

Summary
Decitabine is a DNA‐hypomethylating agent that has been widely applied for the treatment of acute myeloid leukaemia (AML) patients who are elderly or unfit for intensive therapy. Although effective, the complete response rate to decitabine is only around 30% and the overall survival remains poor. Emerging data support that regulation of DNA methylation is critical to control immune cell development, differentiation and activation. We hypothesize that defining how decitabine influences the immune responses in AML will facilitate the development of novel immune‐based leukaemia therapeutics. Here, we performed phenotypic and functional immune analysis on clinical samples from AML patients receiving decitabine treatment and demonstrated a significant impact of decitabine on the immune system. T‐cell expression of inhibitory molecules was upregulated and the ability of CD8 T cells to produce cytokines was decreased upon decitabine treatment. Importantly, in an unbiased comprehensive analysis, we identified a unique immune signature containing a cluster of key immune markers that clearly separate patients who achieved complete remission after decitabine from those who failed to do so. Therefore, this immune signature has a strong predictive value for clinical response. Collectively, our study suggests that immune‐based analyses may predict clinical response to decitabine and provide a therapeutic strategy to improve the treatment of AML.
Keywords: AML, T‐cell exhaustion, immune signature, CD38, decitabine
Despite significant advances in the knowledge of the genetic and cellular processes in leukaemia pathogenesis, successful treatment of acute myeloid leukaemia (AML) remains challenging, with 5‐year survival of only 27·4% according to the National Cancer Institute. The prognosis for elderly patients or patients unfit for intensive chemotherapy is especially poor due to limited treatment options. The median age of AML at diagnosis is 68 years. Clearly, novel effective leukaemia therapy is an urgent unmet need (DeSantis et al, 2014; Noone et al, 2018).
Epigenetic changes caused by increased DNA methylation, particularly in CpG‐rich regions of the genome, are known to be tumourigenic (Dawson & Kouzarides, 2012; Klutstein et al, 2016). This aberrant methylation can be reversed by hypomethylating agents (HMA), which have achieved clinical benefit in treating patients with haematological cancer (Kantarjian et al, 2006; Yang et al, 2006). Decitabine is an important HMA that has been widely applied for AML treatment in clinical practice and is considered standard care as the first line treatment of elderly patients or patients unfit for intensive chemotherapy (Jabbour et al, 2008; Erba, 2015; Welch et al, 2016; Tamamyan et al, 2017). Although effective, the complete response (CR) rate to decitabine is only around 30–40% and even for patients responding to this treatment, the majority develop resistance within 1 year (Blum et al, 2010; Ritchie et al, 2013; Bhatnagar et al, 2014; Khan et al, 2017). As a cytidine analogue, decitabine incorporates into DNA and results in demethylation by covalently trapping DNA methyltransferase (DNMT). In tumour cells, DNA hypomethylation subsequently activates a series of molecular and cellular processes, including upregulation of tumour suppressor genes, cell cycle arrest and tumour apoptosis, which eventually leads to tumour elimination (Mizuno et al, 2001; Schmelz et al, 2005; Stresemann & Lyko, 2008; Tsai et al, 2012). However, knowledge of how decitabine influences cells or organ systems beyond tumour is limited.
Immune surveillance is a key component of the host’s resistance to cancer. Therefore, T‐cell exhaustion and immune suppression are considered crucial mechanisms for tumour progression (Thommen & Schumacher, 2018; McLane et al, 2019). Emerging data support that epigenetic regulation, particularly DNA methylation, is critical to control immune cell development, differentiation and activation (Ladle et al, 2016; Abdelsamed et al, 2017). Therefore, we hypothesized that defining how decitabine influences the immune response in AML patients will reveal key mechanisms involved in refractory/resistant disease and facilitate the development of novel effective leukaemia therapeutics. Here we defined the phenotypic and functional characteristics of peripheral blood mononuclear cells (PBMCs) collected from AML patients undergoing treatment with decitabine. We aimed to determine (i) how decitabine influences the immune system; and (ii) whether the immune status in AML patients predicts the response to decitabine treatment.
Materials and methods
Patient samples
Peripheral blood samples were collected from AML patients from the tissue bank maintained by the Penn State Cancer Institute of Penn State University College of Medicine, Hershey, PA. The study was approved by the Institutional Review Board of Penn State University College of Medicine. Full written informed consent was obtained from all patients. Twenty‐eight peripheral blood samples from 14 patients (6 males and 8 females with a median age of 70 years, range, 52–79 years) with the diagnosis of AML per World Health Organization (WHO) classification were used (Arber et al, 2016). Clinical outcome was evaluable in 12 patients. Response criteria is per the European LeukaemiaNet (ELN) 2017 recommendation (Dohner et al, 2017). Four patients achieved complete remission (CR), 1 CR with incomplete haematological recovery (CRi), 2 partial remission (PR), 3 stable disease (SD) and 2 progressive disease (PD). Clinical characteristics of these patients are summarized in Tables S1 and S2.
Immunofluorescence staining and flow cytometric analysis
Peripheral blood mononuclear cells (PBMCs) were incubated with fixable viability dye eFluorTM 506 (eBioscience, San Diego, CA) in phosphate‐buffered saline for 20 min at 4°C, followed by incubation with directly conjugated monoclonal antibodies (mAbs) for 30 min at 4°C. Transcription factor staining was performed after the surface incubation using transcription factor buffer set (BD Pharmingen, San Jose, CA) according to the manufacturer’s instructions. The cells were washed before flow cytometric analysis. All mAbs used for different panels are listed in Table S3. LSR Fortessa flow cytometer (BD Biosciences, San Jose, CA) was used for data acquisition, and the data analysis was performed using FlowJo Software (Version 10.0, Tree Star, Ashland, OR).
In vitro stimulation and intracellular staining
PBMCs were cultured in RPMI 1640 medium (Corning) with 10% fetal bovine serum and stimulated with anti‐CD3/CD28 (2 and 5 μg/ml, respectively, eBioscience), plus Golgiplug (BD Biosciences) for 5 h. The cells were then stained with anti‐CD3‐BV786, anti‐CD4‐BV711, anti‐CD8‐APC‐H7 (BD Biosciences), and fixed/permeabilized using BD cytofix/cytoperm kit (BD Biosciences) followed by intracellularly staining with anti‐interferon (IFN)‐γ‐allophycocyanin (APC), anti‐tumour necrosis factor (TNF)‐fluorescein isothiocyanate (FITC), anti‐interleukin (IL)‐2 phycoerythrin‐cyanin 7 (PE‐Cy7), anti‐IL‐10‐PE‐CF594 and anti‐transforming growth factor (TGF)‐β‐PE (BD Biosciences). Data acquisition and analysis were as described above.
Statistical analysis
Statistical parameters and statistical significance are reported in the Figures and Figure Legends. A P < 0·05 was considered significant. Data are presented as the median and interquartile range (IQR). Due to small sample size, comparisons of patient characteristics for unpaired data were analysed using Fisher exact test (categorical variables) or Wilcoxon‐rank sum test (continuous variables). For paired data comparisons (i.e., before‐after treatment comparison), Wilcoxon signed‐rank tests were used for analysis. Spearman’s rank correlation coefficients were calculated to evaluate the correlation. In particular, we consider multiple comparison correction over multiple biomarkers. The false discovery rate (FDR) for multiple comparisons is controlled as 0·05 by using the Benjamini and Hochberg approach for P‐value adjustment (Benjamini & Hochberg, 1995). Also, principal component analysis (PCA) was performed using the ggbiplot function from the ggplot2 package in R, and hierarchical clustering and heatmaps were obtained using the pheatmap package. GraphPad Prism (GraphPad Software Inc., San Diego, CA) and R version 3.5.1 were used for statistical calculations and visualization.
Results
Frequencies of peripheral immune cell subsets are minimally altered upon decitabine treatment in AML patients
To determine how decitabine treatment influences the immune composition of patients with AML, we analysed cryopreserved PBMC samples from a cohort of 14 elderly patients with AML collected before and 1 month after initiation of treatment with decitabine. All samples were stained with 9 separate and partially overlapping panels of antibodies (Table S3) followed by flow cytometry analysis. In total, 46 leucocyte markers were used to identify the immune cell populations and dissect the differentiation, phenotypic and functional status of each immune component. As expected, we observed a decrease in blast frequency upon decitabine treatment (data not shown). Among the non‐blast PBMCs, we gated CD4 T cells, CD8 T cells, regulatory T (Treg), natural killer (NK) cells, NKT cells, B cells, dendritic cells (DCs), monocytes and myeloid‐derived suppressor cells (MDSCs) based on well‐recognized marker patterns (Table S4). We found comparable frequencies of these major immune cell components prior to and following decitabine treatment (Fig 1). Therefore, treatment with decitabine did not significantly affect the frequency of major immune components in AML.
Figure 1.
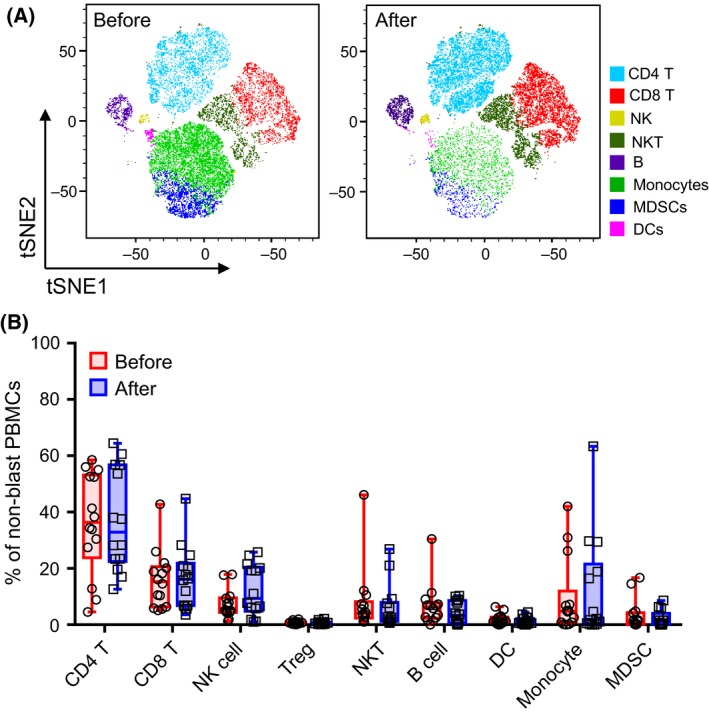
Frequencies of peripheral immune cell subsets are minimally altered upon decitabine treatment in AML patients. Flow‐cytometry analysis was performed on peripheral blood mononuclear cells (PBMCs) collected from patients with acute myeloid leukaemia (n = 14) before and after decitabine treatment. Each immune cell component was gated based on defined markers (listed in the Table S4). (A) Representative tSNE visualization of 40 000 merged events from samples of one patient before and after decitabine treatment. The graph was generated using FlowJo software. Coloured cells were manually annotated based on the defined markers. (B) Box‐and‐whisker plots show the frequency of immune cell subsets in the non‐blast PBMCs before (circle, red) and after (square, blue) decitabine treatment. Each circle or square represents the data of an individual patient (total n = 14). P‐values were calculated using Wilcoxon signed‐rank tests and were corrected for the multiple comparison using the Benjamini‐Hochberg adjustment. DCs, dendritic cells; MDSCs, myeloid‐derived suppressor cells; NK, natural killer cells; NKT, natural killer T cells; PBMCs, peripheral blood mononuclear cells; Treg, T‐regulatory cells; tSNE, t‐distributed stochastic neighbour embedding.
Decitabine treatment significantly impacts the differentiation, phenotype and function of T cells in AML patients
T cells represented the majority cell population in non‐blast PBMCs from AML patients. Decitabine did not significantly alter CD4 or CD8 T cells frequencies (Fig 1). We further examined the effect of decitabine on T cell differentiation. Based on the expression of CD45RA and CCR7, T cells are divided into four subsets: CCR7+CD45RA+ naïve cells (TN), CCR7+CD45RA− central memory (TCM) cells, CCR7−CD45RA− effector memory (TEM) cells and CCR7−CD45RA+ terminally differentiated effector (TEMRA) cells. The majority of CD4 T cells were TN, TCM or TEM, with only a small frequency of TEMRA detected. No significant difference was observed among these CD4 subsets prior versus post‐decitabine treatment (Fig 2A). In contrast, TEMRA represented the largest subset of CD8 T cells, and we found a significant decrease of CD8 TEMRA cells (median = 56·10% vs. 43·00%) upon decitabine treatment (Fig 2B).
Figure 2.
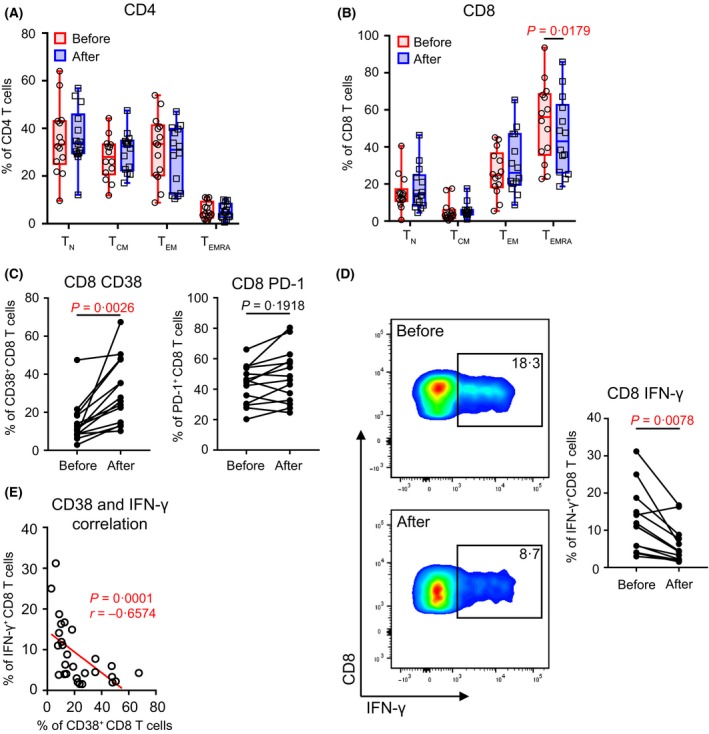
Decitabine treatment significantly impacts the differentiation, phenotype, and function of T cells in acute myeloid leukaemia patients. (A, B) Impact of decitabine on T‐cell differentiation is displayed as box‐and‐whisker plots. The frequencies for each subpopulation (TN, TCM, TEM, or TEMRA) among CD4 (left) and CD8 (right) T cells before (circle, red) and after (square, blue) decitabine treatment are shown. Each spot represents the data from an individual patient (n = 14). (C) Plots of the frequencies of CD38 or PD‐1‐expressing CD8 T cells before and after decitabine treatment. (D) Production of IFN‐γ by CD8 T cells upon in vitro anti‐CD3/anti‐CD28 stimulation. Shown are the representative flow data (left) and plots of summary for data of all patients (right, n = 14). Data from patients before and after decitabine treatment are shown. (E) Correlation between the CD8 T cell production of IFN‐γ and their expression of CD38. For A‐B, P‐values were calculated using Wilcoxon signed‐rank tests. For (C, D), total of 26 immune markers, including activation molecules, co‐stimulatory or inhibitory receptors, and transcriptional factors, were tested on CD8 T cells (Table S5). P‐values were calculated using Wilcoxon signed‐rank tests and were corrected for the multiple comparison using the Benjamini‐Hochberg adjustment. The numbers shown here indicate adjusted P‐values for CD38, PD‐1, and IFN‐γ. No statistical significance was detected in all other markers. For (E), Spearman non‐parametric test was used to analyse correlations. The fitted line was estimated through linear regression. The correlation coefficient r and P‐value are shown. TCM, CCR7+CD45RA− central memory T cells; TEM, CCR7−CD45RA− effector memory T cells; TEMRA, CCR7−CD45RA+ terminally differentiated effector T cells; TN, CCR7+CD45RA+ naïve T cells.
We next performed comprehensive phenotypic and functional analysis of CD8 T cells. A large number of T cell phenotypic markers including activation molecules, co‐stimulatory or inhibitory receptors, and transcriptional factors were assessed. Significant increase of CD38 expression on CD8 T cells upon decitabine treatment was detected (median = 10·83% vs. 26·77%, Fig 2C). Of note, we didn’t observe statistically significant alteration in programmed cell death 1 (PD‐1, also termed PDCD1) expression (Fig 2C). The functional status of T cells was evaluated by cytokine release assays upon in vitro stimulation with anti‐CD3 and anti‐CD28 antibodies. We observed significantly lower numbers of CD8 T cells producing intracellular IFN‐γ in samples of patients post‐decitabine treatment compared with that of paired samples from the same individuals at initial diagnosis (Fig 2D). Notably, there was a strong negative correlation of CD38 expression on CD8 T cells to their production of IFN‐γ (Spearman’s r = −0·6574, P = 0·0001, Fig 2E), indicating an inhibitory regulation of CD38 to T cell function. Of note, we performed analyses of blasts for the surface markers (PD‐L1 [CD274], CD155, V‐set immunoregulatory receptor [VISTA, VSIR], HLA‐A/B/C and HLA‐DR). Significant differences were not observed in samples between prior to versus post‐decitabine treatment (Figure S1).
Taken together, these data demonstrate that decitabine treatment is associated with increased T cell expression of inhibitory receptors and reduced T cell function indicated by decreased cytokine production.
AML patients who responded to decitabine have a distinct immune signature from those who failed decitabine treatment
Given the strong impact of decitabine on T‐cell activity in AML, we hypothesized that the immune status of AML patients associates with clinical response to decitabine. To test our hypothesis, we chose PBMC samples from 12 patients whose clinical outcome was evaluable. Based on ELN 2017 criteria for clinical response (Dohner et al, 2017), 5 patients achieved complete remission (CR) or CR with incomplete haematological recovery (CRi) after decitabine treatment, whereas the other 7 patients failed to achieve CR/CRi, and were defined as responders (CR/CRi) versus non‐responders (no CR/CRi). Two samples were collected from each patient (at initial diagnosis and 1‐month post‐decitabine treatment), so that a total of 24 samples (10 responders and 14 non‐responders) were used in this study. Flow cytometry based immunophenotypic and functional assays were applied to each sample. In an unsupervised PCA for comprehensive immune markers, we observed a distinct pattern between the responders and non‐responders (Fig 3). This encouraging finding suggests a strong association of immune signature with clinical response to decitabine treatment in AML patients.
Figure 3.
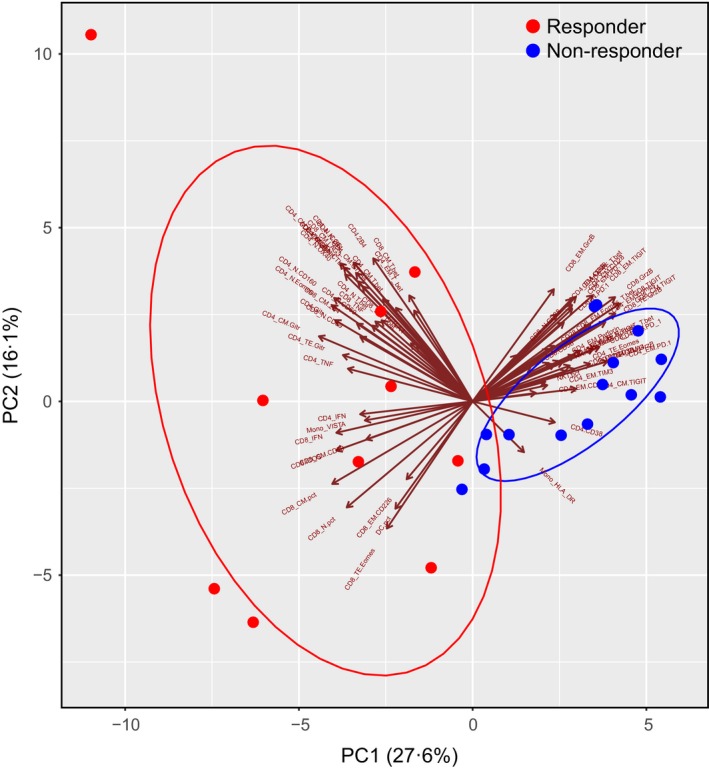
Unsupervised PCA for immune markers showed distinct patterns between the responders and non‐responders to decitabine treatment. Flow cytometry analyses of immune phenotypes and functions were performed on peripheral blood mononuclear cells from responders (n = 10) and non‐responders (n = 14). Seventy‐three parameters including the frequencies of each immune cell population and its subsets, as well as their phenotype, function and transcription factors were analysed through principal component analysis (PCA) algorithms into two components, PC1 and PC2. Cell profiles of responders and non‐responders were superimposed on the PCA plot. The circles exhibit the two clusters of patients. The direction and length of the arrow represents the contribution and degree of a parameter to each component.
High functional T cell status associates with good clinical response to decitabine in AML
We further dissected the association of each immune marker with the clinical response to decitabine. Frequencies of each immune component were compared between responders versus non‐responders. We observed no statistically significant difference in NK, NKT, Treg, B cells, DCs and monocytes (Figure S2A). No difference was detected in the proportion of CD4 or CD8 T cells either. However, when the T cell differentiation status was analysed, we found more TN and TCM but less TEMRA in responders compared with that of non‐responders (Fig 4A). Phenotypic studies showed that responders had a significantly higher frequency of co‐stimulatory molecule inducible T cell costimulatory (ICOS)‐expressing CD8 T cells, whereas the frequency of CD8 T cells expressing inhibitory molecules, such as T cell immunoreceptor with Ig and ITIM domains (TIGIT) and CD38, were trending lower in these patients (Fig 4B). Interestingly, PD‐1 expression on T cells was comparable between responders and non‐responders (Fig 4B). We also examined the expression of Eomesodermin (EOMES), a key transcription factor governing CD8 T‐cell exhaustion, and observed a trend of higher frequency of EOMES‐expressing CD8 T cells in non‐responders (Fig 4C). Importantly, when functional status of T cells was measured by cytokine release upon in vitro stimulation with anti‐CD3 and anti‐CD28, significantly higher IFN‐γ production by CD8 T cells was observed in responders compared with non‐responders (Fig 4D). We did not observe significant difference of other cytokine release, such as TNF‐α and IL‐2 by CD8 T cells (Fig 4D) or CD4 T cells (Figure S2B).
Figure 4.
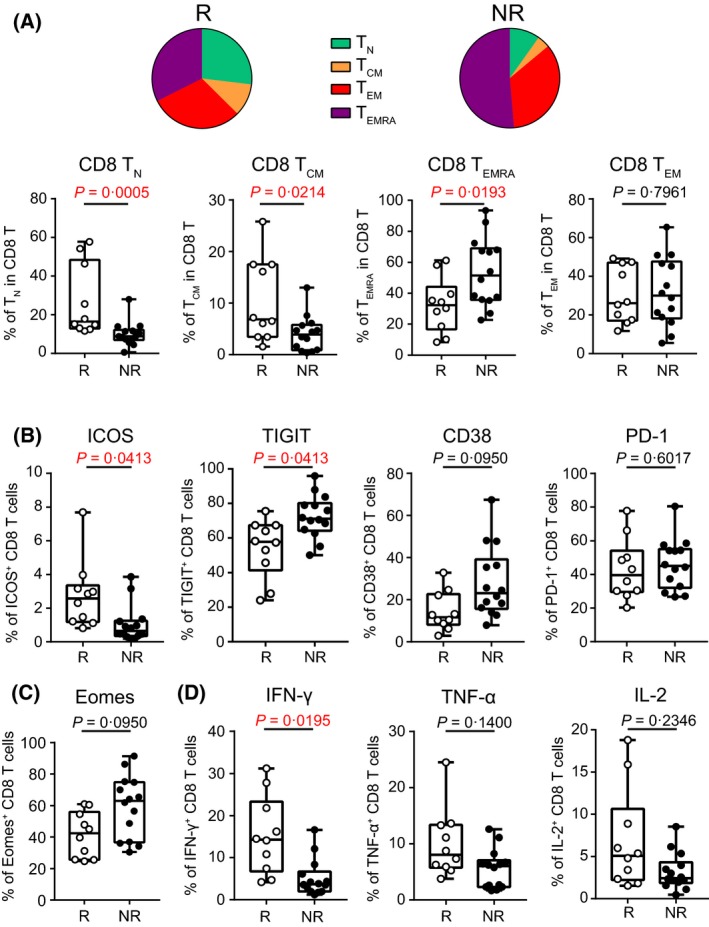
High functional T cell status associates with good clinical response to decitabine in AML. Cell components, phenotypes and functions were compared between the responders (R, n = 10) versus non‐responders (NR, n = 14). (A) Shown are the frequencies of TN, TCM, TEM, TEMRA subsets among CD8 T cells. The pie charts represent the mean values of each subset. The box‐and‐whiskers plots depict the summary of data from all patients. Each spot represents an individual patient. (B) Surface expression of co‐stimulatory or co‐inhibitory molecules on CD8 T cells are shown. (C) Shown is the intracellular expression of transcription factor EOMES on CD8 T cells. (D) Production of IFN‐γ, TNF‐α, and IL‐2 by CD8 T cells upon in vitro stimulation with anti‐CD3 and anti‐CD28 antibodies. For A, P‐values were calculated using Wilcoxon rank‐sum tests. For (B–D), total of 26 immune markers, including activation molecules, co‐stimulatory or inhibitory receptors, and transcriptional factors, were tested on CD8 T cells (Table S5). P‐values were calculated using Wilcoxon rank‐sum tests and were corrected for the multiple comparison using the Benjamini‐Hochberg adjustment. The numbers shown here indicate adjusted P‐values. No statistical significance was detected in other markers. TCM, CCR7+CD45RA− central memory T cells; TEM, CCR7−CD45RA− effector memory T cells; TEMRA, CCR7−CD45RA+ terminally differentiated effector T cells; TN, CCR7+CD45RA+ naïve T cells.
Collectively, our data demonstrate a correlation between phenotypically and functionally active T cells and positive clinical response to decitabine in AML.
Identification of the immune signature that predicts clinical response to decitabine in AML patients
To identify immune markers that may predict clinical response to decitabine, we evaluated data from samples of the 12 AML patients at initial diagnosis (prior to decitabine treatment). Comprehensive immune analyses were performed to identify differences between responders versus non‐responders. In addition to analysing bulk CD4 and CD8 T cells, we further assessed the expression of phenotypic and functional markers on T cells at each differentiation stage (TN, TCM, TEM, and TTERMA). Markers showing a different expression pattern between responders and non‐responders were selected for further analysis. We then performed hierarchical clustering on normalized expression levels of the selected immune markers for each patient. Two major clades were identified by this analysis: one consisted of samples from all the 5 responders; while the other was that from all the 7 non‐responders (Fig 5). This unbiased clustering approach adequately segregated the responders from non‐responders based on the study of samples prior to decitabine therapy, suggesting that the immune signature, composed of a cluster of immune markers, may have important predictive value for the clinical response to decitabine treatment in AML.
Figure 5.
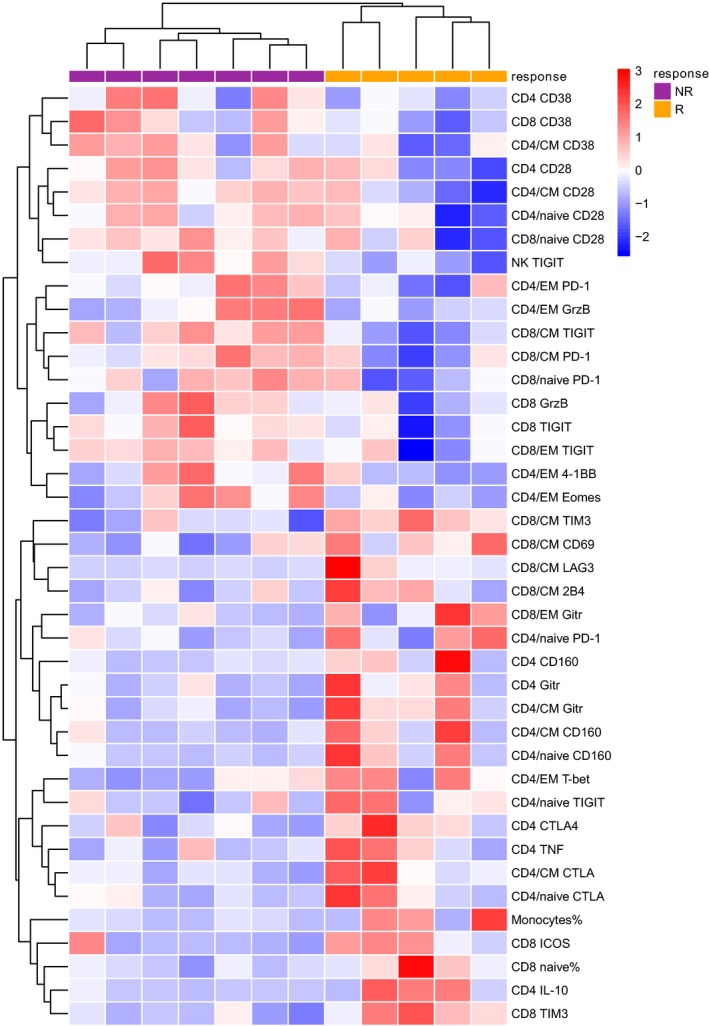
Identification of the immune signature that predicts clinical response to decitabine in AML patient. Flow cytometry analysis was performed on peripheral blood mononuclear cells collected from patients at initial diagnosis (prior to decitabine treatment). Shown is the heat map of immune markers that were normalized to a mean of 0 and standard deviation of 1. Relative over‐expression and under‐expression are assigned here as red and blue colour, respectively. Each column represents one patient sample and each row represents an immune marker that was examined. The dendrograms for markers (rows) and samples (columns) were constructed by hierarchical clustering (Euclidean distance, complete linkage). By clustering, patient responses to decitabine are separated as indicated by the bars at the top [purple and orange for non‐responders (NR) and responders (R), respectively].
Discussion
In our current study, we demonstrated a significant impact of decitabine on the immune system of AML patients. T‐cell expression of inhibitory molecules was upregulated and the ability of CD8 T cells to produce cytokines was decreased upon decitabine treatment. Importantly, in an unbiased comprehensive analysis, we identified a unique immune signature, containing a cluster of key immune markers that clearly separate patients who achieved CR/CRi after decitabine from those who failed to do so. Therefore, this immune signature has a strong predictive value for clinical response to decitabine.
Elegant studies in a mouse model of chronic viral infection demonstrate a critical role for DNA methylation in T‐cell effector function and exhaustion (Scharer et al, 2013; Ahn et al, 2016; Abdelsamed et al, 2017; Ghoneim et al, 2017). Persistent antigen stimulation induces DNA methylation of exhaustion‐associated genes such as IFNG, MYC, TCF7, TBX21 and EOMES in T cells. T cells eventually become fully exhausted, a stage which is not reversible by immune‐check point blockade. Importantly, inhibition of DNA methylation successfully overcomes this barrier in that decitabine followed by PD‐1 blockade effectively reinvigorates the function of exhausted CD8 T cells (Ghoneim et al, 2017). The observation that demethylation improves CD8 T cells response by reversing T‐cell exhaustion is in contrast to our finding that T‐cell function was decreased manifested by reduced production of cytokines upon decitabine treatment in AML patients. DNA methylation is a complex molecular process and highly context‐specific (Schultz et al, 2015). The different regulation under each individual model system or disease status, e.g. mouse model of chronic viral infection versus patients with AML, may explain the discrepancy. In addition, multiple studies have demonstrated that DNA methylation is altered as we age (Issa, 2014; Field et al, 2018). Our study was focused on elderly AML patients who received decitabine as they were unfit for intensive chemotherapy. The average age of patients in our study was 70 years old. The DNA methylation status and regulation might be unique to this population. These observations suggest that understanding the disease or clinical setting‐specific molecular regulation is crucial for the optimal clinical application of novel therapeutic strategies.
We made a striking observation that the frequency of CD38‐expressing T cells was significantly enhanced upon decitabine treatment. This increase was universal among all the different stages of CD4 and CD8 T cells examined (Figure S3). CD38 is expressed on the surface of a variety of immune cells, but its effect on T cells is controversial. Earlier work showed that CD38 is mainly expressed on activated and memory T cells, and engagement of CD38 initiates an intracellular signalling cascade involved in T‐cell activation and proliferation which enhances T‐cell function (Deaglio et al, 2001; Malavasi et al, 2008). Therefore, CD38 has been considered an activation marker for T cells. Most recently, emerging evidence suggests a role for CD38 in T cell exhaustion. In a study of samples collected from human immunodeficiency virus (HIV) patients, Hoffmann et al (2016) demonstrated a strong correlation between expression of CD38 and PD‐1 on CD8 T cells; CD38+ T cells displayed an exhausted functional phenotype by expressing a TBX21 (or T‐bet)dim EOMEShi phenotype; additionally, high expression of CD38 on CD8 T cells was associated with poor viral control in AIDS. In another report, using mouse models of cancer, Chen et al (2018) discovered that CD38 is a novel immune checkpoint in that upregulation of CD38 in the tumour microenvironment significantly inhibits CD8 T‐cell function via adenosine signalling. Consistently, we found a negative impact of CD38 on T cells in AML. Expression of CD38 negatively correlated with cytokine production by CD8 T cells (Fig 2E). In addition, compared with CD38− cells, CD38+ CD8 T cells expressed significantly higher levels of co‐inhibitory molecules including PD‐1, TIGIT, lymphocyte activating 3 (LAG‐3), and CD244 (or 2B4) (Figure S4). Furthermore, high expression of CD38 on both CD8 and CD4 T cells was strongly associated with failure of response to decitabine treatment and thus poor clinical outcome (Fig 4E). These significant findings, together with our observation that decitabine treatment upregulated CD38 on T cells, argue strongly for a novel AML therapeutic approach by adding CD38 inhibition to the decitabine‐based regimen. In fact, given that a portion of AML patients express CD38 on their blasts, targeted treatment by anti‐CD38 antibody may have direct anti‐leukaemia effects in addition to improving immune response.
Targeting PD‐1 for immunotherapy of cancer has achieved great success. Several blockade antibodies to PD‐1 or PD‐L1 have been approved by the US Food and Drug Administration for treatment of a variety of solid tumours. Recent studies demonstrated that expression of molecules in the PD‐1 pathway are enhanced after hypomethylating agent treatment in MDS and AML patients (Yang et al, 2014; Orskov et al, 2015). These observations form a strong rationale for combinational treatment of hypomethylating agents and PD‐1 inhibition in cancer therapy. Several clinical studies, including our own investigator‐initiated trial adding blockade antibodies against PD‐1 to the hypomethylating agents (either decitabine or azacitidine) in treatment of AML, are currently ongoing (NCT02397720, NCT02845297, NCT02953561, NCT03395873). Daver et al. (2018) have recently reported a promising result from an early clinical study treating patients with relapse/refractory AML using combination of azacitidine and nivolumab, a widely used PD‐1‐targeting agent. Interestingly, we observed no statistically significant impact of decitabine on T‐cell expression of PD‐1 in our AML patients, which might be due to the limited sample size in our study. Alternatively, different hypomethylating agents may have distinct effects on PD‐1 expression. Most AML patients in the studies reported by Orskov et al (2015) and Yang et al (2014) received azacitidine, whereas decitabine was applied to all patients in our study.
Upon a comprehensive analysis of immune markers in patients prior to decitabine treatment and their correlation to clinical outcome, we identified an immune signature that predicts complete response to decitabine in AML patients. This is highly significant for clinical practice. Although hypomethylating agents, including decitabine, have been standard care as first line treatment for elderly AML patients who are unfit for intensive chemotherapy, the CR rate is only around 30%. Many patients do not benefit from this single agent treatment. Defining features predicting clinical response is crucial to identify this patient population and triage them to novel or combinational therapy. In fact, there was a trend of higher expression of several immune checkpoints (e.g. CD38, PD‐1, and TIGIT) whereas co‐stimulatory molecules [TNF receptor superfamily member 18 (TNFRSF18, also termed GITR) and ICOS] were lower in non‐responders (Fig 5), indicating that immunotherapy targeting these pathways may be beneficial. Our data is limited by the small sample size, so validation in a large cohort of clinical samples is warranted before clinical applications. Nevertheless, our study provides new insight into the impact of decitabine on the immune response, which will be key for the design of novel clinical trials aiming to improve decitabine‐based treatment in AML.
In summary, our study identified a unique immune signature predicting clinical response to decitabine in AML patients. We demonstrated that high functional CD8 T‐cell status associates with good clinical outcome. In addition, decitabine treatment downregulated T‐cell response. Data from this study not only form a strong rationale for integrating immunotherapy into decitabine treatment, they also provide a practical approach to identify specific patient populations that will probably benefit from this therapeutic strategy.
Author Contributions
C.Z. designed the experiments, performed the research, analysed the results and wrote the manuscript. B.J. performed the research and analysed the results. D.F.C, W.C.E, W.B.R, S.M, S.N, N.S, J.M.S, acquired samples, managed patients and discussed the data. T.D.S and R.J.H reviewed the manuscript. H.Zeng designed the experiments, analysed results and wrote the manuscript; H.Zheng devised the concept, designed the experiments, oversaw the interpretation and presentation of the data and wrote the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Fig S1. Expression of surface markers on circulating AML blasts before and after decitabine treatment.
Fig S2. (A) Box‐and‐whisker plots show the frequency of immune cell subsets in the non‐blast PBMCs between responders (circle, red, n=10) and non‐responders (square, blue, n=14) of decitabine treatment each circle or square represents the data of an individual patient.
Fig S3. The expression of CD38 on each subpopulation of T cells in patients before and after decitabine treatment.
Fig S4. Expression of co‐inhibitory molecules on CD38+ CD8 T cells.
Table S1. Patients’ characteristics.
Table S2. Patients’ molecular genetics.
Table S3. Staining panels for flow cytometry.
Table S4. Identification of immune cell populations.
Table S5. T cell immune markers tested in Figure 2 and Figure 4.
Acknowledgements
This work was supported by the American Society of Hematology Scholar Award Grant, Penn State Cancer Institute Funds, the Penn State University Enhancing Health Initiative and the Kiesendahl Endowment funding. We would like to thank the technical support of Jade Vogel, Nate Sheaffer, and Joe Bednarczyk of the Flow Cytometry Core Facility of the Penn State College of Medicine.
Hui Zeng and Hong Zheng are co‐corresponding authors.
Contributor Information
Hui Zeng, Email: zenghui@ccmu.edu.cn.
Hong Zheng, Email: hzheng@pennstatehealth.psu.edu.
References
- Abdelsamed, H.A. , Moustaki, A. , Fan, Y. , Dogra, P. , Ghoneim, H.E. , Zebley, C.C. , Triplett, B.M. , Sekaly, R.P. & Youngblood, B. (2017) Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. Journal of Experimental Medicine, 214, 1593–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, E. , Youngblood, B. , Lee, J. , Lee, J. , Sarkar, S. & Ahmed, R. (2016) Demethylation of the PD‐1 promoter is imprinted during the effector phase of CD8 T cell exhaustion. Journal of Virology, 90, 8934–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber, D.A. , Orazi, A. , Hasserjian, R. , Thiele, J. , Borowitz, M.J. , Le Beau, M.M. , Bloomfield, C.D. , Cazzola, M. & Vardiman, J.W. (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood, 127, 2391–2405. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. & Hochberg, Y. (1995) Controlling the false discovery rate ‐ a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological), 57, 289–300. [Google Scholar]
- Bhatnagar, B. , Duong, V.H. , Gourdin, T.S. , Tidwell, M.L. , Chen, C. , Ning, Y. , Emadi, A. , Sausville, E.A. & Baer, M.R. (2014) Ten‐day decitabine as initial therapy for newly diagnosed patients with acute myeloid leukemia unfit for intensive chemotherapy. Leukaemia & Lymphoma, 55, 1533–1537. [DOI] [PubMed] [Google Scholar]
- Blum, W. , Garzon, R. , Klisovic, R.B. , Schwind, S. , Walker, A. , Geyer, S. , Liu, S. , Havelange, V. , Becker, H. , Schaaf, L. , Mickle, J. , Devine, H. , Kefauver, C. , Devine, S.M. , Chan, K.K. , Heerema, N.A. , Bloomfield, C.D. , Grever, M.R. , Byrd, J.C. , Villalona‐Calero, M. , Croce, C.M. & Marcucci, G. (2010) Clinical response and miR‐29b predictive significance in older AML patients treated with a 10‐day schedule of decitabine. Proceedings of the National Academy of Sciences of the United States of America, 107, 7473–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Diao, L. , Yang, Y. , Yi, X. , Rodriguez, B.L. , Li, Y. , Villalobos, P.A. , Cascone, T. , Liu, X. , Tan, L. , Lorenzi, P.L. , Huang, A. , Zhao, Q. , Peng, D. , Fradette, J.J. , Peng, D.H. , Ungewiss, C. , Roybal, J. , Tong, P. , Oba, J. , Skoulidis, F. , Peng, W. , Carter, B.W. , Gay, C.M. , Fan, Y. , Class, C.A. , Zhu, J. , Rodriguez‐Canales, J. , Kawakami, M. , Byers, L.A. , Woodman, S.E. , Papadimitrakopoulou, V.A. , Dmitrovsky, E. , Wang, J. , Ullrich, S.E. , Wistuba, I.I. , Heymach, J.V. , Qin, F.X. & Gibbons, D.L. (2018) CD38‐mediated immunosuppression as a mechanism of tumor cell escape from PD‐1/PD‐L1 blockade. Cancer Discovery, 8, 1156–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daver, N. , Garcia‐Manero, G. , Basu, S. , Boddu, P.C. , Alfayez, M. , Cortes, J.E. , Konopleva, M. , Ravandi‐Kashani, F. , Jabbour, E. , Kadia, T. , Nogueras‐Gonzalez, G.M. , Ning, J. , Pemmaraju, N. , DiNardo, C.D. , Andreeff, M. , Pierce, S.A. , Gordon, T. , Kornblau, S.M. , Flores, W. , Alhamal, Z. , Bueso‐Ramos, C. , Jorgensen, J.L. , Patel, K.P. , Blando, J. , Allison, J.P. , Sharma, P. & Kantarjian, H. (2018) Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open‐label, phase II study. Cancer Discovery, 9, 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, M.A. & Kouzarides, T. (2012) Cancer epigenetics: from mechanism to therapy. Cell, 150, 12–27. [DOI] [PubMed] [Google Scholar]
- Deaglio, S. , Mehta, K. & Malavasi, F. (2001) Human CD38: a (r)evolutionary story of enzymes and receptors. Leukemia Research, 25, 1–12. [DOI] [PubMed] [Google Scholar]
- DeSantis, C.E. , Lin, C.C. , Mariotto, A.B. , Siegel, R.L. , Stein, K.D. , Kramer, J.L. , Alteri, R. , Robbins, A.S. & Jemal, A. (2014) Cancer treatment and survivorship statistics, 2014. CA: A Cancer Journal for Clinicians, 64, 252–271. [DOI] [PubMed] [Google Scholar]
- Dohner, H. , Estey, E. , Grimwade, D. , Amadori, S. , Appelbaum, F.R. , Buchner, T. , Dombret, H. , Ebert, B.L. , Fenaux, P. , Larson, R.A. , Levine, R.L. , Lo‐Coco, F. , Naoe, T. , Niederwieser, D. , Ossenkoppele, G.J. , Sanz, M. , Sierra, J. , Tallman, M.S. , Tien, H.F. , Wei, A.H. , Lowenberg, B. & Bloomfield, C.D. (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood, 129, 424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erba, H.P. (2015) Finding the optimal combination therapy for the treatment of newly diagnosed AML in older patients unfit for intensive therapy. Leukemia Research, 39, 183–191. [DOI] [PubMed] [Google Scholar]
- Field, A.E. , Robertson, N.A. , Wang, T. , Havas, A. , Ideker, T. & Adams, P.D. (2018) DNA methylation clocks in aging: categories, causes, and consequences. Molecular Cell, 71, 882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneim, H.E. , Fan, Y. , Moustaki, A. , Abdelsamed, H.A. , Dash, P. , Dogra, P. , Carter, R. , Awad, W. , Neale, G. , Thomas, P.G. & Youngblood, B. (2017) De novo epigenetic programs inhibit PD‐1 blockade‐mediated T cell rejuvenation. Cell, 170, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Pantazis, N. , Martin, G.E. , Hickling, S. , Hurst, J. , Meyerowitz, J. , Willberg, C.B. , Robinson, N. , Brown, H. , Fisher, M. , Kinloch, S. , Babiker, A. , Weber, J. , Nwokolo, N. , Fox, J. , Fidler, S. , Phillips, R. & Frater, J. ; SPARTAC and CHERUB Investigators . (2016) Exhaustion of activated CD8 T cells predicts disease progression in primary HIV‐1 infection. PLoS Pathogens, 12, e1005661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa, J.P. (2014) Aging and epigenetic drift: a vicious cycle. Journal of Clinical Investigation, 124, 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbour, E. , Issa, J.P. , Garcia‐Manero, G. & Kantarjian, H. (2008) Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer, 112, 2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian, H. , Issa, J.P. , Rosenfeld, C.S. , Bennett, J.M. , Albitar, M. , DiPersio, J. , Klimek, V. , Slack, J. , de Castro, C. , Ravandi, F. , Helmer, R. 3rd , Shen, L. , Nimer, S.D. , Leavitt, R. , Raza, A. & Saba, H. (2006) Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer, 106, 1794–1803. [DOI] [PubMed] [Google Scholar]
- Khan, N. , Hantel, A. , Knoebel, R.W. , Artz, A. , Larson, R.A. , Godley, L.A. , Thirman, M.J. , Liu, H. , Churpek, J.E. , King, D. , Odenike, O. & Stock, W. (2017) Efficacy of single‐agent decitabine in relapsed and refractory acute myeloid leukemia. Leukaemia & Lymphoma, 58, 1–7. [DOI] [PubMed] [Google Scholar]
- Klutstein, M. , Nejman, D. , Greenfield, R. & Cedar, H. (2016) DNA methylation in cancer and aging. Cancer Research, 76, 3446–3450. [DOI] [PubMed] [Google Scholar]
- Ladle, B.H. , Li, K.P. , Phillips, M.J. , Pucsek, A.B. , Haile, A. , Powell, J.D. , Jaffee, E.M. , Hildeman, D.A. & Gamper, C.J. (2016) De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8 + T‐cell fate decisions following activation. Proceedings of the National Academy of Sciences of the United States of America, 113, 10631–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavasi, F. , Deaglio, S. , Funaro, A. , Ferrero, E. , Horenstein, A.L. , Ortolan, E. , Vaisitti, T. & Aydin, S. (2008) Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiological Reviews, 88, 841–886. [DOI] [PubMed] [Google Scholar]
- McLane, L.M. , Abdel‐Hakeem, M.S. & Wherry, E.J. (2019) CD8 T Cell Exhaustion during chronic viral infection and cancer. Annual Review of Immunology, 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Mizuno, S. , Chijiwa, T. , Okamura, T. , Akashi, K. , Fukumaki, Y. , Niho, Y. & Sasaki, H. (2001) Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood, 97, 1172–1179. [DOI] [PubMed] [Google Scholar]
- Noone, A.M. , Howlader, N. , Krapcho, M. , Miller, D. , Brest, A. , Yu, M. , Ruhl, J. , Tatalovich, Z. , Mariotto, A. , Lewis, D.R. , Chen, H.S. & Feuer, E.J. (2018) SEER cancer statistics review, 1975–2015. National Cancer Institute, Bethesda, MD: Available at: https://seer.cancer.gov/csr/1975_2015/, based on November 2017 SEER data submission, posted to the SEER web site. [Google Scholar]
- Orskov, A.D. , Treppendahl, M.B. , Skovbo, A. , Holm, M.S. , Friis, L.S. , Hokland, M. & Gronbaek, K. (2015) Hypomethylation and up‐regulation of PD‐1 in T cells by azacytidine in MDS/AML patients: a rationale for combined targeting of PD‐1 and DNA methylation. Oncotarget, 6, 9612–9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, E.K. , Feldman, E.J. , Christos, P.J. , Rohan, S.D. , Lagassa, C.B. , Ippoliti, C. , Scandura, J.M. , Carlson, K. & Roboz, G.J. (2013) Decitabine in patients with newly diagnosed and relapsed acute myeloid leukemia. Leukaemia & Lymphoma, 54, 2003–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer, C.D. , Barwick, B.G. , Youngblood, B.A. , Ahmed, R. & Boss, J.M. (2013) Global DNA methylation remodeling accompanies CD8 T cell effector function. The Journal of Immunology, 191, 3419–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelz, K. , Sattler, N. , Wagner, M. , Lubbert, M. , Dorken, B. & Tamm, I. (2005) Induction of gene expression by 5‐Aza‐2'‐deoxycytidine in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) but not epithelial cells by DNA‐methylation‐dependent and ‐independent mechanisms. Leukemia, 19, 103–111. [DOI] [PubMed] [Google Scholar]
- Schultz, M.D. , He, Y. , Whitaker, J.W. , Hariharan, M. , Mukamel, E.A. , Leung, D. , Rajagopal, N. , Nery, J.R. , Urich, M.A. , Chen, H. , Lin, S. , Lin, Y. , Jung, I. , Schmitt, A.D. , Selvaraj, S. , Ren, B. , Sejnowski, T.J. , Wang, W. & Ecker, J.R. (2015) Human body epigenome maps reveal noncanonical DNA methylation variation. Nature, 523, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stresemann, C. & Lyko, F. (2008) Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. International Journal of Cancer, 123, 8–13. [DOI] [PubMed] [Google Scholar]
- Tamamyan, G. , Kadia, T. , Ravandi, F. , Borthakur, G. , Cortes, J. , Jabbour, E. , Daver, N. , Ohanian, M. , Kantarjian, H. & Konopleva, M. (2017) Frontline treatment of acute myeloid leukemia in adults. Critical Reviews in Oncology Hematology, 110, 20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen, D.S. & Schumacher, T.N. (2018) T cell dysfunction in cancer. Cancer Cell, 33, 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, H.C. , Li, H. , Van Neste, L. , Cai, Y. , Robert, C. , Rassool, F.V. , Shin, J.J. , Harbom, K.M. , Beaty, R. , Pappou, E. , Harris, J. , Yen, R.W. , Ahuja, N. , Brock, M.V. , Stearns, V. , Feller‐Kopman, D. , Yarmus, L.B. , Lin, Y.C. , Welm, A.L. , Issa, J.P. , Minn, I. , Matsui, W. , Jang, Y.Y. , Sharkis, S.J. , Baylin, S.B. & Zahnow, C.A. (2012) Transient low doses of DNA‐demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell, 21, 430–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, J.S. , Petti, A.A. , Miller, C.A. , Fronick, C.C. , O'Laughlin, M. , Fulton, R.S. , Wilson, R.K. , Baty, J.D. , Duncavage, E.J. , Tandon, B. , Lee, Y.S. , Wartman, L.D. , Uy, G.L. , Ghobadi, A. , Tomasson, M.H. , Pusic, I. , Romee, R. , Fehniger, T.A. , Stockerl‐Goldstein, K.E. , Vij, R. , Oh, S.T. , Abboud, C.N. , Cashen, A.F. , Schroeder, M.A. , Jacoby, M.A. , Heath, S.E. , Luber, K. , Janke, M.R. , Hantel, A. , Khan, N. , Sukhanova, M.J. , Knoebel, R.W. , Stock, W. , Graubert, T.A. , Walter, M.J. , Westervelt, P. , Link, D.C. , DiPersio, J.F. & Ley, T.J. (2016) TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. New England Journal of Medicine, 375, 2023–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, A.S. , Doshi, K.D. , Choi, S.W. , Mason, J.B. , Mannari, R.K. , Gharybian, V. , Luna, R. , Rashid, A. , Shen, L. , Estecio, M.R. , Kantarjian, H.M. , Garcia‐Manero, G. & Issa, J.P. (2006) DNA methylation changes after 5‐aza‐2'‐deoxycytidine therapy in patients with leukemia. Cancer Research, 66, 5495–5503. [DOI] [PubMed] [Google Scholar]
- Yang, H. , Bueso‐Ramos, C. , DiNardo, C. , Estecio, M.R. , Davanlou, M. , Geng, Q.R. , Fang, Z. , Nguyen, M. , Pierce, S. , Wei, Y. , Parmar, S. , Cortes, J. , Kantarjian, H. & Garcia‐Manero, G. (2014) Expression of PD‐L1, PD‐L2, PD‐1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia, 28, 1280–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Expression of surface markers on circulating AML blasts before and after decitabine treatment.
Fig S2. (A) Box‐and‐whisker plots show the frequency of immune cell subsets in the non‐blast PBMCs between responders (circle, red, n=10) and non‐responders (square, blue, n=14) of decitabine treatment each circle or square represents the data of an individual patient.
Fig S3. The expression of CD38 on each subpopulation of T cells in patients before and after decitabine treatment.
Fig S4. Expression of co‐inhibitory molecules on CD38+ CD8 T cells.
Table S1. Patients’ characteristics.
Table S2. Patients’ molecular genetics.
Table S3. Staining panels for flow cytometry.
Table S4. Identification of immune cell populations.
Table S5. T cell immune markers tested in Figure 2 and Figure 4.