

Abstract
The induction of polyarthritis and polyarthralgia is a hallmark of arthritogenic alphavirus infections, with an exceptionally higher morbidity observed with chikungunya virus (CHIKV). While the mechanisms underlying these incapacitating acute symptoms remain partially understood, the progression to chronic conditions in some cases remains unanswered. The highly pro‐inflammatory nature of alphavirus disease has suggested the involvement of virus‐specific, joint‐infiltrating Th1 cells as one of the main pathogenic mediators of CHIKV‐induced joint pathologies. This review summarizes the role of cell‐mediated immune responses in CHIKV pathogenesis, with a specific focus on pro‐inflammatory Th1 responses in the development of CHIKV joint inflammation. Furthermore, due to the explosive nature of arthritogenic alphavirus outbreaks and their recent expansion across the world, co‐infections with other highly prevalent pathogens such as malaria are likely to occur but the pathological outcomes of such interactions in humans are unknown. This review will also discuss the potential impact of malaria co‐infections on CHIKV pathogenesis and their relevance in alphavirus control programs in endemic areas.
Keywords: CD4+ T cell, chikungunya virus, co‐infection, malaria
1. INTRODUCTION
Arthritogenic alphaviruses are a group of clinically relevant enveloped positive sense, single‐stranded RNA viruses that belong to the family Togaviridae. 1 These viruses have been linked to the development of acute and persistent arthritic conditions in human populations.2, 3 They are mainly transmitted by Aedes and Culex mosquitoes which confer them wide global distributions.4, 5 Arthritogenic alphaviruses are typically referred as “Old World alphaviruses” and comprise of chikungunya virus (CHIKV, widely distributed in the tropics), O'nyong‐nyong virus (ONNV, restricted to Africa), Mayaro virus (MAYV, endemic to Central and South America), Barmah Forest virus (BFV, confined to Australia), Ross River virus (RRV, reported in Australia, Papua New Guinea, and islands of the South Pacific region), and Sindbis virus (SINV, distributed in Africa, Middle East, Europe, and Australasia).5
In humans, arthritogenic alphavirus infection typically causes a febrile illness characterized by high viremia, maculopapular skin rash, muscle pain, hallmark debilitating polyarthralgia, polyarthritis with or without effusions, and in some cases lymphadenopathy.3, 6 The virus incubation period prior to the clinical manifestations depends on the alphavirus species. Typically, it is relatively short with an average of 7‐9 days.2 The disease is self‐limiting and usually resolves within 2 weeks, but chronic pathologies such as polyarthritis may develop, which could last from months to years.7 Neurological complications are rare, but recent reports have suggested that serious clinical forms of CHIKV disease could compromise brain tissues leading to permanent neurological damage.8, 9, 10, 11
Among the arthritogenic alphaviruses, research on CHIKV was the most extensive owing to the global epidemics since 2005.12 The availability of mouse models that captures major features of human disease have generated a wealth of information.13, 14 These studies have yielded important evidence on the involvement of host immune responses in the development of alphavirus arthritides. CHIKV infections trigger a strong immune response characterized by the release of pro‐inflammatory cytokines and chemokines,15, 16, 17 followed by the activation and trafficking of myeloid and lymphoid cells to affected tissues,18, 19 leading to joint swelling. While these immune signatures have been identified, the interplay between these factors underlying the development of acute and chronic forms of arthritis remains elusive.
The striking similarities between CHIKV arthritic disease and rheumatoid arthritis (RA) at the transcriptomic and cytokine/chemokine levels suggested the potential involvement of common causative agents.20 In fact, two CD4+ effector T cell subsets: Th1 and Th17, have been implicated in the development of RA.21, 22, 23, 24 Th1 cells typically orchestrate cell‐mediated responses against intracellular pathogens through the release of signature cytokines such as IFNγ and IL‐2,25, 26, 27 whereas IL‐17‐secreting Th17 cells have been linked to autoimmunity and neutrophil recruitment to the site of infection.28, 29 This prompted the hypothesis that CHIKV‐induced inflammation could be also mediated by pathogenic CD4+ T cell responses.
2. ROLE OF CELL‐MEDIATED IMMUNITY IN THE DEVELOPMENT OF CHIKV‐INDUCED INFLAMMATION
2.1. Pro‐inflammatory immune mediators induced upon CHIKV infection
Inflammatory cytokines such as IFNγ, IFNα, IL‐2, IL‐2R, IL‐6, IL‐7, IL‐12, IL‐15, IL‐17, and IL‐18 have been shown to be upregulated during acute CHIKF.17 Moreover, high levels of IL‐15 (a T‐cell growth factor),30 IL‐2R (produced upon T cell activation),31 CXCL9 and CXCL10 (chemokines that bind to CXCR3 primarily expressed on activated T lymphocytes)32 suggested the involvement of T cell responses during the acute phase of disease. Transcriptomics analysis in CHIKV mouse models revealed overlapping pro‐inflammatory gene expression signatures with RA patients.20 Similarly, canonical pathways analysis showed shared patterns involving monocyte/macrophages, NK cell, B cell, and T cell signaling.20 Among T cells, CD4+ helper T cells have been associated with acute CHIKF and RA. It has been shown that CHIKV infection triggers strong IFNγ‐producing CD4+ T cell responses (Th1).13 This subset was also reported in the synovium of a patient displaying chronic CHIKV‐induced inflammation.18 Similarly, Th1‐polarized cells have been shown to preferentially accumulate in RA joints.21 Collectively, these observations supported the idea that CHIKV‐induced joint swelling and RA could be mediated by pathogenic host immune responses in a similar fashion.
2.2. Infiltration of innate immune cells into swollen joints
Patrolling monocytes and tissue‐resident macrophages are part of the first line of defense upon viral infection. These specialized phagocytic cells play the role of first responders against a wide range of pathogens and, upon activation, release immune modulators such as TNFα, IL‐1β, and IL‐6 which trigger localized inflammation.33 Macrophages and monocytes are one of the first immune subsets identified in the synovial tissue cellular infiltrate of CHIKV‐infected patients.18, 34 In line with this observation, mouse studies of CHIKV and RRV suggested that the monocytes/macrophages subset represents an important fraction of the cellular infiltrate in mouse swollen joints.13, 35, 36, 37 One of the first clues on the functional role of macrophages in CHIKV‐induced inflammation was reported by Rulli et al.38 In this study, treatment with Bindarit, an anti‐inflammatory small molecule that modulates the NFκB pathway and inhibits, among others, the synthesis of monocyte chemotactic protein (MCP)‐1,39 ameliorated CHIKV‐induced joint swelling in mice. In a parallel study, targeted depletion of macrophages by clodronate liposomes treatment in CHIKV‐infected mice yielded a similar outcome.13 Although these results indicate that monocytes and macrophages might contribute to the CHIKV inflammatory pathology, a separate study suggested that infection in mice deficient of CCR2, a receptor involved in monocyte chemotaxis, resulted in aggravated and prolonged swelling manifestation due to an increased and persistent neutrophil infiltration.40 Taken together, these observations suggest a model in which monocytes and macrophages could act as a double‐edged sword by mediating localized inflammation while restricting excessive infiltration of neutrophils in infected joints.41
Neutrophils and NK cells have been also identified in inflamed tissues of CHIKF patients and in the swollen footpads of CHIKV‐infected mice.13, 18, 19 Increased levels of powerful neutrophil chemoattractants such as CXCL1 and CXCL2, and MPO (myeloperoxidase) have been observed in mouse infected joints at the peak of inflammation.19, 40 Supporting these observations, massive neutrophil infiltration has been linked to an exacerbated swelling pathology in mouse models.19, 40 In a similar fashion, NK cells presence in inflamed tissues is believed to play an important role in CHIKV pathology by exerting direct antiviral activity through cytotoxic mechanisms and by producing IFNγ to enhance both innate and adaptive immune responses.4, 42, 43 Upregulation of IL‐12, a potent NK cell stimulator, has been reported in joints of virus‐infected mice20 suggesting the involvement of activated NK cells. Moreover, a rapid expansion of NK cells and increased cytotoxic activity during the acute phase of CHIKF have been observed in a patient cohort.43, 44 Some insights on the function of NK cells in CHIKV‐induced swelling have also been observed in one of our studies.16 Particularly, the more severe early acute joint swelling induced by La Réunion LR2006 OPY1 isolate as compared to the Caribbean CNR20235 isolate was associated with a higher and intensive NK cell activity during the early stages of the immune response in mouse joints. Importantly, the functional role of NK cells in early acute CHIKV‐induced swelling was demonstrated by depletion experiments in LR2006 OPY1 infection.16 These results suggest that NK cells are important mediators of CHIKV‐induced joint swelling likely through the activation of myeloid cells which release inflammatory mediators such us IL‐6,45 leading to vascular leakage and edema.
2.3. Involvement of T cells in CHIKV‐induced inflammation
The hypothetical function of T cells during CHIKV infection was initially postulated based on their proliferation and activation profiles in CHIKV‐infected patients. Activated peripheral CD4+ and CD8+ T cell levels were found to be elevated in CHIKF patients from the 2006 to 2007 outbreaks in La Reunion Island.18 Moreover, functional characterization of these cells further revealed the ability to recognize CHIKV‐derived peptides and to produce high levels of IFNγ thus suggesting the engagement of Th1‐biased immune responses upon CHIKV infection.18
In further support of these observations, immune profiling of peripheral blood of patients from the Gabonese CHIKV outbreak of 200746 revealed a strong early activation and proliferation of CD8+ T cells followed by the engagement of CD4+ T cell responses at a later point. These results supported a model in which the early expansion of CD8 T+ cells was probably required for clearance of virus‐infected cells, whereas the subsequent proliferation of helper T cells was needed to support the proper development of antiviral humoral responses. In support of this model, studies in SINV and RVV previously showed that CD8+ T cells were involved in limiting SINV replication in the central nervous system47 and eliminating RRV‐infected macrophages in vitro.48
2.4. CD4+ but not CD8+ T cells are pathogenic mediators of CHIKV‐induced joint swelling
The definitive roles of different T cell subsets were shown through depletion studies in mouse models. Subset specific depletion of CD4+ T cells but not CD8+ T cells abolished the major peak of joint swelling during CHIKV infection with no impact on viral tropism, demonstrating the pathogenic role of CD4+ T cells.13 In addition, adoptive transfer of CD4+ T cells isolated from CHIKV‐infected donors hastened and aggravated peak joint swelling in CHIKV‐infected TCR−/− mice as compared to naive donors, demonstrating that this process is mediated by virus‐specific CD4+ T cells.49 CHIKV proteome‐wide screening assays identified nsP1‐P4‐2 (145‐162 aa, non‐structural protein 1) and E2EP3 (2800‐2818 aa, E2 glycoprotein) as dominant mouse CD4+ T cell epitopes.50 The transfer of primary CD4+ T cell line specific for nsP1‐P4‐2 and E2EP3 generated through a prime/boost strategy, that predominantly express IFNγ, partially recapitulated joint swelling in TCR‐/‐ mice, further supporting the pathogenic role of Th1 responses.49
On the other hand, the protective role of CD8+ T cells was ruled out based on experimental findings. Firstly, infection of CD8 deficient mice or CD8+ T cell‐depleted mice did not alter joint pathology or reduce viral load in the blood and tissues.51 Next, adoptively transferred CHIKV‐specific CD8+ T cells induced upon live‐attenuated CHIKV vaccination failed to control CHIKV infection in mice.52 Although CD8+ T cells have been linked to protection against RRV48 and SINV,47 findings in CHIKV suggest that CD8+ T cells play different roles in combating alphavirus infections.
2.5. Th1 cytokines and CD4+ T cells interplay during CHIKV infection
IFNγ is the main effector cytokine produced by polarized Th1 CD4+ T cells and is needed for the control of intracellular infections caused by viruses, bacteria, and protozoa.26, 27, 53, 54, 55 It is also believed to play a central role on cell‐mediated immunity by upregulating phagocytic and microbial killing capabilities in monocytes/macrophages56, 57, 58, 59 as wells as orchestrating, together with TNFα, the recruitment of mononuclear cells to the site of the infection.60, 61
CHIKV infection in the absence of IFNγ has provided inconclusive results. In Teo et al49, 51 infection of IFNγ‐deficient (IFNγ−/−) mice resulted in a slight increase in viremia, footpad viral load, and footpad swelling, suggesting a possible antiviral role for this cytokine as previously observed in other alphaviruses.62 This is partially corroborated by a recent work63 where CHIKV infection of IFNγ−/− mice led to higher viremia but a marginal reduction in footpad swelling. Conversely, Nakaya et al20 reported that CHIKV‐infected IFNγ−/− animals displayed markedly reduced joint swelling with little effect on viremia. These discrepancies might be due to differential pathogenicity derived from the use of distinct CHIKV isolates: SGP11 in Ref. [49, 51] and LR2006‐OPY1 in Ref. [20, 63].
The contribution of IFNγ to CHIKV‐induced inflammation remains unclear, however, recent studies have suggested that CD4+ T cells might mediate CHIKV arthritic disease through the secretion of proteins other than IFNγ. RNA‐Seq analysis of CHIKV‐infected mouse footpads at the peak of swelling revealed highly induction of Granzyme A,63 a serine protease produced by NK cells, CD8+ T cells, and Th1 CD4+ T cells.64, 65, 66 Furthermore, deficiency of Granzyme A (GzmA−/−) or treatment with Serpinb6b—a Granzyme A inhibitor67—in mice abolished CHIKV‐induced joint swelling without affecting viremia. Collectively, these results suggest that Th1 cells could be mediating virus‐induced swelling through Granzyme A but not IFNγ secretion. Further adoptive transfer experiments using CD4+ T cells from Granzyme A deficient mice into CHIKV‐infected TCR−/− recipients are needed to confirm that infiltrating CHIKV‐specific Th1 cells mediate swelling through Granzyme A (Figure 1).
Figure 1.
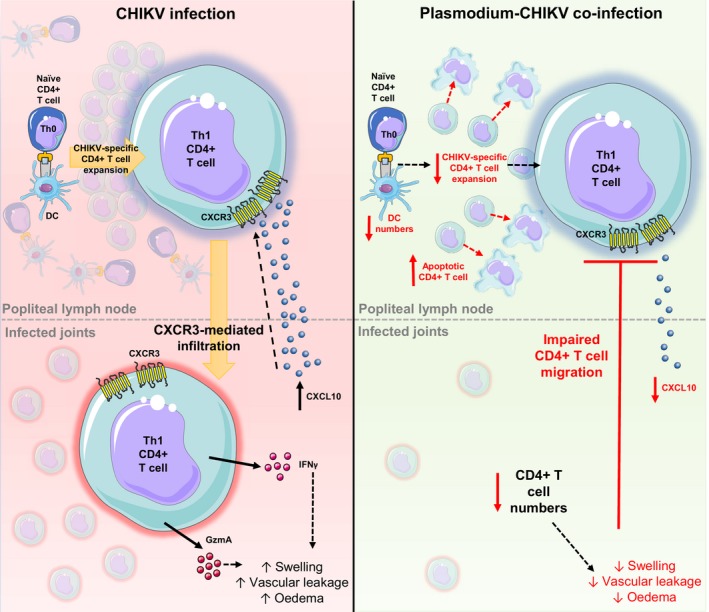
Malaria co‐infection impairs CHIKV‐specific CD4+ T cell responses in the pLN‐footpad axis. In CHIKV‐infected mice, virus‐specific CD4+ T cells multiply in the popliteal lymph node (pLN) and acquire a Th1 phenotype. Th1 cells migrate to infected tissues through a CXCR3‐mediated mechanism and potentially drive footpad swelling by releasing cytokines such as INFγ and GzmA. Upon co‐infection, a drop in the numbers of conventional dendritic cells (DCs) in the pLN is associated with diminished expansion of pathogenic CD4+ T cells. Moreover, malaria infections supress CHIKV‐specific CD4+ T cell responses by inducing early apoptosis of pLN CD4+ T cells and affecting CXCR3‐mediated joint infiltration thus leading to suppression of joint swelling. CXCL10: C‐X‐C motif chemokine 10, INFγ: interferon gamma, GzmA: granzyme A. This figure contain modified images from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/
3. CO‐INFECTION WITH PLASMODIUM PARASITES MODULATES CHIKV PATHOLOGIES
The CHIKV outbreaks in Asia, the Indian Ocean islands and its recent introduction to the Americas and the Caribbean made clear the ability of arthritogenic alphaviruses to rapidly spread and cause epidemics on a global scale. This has been fueled by genomic mutations favoring virus ecological fit to new mosquito vectors68, 69, 70, 71 and an exponential increase in international travelers to and from developing economies.12, 72, 73, 74, 75 Importantly, the expansion of CHIKV distribution and the establishment of local transmission hotspots in newly colonized areas pose a high risk of co‐infections with other highly prevalent tropical diseases. A number of epidemiological studies have reported co‐infections of CHIKV with pathogens such as Zika virus (ZIKV), dengue virus (DENV), and malaria parasites in humans76, 77, 78, 79, 80, 81, 82, 83, 84, 85 although the pathological outcomes of these co‐infections have not been clearly defined. Particularly, different clinical reports from African malaria cohorts86, 87, 88, 89 suggested the presence of CHIKV infections. Furthermore, the highly pro‐inflammatory profiles of both diseases—including the detrimental contribution of T cell‐mediated immune responses to the pathologies—and the immunosuppressive nature of malaria infections speculate that immune modulation might occur.
3.1. Plasmodium‐Chikungunya co‐infections do occur in endemic areas
Although CHIKV‐malaria co‐infections in humans have been shown to occur in the African continent, there was a lack of evidence of such infections in Southeast Asia. However, anti‐CHIKV IgM and IgG antibodies detected in 36.5% and 88.5%, respectively, in patients diagnosed with acute P. vivax from Thailand suggested both pre‐exposure and ongoing CHIKV infection.90 CHIKV‐Plasmodium co‐infections are therefore not only restricted to Africa and are likely to occur in areas of co‐circulation in Asia and Latin America. Moreover, although CHIKV and Plasmodium parasites are known to be vectored by Aedes and Anopheles mosquitoes, respectively, entomological studies have reported the existence of CHIKV‐infected Anopheles populations that might contribute to viral transmission.91, 92 These reports suggest an increasing likelihood of concurrent co‐infections by CHIKV and Plasmodium in endemic areas.
There are well‐established rodent malaria models such as Plasmodium berghei ANKA (PbA) and P. yoelii 17X (Py17x). In the lethal experimental cerebral malaria (ECM)93, 94, 95 model, PbA‐infected animals succumb during the first week postinfection due to neurological complications. The P. yoelii 17X (Py17x) mouse model is a non‐lethal, self‐resolving infection used in studies of acquired immunity against the parasite.96 These experimental models would be ideal to study co‐infections with CHIKV.
3.2. Immune modulation of CHIKV innate responses by Plasmodium co‐infections
Immunosuppression has been well‐documented for blood‐stage Plasmodium infections in both human97, 98, 99, 100, 101, 102, 103 and animal models.104, 105, 106, 107, 108, 109, 110, 111, 112, 113 Furthermore, collective evidence has suggested that Plasmodium‐associated immunosuppression increases the host susceptibility to secondary infections potentially leading to more complicated pathologies.114, 115, 116, 117, 118 Pre‐infection with murine malaria strains for 4 days followed by subsequent inoculation with CHIKV (early sequential co‐infection) or concurrent CHIKV‐Plasmodium co‐infection markedly reduced the development of virus‐induced joint swelling.119 Moreover, sequential co‐infection but not concurrent co‐infection abolished viremia in infected animals. Although one might intuitively think that Plasmodium‐CHIKV co‐infections would worsen the pathological outcome, our observations suggested a protective effect of malaria on CHIKV‐induced pathologies. Moreover, the precise timing of pathogen inoculation is essential to determine the immunomodulation outcome.
The reduction of CHIKV‐induced footpad swelling in co‐infected animals was associated with lesser numbers of joint‐infiltrating innate pathogenic subsets such as neutrophils, NK cells, and inflammatory monocytes.90, 119 Reduced edema, muscle necrosis, and vascular leakage were also observed suggesting generalized suppression of signature inflammatory responses induced by CHIKV95, 122 (Figure 2). Of note, blood‐stage malaria has been linked to dysregulation in the motility of different inflammatory immune subsets. For example, P. vivax controlled human malaria infection has been shown to induce the depletion of neutrophil populations coupled to the downregulation of chemokine receptors CRXCR1, CXCR2, CCR3, and the growth factor receptor CSF3R responsible of neutrophil maturation and survival.120 Furthermore, an in vitro study reported that parasite‐derived antigens such as P. falciparum merozoite surface protein 1 (PfMSP‐1) impairs neutrophil chemotaxis through the blockade of NFκB signaling.121 Reduced expression of cytoadhesion molecules such as CD11b, CD11c, and CD18 and diminished diapedesis and responsiveness to chemotactic factors (ie, TNFα or MCP‐1) have also been reported in monocytes upon Plasmodium infection.122, 123 Complementary studies are needed to demonstrate whether the observed reduction of the innate cellular infiltrate in joints of co‐infected animals is due to malaria‐associated impaired chemotaxis and dysregulation of the chemokine network in infected tissues.
Figure 2.
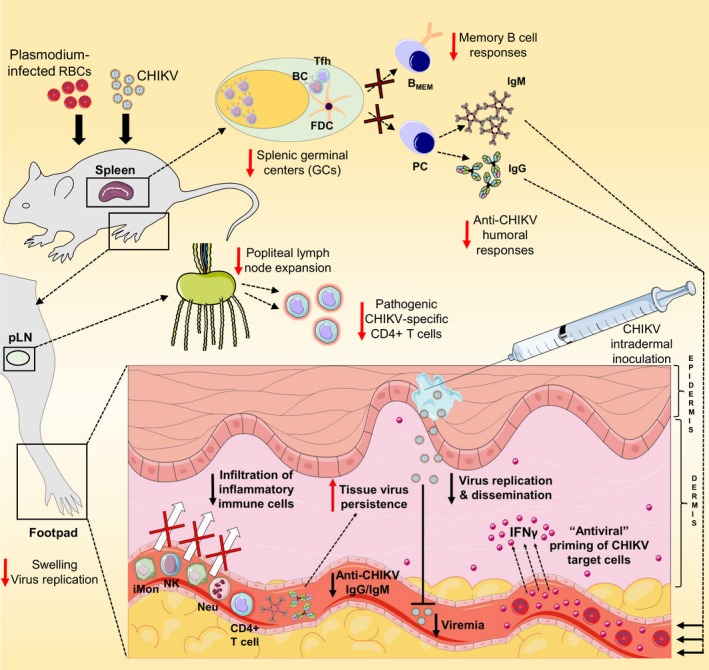
Regulation of CHIKV immune responses by malaria infections. Murine malaria infections lead to the dysregulation of splenic germinal center (GC) responses resulting in impaired generation of memory B cells and reduced CHIKV‐specific IgG/IgM serum titers associated with delayed viral clearance in infected tissues. The expansion of popliteal lymph nodes is also affected in co‐infected animals (see Figure 1). Locally in the footpads, the antiviral affects of INFγ induced by pre‐existing malaria infections limit CHIKV replication and dissemination resulting in reduced viremia. Moreover, suppression of CHIKV‐induced swelling is associated with diminished infiltration of inflammatory innate subsets and CD4+ T cells into infected tissues. BC, B cell; Tfh, follicular helper T cell; FDC, follicular dendritic cell; BMEM, memory B cell; PC, plasma cell; IgG, immunoglobulin G; IgM, immunoglobulin M; iMon, inflammatory monocytes; NK, natural killer cells; Neu, neutrophils; INFγ, interferon gamma. This figure contain modified images from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. http://smart.servier.com/
The abrogation of viremia only in animals pre‐infected with malaria (4 days prior CHIKV inoculation) suggested that immune mediators produced upon parasite infection could be responsible for the control of CHIKV dissemination. Human and mouse studies have reported early induction of IFNγ upon Plasmodium infections124, 125, 126 and an antiviral role of this cytokine has been also proposed during CHIKV infections.51, 63 In the context of sequential Plasmodium‐CHIKV co‐infection, the abrogation of viremia was reverted in mice lacking IFNγ and in wildtype mice treated with IFNγ neutralizing antibodies.119 The induced IFNγ by pre‐existing Plasmodium infection could exert an antiviral activity through the priming of primary CHIKV targets such as fibroblasts, myocytes, endothelial cells, and macrophages during the early stages of infection13, 127, 128 (Figure 2). Particularly, endothelial cells and fibroblasts have been shown to display antiviral ability upon IFNγ stimulation by interfering with virus gene transcription129 and by increasing their responsiveness to viral nucleic acids.130 Transcriptomic studies would be valuable to identify the antiviral pathways triggered by IFNγ exposure in different CHIKV‐target subsets at the site of infection.
Plasmodium inoculation during an ongoing CHIKV infection modified the outcome of murine malaria. Co‐infection in mice pre‐infected with CHIKV for 4 days with either PbA or Py17x did not affect the development of footpad swelling or viremia. In contrast, ongoing CHIKV infection exacerbated Plasmodium‐induced pathology by increasing the parasite load in the blood at the later stage of the disease. The availability of type I interferon induced by a pre‐existing CHIKV infection16, 17 might be responsible of the increase in parasitemia. Supporting this hypothesis, a recent mouse study reported that blockade of type I interferon signaling resulted in a better parasite control due to the establishment of robust antimalarial humoral responses reflected in increased Tfh numbers, GC reactions, and Plasmodium‐specific antibody titers.131, 132 Finally, the re‐challenge of recovered animals previously infected with either malaria or CHIKV with their respective heterologous pathogen did not affect the normal development of any of their individual pathologies.119
3.3. Immune modulation of CHIKV T cell responses by Plasmodium co‐infections
Interestingly, the marked reduction of footpad swelling in co‐infected animals at 6 days post‐CHIKV infection (dpi) revealed diminished infiltration of virus‐specific CD4+ T cells in infected joints.119 Furthermore, co‐infection in LTα−/− animals (devoid of lymph nodes (LNs)), but not splenectomized mice, recovered the major peak of footpad inflammation suggesting that LN is the main secondary lymphoid organ of immune regulation.90, 119
Immune profiling of the popliteal LN (pLN), the nearest draining LN to the site of infection, from the concurrent co‐infected animals revealed decreased numbers of total and CHIKV‐specific CD4+ T cells.119 Concordantly, increased apoptotic CD4+ T cells in the pLN at 5 and 6 dpi were detected. This was supported by previous findings where apoptosis in lymph nodes and secondary lymphoid organs upon malaria infection was described.133, 134 In addition, it is also known that blood‐stage malaria infections suppress dendritic cell (DC) responses by affecting DC maturation,142, 143, 144 reducing surface expression of MHC‐II142, 145, 146, 147 and co‐stimulatory molecule CD86,146 inducing DC apoptosis148, 149 and impairing their ability to process and present parasite‐derived antigens to T cells.150, 151 In line with this, reduced numbers of conventional DCs in the pLN of co‐infected mice were also observed, suggesting that reduced expansion of CD4+ T cells might be due in part to the drop in DCs numbers associated with malaria.135, 136, 137, 138 Further research is needed to determine whether DCs ability to process and present CHIKV‐derived antigens (signal 1) and to co‐stimulate (signal 2) naive CD4+ T cell in the pLN is affected upon co‐infection. Similarly, whether Plasmodium infection alter the phagocytic and migratory potential of DCs, and other APCs, at the site of CHIKV inoculation remains to be explored.
Impaired expansion and early apoptosis of CD4+ T cells in draining lymph nodes in co‐infected mice could only partially explain the suppression of joint swelling. Virus‐specific CD4+ T cells in the pLN was only suppressed by ~50%, but CD4+ T cells detection in the co‐infected animals' footpad was almost completely abolished.119 Using adoptive transfer of CSFE‐labeled CD4+ T cells isolated from CHIKV‐infected mice, it was shown that the migratory potential of CD4+ T cells into the infected footpad was also abolished by co‐infection. This blockage of migration was mediated by the suppression of Th1‐associated chemokines such as RANTES, MIP‐1α, MIP‐1β, and CXCL10 in the footpad. Furthermore, in vivo blockade experiments showed that Th1 chemokine receptor CXCR3 and not CCR5 is functionally important for CD4+ T cell‐mediated CHIKV inflammation and that CXCR3‐mediated chemotaxis was impaired during co‐infection.119
Taken together, these results suggest that the expansion, survival, and trafficking of pathogenic CHIKV‐specific CD4+ T cells in the pLN/footpad axis are hampered by co‐infections with Plasmodium parasites (Figure 1). Additionally, the contribution of T cell anergy139, 140 and exhaustion141, 142, 143, 144 extensively reported upon severe malaria infections remain to be explored.
3.4. Immune modulation of CHIKV B cell responses by Plasmodium co‐infections
B cell immune responses are altered during malaria. This has been proposed as a mechanism to avoid the establishment of efficient and long‐lasting humoral responses.145 Notably, the development of naturally acquired immunity against Plasmodium is very slow and might take several years146, 147, 148, 149 and is associated with a very low frequency of Plasmodium‐specific memory B cells detected in populations from malaria endemic areas.150, 151 Studies using murine and non‐human primate models107, 132, 146 have suggested impaired induction and maturation of germinal centers (GCs) thus leading to the production of low‐affinity, short‐lived antibody responses and the development of limited B cell memory. Such immune regulation could have important implications on the generation of antibodies against heterologous pathogens upon co‐infection.
CHIKV‐neutralizing antibody responses are known to play a pivotal role in the control of CHIKV infection. Lum et al152 reported that CHIKV infection in mice lacking functional B cells resulted in high‐level viremia that lasted for more than a year. Notably, co‐infection experiments (in both sequential and concurrent settings) revealed delayed virus clearance from footpad tissues.119 In further support of this, circulating levels of CHIKV‐specific IgM and IgG and their virus‐neutralizing capacities were found to be reduced at 15 dpi upon concurrent co‐infection. Complementary experiments allowed us to identify the spleen as the main site of B cell dysregulation as co‐infection in splenectomized mice led to similar CHIKV‐specific antibody levels and tissue virus clearance at the later time‐points of the disease than non‐splenectomized animals.119 This could be due to delay in the induction of GC reactions and a reduced number of GC‐dependent CD73+ memory B cells in the co‐infected animals119 (Figure 2). Although the precise mechanisms governing impairment of CHIKV antibody responses upon malaria co‐infection are still unknown, the answer might lay on the impact of Plasmodium infections on T cell‐dependent enhancement of B cell responses. Millington et al138 reported that helper T cells activated by hemozoin‐loaded DCs displayed reduced migration to B cell compartments in lymphoid‐organ follicles leading to defective B cell expansion and antibody production in mice. Similarly, severe malaria infections in mouse models were linked to impaired T follicular helper cells (Tfh) maturation and dysregulation of GC responses.153
4. CONCLUDING REMARKS
The unique protective effects exerted by Plasmodium infections against CHIKV pathologies might have relevant implications in the control of both illnesses in endemic areas. Specific interventions aimed at replicating disease‐protective mechanisms, will provide new therapeutic approaches and/or adjunct therapies to antiviral or antimalarial treatment.
At the epidemiological level, co‐infections by CHIKV and Plasmodium parasites can be underestimated in human populations due to the immunosuppressive nature of malaria which can result in the development of asymptomatic CHIKV infections without compromised joints. The absence of alphavirus screening in confirmed malaria cases might also contribute to a higher burden of asymptomatic CHIKV carriers.87 On the other hand, reduced viremia upon co‐infection could impact CHIKV transmission dynamics by reducing the number of successfully infected mosquitoes after a blood meal. These observations highlight the need for the incorporation of alphavirus diagnosis in malaria intervention programs.
Lastly, O'nyong‐nyong virus (ONNV), an emerging arthritogenic alphavirus known to cause similar clinical manifestations than CHIKV,154, 155 is transmitted in the African sub‐continent by two main malaria vectors, Anopheles gambiae and A. funestus, favoring a higher likelihood of co‐infections than CHIKV. A similar situation could arise in Latin America, where emerging Mayaro Virus (MAYV) has been recently shown to infect Anopheles mosquitoes suggesting that co‐infections with malaria could happen.156 It is therefore crucial to understand the transmission dynamics, pathological implications and burden of alphavirus‐Plasmodium co‐infections for the future development of improved diagnostics and treatment strategies.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
ACKNOWLEDGEMENTS
This work was funded by SIgN, Agency for Science, Technology and Research (A*STAR) core grant.
Torres‐Ruesta A, Teo T‐H, Chan Y‐H, Rénia L, Ng LFP. Pathogenic Th1 responses in CHIKV‐induced inflammation and their modulation upon Plasmodium parasites co‐infection. Immunol Rev. 2020;294:80–91. 10.1111/imr.12825
This article is part of a series of reviews covering Inflammatory Arthritis appearing in Volume 294 of Immunological Reviews.
REFERENCES
- 1. Atkins GJ. The pathogenesis of Alphaviruses. ISRN Virology. 2013;2013:22. [Google Scholar]
- 2. Suhrbier A, Jaffar‐Bandjee MC, Gasque P. Arthritogenic alphaviruses–an overview. Nat Rev Rheumatol. 2012;8(7):420‐429. [DOI] [PubMed] [Google Scholar]
- 3. Mostafavi H, Abeyratne E, Zaid A, Taylor A. Arthritogenic alphavirus‐induced immunopathology and targeting host inflammation as a therapeutic strategy for alphaviral disease. Viruses. 2019;11(3):290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tanabe ISB, Tanabe ELL, Santos EC, et al. Cellular and molecular immune response to Chikungunya virus infection. Front Cell Infect Microbiol. 2018;8:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Powers AM, Brault AC, Shirako Y, et al. Evolutionary relationships and systematics of the alphaviruses. J Virol. 2001;75(21):10118‐10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mejia CR, Lopez‐Velez R. Tropical arthritogenic alphaviruses. Reumatol Clin. 2018;14(2):97‐105. [DOI] [PubMed] [Google Scholar]
- 7. Zaid A, Gerardin P, Taylor A, Mostafavi H, Malvy D, Mahalingam S. Chikungunya arthritis: implications of acute and chronic inflammation mechanisms on disease management. Arthritis Rheumatol. 2018;70(4):484‐495. [DOI] [PubMed] [Google Scholar]
- 8. Daginawala HatimF, Chandak NitinH, Kashyap RajpalS, et al. Neurological complications of Chikungunya virus infection. Neurol India. 2009;57(2):177‐180. [DOI] [PubMed] [Google Scholar]
- 9. Mahto SK, Gupta PK, Singh A, Meena RC. Atypical neurological manifestations of chikungunya fever: two case reports. Indian J Crit Care Med. 2018;22(4):306‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barr KL, Khan E, Farooqi JQ, et al. Evidence of Chikungunya virus disease in pakistan since 2015 with patients demonstrating involvement of the central nervous system. Front Public Health. 2018;6:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sa PKO, Nunes MDM, Leite IR, et al. Chikungunya virus infection with severe neurologic manifestations: report of four fatal cases. Rev Soc Bras Med Trop. 2017;50(2):265‐268. [DOI] [PubMed] [Google Scholar]
- 12. Wahid B, Ali A, Rafique S, Idrees M. Global expansion of chikungunya virus: mapping the 64‐year history. Int J Infect Dis. 2017;58:69‐76. [DOI] [PubMed] [Google Scholar]
- 13. Gardner J, Anraku I, Le TT, et al. Chikungunya virus arthritis in adult wild‐type mice. J Virol. 2010;84(16):8021‐8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan YH, Lum FM, Ng LFP. Limitations of current in vivo mouse models for the study of Chikungunya virus pathogenesis. Med Sci (Basel). 2015;3(3):64‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ng LF, Chow A, Sun YJ, et al. IL‐1beta, IL‐6, and RANTES as biomarkers of Chikungunya severity. PLoS ONE. 2009;4(1):e4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Teo TH, Her Z, Tan JJ, et al. Caribbean and La Reunion Chikungunya virus isolates differ in their capacity to induce proinflammatory Th1 and NK cell responses and acute joint pathology. J Virol. 2015;89(15):7955‐7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Teng T‐S, Kam YW, Lee B, et al. A systematic meta‐analysis of immune signatures in patients with acute Chikungunya virus infection. J Infect Dis. 2015;211(12):1925‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoarau JJ, Jaffar Bandjee M‐C, Krejbich Trotot P, et al. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J Immunol. 2010;184(10):5914‐5927. [DOI] [PubMed] [Google Scholar]
- 19. Her Z, Teng TS, Tan JJL, et al. Loss of TLR3 aggravates CHIKV replication and pathology due to an altered virus‐specific neutralizing antibody response. EMBO Mol Med. 2015;7(1):24‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakaya HI, Gardner J, Poo YS, Major L, Pulendran B, Suhrbier A. Gene profiling of Chikungunya virus arthritis in a mouse model reveals significant overlap with rheumatoid arthritis. Arthritis Rheum. 2012;64(11):3553‐3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamada H, Nakashima Y, Okazaki K, et al. Preferential accumulation of activated Th1 cells not only in rheumatoid arthritis but also in osteoarthritis joints. J Rheumatol. 2011;38(8):1569‐1575. [DOI] [PubMed] [Google Scholar]
- 22. Bazzazi H, Aghaei M, Memarian A, Asgarian‐Omran H, Behnampour N, Yazdani Y. Th1‐Th17 ratio as a new insight in Rheumatoid Arthritis disease. Iran J Allergy Asthma Immunol. 2018;17(1):68‐77. [PubMed] [Google Scholar]
- 23. Mellado M, Martinez‐Munoz L, Cascio G, Lucas P, Pablos JL, Rodriguez‐Frade JM. T cell migration in Rheumatoid Arthritis. Front Immunol. 2015;6:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burmester GR, Feist E, Dorner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10(2):77‐88. [DOI] [PubMed] [Google Scholar]
- 25. Sallusto F, Lanzavecchia A, Mackay CR. Chemokines and chemokine receptors in T‐cell priming and Th1/Th2‐mediated responses. Immunol Today. 1998;19(12):568‐574. [DOI] [PubMed] [Google Scholar]
- 26. Liew FY. T(H)1 and T(H)2 cells: a historical perspective. Nat Rev Immunol. 2002;2(1):55‐60. [DOI] [PubMed] [Google Scholar]
- 27. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112(5):1557‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sandquist I, Kolls J. Update on regulation and effector functions of Th17 cells. F1000Res. 2018;7:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL‐17 and Th17 cells. Annu Rev Immunol. 2009;27:485‐517. [DOI] [PubMed] [Google Scholar]
- 30. Giri JG, Anderson DM, Kumaki S, Park LS, Grabstein KH, Cosman D. IL‐15, a novel T cell growth factor that shares activities and receptor components with IL‐2. J Leukoc Biol. 1995;57(5):763‐766. [DOI] [PubMed] [Google Scholar]
- 31. Rubin LA, Kurman CC, Fritz ME, et al. Soluble interleukin 2 receptors are released from activated human lymphoid cells in vitro. J Immunol. 1985;135(5):3172‐3177. [PubMed] [Google Scholar]
- 32. Qin S, Rottman JB, Myers P, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101(4):746‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prame Kumar K, Nicholls AJ, Wong CHY. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018;371(3):551‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ozden S, Huerre M, Riviere J‐P, et al. Human muscle satellite cells as targets of Chikungunya virus infection. PLoS ONE. 2007;2(6):e527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lidbury B, Rulli N, Suhrbier A, et al. Macrophage‐derived proinflammatory factors contribute to the development of arthritis and myositis after infection with an arthrogenic alphavirus. J Infect Dis. 2008;197(11):1585‐1593. [DOI] [PubMed] [Google Scholar]
- 36. Lidbury BA, Simeonovic C, Maxwell GE, Marshall ID, Hapel AJ. Macrophage‐induced muscle pathology results in morbidity and mortality for Ross River virus‐infected mice. J Infect Dis. 2000;181(1):27‐34. [DOI] [PubMed] [Google Scholar]
- 37. Morrison TE, Whitmore AC, Shabman RS, Lidbury BA, Mahalingam S, Heise MT. Characterization of Ross River virus tropism and virus‐induced inflammation in a mouse model of viral arthritis and myositis. J Virol. 2006;80(2):737‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rulli NE, Rolph MS, Srikiatkhachorn A, Anantapreecha S, Guglielmotti A, Mahalingam S. Protection from arthritis and myositis in a mouse model of acute chikungunya virus disease by bindarit, an inhibitor of monocyte chemotactic protein‐1 synthesis. J Infect Dis. 2011;204(7):1026‐1030. [DOI] [PubMed] [Google Scholar]
- 39. Mora E, Guglielmotti A, Biondi G, Sassone‐Corsi P. Bindarit: an anti‐inflammatory small molecule that modulates the NFkappaB pathway. Cell Cycle. 2012;11(1):159‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poo YS, Nakaya H, Gardner J, et al. CCR2 deficiency promotes exacerbated chronic erosive neutrophil‐dominated chikungunya virus arthritis. J Virol. 2014;88(12):6862‐6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lum FM. Host adaptive immunity modulates Chickungunya virus disease manifestation. Singapore: National University of Singapore; 2015. [Google Scholar]
- 42. Maucourant C, Petitdemange C, Yssel H, Vieillard V. Control of acute Arboviral infection by Natural Killer Cells. Viruses. 2019;11(2):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petitdemange C, Becquart P, Wauquier N, et al. Unconventional repertoire profile is imprinted during acute Chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog. 2011;7(9):e1002268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thanapati S, Das R, Tripathy AS. Phenotypic and functional analyses of NK and NKT‐like populations during the early stages of chikungunya infection. Front Microbiol. 2015;6:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rouzaire P, Luci C, Blasco E, et al. Natural killer cells and T cells induce different types of skin reactions during recall responses to haptens. Eur J Immunol. 2012;42(1):80‐88. [DOI] [PubMed] [Google Scholar]
- 46. Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi‐Mbiguino A, Leroy EM. The acute phase of Chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J Infect Dis. 2011;204(1):115‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Binder GK, Griffin DE. Interferon‐gamma‐mediated site‐specific clearance of alphavirus from CNS neurons. Science. 2001;293(5528):303‐306. [DOI] [PubMed] [Google Scholar]
- 48. Linn ML, Mateo L, Gardner J, Suhrbier A. Alphavirus‐specific cytotoxic T lymphocytes recognize a cross‐reactive epitope from the capsid protein and can eliminate virus from persistently infected macrophages. J Virol. 1998;72(6):5146‐5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Teo T‐H, Chan YH, Lee WWL, et al. Fingolimod treatment abrogates chikungunya virus‐induced arthralgia. Sci Transl Med. 2017;9(375):eaal1333. [DOI] [PubMed] [Google Scholar]
- 50. Kam YW, Lum FM, Teo TH, et al. Early neutralizing IgG response to Chikungunya virus in infected patients targets a dominant linear epitope on the E2 glycoprotein. EMBO Mol Med. 2012;4(4):330‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Teo TH, Lum FM, Claser C, et al. A pathogenic role for CD4+ T cells during Chikungunya virus infection in mice. J Immunol. 2013;190(1):259‐269. [DOI] [PubMed] [Google Scholar]
- 52. Chu H, Das SC, Fuchs JF, et al. Deciphering the protective role of adaptive immunity to CHIKV/IRES a novel candidate vaccine against Chikungunya in the A129 mouse model. Vaccine. 2013;31(33):3353‐3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383(6603):787‐793. [DOI] [PubMed] [Google Scholar]
- 54. Jankovic D, Feng CG. CD4+ T cell differentiation in infection: amendments to the Th1/Th2 axiom. Front Immunol. 2015;6:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+ T cells: differentiation and functions. Clin Dev Immunol. 2012;2012:925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paludan SR. Interleukin‐4 and interferon‐gamma: the quintessence of a mutual antagonistic relationship. Scand J Immunol. 1998;48(5):459‐468. [DOI] [PubMed] [Google Scholar]
- 57. Wu C, Xue Y, Wang P, et al. IFN‐gamma primes macrophage activation by increasing phosphatase and tensin homolog via downregulation of miR‐3473b. J Immunol. 2014;193(6):3036‐3044. [DOI] [PubMed] [Google Scholar]
- 58. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon‐gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75(2):163‐189. [DOI] [PubMed] [Google Scholar]
- 59. Held TK, Weihua X, Yuan L, Kalvakolanu DV, Cross AS. Gamma interferon augments macrophage activation by lipopolysaccharide by two distinct mechanisms, at the signal transduction level and via an autocrine mechanism involving tumor necrosis factor alpha and interleukin‐1. Infect Immun. 1999;67(1):206‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cuff CA, Schwartz J, Bergman CM, Russell KS, Bender JR, Ruddle NH. Lymphotoxin α3 induces chemokines and adhesion molecules: insight into the role of LTα in inflammation and lymphoid organ development. J Immunol. 1998;161(12):6853‐6860. [PubMed] [Google Scholar]
- 61. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon‐gamma. Annu Rev Immunol. 1997;15:749‐795. [DOI] [PubMed] [Google Scholar]
- 62. Ryman KD, Meier KC, Gardner CL, Adegboyega PA, Klimstra WB. Non‐pathogenic Sindbis virus causes hemorrhagic fever in the absence of alpha/beta and gamma interferons. Virology. 2007;368(2):273‐285. [DOI] [PubMed] [Google Scholar]
- 63. Wilson JAC, Prow NA, Schroder WA, et al. RNA‐Seq analysis of chikungunya virus infection and identification of granzyme A as a major promoter of arthritic inflammation. PLoS Pathog. 2017;13(2):e1006155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hidalgo LG, Einecke G, Allanach K, Halloran PF. The transcriptome of human cytotoxic T cells: similarities and disparities among allostimulated CD4+ CTL, CD8+ CTL and NK cells. Am J Transplant. 2008;8(3):627‐636. [DOI] [PubMed] [Google Scholar]
- 65. Takeuchi A, Saito T. CD4 CTL, a cytotoxic subset of CD4+ T cells their differentiation and function. Front Immunol. 2017;8:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Russell JH, Ley TJ. Lymphocyte‐mediated cytotoxicity. Annu Rev Immunol. 2002;20:323‐370. [DOI] [PubMed] [Google Scholar]
- 67. Kaiserman D, Stewart SE, Plasman K, Gevaert K, Van Damme P, Bird PI. Identification of Serpinb6b as a species‐specific mouse granzyme A inhibitor suggests functional divergence between human and mouse granzyme A. J Biol Chem. 2014;289(13):9408‐9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tsetsarkin KA, McGee CE, Volk SM, Vanlandingham DL, Weaver SC, Higgs S. Epistatic roles of E2 glycoprotein mutations in adaption of chikungunya virus to Aedes albopictus and Ae. aegypti mosquitoes. PLoS ONE. 2009;4(8):e6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tsetsarkin KA, Chen R, Weaver SC. Interspecies transmission and chikungunya virus emergence. Curr Opin Virol. 2016;16:143‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. A single mutation in Chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007;3(12):e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schuffenecker I, Iteman I, Michault A, et al. Genome microevolution of Chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006;3(7):e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lwande OW, Obanda V, Bucht G, et al. Global emergence of Alphaviruses that cause arthritis in humans. Infect Ecol Epidemiol. 2015;5:29853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Panning M, Grywna K, van Esbroeck M, Emmerich P, Drosten C. Chikungunya fever in travelers returning to Europe from the Indian Ocean region, 2006. Emerg Infect Dis. 2008;14(3):416‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen LH, Wilson ME. Dengue and Chikungunya infections in travelers. Curr Opin Infect Dis. 2010;23(5):438‐444. [DOI] [PubMed] [Google Scholar]
- 75. Johansson MA, Powers AM, Pesik N, Cohen NJ, Staples JE. Nowcasting the spread of Chikungunya virus in the Americas. PLoS ONE. 2014;9(8):e104915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Salam N, Mustafa S, Hafiz A, Chaudhary AA, Deeba F, Parveen S. Global prevalence and distribution of coinfection of malaria, dengue and chikungunya: a systematic review. BMC Public Health. 2018;18(1):710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Raut CG, Rao NM, Sinha DP, Hanumaiah H, Manjunatha MJ. Chikungunya, dengue, and malaria co‐infection after travel to Nigeria. India. Emerg Infect Dis. 2015;21(5):908‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kinimi E, Shayo MJ, Patrick BN, et al. Evidence of Chikungunya virus infection among febrile patients seeking healthcare in selected districts of Tanzania. Infect Ecol Epidemiol. 2018;8(1):1553460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vogels CBF, Ruckert C, Cavany SM, Perkins TA, Ebel GD, Grubaugh ND. Arbovirus coinfection and co‐transmission: a neglected public health concern? PLoS Biol. 2019;17(1):e3000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Savargaonkar D, Sinha S, Srivastava B, et al. An epidemiological study of dengue and its coinfections in Delhi. Int J Infect Dis. 2018;74:41‐46. [DOI] [PubMed] [Google Scholar]
- 81. Villamil‐Gómez WE, Rodríguez‐Morales AJ, Uribe‐García AM, et al. Zika, Dengue, and Chikungunya co‐infection in a pregnant woman from Colombia. Int J Infect Dis. 2016;51:135‐138. [DOI] [PubMed] [Google Scholar]
- 82. Furuya‐Kanamori L, Liang S, Milinovich G, et al. Co‐distribution and co‐infection of Chikungunya and dengue viruses. BMC Infect Dis. 2016;16:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kaur M, Singh K, Sidhu SK, et al. Coinfection of Chikungunya and Dengue viruses: a serological study from North Western region of Punjab. India. J Lab Physicians. 2018;10(4):443‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Singh J, Dinkar A, Singh RG, Siddiqui MS, Sinha N, Singh SK. Clinical profile of dengue fever and coinfection with Chikungunya. Ci Ji Yi Xue Za Zhi. 2018;30(3):158‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Waggoner J, Brichard J, Mutuku F, et al. Malaria and Chikungunya detected using molecular diagnostics among febrile kenyan children. Open Forum Infect Dis. 2017;4(3):ofx110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sow A, Loucoubar C, Diallo D, et al. Concurrent malaria and arbovirus infections in Kedougou, southeastern Senegal. Malar J. 2016;15:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Baba M, Logue CH, Oderinde B, et al. Evidence of arbovirus co‐infection in suspected febrile malaria and typhoid patients in Nigeria. J Infect Dev Ctries. 2013;7(1):51‐59. [DOI] [PubMed] [Google Scholar]
- 88. Ayorinde AF, Oyeyiga AM, Nosegbe NO, Folarin OA. A survey of malaria and some arboviral infections among suspected febrile patients visiting a health centre in Simawa, Ogun State. Nigeria. J Infect Public Health. 2016;9(1):52‐59. [DOI] [PubMed] [Google Scholar]
- 89. Tazeen A, Abdullah M, Hisamuddin M, et al. Concurrent infection with plasmodium vivax and the Dengue and Chikungunya viruses in a paediatric patient from New Delhi, India in 2016. Intervirology. 2017;60(1–2):48‐52. [DOI] [PubMed] [Google Scholar]
- 90. Teo TH. Host Immune modulation during Malaria Parasites and Chikungunya virus coinfection in experimental models . Singapore: National University of Singapore;2015. [Google Scholar]
- 91. Yadav P, Gokhale MD, Barde PV, Singh DK, Mishra AC, Mourya DT. Experimental transmission of Chikungunya virus by Anopheles stephensi mosquitoes. Acta Virol. 2003;47(1):45‐47. [PubMed] [Google Scholar]
- 92. Diallo D, Sall AA, Buenemann M, et al. Landscape ecology of sylvatic Chikungunya virus and mosquito vectors in southeastern Senegal. PLoS Negl Trop Dis. 2012;6(6):e1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Howland SW, Claser C, Poh CM, Gun SY, Renia L. Pathogenic CD8+ T cells in experimental cerebral malaria. Semin Immunopathol. 2015;37(3):221‐231. [DOI] [PubMed] [Google Scholar]
- 94. de Oca MM, Engwerda C, Haque A. Plasmodium berghei ANKA (PbA) infection of C57BL/6J mice: a model of severe malaria. Methods Mol Biol. 2013;1031:203‐213. [DOI] [PubMed] [Google Scholar]
- 95. Engwerda C, Belnoue E, Gruner AC, Renia L. Experimental models of cerebral malaria. Curr Top Microbiol Immunol. 2005;297:103‐143. [PubMed] [Google Scholar]
- 96. Weiss WR, Good MF, Hollingdale MR, Miller LH, Berzofsky JA. Genetic control of immunity to Plasmodium yoelii sporozoites. J Immunol. 1989;143(12):4263‐4266. [PubMed] [Google Scholar]
- 97. Ho M, Webster HK, Looareesuwan S, et al. Antigen‐specific immunosuppression in human malaria due to Plasmodium falciparum. J Infect Dis. 1986;153(4):763‐771. [DOI] [PubMed] [Google Scholar]
- 98. Wyler DJ, Brown J. Malaria antigen‐specific T‐cell responsiveness during infection with Plasmodium falciparum. Clin Exp Immunol. 1977;29(3):401‐407. [PMC free article] [PubMed] [Google Scholar]
- 99. Riley EM, Jobe O, Whittle HC. CD8+ T cells inhibit Plasmodium falciparum‐induced lymphoproliferation and gamma interferon production in cell preparations from some malaria‐immune individuals. Infect Immun. 1989;57(4):1281‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Riley EM, Jobe O, Blackman M, Whittle HC, Greenwood BM. Plasmodium falciparum schizont sonic extracts suppress lymphoproliferative responses to mitogens and antigens in malaria‐immune adults. Infect Immun. 1989;57(10):3181‐3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hviid L, Theander TG, Abu‐Zeid YA, et al. Loss of cellular immune reactivity during acute Plasmodium falciparum malaria. FEMS Microbiol Immunol. 1991;3(4):219‐227. [DOI] [PubMed] [Google Scholar]
- 102. Theander TG, Bygbjerg IC, Andersen BJ, Jepsen S, Kharazmi A, Odum N. Suppression of parasite‐specific response in Plasmodium falciparum malaria. A longitudinal study of blood mononuclear cell proliferation and subset composition. Scand J Immunol. 1986;24(1):73‐81. [DOI] [PubMed] [Google Scholar]
- 103. Chemtai AK, Okelo GB. Suppression of T‐cell proliferative response in Plasmodium falciparum malaria patients–preliminary results. East Afr Med J. 1989;66(12):787‐791. [PubMed] [Google Scholar]
- 104. Wangoo A, Ganguly NK, Mahajan RC. Immunosuppression in murine malaria: suppressor role of macrophages and their products during acute and chronic Plasmodium berghei infection. APMIS. 1990;98(5):407‐414. [PubMed] [Google Scholar]
- 105. Greenwood BM, Playfair JH, Torrigiani G. Immunosuppression in murine malaria. I. General characteristics. Clin Exp Immunol. 1971;8(3):467‐478. [PMC free article] [PubMed] [Google Scholar]
- 106. McBride JS, Micklem HS, Ure JM. Immunosuppression in murine malaria. I. Response to type III pneumococcal polysaccharide. Immunology. 1977;32(5):635‐644. [PMC free article] [PubMed] [Google Scholar]
- 107. Greenwood BM, Brown JC, De Jesus DG, Holborow EJ. Immunosuppression in murine malaria. II. The effect on reticulo‐endothelial and germinal centre function. Clin Exp Immunol. 1971;9(3):345‐354. [PMC free article] [PubMed] [Google Scholar]
- 108. McBride JS, Micklem HS. Immunosuppression in murine malaria. II. The primary response to bovine serum albumin. Immunology. 1977;33(2):253‐259. [PMC free article] [PubMed] [Google Scholar]
- 109. Strambachova‐McBride J, Micklem HS, Immunosuppression in murine malaria. III. Induction of tolerance and of immunological memory by soluble bovine serum albumin. Immunology. 1979;36(3):607‐614. [PMC free article] [PubMed] [Google Scholar]
- 110. Strambachová‐Mcbride J, Micklem HS. Immunosuppression in murine malaria. IV. The secondary response to bovine serum albumin. Parasite Immunol. 1979;1(2):141‐157. [DOI] [PubMed] [Google Scholar]
- 111. Khansari N, Segre M, Segre D. Immunosuppression in murine malaria: a soluble immunosuppressive factor derived from Plasmodium berghei‐infected blood. J Immunol. 1981;127(5):1889‐1893. [PubMed] [Google Scholar]
- 112. Mahajan RC, Ganguly NK, Thadani M, Prasad RN. Immunosuppression in murine malaria: role of activated macrophages. Aust J Exp Biol Med Sci. 1986;64(Pt 1):63‐66. [DOI] [PubMed] [Google Scholar]
- 113. Barker LR, Powers KG. Immunosuppression in rodent malaria. Effect upon recovery and antibody response. Am J Trop Med Hyg. 1971;20(3):389‐393. [DOI] [PubMed] [Google Scholar]
- 114. Mabey DC, Brown A, Greenwood BM. Plasmodium falciparum malaria and Salmonella infections in Gambian children. J Infect Dis. 1987;155(6):1319‐1321. [DOI] [PubMed] [Google Scholar]
- 115. McGregor IA, Barr M. Antibody response to tetanus toxoid inoculation in malarious and non‐malarious Gambian children. Trans R Soc Trop Med Hyg. 1962;56(5):364‐367. [Google Scholar]
- 116. Whittle HC, Brown J, Marsh K, et al. T‐cell control of Epstein‐Barr virus‐infected B cells is lost during P. falciparum malaria. Nature. 1984;312(5993):449‐450. [DOI] [PubMed] [Google Scholar]
- 117. Cunnington AJ, de Souza JB, Walther M, Riley EM. Malaria impairs resistance to Salmonella through heme‐ and heme oxygenase‐dependent dysfunctional granulocyte mobilization. Nat Med. 2011;18(1):120‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Takem EN, Roca A, Cunnington A. The association between malaria and non‐typhoid Salmonella bacteraemia in children in sub‐Saharan Africa: a literature review. Malar J. 2014;13:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Teo TH, Lum FM, Ghaffar K, et al. Plasmodium co‐infection protects against chikungunya virus‐induced pathologies. Nat Commun. 2018;9(1):3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Vallejo AF, Read RC, Arevalo‐Herrera M, Herrera S, Elliott T, Polak ME. Malaria systems immunology: plasmodium vivax induces tolerance during primary infection through dysregulation of neutrophils and dendritic cells. J Infect. 2018;77(5):440‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Waisberg M, Cerqueira GC, Yager SB, et al. Plasmodium falciparum merozoite surface protein 1 blocks the proinflammatory protein S100P. Proc Natl Acad Sci USA. 2012;109(14):5429‐5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Skorokhod OA, Barrera V, Heller R, et al. Malarial pigment hemozoin impairs chemotactic motility and transendothelial migration of monocytes via 4‐hydroxynonenal. Free Radic Biol Med. 2014;75:210‐221. [DOI] [PubMed] [Google Scholar]
- 123. Mandala WL, Msefula CL, Gondwe EN, Drayson MT, Molyneux ME, MacLennan CA. Monocyte activation and cytokine production in Malawian children presenting with P. falciparum malaria. Parasite Immunol. 2016;38(5):317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. King T, Lamb T. Interferon‐gamma: the Jekyll and Hyde of Malaria. PLoS Pathog. 2015;11(10):e1005118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. De Souza JB, Williamson KH, Otani T, Playfair JH. Early gamma interferon responses in lethal and nonlethal murine blood‐stage malaria. Infect Immun. 1997;65(5):1593‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Artavanis‐Tsakonas K, Riley EM. Innate immune response to malaria: rapid induction of IFN‐gamma from human NK cells by live Plasmodium falciparum‐infected erythrocytes. J Immunol. 2002;169(6):2956‐2963. [DOI] [PubMed] [Google Scholar]
- 127. Carpentier KS, Morrison TE. Innate immune control of alphavirus infection. Curr Opin Virol. 2018;28:53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kang S, Brown HM, Hwang S. Direct antiviral mechanisms of interferon‐gamma. Immune Netw. 2018;18(5):e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Milligan S, Robinson M, O'Donnell E, Blackbourn DJ. Inflammatory cytokines inhibit Kaposi's sarcoma‐associated herpesvirus lytic gene transcription in in vitro‐infected endothelial cells. J Virol. 2004;78(5):2591‐2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Costa‐Pereira AP, Williams TM, Strobl B, Watling D, Briscoe J, Kerr IM. The antiviral response to gamma interferon. J Virol. 2002;76(18):9060‐9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zander RA, Guthmiller JJ, Graham AC, et al. Type I interferons induce T regulatory 1 responses and restrict humoral immunity during experimental malaria. PLoS Pathog. 2016;12(10):e1005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sebina I, James KR, Soon MSF, et al. IFNAR1‐signalling obstructs ICOS‐mediated humoral immunity during non‐lethal blood‐stage plasmodium infection. PLoS Pathog. 2016;12(11):e1005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Sanchez‐Torres L, Rodriguez‐Ropon A, Aguilar‐Medina M, Favila‐Castillo L. Mouse splenic CD4+ and CD8+ T cells undergo extensive apoptosis during a Plasmodium chabaudi chabaudi AS infection. Parasite Immunol. 2001;23(12):617‐626. [DOI] [PubMed] [Google Scholar]
- 134. Helmby H, Jonsson G, Troye‐Blomberg M. Cellular changes and apoptosis in the spleens and peripheral blood of mice infected with blood‐stage Plasmodium chabaudi chabaudi AS. Infect Immun. 2000;68(3):1485‐1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pinzon‐Charry A, Woodberry T, Kienzle V, et al. Apoptosis and dysfunction of blood dendritic cells in patients with falciparum and vivax malaria. J Exp Med. 2013;210(8):1635‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Woodberry T, Minigo G, Piera KA, et al. Low‐level Plasmodium falciparum blood‐stage infection causes dendritic cell apoptosis and dysfunction in healthy volunteers. J Infect Dis. 2012;206(3):333‐340. [DOI] [PubMed] [Google Scholar]
- 137. Millington OR, Gibson VB, Rush CM, et al. Malaria impairs T cell clustering and immune priming despite normal signal 1 from dendritic cells. PLoS Pathog. 2007;3(10):1380‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Millington OR, Di Lorenzo C, Phillips RS, Garside P, Brewer JM. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin‐induced failure of dendritic cell function. J Biol. 2006;5(2):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Theander TG, Bygbjerg IC, Jacobsen L, Jepsen S, Larsen PB, Kharazmi A. Low parasite specific T cell response in clinically immune individuals with low grade Plasmodium falciparum parasitaemia. Trans R Soc Trop Med Hyg. 1986;80(6):1000‐1001. [DOI] [PubMed] [Google Scholar]
- 140. Martini F, Paglia MG, Montesano C, et al. V gamma 9V delta 2 T‐cell anergy and complementarity‐determining region 3‐specific depletion during paroxysm of nonendemic malaria infection. Infect Immun. 2003;71(5):2945‐2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Illingworth J, Butler NS, Roetynck S, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol. 2013;190(3):1038‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wykes MN, Horne‐Debets JM, Leow CY, Karunarathne DS. Malaria drives T cells to exhaustion. Front Microbiol. 2014;5:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Shankar EM, Vignesh R, Dash AP. Recent advances on T‐cell exhaustion in malaria infection. Med Microbiol Immunol. 2018;207(3–4):167‐174. [DOI] [PubMed] [Google Scholar]
- 144. Frimpong A, Kusi KA, Adu‐Gyasi D, Amponsah J, Ofori MF, Ndifon W. Phenotypic evidence of T cell exhaustion and senescence during symptomatic plasmodium falciparum Malaria. Front Immunol. 2019;10:1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Beeson JG, Kurtovic L, Dobaño C, et al. Challenges and strategies for developing efficacious and long‐lasting malaria vaccines. Sci Transl Med. 2019;11(474):eaau1458. [DOI] [PubMed] [Google Scholar]
- 146. Silveira ELV, Dominguez MR, Soares IS. To B or not to B: understanding B cell responses in the development of malaria infection. Front Immunol. 2018;9:2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ly A, Hansen DS. Development of B cell memory in Malaria. Front Immunol. 2019;10:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hviid L, Barfod L, Fowkes FJ. Trying to remember: immunological B cell memory to malaria. Trends Parasitol. 2015;31(3):89‐94. [DOI] [PubMed] [Google Scholar]
- 149. Scholzen A, Sauerwein RW. How malaria modulates memory: activation and dysregulation of B cells in Plasmodium infection. Trends Parasitol. 2013;29(5):252‐262. [DOI] [PubMed] [Google Scholar]
- 150. Weiss GE, Traore B, Kayentao K, et al. The Plasmodium falciparum‐specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. 2010;6(5):e1000912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Nogaro SI, Hafalla JC, Walther B, et al. The breadth, but not the magnitude, of circulating memory B cell responses to P. falciparum increases with age/exposure in an area of low transmission. PLoS ONE. 2011;6(10):e25582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lum FM, Teo TH, Lee WW, Kam YW, Renia L, Ng LF. An essential role of antibodies in the control of Chikungunya virus infection. J Immunol. 2013;190(12):6295‐6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ryg‐Cornejo V, Ioannidis L, Ly A, et al. Severe Malaria infections impair germinal center responses by inhibiting T follicular helper cell differentiation. Cell Rep. 2016;14(1):68‐81. [DOI] [PubMed] [Google Scholar]
- 154. Rezza G, Chen R, Weaver SC. O'nyong‐nyong fever: a neglected mosquito‐borne viral disease. Pathog Glob Health. 2017;111(6):271‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Powers AM, Brault AC, Tesh RB, Weaver SC. Re‐emergence of Chikungunya and O'nyong‐nyong viruses: evidence for distinct geographical lineages and distant evolutionary relationships. J Gen Virol. 2000;81(Pt 2):471‐479. [DOI] [PubMed] [Google Scholar]
- 156. Brustolin M, Pujhari S, Henderson CA, Rasgon JL. Anopheles mosquitoes may drive invasion and transmission of Mayaro virus across geographically diverse regions. PLoS Negl Trop Dis. 2018;12(11):e0006895. [DOI] [PMC free article] [PubMed] [Google Scholar]