

Abstract
Chemokines bind to membrane-spanning chemokine receptors, which signal through G proteins and promote cell migration. However, the atypical chemokine receptor 3 (ACKR3) does not appear to couple to G proteins and instead of directly promoting cell migration, it regulates the extracellular concentration of chemokines that it shares with G protein-coupled receptors (GPCRs) CXCR3 and CXCR4, thereby influencing the migration and signaling responses of these receptors. How these receptors bind their ligands is important for understanding these different processes. Here, we applied association and dissociation kinetic measurements coupled to β-arrestin recruitment assays to investigate ACKR3:chemokine interactions. Our results showed that CXCL12 binding is unusually slow and driven by the interplay between multiple binding epitopes. We also found that the N-terminus of the receptor played a key role in chemokine binding and activation by preventing chemokine dissociation. It has been previously hypothesized that chemokines initially bind receptors through interactions between the globular domain of the chemokine and the receptor N-terminus, which then guides the chemokine N-terminus into the transmembrane pocket of the receptor to initiate signaling. On the basis of our kinetic data, we propose an alternative mechanism in which the N-terminus of the chemokine initially forms interactions with the extracellular loops and transmembrane pocket of the receptor followed by the receptor N-terminus wrapping around the core of the chemokine to prolong its residence time. These data provide insight into how ACKR3 competes and cooperates with canonical GPCRs in its function as a scavenger receptor.
Introduction
Chemokines regulate cell migration and other physiological processes by binding to and activating chemokine receptors in the cell membrane (1, 2). For the majority of the 20 human chemokine receptors, chemokine binding leads to G protein-mediated activation of downstream signaling pathways. Phosphorylation by G protein receptor kinases (GRKs) and recruitment of β-arrestins often follow and trigger additional signaling pathways as well as receptor desensitization and internalization. However, some chemokine receptors do not signal through G proteins and are therefore referred to as atypical chemokine receptors (ACKRs) (3). ACKR3 (also known as CXCR7), is one such ACKR that interacts with CXCL11 and CXCL12, chemokines shared by the G protein-coupled receptors CXCR4 and CXCR3, respectively (4, 5). In contrast to most canonical chemokine receptors that promote cell migration, one of the main functions of ACKR3 is to regulate extracellular chemokine concentrations by ligand scavenging, which in turn maintains responsiveness of cells expressing CXCR4 (6, 7). Despite its unique pharmacology, ACKR3 appears structurally similar to the canonical G protein-coupled chemokine receptors (8); thus, it is unclear how it competes with CXCR4 and CXCR3 for their mutual ligands.
Similar to other Class A GPCRs, the three-dimensional structure of chemokine receptors consists of seven transmembrane (7TM) helices, flanked by an extended extracellular N-terminus and intracellular C-terminus and connected by extracellular and intracellular loops (9–12). Chemokines are small soluble proteins comprised of a disordered N-terminus appended to a globular core that is stabilized by disulfide bonds. The interaction interface between chemokines and their receptors has historically been described in terms of two chemokine recognition sites (CRS) (2, 13–15): CRS1 includes interactions between the core of the chemokine and the N-terminus of the receptor while CRS2 involves the orthosteric pocket and extracellular loops (ECLs) of the receptor and the N-terminus of the chemokine (Fig 1A) (16). Crystal structures of CXCR4 in complex with the viral chemokine vMIP-II (17), US28 in complex with CX3CL1 (18) and CCR5 in complex with an antagonist version of CCL5 (19) confirmed this model but also showed that chemokines have a continuous interaction interface with the receptor in the region between CRS1 and 2, which was consequently termed CRS1.5 (16, 17). However, the crystal structures all lack information about the distal N-terminus of the receptor since this region was not resolved in any of the structures. Using radiolytic footprinting experiments to define features of the ACKR3:CXCL12 complex, we recently uncovered an additional point of contact between this region (residues 2–7 of ACKR3) and the β1 strand of CXCL12 (8), and in analogy with previous nomenclature, this region is referred to as CRS0.5 (Fig 1A). Mutagenesis studies in combination with binding and functional assays suggest that the receptor N-terminus generally contributes primarily to chemokine binding while CRS2 interactions in the receptor binding pocket are important for both binding affinity and receptor activation (2, 13). Nuclear magnetic resonance (NMR) studies show that soluble peptides corresponding to receptor N-termini are able to interact with chemokines (20) and that in the case of CXCR4, the chemokine can remain bound to the full-length receptor even in the presence of a small molecule in the orthosteric pocket (21). Based in part on these observations, the receptor N-terminus has been hypothesized to act as an initial interface for chemokine docking, which anchors the chemokine and allows the chemokine N-terminus to form interactions within the orthosteric pocket of the receptor in a two-step mechanism (13, 14, 21). However, there is little experimental data that supports or refutes this model, especially regarding the order in which interactions are formed and their relative importance for chemokine association and dissociation. More generally, the role that the receptor N-terminus plays in chemokine binding and how interactions involving the receptor N-terminus influence those in the receptor binding pocket remain to be fully elucidated.
Fig. 1. Arrestin recruitment of N-terminally truncated ACKR3 variants.
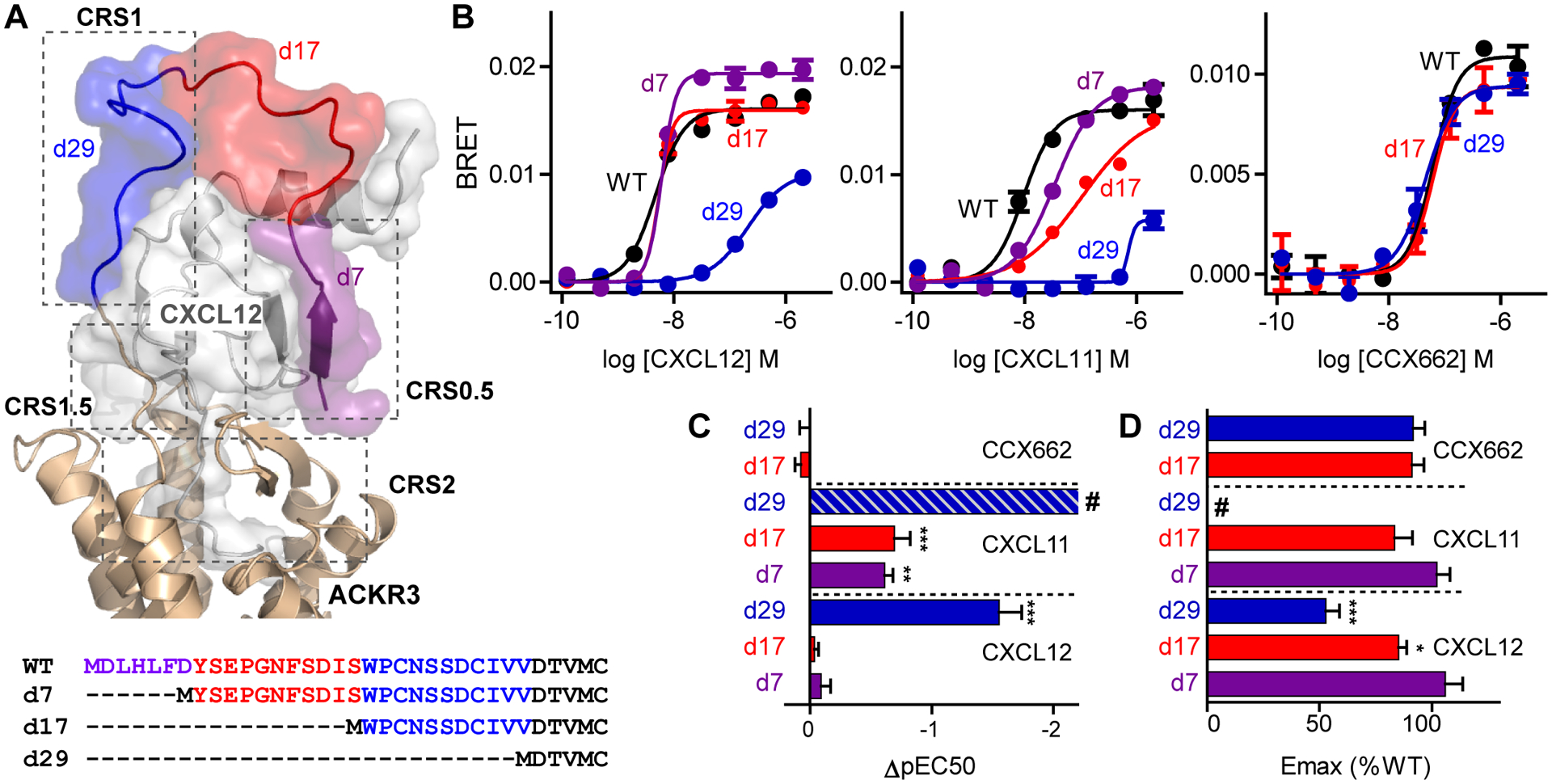
(A) Extracellular portion of the ACKR3:CXCL12 complex from experimentally-driven molecular model (8) and sequences of ACKR3 truncation mutants. Truncated regions are highlighted according to the color scheme in the model. (B) Representative examples of recruitment of GFP10-β-arrestin-2 to ACKR3-Rluc3 variants stimulated with different concentrations of CXCL12WT, CXCL11 and CCX662 measured by BRET. Each point corresponds to the average and standard error of two measurements. (C and D) Potency (C) and efficacy (D) of ligand-induced β-arrestin-2 recruitment determined relative to ACKR3WT from fitting of dose response curves. pEC50 and Emax values for truncation mutants were determined as ΔpEC50=pEC50,mutant-pEC50,WT and %Emax =Emax,mutant/Emax,WT × 100. Data are mean and standard error of three or more experiments. Significantly lowered potency or efficacy relative to ACKR3WT with the same ligand is noted: *P<0.05, **P<0.01, ***P<0.001 from one-way ANOVA with Dunnett’s multiple comparison test. # in (C) indicates that CXCL11-induced arrestin recruitment by ACKR3d29 could not be accurately fit and ΔpEC50 was estimated from the raw data to be >−2.2.
Here, we characterized the contribution of the N-terminus to ACKR3 binding and activation by using receptor truncations, chemokine mutants and ligand association/dissociation kinetic experiments. The data suggest a modified mechanism for the role of the receptor N-terminus in chemokine binding that differs from the previously hypothesized mechanism. Although there are no “hot spots” of interaction in the N-terminus, it plays an important role in modulating the residence time of the chemokine on the receptor. The data also suggest an interdependency of interactions involving the receptor N-terminus with those in the receptor binding pocket, and that at least in the case of ACKR3:CXCL12, the chemokine first engages the orthosteric pocket, which is followed by interactions with the receptor N-terminus. The kinetic mechanisms by which ACKR3 binds CXCL12 show key differences from CXCR4:CXCL12 that can be reconciled with its function as a scavenger receptor.
Results
Truncation of the receptor N-terminus affected its activation by chemokine but not by a small-molecule agonist
To test the contribution of the ACKR3 N-terminus to chemokine binding and activation, three N-terminal truncation mutants of ACKR3 were generated. Truncation of the first 29 residues (ACKR3d29) removed both CRS1 and CRS0.5 regions as well as the disulfide between residue 21 and 26 (8); truncation of the first 17 residues (ACKR3d17) removed CRS0.5 and part of the CRS1 region; and truncation of the first seven residues (ACKR3d7) retained all interactions except those in the CRS0.5 region (Fig 1A). To determine the effect of the truncations on receptor function, we then tested the ability of the mutant receptors expressed in human embryonic kidney (HEK) 293T cells with Renilla luciferase 3 (Rluc3) fused to their C-termini to recruit green fluorescence protein 10 (GFP10)-β-arrestin-2 using a bioluminescence resonance energy transfer (BRET) based assay. Stimulation with CXCL12WT, ACKR3d7 and ACKR3d17 recruited arrestin with the same potency as ACKR3WT and only a slightly lowered efficacy (Fig 1B, C and D). By contrast, truncation of the first 29 residues of ACKR3 lead to a significantly lowered potency and efficacy of CXCL12-induced arrestin recruitment indicating that the N-terminus, specifically the region between residues 17–29, was important for receptor function.
CXCL11 showed important differences from CXCL12: its ability to activate ACKR3 was affected by all three truncated receptor variants (Fig 1B, C and D). ACKR3d7 and ACKR3d17 had a significantly lowered potency compared to the wild type receptor for CXCL11-induced βarrestin-2 recruitment, while ACKR3d29 was severely impaired and only recruited β-arrestin-2 at the highest concentration of CXCL11, precluding an accurate determination of potency and efficacy values. These data underscore the strong dependence of CXCL11 on the ACKR3 N-terminus for receptor activation.
By contrast to the chemokines, the small molecule CCX662 (22), predicted to bind in the orthosteric pocket, recruited β-arrestin-2 with similar potencies and efficacies by activating ACKR3WT, ACKR3d17 and ACKR3d29 (Fig 1B, C and D). Furthermore, the efficacies approached that observed for activation of ACKR3WT by CXCL12WT (fig S1A, B). This result confirmed that the receptor can be activated almost fully even in the absence of the N-terminus and by a small molecule agonist. It also suggested that all three ACKR3 variants are equally expressed at the surface, although this could not be directly tested due to a lack of antibodies against ACKR3d17 and ACKR3d29 and the need to keep the receptor N-terminus free from affinity tags (fig S2A, B, C and D).
Together, these results showed that both CXCL11 and CXCL12 relied on interactions with the receptor N-terminus for their function. The key interactions with CXCL12 appeared to be concentrated to residues 17–29 whereas CXCL11 required the full N-terminus of the receptor (Fig 1B, C and D).
The kinetics of CXCL12 binding to ACKR3 was unusually slow
To further probe the role of the ACKR3 N-terminus on its interaction with chemokine we characterized the binding of CXCL11 and CXCL12 to ACKR3WT and the truncation mutants ACKR3d17 and ACKR3d29. Ligand binding to chemokine receptors is typically measured using equilibrium experiments to obtain IC50 or in some cases Kd values. Although informative, these numbers do not provide information on association and dissociation rates of ligands, which are important for a complete understanding of the binding mechanisms and the effects of mutations. We therefore sought to develop assays to determine association and dissociation rates for chemokine binding to receptors.
Chemokine off-rates were measured by co-expressing ACKR3WT with hemagglutinin (HA)tagged chemokines expressed in Spodoptera frugiperda Sf9 cells. The chemokines were then labeled with Fluorescein isothiocyanate (FITC)-conjugated antibody against HA, and following the addition of a large excess of the small molecule CCX777 (8, 23) to prevent re-association of the chemokine, time-dependent chemokine dissociation was followed by flow cytometry. CXCL12WT had a dissociative half-life of 102 ± 18 min (average ± standard error of n=6 independent measurements, represented in Fig 2A), which is longer than what has been reported for CCR2:CCL2 (21 min) and CXCR4:CXCL12 (1.4 min) (24, 25). For example, when assayed by surface plasmon resonance (SPR) (25), CXCL12 had an approximately 70-fold faster off-rate from CXCR4 than that observed for ACKR3, which was greater than the approximately 10-fold difference in affinity between the receptors typically reported from competition binding experiments (Table S1). When tested in our assays, CXCL11 had a faster dissociation rate than CXCL12. In fact, using our current experimental setup the dissociation was too fast to accurately quantify. These results highlight another substantial difference between the two native ligands, CXCL11 and CXCL12, in their interaction with ACKR3.
Fig. 2. Kinetics of CXCL12 binding to ACKR3.
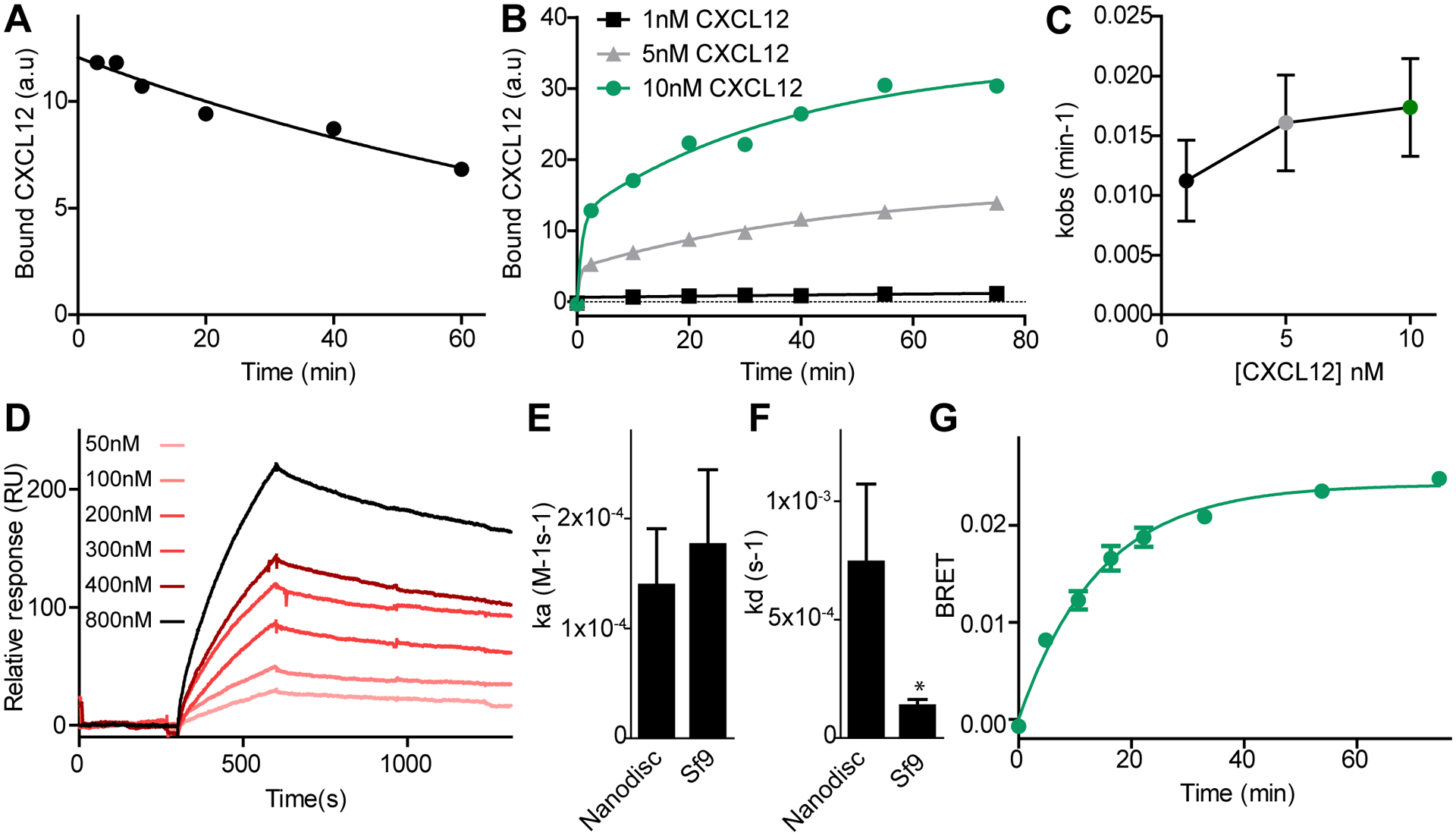
(A) Dissociation of CXCL12WT was measured by flow cytometry by monitoring the decrease in geometric mean of FITC fluorescence after addition of the small molecule ligand CCX777 to live Sf9 cells co-expressing ACKR3WT and HA-tagged CXCL12WT bound to FITC-conjugated antibody. (B) Association of CXCL12WT to ACKR3WT after adding HA-tagged CXCL12WT complexed with FITC-conjugated antibody against HA to cells expressing ACKR3WT. (C) kobs values for the slow phase of CXCL12WT association to ACKR3WT at different ligand concentrations determined from fitting the data to a two component exponential equation. (D) Binding of ACKR3 in nanodiscs to immobilized CXCL12 measured by SPR. (E and F) Association (E) and dissociation rate constants (F) were determined for binding of ACKR3 in nanodiscs to immobilized CXCL12 detected by SPR, and for CXCL12 binding to ACKR3 in live Sf9 cells detected by flow cytometry. The asterisk indicates that the dissociation rate is slower in Sf9 cells as determined from unpaired t-test with p < 0.05. (G) Kinetics of β-arrestin-2 recruitment to ACKR3WT in HEK293T cells followed by BRET. Curves in (A), (B), (D) and (G) are representative examples of six (A) or three (B, D and G) independent results, and each point or bar in (C), (E) and (F) are means and standard errors of three or more experiments.
To assay chemokine association, HA-tagged CXCL12WT was again labeled with a FITC-conjugated anti-HA antibody. FITC-labeled CXCL12WT was then added to ACKR3WT-expressing Sf9 cells and association was followed using flow cytometry (fig S3A). Fitting of the association data required a multiple component exponential due to an initial fast phase corresponding to 35 ± 4 % that was too rapid to quantify using the current methods (fig S3B). This fast phase was followed by a slower phase representing the remaining 65% of the data, which was characterized by an observed rate (kobs) of 0.019 ± 0.004 min−1 when 10 nM CXCL12WT was used in the experiment (Fig 2B). Based on the measured off-rate and the expression for kobs of a ligand interaction with a single state receptor, this equated to an on-rate of 1.8 ± 0.7 × 104 M−1 s−1, which is approximately 25-fold slower than what has been measured for CXCR4 (25).
In standard bimolecular binding reactions, kobs is expected to increase linearly with increasing ligand concentration. From the association data (Fig 2C) it is not clear if CXCL12WT binding follows this ideal behavior since the cell-based experiments did not allow for a sufficiently wide range of chemokine concentrations to test this due to high receptor-independent binding of chemokines to cells at CXCL12 concentrations above 10 nM (likely due to interactions with glycosaminoglycans (26)). To determine if the binding constants were indicative of a pseudo first-order interaction and to confirm the slow kinetics of CXCL12 binding, we therefore utilized SPR. C-terminally biotinylated CXCL12 (CXCL12-biotin) was immobilized on a SPR chip and the binding of purified ACKR3 reconstituted into nanodiscs (27) was followed at different concentrations of receptor (Fig 2D). Fitting of the binding kinetics suggested that the SPR data followed a pseudo first-order kinetic model with a single exponential being sufficient to fit the association (fig S4A). Association of ACKR3 to CXCL12-biotin occurred on a time scale similar to the dominant association phase seen in cells (Fig 2E), whereas dissociation of ACKR3 in nanodiscs was faster than that detected by the cell-based methods (Fig 2F). The faster dissociation rate was likely an effect of biotin interfering with CXCL12 binding to receptor as previously observed for S6-tagged CXCL12 binding to CXCR4 (28). Consistent with this hypothesis, the midpoints of thermal unfolding (Tm) of the purified ACKR3:CXCL12-biotin complex was reduced relative to ACKR3:CXCL12 (fig S4B). Equivalent SPR experiments with ACKR3 reconstituted into n-dodecyl-β-D-maltopyranoside (DDM):cholesteryl hemisuccinate (CHS) detergent micelles showed no differences in association or dissociation constants compared to the results in nanodics (fig S4C–E).
A single-exponential function was sufficient to fit receptor:chemokine association in the SPR experiments whereas association in Sf9 cells fit best to a two-component model. A plausible reason for this difference could be the fact that receptors in cell membranes exist in different environments, which may affect chemokine association rates. The presence of cell surface glycosaminoglycans, which are well-known to bind chemokines (26), may also contribute to apparent inhomogeneity. Also, 7TM receptors are known to be inherently dynamic and even in the absence of ligand populate multiple active and inactive states (29), which can have different ligand association rates (30) and could contribute to the two-phase behavior of CXCL12 binding to ACKR3 in Sf9 cells. This effect might be less prominent in the SPR experiments since these experiments are done at higher concentrations and with purified receptors in solution binding to immobilized chemokines, in contrast to the experiments in Sf9 cells where soluble chemokines bind receptors immobilized in the cell membrane. A two-phase behavior similar to that observed for CXCL12 binding to ACKR3 in Sf9 cells has been observed for live cell ligand binding to the peptide-binding class B GPCR, Parathyroid Hormone 1 Receptor (PTH1R), in mammalian cells (31). In the case of PTH1R, this behavior was explained by an initial binding step followed by a conformational rearrangement of the receptor:peptide complex. Since our binding experiments do not distinguish different bound states, it is unlikely that a similar model explains the two-phase behavior of ACKR3.
Together, the data showed that the time scale of CXCL12WT binding to ACKR3WT was similar in three different membrane-mimicking systems (Sf9 cells, nanodiscs and DDM/CHS micelles), with two different experimental setups (flow cytometry and SPR) and with detection of receptor binding to immobilized chemokine or chemokine binding immobilized receptor. Based on these results we concluded that the slow binding kinetics was an inherent feature of the ACKR3:CXCL12 interaction.
To further determine if the measured binding kinetics are relevant in the context of CXCL12 activation of ACKR3 we tested the rate of arrestin association to the receptor using time-resolved BRET experiments where arrestin association was quantified at different time points after chemokine addition (Fig 2G). When fit to a single exponential, the kobs for arrestin recruitment to ACKR3WT induced by 10 nM CXCL12WT was 3-fold faster than the observed kobs of the slow phase of binding to Sf9 cells at the same concentration. The faster rate could be explained by the higher temperature used in the BRET experiments since HEK293T cells were maintained at 37°C during chemokine association and the Sf9 cells and SPR experiments were performed at ambient temperature. Nevertheless, comparison of the kobs values for arrestin association and CXCL12 binding showed that arrestin recruitment happened on similar time scale to CXCL12 association, suggesting that chemokine binding could be rate-limiting for arrestin association.
ACKR3 is known to have an approximately 10-fold higher affinity for CXCL12 than CXCR4 (Table S1); thus these results suggest that hidden within this relatively modest affinity difference are more significant differences in binding kinetics where ACKR3 has a slower CXCL12 association rate than CXCR4 but the even slower dissociation rate makes it a higher affinity binder.
N-terminal truncations of ACKR3 affected chemokine dissociation but not association
To test the role of the receptor N-terminus in ligand binding we examined truncated versions of ACKR3 for their ability to bind ligands in stability and kinetic association/dissociation experiments. Stable expression of the truncated receptors required fusion of thermostabilized apocytochrome b562 (bril) to the N-terminus of ACKR3, the addition of which did not affect the dissociation rate or fold of the receptor (fig S5A, B, C and D). To confirm proper folding of the truncated constructs, receptor:ligand complexes were purified from Sf9 cells (Fig 3A) and tested in thermostability assays (32). Bril-ACKR3WT, bril-ACKR3d17 and bril-ACKR3d29 had identical Tm when in complex with the small molecule CCX662 (Fig 3B). This confirmed the structural integrity of the truncated complexes and was in perfect agreement with CCX662-induced arrestin recruitment experiments (Fig 1B, C and D). In complex with the CXCL12 mutant CXCL12LRHQ, which was selected from a phage display library (33) and has the three first residues in its sequence replaced by Leu-Arg-His-Gln, bril-ACKR3d29 had a lower Tm compared to the other ACKR3 variants (Fig 3B). This is again consistent with the data described above (Fig 1B, C and D) where ACKR3d29 had a lower potency and efficacy of arrestin recruitment when stimulated with CXCL12WT. CXCL12WT association experiments (Fig 3C) showed that the population of the slow receptor binding phase was slightly higher for bril-ACKR3d29 compared to bril-ACKR3WT (78% vs 65%, Fig 3D) but there was no significant difference between bril-ACKR3WT and bril-ACKR3d17 or bril-ACKR3d29 in the rate of the slow association phase (Fig 3E). Consistent with the binding data, time-resolved BRET experiments with 10 nM CXCL12 (Fig 3F) showed no significant differences in the rate of arrestin recruitment due to the N-terminal truncations (Fig 3G). In contrast, bril-ACKR3d29 had a 30-fold faster chemokine dissociation rate from receptors in Sf9 cells compared to bril-ACKR3WT (Fig 3H and I). These data demonstrated that the main effect of the N-terminal truncation on arrestin recruitment to ACKR3d29 can be attributed to an increased dissociation rate from the receptor. This result also suggests that the role of residues 18–29 of ACKR3 is primarily to prevent chemokine dissociation after the chemokine has engaged the receptor (Fig 3J).
Fig. 3. Ligand binding kinetics of truncated ACKR3 variants.
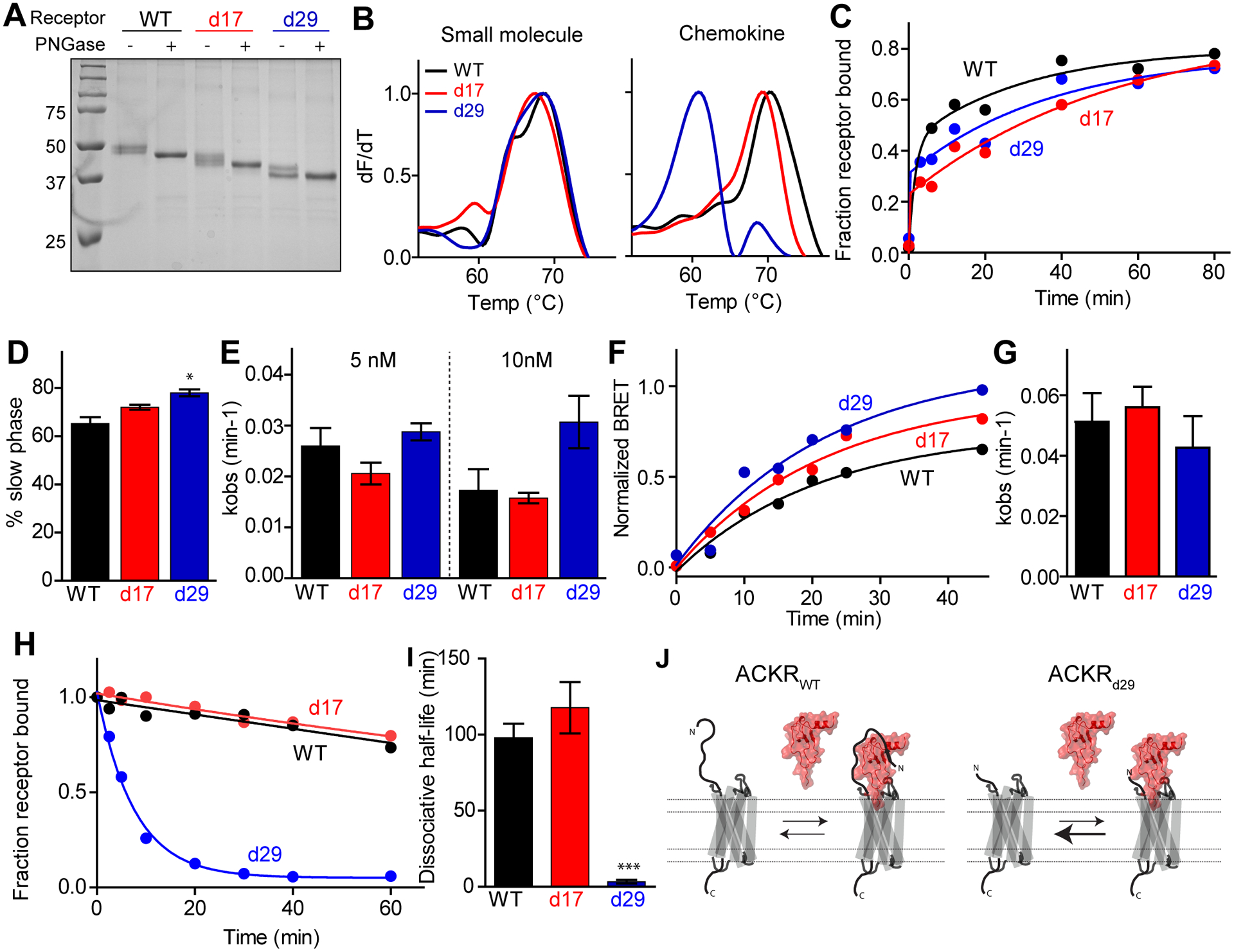
(A) Representative example of SDS-PAGE of purified bril-ACKR3WT, bril-ACKR3d17 and bril-ACKR3d29. Addition of PNGase F deglycosylates the receptor. (B) Representative example of thermal unfolding of ACKR3 variants in complex with CCX662 and CXCL12LRHQ measured using CPM fluorescence (32). (C) Representative example of CXCL12WT association to ACKR3 variants at 5 nM chemokine detected by flow cytometry. (D) Percent of the association curves corresponding to the slower phase of chemokine association determined from fitting association curves at 10 nM CXCL12WT to a two-phase exponential equation. Bars represent the average and standard errors of three or more experiments. ACKR3d29 has a larger slow component than ACKR3WT (P<0.05 as determined from one-way anova with Dunnett’s multiple comparison test). (E) Mean and standard errors from three or more measurements of kobs values for the slow phase in (D). (F) Representative example of arrestin recruitment to ACKR3 variants determined from BRET experiments after addition of 10 nM CXCL12WT. (G) Average and standard errors of kobs values from fitting three time-resolved BRET experiments to single exponential equations. (H) Representative example of CXCL12WT dissociation from ACKR3:CXCL12 complexes in Sf9 cells. (I) Mean and standard errors of CXCL12WT dissociative half-life determined from fitting three or more dissociation curves to a single-phase exponential equation. The dissociative half-life of bril-ACKR3d29 is significantly shorter than ACKR3WT (P<0.001 as determined from one-way anova with Dunnett’s multiple comparison test). (J) Schematic representation of CXCL12 binding equilibria for ACKR3WT and ACKR3d29 highlighting the faster dissociation rate but unchanged association rate of CXCL12 binding to the truncated receptor.
The impact of ACKR3 N-terminal truncation was modulated by chemokine dissociation rates
Compared to other chemokine receptors, point mutations of the ACKR3 N-terminus have been reported to have relatively minor effects on the ability of the receptor to be activated by CXCL12 and CXCL11(34, 35). Our data showing that N-terminal truncations of ACKR3 had major effects on both CXCL11 and CXCL12 activation was therefore surprising. Of the two chemokines, CXCL11-mediated β-arrestin-2 recruitment to ACKR3 was more sensitive to mutations (34) and truncations of the receptor N-terminus than recruitment induced by CXCL12 (Fig 1B, C and D). Additionally, CXCL11 had a faster off-rate from the receptor than CXCL12, too fast to measure with our assay. Based on these observations, we hypothesized that the dissociation rate of a chemokine may affect how sensitive its potency and efficacy of activation are to N-terminal receptor truncations. To test this hypothesis, we compared binding kinetics and β-arrestin recruitment of CXCL12WT to the single point mutant CXCL12P2G (14) and to CXCL12LRHQ (33). These two variants only differ from CXCL12WT in the distal N-terminal region of the chemokine that interacts in the CRS2 region (orthosteric pocket and extracellular loops) of the receptor. Thus, the interactions that the first 29 residues of ACKR3 make with the chemokines are identical and far separated from the ACKR3:CXCL12 interactions that are unique to the respective chemokine (Fig 4A). It has been previously shown that all three CXCL12 variants are high affinity ligands of ACKR3 with IC50 values in the pM to low nM range (33). However, preliminary co-expression experiments suggested different binding kinetics for the three chemokines since CXCL12WT and CXCL12LRHQ could be co-purified with ACKR3 from Sf9 cells (27) while CXCL12P2G could not, presumably because it rapidly dissociated from the receptor.
Fig. 4. ACKR3 binding kinetics and arrestin recruitment of CXCL12 mutants.
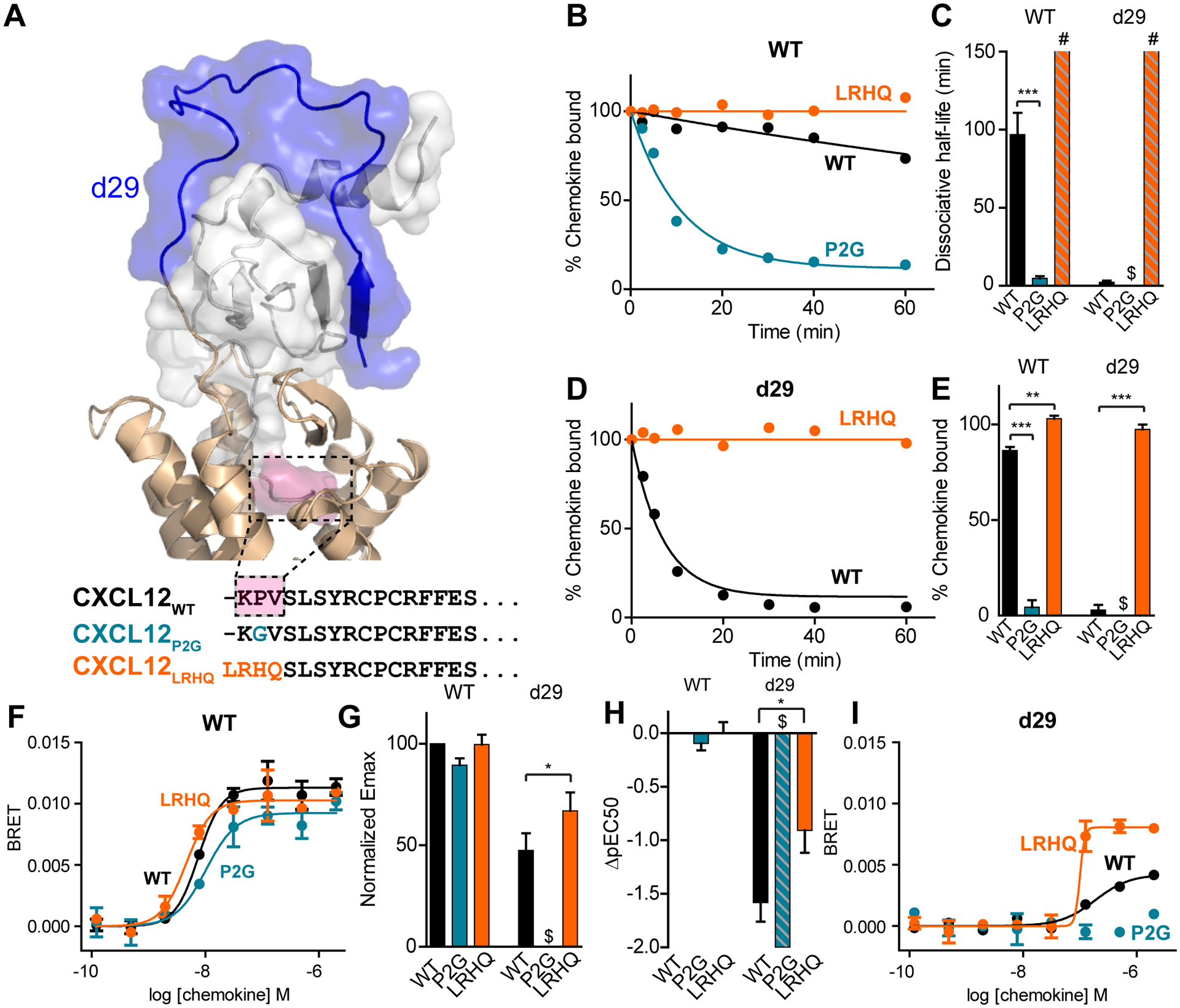
(A) Extracellular portion of ACKR3:CXCL12 model and N-terminal sequences of CXCL12 variants. Residues in CXCL12WT that differ between the three mutants are highlighted in pink in the model. (B) Representative curves for CXCL12 dissociation from bril-ACKR3WT detected by flow cytometry. (C) Means and standard errors from three or more experiments of dissociative half-lives determined from fitting dissociation curves to a single exponential. The dissociative half-life of CXCL12LRHQ was too slow to quantify but was estimated to be longer than 150 min (highlighted by # in figure). Binding of CXCL12P2G to ACKR3d29 was too low to quantify a dissociation rate (highlighted by $ in figure). (D) Representative CXCL12 dissociation curves from bril-ACKR3d29 detected by flow cytometry. (E) Means and standard errors of the percent specific chemokine binding remaining 20 min after the start of dissociation for three or more experiments. CXCL12LRHQ has a higher fraction of chemokine bound than CXCL12WT for both bril-ACKR3WT and bril-ACKR3d29 and CXCL12P2G has a lower fraction bound for bril-ACKR3WT. Binding of CXCL12P2G to ACKR3d29 was too low to quantify (highlighted by $ in figure). (F) Representative dose-response curves for β-arrestin-2 recruitment to ACKR3WT and ACKR3d29. (G) Emax normalized to ACKR3WT with CXCL12WT (%Emax =Emax,mutant/Emax,WT × 100). Each bar represents the average and standard errors of three or more experiments. (H) Mean and standard errors of pEC50 relative to ACKR3WT with CXCL12WT (ΔpEC50=pEC50,mutant-pEC50,WT). CXCL12P2G-mediated recruitment of arrestin to ACKR3d29 was barely detectable and the ΔpEC50 was estimated to be less than −2 (highlighted by $ in G and H). (I) Representative dose-response curves for β-arrestin-2 recruitment to ACKR3WT and ACKR3d29. Significant differences for CXCL12P2G and CXCL12LRHQ compared to CXCL12WT are noted: *P<0.05, **P<0.01, ***P<0.001 from one-way ANOVA with Dunnett’s multiple comparison test.
To quantify these potential differences we tested the mutant chemokines with bril-ACKR3WT and bril-ACKR3d29 in dissociation experiments. CXCL12WT, CXCL12P2G and CXCL12LRHQ had significantly different off-rates from bril-ACKR3WT ranging from CXCL12P2G, which had a dissociative half-life of 7.2 ± 1.2 min (~15-fold faster than ACKR3WT) to CXCL12LRHQ, which had no measurable dissociation during the time scale of the experiment (Fig 4B, C). Differences in dissociation rates were even further enhanced with the truncated receptor, bril-ACKR3d29. CXCL12LRHQ was fully bound even after 60 min competition with unlabeled ligand while we were unable to determine detectable binding of CXCL12P2G due to its rapid dissociation rate (Fig 4D and E).
Despite differences in dissociation rates from the receptor, all three chemokine mutants induced β-arrestin recruitment to ACKR3WT with similar efficacy and potency (Fig 4F, G and H). This was consistent with the idea that the exact sequence of the chemokine N-terminus is not crucial for ACKR3 activation (35), which contrasts with most canonical G protein-coupled chemokine receptor:chemokine pairs where specific interactions with the chemokine N-terminus play a crucial role in signaling (16). However, significant differences in the ability to promote β-arrestin-2 recruitment to the truncated receptor ACKR3d29 was observed for the three chemokines (Fig 4G, H and I). CXCL12LRHQ recruited β-arrestin-2 to the truncated receptor with higher efficacy and potency than the other chemokines, including CXCL12WT, while CXCL12P2G was a very weak agonist that was only able to recruit a small amount of β-arrestin-2 even at 2 μM of chemokine (fig S6).
Thus, the chemokine with the slowest dissociation rate was also the one least impacted by the N-terminal truncation of the receptor in agreement with the hypothesis that there is a correlation between a fast chemokine off-rate and sensitivity to N-terminal receptor truncation and mutations. This further suggests that the residence time of agonists on ACKR3 affects their ability to recruit β-arrestin-2.
Discussion
On the basis of previous studies, it has been proposed that the chemokine receptor N-terminus acts as a landing pad for the core of the chemokine, which then orients the chemokine N-terminus to activate the receptor through interactions in the orthosteric pocket (13, 14). However, the order of binding has never been experimentally demonstrated. Based on our studies of CXCL12 binding to ACKR3, we propose a different model where interactions are initially formed between the N-terminus of the chemokine and the orthosteric pocket and extracellular loops of the receptor (CRS1.5/CRS2 region in Fig 1A). This initial interaction is followed by the N-terminus of the receptor wrapping around the chemokine to form the CRS0.5/CRS1 interactions and anchoring the chemokine to the receptor (Fig 5).
Fig. 5. Model of the mechanism of chemokine interaction with the receptor.
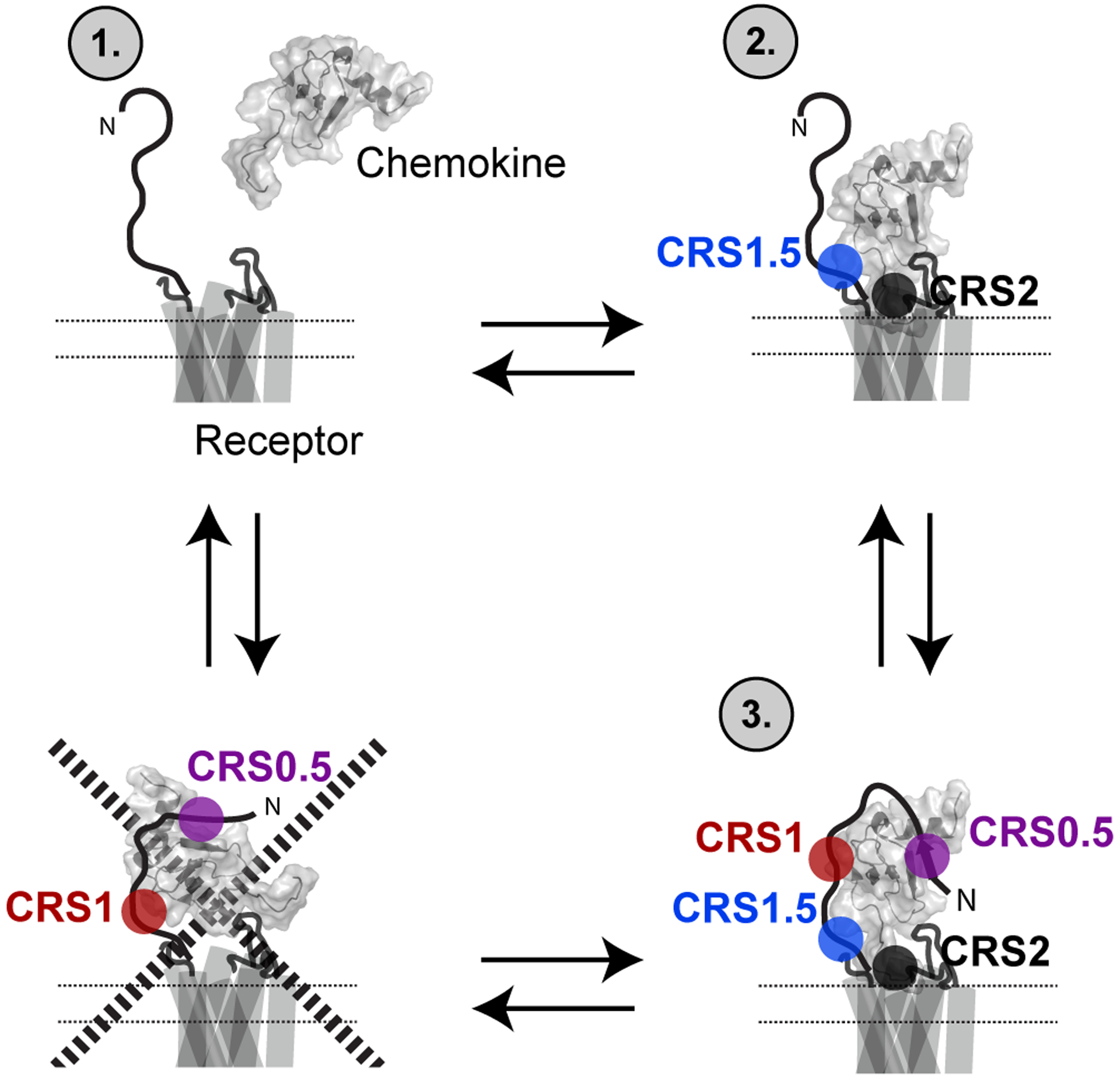
Experiments with chemokine mutations (CXCL12P2G and CXCL12LRHQ) and receptor truncations (Δ2–29) suggested that the chemokine initially engages with the CRS1.5 and CRS2 of the receptor followed by formation of additional interactions with the CRS0.5 and CRS1 epitopes. Initial docking of the receptor N-terminus with the core of the chemokine (bottom left corner) is not a key step in the formation of the fully engaged receptor:chemokine complex.
Our results showed that chemokine binding is driven by the interplay between different epitopes (CRS0.5/1/1.5/2) on the receptor and that the contribution from these epitopes cannot be completely uncoupled. The ACKR3 N-terminus had a critical role in chemokine binding and activation. However, this role is not only dependent on the specific “CRS1” interactions but rather on the total sum of interactions that the chemokine makes with other regions of the receptor. These observations suggest that truncations and mutations of the receptor or chemokine N-termini may affect the potency and efficacy of activation by the chemokine but only if the truncated or mutated region contributes a large enough fraction of the total binding energy. CXCL11-induced ACKR3 activation was more sensitive than CXCL12-induced activation to N-terminal receptor mutations (34) and truncations. According to our model, CXCL11 does not necessarily make more and/or stronger interactions with the distal N-terminus of the receptor, but those interactions represent a larger portion of the total interaction energy. Similarly, CXCL12 binding and activation of ACKR3 is less sensitive to point mutations in the receptor N-terminus than CXCL12 binding and activation of CXCR4 (34, 35). It was therefore suggested that, as opposed to CXCR4, interactions with the ACKR3 N-terminus is not an essential determinant of CXCL11- and CXCL12-induced receptor activation (35). Our data showed that the ACKR3 N-terminus plays a key role in chemokine binding and activation and suggest that the reason for ACKR3’s reduced sensitivity to mutations is that CXCL12 has a much slower off-rate from ACKR3 than from CXCR4.
Biased signaling where ligands or receptor variants selectively activate one signaling pathway over another is an emerging concept (36). Our observations suggest a potential mechanism for how CRS0.5/1 interactions could control receptor activation of different signaling pathways. A chemokine with fast dissociation rate may not have a long enough residence time on the receptor to allow for G protein kinase (GRK) phosphorylation followed by β-arrestin recruitment. Thus, changes in chemokine residence times may alter the coupling to different intracellular adaptor proteins and be a source of apparent signaling bias. This would be analogous to the dopamine D2 receptor where apparent signaling bias is affected by agonist dissociation rates (37). In this study, we utilized β-arrestin-2 recruitment as a measure of receptor activation. However, recent studies have shown differences in intracellular trafficking between CXCL11- and CXCL12-activated ACKR3, potentially due to interaction with other adaptor proteins than arrestins (38). In this case and in the case of other receptors, intracellular effector proteins may be differentially affected by changes in ligand lifetimes on receptors.
Although our data suggests that the main contribution of the N-terminus is to prevent the chemokine from dissociating from the receptor, it should also be noted that CXCL12LRHQ has a lower efficacy for ACKR3d29 than for ACKR3WT even with its very long dissociative half-life (longer than that of CXCL12WT from ACKR3WT). Thus, even if ACKR3d29 can be fully activated by a small molecule agonist, there must be additional interactions within the receptor:chemokine complex that are needed for full activation of the receptor and that are not formed when the chemokine binds the truncated receptor.
The results presented here are likely to also apply to other chemokines and chemokine receptors. Mutational studies have shown that chemokines rely to different extents on CRS2 interactions versus interactions involving the receptor N-termini (CRS0.5/1/1.5) (2). For example, N-terminally truncated CCL2 is a high affinity CCR2 antagonist (39). On the other hand, N-terminal truncations significantly affect the ability of CXCL12 to bind as well as activate CXCR4 (40). Our efforts were focused on ACKR3 and CXCL12, which comprise a CXC receptor:CXC chemokine pair and are likely not representative of the whole family of receptors and chemokines. Thus, more data with other receptors and chemokines would further clarify the role of the receptor N-terminus in chemokine binding. Still, previous results have shown similarities between the ACKR3:CXCL12 complex and the same chemokine in complex with CXCR4. For example, several residues that affect CXCR4 chemokine binding and activation are also implicated in CXCL12 activation of ACKR3 (8, 41).
Post-translational modifications of the N-terminus have been identified for a number of different chemokine receptors. A notable example of this is Tyr sulfation that occurs for multiple receptors (42) and can have significant effects on chemokine binding affinity (43). Based on our model, sulfation likely reduces the dissociation rates of chemokine. In analogy with the effects of N-terminal truncation of ACKR3 on CXCL11 and CXCL12, this modification could therefore serve as a regulatory mechanism that impacts signaling, with different effects on specific chemokines that bind to the same receptor.
Both association and dissociation rates of CXCL12 binding to ACKR3 are slower than what has been reported for binding to CXCR4. One of the proposed roles of ACKR3 is to regulate CXCR4 by controlling the extracellular concentration of CXCL12 (6, 7). Based on its higher affinity for CXCL12, it was believed that the chemokine would preferentially bind ACKR3 over CXCR4 (44). However, while this may be observed in equilibrium experiments, the kinetic differences between the two receptors, with the CXCR4 on- and off-rates being considerably faster than those for ACKR3, could make CXCR4 kinetically favored for CXCL12 binding. In such a hypothetical scenario CXCL12 may initially bind CXCR4, leading to G protein-coupled signaling but eventually favor ACKR3 binding, which would lead to chemokine internalization and degradation. Thus, differences in their binding kinetics could be of fundamental importance for the interplay between ACKR3 and CXCR4 in vivo.
Materials and Methods
Cloning
A pFastBac1 vector containing residues 2–332 of human ACKR3 with a N-terminal GP64 promoter, HA signal sequence and C-terminal FLAG and 10x His tags was utilized for baculovirus expression (27). For expression in HEK293T cells, human ACKR3 was cloned into a pcDNA vector containing the Rluc3 gene (a kind gift from Nikolaus Heveker, Universite de Montreal, Montreal, QC, Canada) at the C terminus of the receptor. Constructs where 7–29 residues were removed from the receptor N-terminus were obtained using the SLIM protocol (45). For chemokine expression in Sf9 cells CXCL11 and CXCL12 with their native signal sequences and C-terminal HA tags were cloned into a pFastBac1 vector containing a polyhedrin (polH) promoter. CXCL12P2G and CXCL12LRHQ mutant constructs were produced by site directed mutagenesis using standard quick-change methods.
Ligands
Recombinant CXCL11, CXCL12WT, CXCL12P2G and CXCL12LRHQ were expressed and purified from E. coli by slight variations of the protocols previously described (8, 46). Small molecule ACKR3 ligands CCX662 and CCX777 were a gift from Chemocentryx (23).
BRET-based β-arrestin-2 recruitment assays
HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Cells were transfected in six-well plates with 0.05–0.125 μg of ACKR3 DNA and 1 μg of a pcDNA vector containing GFP10-β-arrestin-2 (kind gift from Nikolaus Heveker, Universite de Montreal). 48 hours after transfection the cells were washed and resuspended in PBS buffer containing 0.1 % glucose and transferred to 96-well white clearbottom tissue culture assay plates (BD Falcon) (100,000 cells per well). For dose-response experiments the plates were incubated for 40 min at 37°C and then stimulated for 20 min with different concentrations of ligand. β-arrestin-2 expression was quantified by measuring GFP10 fluorescence emission at 510 nm after excitation at 400 nm. The bottom of the plate was covered and luciferase substrate (Deep Blue C) was added to a final concentration of 5 μM. BRET was measured on a VictorX Light multilabel plate reader (Perkin-Elmer Life Sciences) as the ratio of GFP10 emission at 515 nm to Rluc3 emission at 410 nm. Each point was recorded in duplicate, average BRET values were plotted against log [ligand] and the resulting dose-response curves were fit using non-linear fitting in GraphPad Prism to obtain Emax and pEC50 values. For time-resolved BRET experiments cells were incubated for 40 min and then stimulated with 10 nM chemokine for times ranging from 4–70 min before measuring BRET. Averaged BRET values from duplicate measurements were plotted as a function of time and the association rates were determined from non-linear fitting to single exponential functions.
Sf9 cell expression
ACKR3 was expressed in Sf9 cells as previously described (27). Briefly, cells were cultured in ESF921 media (Expression Systems) at 27°C with shaking at 140 rpm. The Bac-to-Bac Baculovirus Expression System (Invitrogen) was used to produce baculovirus containing the different ACKR3 and CXCL12 variants as previously described (27). Briefly, recombinant bacmids were incubated with 3 μL of Xtreme Gene Transfection Reagent (Roche) and 100 μL of transfection medium (Expression Systems) for 30 minutes. The mixture was added to 2.5 mL of Sf9 cells at a density of 1.3 × 106 cells mL−1 and the cells were incubated for 96 h with 300 rpm shaking at 27°C. Cells were pelleted using centrifugation and 400 μL of the resulting supernatant (P0 stock) was used to transfect 40 mL of Sf9 cells at a density of 2.5 × 106 cells mL−1. After 48 h the cells were centrifuged and the resulting supernatant was stored at 4°C until further use (P1 stock). Virus titers were determined using flow cytometry staining with a phycoerythrin (PE)conjugated GP64 antibody. To initiate protein expression P1 virus corresponding to ACKR3 alone or ACKR3 and CXCL12 (co-expression) was added to Sf9 cells at a density of ~2.5 × 106 cells mL−1 at a multiplicity of infection of 5. Cells were grown for 48h at 27°C with shaking at 140 rpm prior to experiments.
Binding experiments
For association experiments, Sf9 cells expressing ACKR3 were washed twice in assay buffer (20 mM phosphate buffered saline (PBS), 0.5% bovine serum albumin) and resuspended to a density of 3 × 106 cells mL−1. 10 μL (30,000 cells) of cells were transferred to a 96-well assay plate and 2 μL from a 0.05 mg mL−1 of the viability dye 7-amino-actinomycin D (7AAD) and 68 μL of room temperature assay buffer was added to each well. Purified HA-tagged CXCL12WT was incubated with a 1.3-fold molar excess of FITC-conjugated anti-HA antibody (Sigma-Aldrich, H7411) for 20 min. The CXCL12:antibody stock was then combined with assay buffer to make 5, 25 and 50 nM (5x) solutions, which were added to a final concentration of 1, 5 or 10 nM in a final volume of 100 μL. To quantify non-specific binding of CXCL12WT, 20 μM of the small molecule ligand CCX777 was added to cells prior to addition of chemokine. Cells were fixed at different time points with a 0.67 % (w/v) final concentration of paraformaldehyde.
For dissociation experiments 10 μL of Sf9 cells (30,000 cells) co-expressing ACKR3 and HA-tagged chemokine were incubated with FITC-conjugated anti-HA antibody (final antibody dilution 200x from 1 mg mL−1 stock) and 1 μL of 7AAD solution (from 0.05 mg mL−1 stock). After 20 min incubation, assay buffer containing 20 μM CCX777 was added and cells were fixed at different time points with 0.67 % (w/v) of paraformaldehyde (PFA). Non-specific binding was measured by expressing respective HA-tagged chemokine in the absence of ACKR3. Total receptor expression was measured by incubating 10 μL of cells with FITC-conjugated anti-FLAG antibody and 0.0075% (v/v) Triton X-100 and acquiring FITC fluorescence. Chemokine binding was quantified from FITC fluorescence using a Guava easyCyte flow cytometer (Millipore). Data was analyzed using FlowJo. The geometric mean of FITC fluorescence for live cells (selected from 7AAD fluorescence) was plotted against time and non-specific binding was subtracted. The resulting specific binding was fit using exponential functions in GraphPad Prism.
Thermostability assays
ACKR3 was purified from Sf9 membranes as previously described (8, 27). Briefly, pellets corresponding to 40 mL of Sf9 cells expressing different ACKR3 variants (bril-ACKR3WT, bril-ACKR3d17 and bril-ACKR3d29) were thawed and washed with low and high salt buffers (10 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl and 0 or 1 M NaCl). Washed membranes were solubilized with buffer containing 50 mM HEPES, pH 7.5, 400 mM NaCl, 0.75/0.15% (w/v) DDM/CHS and 50 μM CCX777 and receptor was purified by affinity chromatography using Talon IMAC resin (Teknova). Eluted protein was buffer exchanged into 50 mM HEPES pH 7.5, 10% glycerol, 150 mM NaCl, 0.025/0.005 % (w/v) DDM/CHS (exchange buffer) and stored at 4°C for up to two weeks. Receptor glycosylation was removed by incubation over night with PNGaseF (NEB). For SDS-PAGE analysis ~6 μg of receptor was incubated with SDS-PAGE sample buffer and separated on 10% Tris-tricine polyacrylamide gels. For thermostability assays, 0.3 μM of ACKR3 and 2.5 μM of 7-diethylamino-3-(40-maleimidylphenyl)-4-methylcoumarin (CPM) was incubated for 15 min in exchange buffer. Thermostability was determined using a RotorGeneQ 6-plex RT-PCR instrument (Qiagen) from the increase in CPM fluorescence (excitation 365 nm, emission 460 nm) when the sample temperature was ramped from 25–95 °C.
SPR
SPR experiments were performed on a BIAcore 3000 machine (GE Healtcare) using a CM5 chip (GE Healthcare) and a method adapted from Dyer et al (26). Following equilibration with running buffer (10 mM HEPES, 150 mM NaCl, 3 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4) two flow cells on the CM5 chip were activated with a 1:1 mix of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (0.1 M) and N-hydroxy-succinimide (NHS) (0.2 M) (300 μl in total). Neutravidin (Invitrogen) was then flowed over the activated surface (0.2 mg mL−1 in 20 mM sodium acetate, pH 6.0) until surface saturation was reached, followed by surface deactivation with ethanolamine. Both flow cells were then thoroughly washed to remove non-bound neutravidin using regeneration buffer (0.1 M glycine, 1 M NaCl, 0.1% Tween-20, pH 9.5). Biotinylated CXCL12 (46) was then flowed over one flow cell of the chip at 10 μL min−1 (10 μg mL−1 in 10 mM HEPES, 150 mM NaCl, pH 7.4) until surface saturation was reached followed by washing with regeneration buffer. A paired flow cell with neutravidin, but without CXCL12, was prepared at the same time for reference subtraction to analyze specific interactions with immobilized CXCL12.
ACKR3 purified in DDM/CHS (see thermostability assays section for details) was reconstituted into membrane scaffolding protein (MSP) 1E3D1 nanodiscs as described previously (27). Briefly, receptor was mixed in cholate buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 25mM sodium cholate) with MSP1E3D1 protein, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPC/POPG, molar ratio 3:2) lipids at a 0.1:1:110 molar ratio of ACKR3:MSP:lipid. Cholate was removed using biobeads and nanodiscs were purified by size exclusion chromatography. Protein-containing nanodiscs were separated from empty nanodiscs by binding to Talon resin and eluted by 250 mM imidazole. The final sample was obtained after buffer exchange into 25 mM HEPES pH 7.5, 150 mM NaCl using spin concentrators.
Pilot experiments showed that flow rate had no effect upon the interaction, or associated residual plots. Experiments were undertaken at a fast flow rate of 40 μL min−1; thus, ensuring there were limited mass transfer mediated effects in these experiments. Pilot experiments demonstrated that the regeneration buffer used removed all of the bound material and had no effect on subsequent interaction analyses.
Interaction experiments were undertaken by flowing the indicated concentrations of ACKR3 (in 10 mM HEPES, 150 mM NaCl, 3 mM EDTA, pH 7.4 for nanodisc samples, same buffer containing 0.025/0.005% (w/v) DDM/CHS for experiments in micelles) over both flow cells with the non-specific interaction signal (no CXCL12) being subtracted from the specific interaction signal (with CXCL12). Due to subtle mis-matches between the assay buffer and the ACKR3-containing buffer the curves produced by injecting ACKR3 buffer alone was also subtracted from the raw data. The surface was regenerated between ACKR3 injections using regeneration buffer. To ensure that the signal produced was from ACKR3 specifically, receptor-free nanodiscs were also flowed over the surface at matching concentrations, producing no positive signal.
Curves generated as above were then analyzed using the BIAevaluation software (GE Healthcare) using a 1:1 binding model. The dissociation phase of curves measured at different ACKR3 concentrations were fit to a global dissociation constant (kd), which was then used to fit the association phase. Initially, local association constants (ka) for each chemokine concentrations were obtained to confirm that the binding reaction followed pseudo-first order kinetics. To obtain a single ka value for the experiment curves at different chemokine concentrations were then fit globally.
Supplementary Material
Fig. S1. β-arrestin recruitment to ACKR3WT induced by CCX662 and CXCL11 in BRET experiments.
Fig. S2. Expression of ACKR3 variants in HEK cells.
Fig. S3. Kinetics of association of CXCL12 with ACKR3 on live Sf9 cells.
Fig. S4. SPR in nanodiscs and detergent micelles.
Fig. S5. N-terminal cytochrome b562-RIL fusion protein did not affect CXCL12 dissociation rate or folding of ACKR3.
Fig S6. BRET signal increase after addition of 2 μM chemokine for ACKR3WT and ACKR3d29.
Table S1. IC50 values (nM) for equilibrium binding studies reported in literature.
Acknowledgements:
The authors thank Catherina Salanga and Bryan Stephens for providing purified CXCL11 and CXCL12 protein, Chemocentryx for the small molecule compounds CCX662 and CCX777 and Nikolaus Heveker (Universite de Montreal) for providing vectors for BRET experiments.
Funding: This work was supported by grants from the NIH to TMH (U01 GM094612, R21 AI121918, R01 GM117424, R01 AI118985). MG was supported by a Robertson Foundation/Cancer Research Institute Irvington Postdoctoral Fellowship.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and notes
- 1.Griffith JW, Sokol CL, and Luster AD. 2014. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 32: 659–702. [DOI] [PubMed] [Google Scholar]
- 2.Allen SJ, Crown SE, and Handel TM. 2007. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol 25: 787–820. [DOI] [PubMed] [Google Scholar]
- 3.Nibbs RJ, and Graham GJ. 2013. Immune regulation by atypical chemokine receptors. Nat Rev Immunol 13: 815–829. [DOI] [PubMed] [Google Scholar]
- 4.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, and Schall TJ. 2006. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203: 2201–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, and Lefkowitz RJ. 2010. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A 107: 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luker KE, Lewin SA, Mihalko LA, Schmidt BT, Winkler JS, Coggins NL, Thomas DG, and Luker GD. 2012. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene 31: 4750–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe P, Mueller W, Schütz D, MacKay F, Thelen M, Zhang P, and Stumm R. 2014. CXCR7 prevents excessive CXCL12-mediated downregulation of CXCR4 in migrating cortical interneurons. Development 141: 1857–1863. [DOI] [PubMed] [Google Scholar]
- 8.Gustavsson M, Wang L, van Gils N, Stephens BS, Zhang P, Schall TJ, Yang S, Abagyan R, Chance MR, Kufareva I, and Handel TM. 2017. Structural basis of ligand interaction with atypical chemokine receptor 3. Nat Commun 8: 14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q, and Wu B. 2013. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341: 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, and Stevens RC. 2010. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Qin L, Zacarías NV, de Vries H, Han GW, Gustavsson M, Dabros M, Zhao C, Cherney RJ, Carter P, Stamos D, Abagyan R, Cherezov V, Stevens RC, IJzerman AP, Heitman LH, Tebben A, Kufareva I, and Handel TM. 2016. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540: 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oswald C, Rappas M, Kean J, Doré AS, Errey JC, Bennett K, Deflorian F, Christopher JA, Jazayeri A, Mason JS, Congreve M, Cooke RM, and Marshall FH. 2016. Intracellular allosteric antagonism of the CCR9 receptor. Nature 540: 462–465. [DOI] [PubMed] [Google Scholar]
- 13.Scholten DJ, Canals M, Maussang D, Roumen L, Smit MJ, Wijtmans M, de Graaf C, Vischer HF, and Leurs R. 2012. Pharmacological modulation of chemokine receptor function. Br J Pharmacol 165: 1617–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, and Clark-Lewis I. 1997. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J 16: 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanpain C, Doranz BJ, Bondue A, Govaerts C, De Leener A, Vassart G, Doms RW, Proudfoot A, and Parmentier M. 2003. The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J Biol Chem 278: 5179–5187. [DOI] [PubMed] [Google Scholar]
- 16.Kufareva I, Gustavsson M, Zheng Y, Stephens BS, and Handel TM. 2017. What Do Structures Tell Us About Chemokine Receptor Function and Antagonism? Annu Rev Biophys 46: 175–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, Abagyan R, Stevens RC, and Handel TM. 2015. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347: 1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, and Garcia KC. 2015. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347: 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Y, Han GW, Abagyan R, Wu B, Stevens RC, Cherezov V, Kufareva I, and Handel TM. 2017. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 46: 1005–1017 e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziarek JJ, Kleist AB, London N, Raveh B, Montpas N, Bonneterre J, St-Onge G, DiCosmo-Ponticello CJ, Koplinski CA, Roy I, Stephens B, Thelen S, Veldkamp CT, Coffman FD, Cohen MC, Dwinell MB, Thelen M, Peterson FC, Heveker N, and Volkman BF. 2017. Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kofuku Y, Yoshiura C, Ueda T, Terasawa H, Hirai T, Tominaga S, Hirose M, Maeda Y, Takahashi H, Terashima Y, Matsushima K, and Shimada I. 2009. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J Biol Chem 284: 35240–35250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walters MJ, Ebsworth K, Berahovich RD, Penfold ME, Liu SC, Al Omran R, Kioi M, Chernikova SB, Tseng D, Mulkearns-Hubert EE, Sinyuk M, Ransohoff RM, Lathia JD, Karamchandani J, Kohrt HE, Zhang P, Powers JP, Jaen JC, Schall TJ, Merchant M, Recht L, and Brown JM. 2014. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br J Cancer 110: 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Fan P, Gleason MM, Jaen JC, Li L, McMahon JP, Powers J, Zeng Y, and Zhang P. 2010. Modulators of CXCR7. U. S. Patent, ed. Chemocentryx, Inc., United States. [Google Scholar]
- 24.Zweemer AJ, Nederpelt I, Vrieling H, Hafith S, Doornbos ML, de Vries H, Abt J, Gross R, Stamos D, Saunders J, Smit MJ, Ijzerman AP, and Heitman LH. 2013. Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Mol Pharmacol 84: 551–561. [DOI] [PubMed] [Google Scholar]
- 25.Vega B, Muñoz LM, Holgado BL, Lucas P, Rodríguez-Frade JM, Calle A, Rodríguez-Fernández JL, Lechuga LM, Rodríguez JF, Gutiérrez-Gallego R, and Mellado M. 2011. Technical advance: Surface plasmon resonance-based analysis of CXCL12 binding using immobilized lentiviral particles. J Leukoc Biol 90: 399–408. [DOI] [PubMed] [Google Scholar]
- 26.Dyer DP, Salanga CL, Volkman BF, Kawamura T, and Handel TM. 2016. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 26: 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gustavsson M, Zheng Y, and Handel TM. 2016. Production of Chemokine/Chemokine Receptor Complexes for Structural Biophysical Studies. Methods Enzymol 570: 233–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawamura T, Stephens B, Qin L, Yin X, Dores MR, Smith TH, Grimsey N, Abagyan R, Trejo J, Kufareva I, Fuster MM, Salanga CL, and Handel TM. 2014. A general method for site specific fluorescent labeling of recombinant chemokines. PLoS One 9: e81454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manglik A, and Kobilka B. 2014. The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr Opin Cell Biol 27: 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeVree BT, Mahoney JP, Velez-Ruiz GA, Rasmussen SG, Kuszak AJ, Edwald E, Fung JJ, Manglik A, Masureel M, Du Y, Matt RA, Pardon E, Steyaert J, Kobilka BK, and Sunahara RK. 2016. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535: 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castro M, Nikolaev VO, Palm D, Lohse MJ, and Vilardaga JP. 2005. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc Natl Acad Sci U S A 102: 16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexandrov AI, Mileni M, Chien EY, Hanson MA, and Stevens RC. 2008. Microscale fluorescent thermal stability assay for membrane proteins. Structure 16: 351–359. [DOI] [PubMed] [Google Scholar]
- 33.Hanes MS, Salanga CL, Chowdry AB, Comerford I, McColl SR, Kufareva I, and Handel TM. 2015. Dual targeting of the chemokine receptors CXCR4 and ACKR3 with novel engineered chemokines. J Biol Chem 290: 22385–22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benredjem B, Girard M, Rhainds D, St-Onge G, and Heveker N. 2017. Mutational Analysis of Atypical Chemokine Receptor 3 (ACKR3/CXCR7) Interaction with Its Chemokine Ligands CXCL11 and CXCL12. J Biol Chem 292: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szpakowska M, Nevins AM, Meyrath M, Rhainds D, D’huys T, Guité-Vinet F, Dupuis N, Gauthier PA, Counson M, Kleist A, St-Onge G, Hanson J, Schols D, Volkman BF, Heveker N, and Chevigné A. 2018. Different contributions of chemokine N-terminal features attest to a different ligand binding mode and a bias towards activation of ACKR3/CXCR7 compared with CXCR4 and CXCR3. Br J Pharmacol 175: 1419–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rankovic Z, Brust TF, and Bohn LM. 2016. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 26: 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, Javitch JA, Christopoulos A, and Lane JR. 2016. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7: 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montpas N, St-Onge G, Nama N, Rhainds D, Benredjem B, Girard M, Hickson G, Pons V, and Heveker N. 2018. Ligand-specific conformational transitions and intracellular transport are required for atypical chemokine receptor 3-mediated chemokine scavenging. J Biol Chem 293: 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jarnagin K, Grunberger D, Mulkins M, Wong B, Hemmerich S, Paavola C, Bloom A, Bhakta R, Diehl F, Freedman R, McCarley D, Polsky I, Ping-Tsou A, Kosaka A, and Handel TM. 1999. Identification of surface residues of the monocyte chemotactic protein 1 that affect signaling through the receptor CCR2. Biochemistry 38: 16167–16177. [DOI] [PubMed] [Google Scholar]
- 40.McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, and Overall CM. 2001. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem 276: 43503–43508. [DOI] [PubMed] [Google Scholar]
- 41.Wescott MP, Kufareva I, Paes C, Goodman JR, Thaker Y, Puffer BA, Berdougo E, Rucker JB, Handel TM, and Doranz BJ. 2016. Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, Louie S, Hsu W, Yu KM, Nicholas HB, and Rosenquist GL. 2008. Tyrosine sulfation is prevalent in human chemokine receptors important in lung disease. Am J Respir Cell Mol Biol 38: 738–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ludeman JP, and Stone MJ. 2014. The structural role of receptor tyrosine sulfation in chemokine recognition. Br J Pharmacol 171: 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coggins NL, Trakimas D, Chang SL, Ehrlich A, Ray P, Luker KE, Linderman JJ, and Luker GD. 2014. CXCR7 controls competition for recruitment of β-arrestin 2 in cells expressing both CXCR4 and CXCR7. PLoS One 9: e98328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu J, Tillett D, Dawes IW, and March PE. 2008. Site-directed, Ligase-Independent Mutagenesis (SLIM) for highly efficient mutagenesis of plasmids greater than 8kb. J Microbiol Methods 73: 195–198. [DOI] [PubMed] [Google Scholar]
- 46.Allen SJ, Hamel DJ, and Handel TM. 2011. A rapid and efficient way to obtain modified chemokines for functional and biophysical studies. Cytokine 55: 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffmann F et al. , Rapid uptake and degradation of CXCL12 depend on CXCR7 carboxyl-terminal serine/threonine residues. J Biol Chem 287, 28362–28377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wijtmans M et al. , Synthesis, modeling and functional activity of substituted styrene-amides as small-molecule CXCR7 agonists. Eur J Med Chem 51, 184–192 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Canals M et al. , Ubiquitination of CXCR7 controls receptor trafficking. PLoS One 7, e34192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zabel BA et al. , Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol 183, 3204–3211 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Balabanian K et al. , The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 280, 35760–35766 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. β-arrestin recruitment to ACKR3WT induced by CCX662 and CXCL11 in BRET experiments.
Fig. S2. Expression of ACKR3 variants in HEK cells.
Fig. S3. Kinetics of association of CXCL12 with ACKR3 on live Sf9 cells.
Fig. S4. SPR in nanodiscs and detergent micelles.
Fig. S5. N-terminal cytochrome b562-RIL fusion protein did not affect CXCL12 dissociation rate or folding of ACKR3.
Fig S6. BRET signal increase after addition of 2 μM chemokine for ACKR3WT and ACKR3d29.
Table S1. IC50 values (nM) for equilibrium binding studies reported in literature.