of grade 3–4 hypertriglyceridemia in the proof-of-concept and phase 2 clinical trials. This was also corroborated in animal studies with GS-0976 and appears to be a class effect since other ACC inhibitors also cause hypertriglyceridemia in animals and humans35. Restricting GS-0976 treatment to subjects with TG < 250 mg/dL at baseline could mitigate the development of grade 3–4 hypertriglyceridemia and has been implemented in the ATLAS phase 2 trial. The increase in TG tends to be transient and may improve spontaneously although the compensatory mechanisms are not clearly delineated. Given that the mechanism for hypertriglyceridemia is related to inhibition of PPARα and reduced levels of PUFAs, fibrates and fish oil represent attractive management strategies.
-
Effects on Glucose Homeostasis:
Decreasing DNL by ACC1 inhibition leads to reductions in hepatic DAG content22,34, which in turn leads to reductions in PKCε translocation to the plasma membrane and decreased insulin receptor kinase activity thereby increasing hepatic insulin sensitivity by increasing insulin-stimulated hepatic glycogen synthesis,51. Conversely increasing fatty oxidation by ACC2 inhibition can result in increased accumulation of mitochondrial acetyl-CoA, which in turn can activate pyruvate carboxylase leading to increased hepatic gluconeogenesis52. In preclinical studies, these effects oppose each other leading to minimal effects of GS-0976 on whole body glucose homeostasis. Ongoing clinical studies are examining whether GS-0976 has similar effects on hepatic glucose metabolism in humans with NAFLD.
-
Competitive therapeutic landscape:
Multiple drugs with potent anti-steatotic action and beneficial effects on lipids and cardiovascular outcomes may prove to be more efficacious than GS-097623. These drugs can be broadly divided into two classes:- Drugs that are being developed specifically for NASH including thyroid hormone receptor-β agonists and aramchol.
- Drugs that already are approved to treat T2D and have beneficial effects on hepatic steatosis and cardiovascular outcomes including glucagon-like peptide-1 (GLP-1) receptor agonists and sodium-glucose cotransporter-2 (SGLT2) inhibitors.
In conclusion, currently available preclinical and clinical data support the development of GS-0976 as part of combination therapy for NASH to decrease hepatic steatosis, lipotoxicity, and their downstream deleterious effects. Several safety issues remain including the class-effect on plasma triglycerides and the potential to worsen cardiovascular outcomes and impair glucose homeostasis.
Funding
This paper was supported by grants from the United States Public Health Service: R01 DK113984, R01 DK114793, DK116774, DK119968, P30 DK45735, Gilead Sciences and The Novo Nordisk Foundation Center for Basic Metabolic Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH.
Footnotes
Declaration of Interest
GI Shulman receives investigator-initiated support from Merck, AstraZeneca and Gilead Sciences (the maker of GS-0976) and he serves on Scientific Advisory Boards for Merck, AstraZeneca, Gilead Sciences, Aegerion Pharmaceuticals, Novo Nordisk, iMetabolic Biopharma and Janseen Research and Development. N Alkhouri, E Lawitz, M Noureddin, and R DeFronz receive research funding from, and serve on Scientific Advisory Board for Gilead Sciences (the maker of GS-0976). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
One reviewer received study support and provided consultation services for Gilead Sciences. One reviewer has served as a consultant for Astra Zeneca, Pfizer, AMGEN, Sanofi, Camp4, Medacorp, foresite labs, Akcea, Ionis, and Celgene.
Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. [DOI] [PubMed] [Google Scholar]
- 2.Setiawan VW, Stram DO, Porcel J, Lu SC, Le Marchand L, Noureddin M. Prevalence of chronic liver disease and cirrhosis by underlying cause in understudied ethnic groups: The multiethnic cohort. Hepatology. 2016;64(6):1969–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.•.Kabbany MN, Conjeevaram Selvakumar PK, Watt K, et al. Prevalence of Nonalcoholic Steatohepatitis-Associated Cirrhosis in the United States: An Analysis of National Health and Nutrition Examination Survey Data. The American journal of gastroenterology. 2017;112(4):581–587. [DOI] [PubMed] [Google Scholar]; Important study that provided evidence for the disease burden of NASH as a leading cause for advanced liver disease in the United States.
- 4.Noureddin M, Vipani A, Bresee C, et al. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. The American journal of gastroenterology. 2018;113(11):1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cusi K, Orsak B, Bril F, et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med. 2016;165(5):305–315. [DOI] [PubMed] [Google Scholar]
- 6.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New England journal of medicine. 2010;362(18):1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328–357. [DOI] [PubMed] [Google Scholar]
- 8.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Younossi Z, Ratziu V, Loomba R, et al. Positive Results from REGENERATE: A Phase 3 International, Randomized, Placebo-Controlled Study Evaluating Obeticholic Acid Treatment for NASH. Hepatology. 2019;70(1):Abstract GS-06. [Google Scholar]
- 10.Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert review of gastroenterology & hepatology. 2009;3(4):445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abulizi A, Perry RJ, Camporez JPG, et al. A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017;31(7):2916–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347(6227):1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 2018;283(4):356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. The Journal of clinical investigation. 2005;115(5):1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.•.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study established the role of increased de novo lipogenesis in the liver as a a major source of hepatic steatosis in subjects with NAFLD.
- 16.Vatner DF, Majumdar SK, Kumashiro N, et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(4):1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cellular and molecular life sciences : CMLS. 2005;62(16):1784–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KH. Regulation of mammalian acetyl-coenzyme A carboxylase. Annual review of nutrition. 1997;17:77–99. [DOI] [PubMed] [Google Scholar]
- 19.Brownsey RW, Zhande R, Boone AN. Isoforms of acetyl-CoA carboxylase: structures, regulatory properties and metabolic functions. Biochemical Society transactions. 1997;25(4):1232–1238. [DOI] [PubMed] [Google Scholar]
- 20.Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl-CoA carboxylase 2. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(4):1444–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abu-Elheiga L, Jayakumar A, Baldini A, Chirala SS, Wakil SJ. Human acetyl-CoA carboxylase: characterization, molecular cloning, and evidence for two isoforms. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(9):4011–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. The Journal of clinical investigation. 2006;116(3):817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samuel VT, Shulman GI. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018;27(1):22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunkeler M, Stuttfeld E, Hagmann A, Imseng S, Maier T. The dynamic organization of fungal acetyl-CoA carboxylase. Nature communications. 2016;7:11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim CW, Moon YA, Park SW, Cheng D, Kwon HJ, Horton JD. Induced polymerization of mammalian acetyl-CoA carboxylase by MIG12 provides a tertiary level of regulation of fatty acid synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(21):9626–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fullerton MD, Galic S, Marcinko K, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nature medicine. 2013;19(12):1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abu-Elheiga L, Oh W, Kordari P, Wakil SJ. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(18):10207–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.•.Abu-Elheiga L, Wu H, Gu Z, Bressler R, Wakil SJ. Acetyl-CoA carboxylase 2−/− mutant mice are protected against fatty liver under high-fat, high-carbohydrate dietary and de novo lipogenic conditions. The Journal of biological chemistry. 2012;287(15):12578–12588. [DOI] [PMC free article] [PubMed] [Google Scholar]; Experimental evidence for the effect of genetic ACC2 inhibition on improving diet-induced liver steatosis in vivo.
- 29.Olson DP, Pulinilkunnil T, Cline GW, Shulman GI, Lowell BB. Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(16):7598–7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoehn KL, Turner N, Swarbrick MM, et al. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab. 2010;11(1):70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao J, DeMayo FJ, Li H, et al. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(22):8552–8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.•.Tong L, Harwood HJ Jr. Acetyl-coenzyme A carboxylases: versatile targets for drug discovery. Journal of cellular biochemistry. 2006;99(6):1476–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]; Important review on ACC enzymes as therapeutic targets for the metabolic syndrome and its complications.
- 33.Harriman G, Greenwood J, Bhat S, et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(13):E1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goedeke L, Bates J, Vatner DF, et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology. 2018;68(6):2197–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim CW, Addy C, Kusunoki J, et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017;26(3):576. [DOI] [PubMed] [Google Scholar]
- 36.Stiede K, Miao W, Blanchette HS, et al. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: A randomized, double-blind, crossover study. Hepatology. 2017;66(2):324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawitz EJ, Coste A, Poordad F, et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2018;16(12):1983–1991 e1983. [DOI] [PubMed] [Google Scholar]
- 38.•.Loomba R, Kayali Z, Noureddin M, et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2018;155(5):1463–1473 e1466. [DOI] [PMC free article] [PubMed] [Google Scholar]; Phase 2 trial demonstratign the efficacy of GS-0976 on reducing liver fat and other markers of liver injury in patients with NAFLD.
- 39.Patel J, Bettencourt R, Cui J, et al. Association of noninvasive quantitative decline in liver fat content on MRI with histologic response in nonalcoholic steatohepatitis. Therapeutic advances in gastroenterology. 2016;9(5):692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lepropre S, Kautbally S, Octave M, et al. AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood. 2018;132(11):1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2015;149(2):389-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ekstedt M, Hagstrom H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547–1554. [DOI] [PubMed] [Google Scholar]
- 43.Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2015;13(4):643–654 e641–649; quiz e639–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harrison SA, Abdelmalek MF, Caldwell S, et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology. 2018;155(4):1140–1153. [DOI] [PubMed] [Google Scholar]
- 45.Bates J, Hollenback D, Zagorska A, et al. Combination of ASK1 and ACC Inhibitors Increases Efficacy in Rodent Models of NASH. Hepatology. 2017;66(S1):Abstract 425. [Google Scholar]
- 46.Mittal S, El-Serag HB, Sada YH, et al. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2016;14(1):124–131 e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohamad B, Shah V, Onyshchenko M, et al. Characterization of hepatocellular carcinoma (HCC) in non-alcoholic fatty liver disease (NAFLD) patients without cirrhosis. Hepatology international. 2016;10(4):632–639. [DOI] [PubMed] [Google Scholar]
- 48.Noureddin M, Rinella ME. Nonalcoholic Fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clinics in liver disease. 2015;19(2):361–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.•.Lally JSV, Ghoshal S, DePeralta DK, et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019;29(1):174–182 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]; Experimental evidence of the role of ACC inhibition as a potential therapeutic target for liver cancer.
- 50.Bansal S, Berk M, Alkhouri N, Partrick DA, Fung JJ, Feldstein A. Stearoyl-CoA desaturase plays an important role in proliferation and chemoresistance in human hepatocellular carcinoma. The Journal of surgical research. 2014;186(1):29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98(4):2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perry RJ, Camporez JG, Kursawe R, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160(4):745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]