

SUMMARY
SF3B1, which encodes an essential spliceosomal protein, is frequently mutated in myelodysplastic syndromes (MDS) and many cancers. However, the defect of mutant SF3B1 is unknown. Here, we analyzed RNA-sequencing data from MDS patients and confirmed that SF3B1 mutants use aberrant 3′ splice sites. To elucidate the underlying mechanism, we purified complexes containing either wild-type or the hotspot K700E mutant SF3B1, and found that levels of a poorly studied spliceosomal protein, SUGP1, were reduced in mutant spliceosomes. Strikingly, SUGP1 knockdown completely recapitulated the splicing errors, whereas SUGP1 overexpression drove the protein, which our data suggests plays an important role in branchsite recognition, into the mutant spliceosome and partially rescued splicing. Other hotspot SF3B1 mutants showed similar altered splicing and diminished interaction with SUGP1. Our study demonstrates that SUGP1 loss is a common defect of spliceosomes with disease-causing SF3B1 mutations and, since this defect can be rescued, suggests possibilities for therapeutic intervention.
In Brief
Zhang et al. report that SF3B1 mutations found in myelodysplastic syndromes and many cancers disrupt interaction with splicing factor SUGP1 during branchsite recognition, leading to aberrant use of upstream branch points and cryptic 3′ splice sites during RNA splicing. This defect in splicing due to cancer-causing SF3B1 mutations is rescuable.
Graphical Abstract
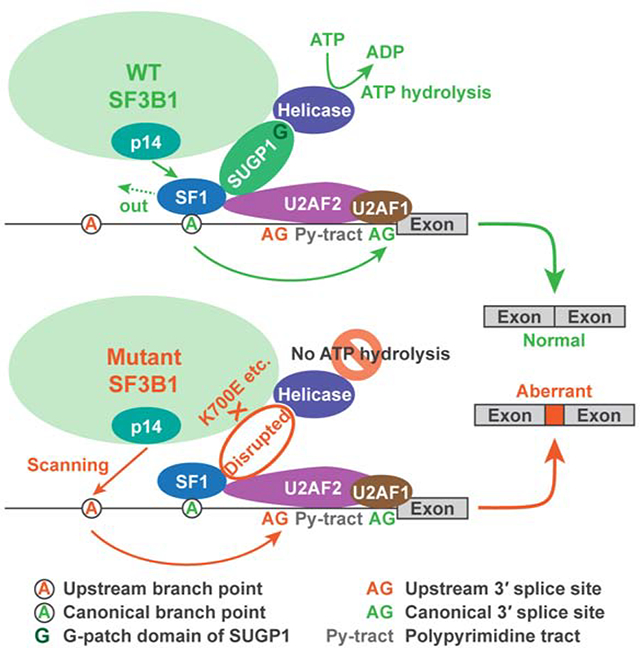
INTRODUCTION
One of the most surprising and exciting findings from the sequencing of many cancer genomes in recent years has been the identification of frequent mutations in genes encoding splicing factors, such as SF3B1 (splicing factor 3b subunit 1), SRSF2 (serine and arginine rich splicing factor 2), and U2AF1 (U2 small nuclear RNA auxiliary factor 1) (Garraway and Lander, 2013; Yoshida and Ogawa, 2014; Yoshida et al., 2011). Despite rapid advances in research on their roles in misregulation of RNA splicing and in pathogenesis (Alsafadi et al., 2016; Darman et al., 2015; DeBoever et al., 2015; Ilagan et al., 2015; Kim et al., 2015; Komeno et al., 2015; Shiozawa et al., 2018; Shirai et al., 2015; Wang et al., 2016; Zhang et al., 2015), no therapeutic interventions have been developed for any of the cancers harboring splicing factor mutations. Among the splicing genes, SF3B1 is the most frequently mutated. Mutations in SF3B1 occur in about one third of patients with myelodysplastic syndromes (MDS) overall and in more than 80% of patients with the subtypes of MDS with ring sideroblasts (Haferlach et al., 2014; Papaemmanuil et al., 2011, 2013; Yoshida et al., 2011). SF3B1 mutations have been found also in chronic lymphocytic leukemia (CLL), uveal melanoma, as well as breast and many other cancers (Biankin et al., 2012; Ellis et al., 2012; Harbour et al., 2013; Kandoth et al., 2013; Landau et al., 2013; Seiler et al., 2018a). Although there have been reports characterizing RNA missplicing induced by SF3B1 mutations (Alsafadi et al., 2016; Darman et al., 2015; DeBoever et al., 2015; Kesarwani et al., 2017), the defect of the mutant SF3B1 spliceosome is still unknown.
The human spliceosome is a large complex that assembles from five snRNAs (small nuclear RNAs) and more than a hundred associated proteins (Wahl et al., 2009). Assembly starts with the binding of U1 snRNP (small nuclear ribonucleoprotein) to the 5′ splice site (ss) at the 5′-end of the intron, followed by the binding of SF1 (splicing factor 1) to the branchsite at the 3′-end of the intron in cooperation with U2AF (the U2AF2/U2AF1 heterodimer) that binds to the polypyrimidine tract and the 3′ss. The U2 snRNP complex is then recruited and its RNA component, U2 snRNA, interacts with the branchsite through base pairing in an ATP-dependent manner, leading to displacement of SF1 from the branchsite by p14, the branchsite binding protein in the U2 snRNP complex (Will and Luhrmann, 2011). Subsequently, the U4/U5/U6 tri-snRNP is assembled, leading to formation of spliceosomal complex B. After extensive structural rearrangements, the spliceosome is activated and catalyzes two sequential transesterification reactions, with the first one covalently-linking the branch point (BP) to the 5′-end of the intron, and the second one joining the two flanking exons (Wahl et al., 2009).
SF3B1 functions by serving as a core component of U2 snRNP, which is critical for branchsite recognition and for the early stages of spliceosome assembly (Will and Luhrmann, 2011). The N-terminus of SF3B1 has multiple U2AF2 binding sites (Thickman et al., 2006), which may facilitate localizing U2 snRNP to the vicinity of the branchsite. The C-terminal two-thirds consists largely of 20 tandem HEAT (Huntingtin, elongation factor 3, a subunit of protein phosphatase 2A, PI3 kinase target of rapamycin 1) repeats that form rod-like helical structures, providing a major scaffold within U2 snRNP for interactions with other SF3b subunits, including p14 (Cretu et al., 2016). Because of the complexity and highly dynamic nature of protein-protein, protein-RNA, and RNA-RNA interactions involved in spliceosome assembly, it has been a great challenge to identify the defect caused by SF3B1 mutations.
Almost all mutations in SF3B1 are located in the HEAT domain and are heterozygous missense substitutions with the absence of nonsense mutations, suggesting that the mutations likely affect specific function(s) related to this domain rather than cause a complete loss of SF3B1 function. Of the over 40 different residues found to be mutated in the HEAT domain, 33 are in HEAT repeats H4–H7 (Cretu et al., 2016), further suggesting that the defect is limited. Nine residues in H4–H8 (including the most frequently mutated residue, K700) are clustered in a small area and are exposed to solvent, suggesting that the mutations may disrupt SF3B1 interactions with other spliceosomal protein(s) rather than with SF3b subunits (Cretu et al., 2016). A study in yeast suggested that this mutated area affects interaction with the N-terminal region of Prp5 (Tang et al., 2016). However, this region is not conserved in humans, and the human SF3B1 K700 residue is not conserved in budding yeast. Therefore, it is still unknown how SF3B1 mutations affect protein interactions in the spliceosome in human diseases.
In this study, we identified the defect of the mutant SF3B1 spliceosome responsible for the errors in splicing. We used affinity purification to isolate spliceosomal complexes associated with either wild-type (WT) or the hotspot K700E mutant SF3B1. We identified the proteins by mass spectrometry and found that levels of the spliceosomal protein SUGP1 (SURP and G-patch domain containing 1) were greatly reduced in the mutant SF3B1 spliceosome. We then show that knockdown (KD) of SUGP1, but not a large number of other SF3B1-associated proteins, or overexpression of a specific dominant negative mutant of SUGP1 with a mutated G-patch domain, completely recapitulated the splicing errors induced by mutant SF3B1. Furthermore, we show that overexpressed SUGP1 can associate with the mutant spliceosome and partially rescue the splicing defects. Finally, we show that several additional hotspot mutations in SF3B1 behave the same way as K700E. Our study thus establishes that loss of SUGP1 is a common defect of spliceosomes with cancer-associated SF3B1 mutations, that the loss solely accounts for the splicing errors caused by SF3B1 mutations, and that this defect is potentially amenable to rescue, providing a molecular basis for developing treatments for mutant SF3B1 cancers.
RESULTS
SF3B1 Mutations Lead to Use of Upstream Cryptic 3′ Splice Sites
To gain insights into the splicing defect of the mutant SF3B1 spliceosome in cancer, we began by analyzing RNA-sequencing data from MDS patient samples. Because SF3B1 is involved in branchsite recognition, which is important for selection of the 3′ss, we focused our analysis only on 3′ss usage, to facilitate identification of primary (direct) splicing changes and not secondary (indirect) effects. To this end, we performed a genome-wide analysis of aberrant 3′ss with enhanced sensitivity using computational approaches derived from the method of DeBoever et al. (2015). The analysis was applied to six MDS patients harboring SF3B1 mutations in the HEAT domain (three K700E, two D781G, and one H662D) and nine MDS patients carrying WT SF3B1. Using a cutoff q value less than 0.05, we identified 1,145 novel (i.e., not annotated previously) cryptic 3′ss more frequently used in mutant SF3B1 samples and 186 cryptic 3′ss in WT SF3B1 samples (Figure 1A). The cryptic 3′ss used in WT SF3B1 samples are widely distributed around the corresponding canonical 3′ss, whereas the great majority of the cryptic 3′ss used in mutant SF3B1 samples are located ~10–30 nt upstream of canonical 3′ss (Figure 1A), consistent with previous observations in CLL, uveal melanoma, and breast cancer (Alsafadi et al., 2016; Darman et al., 2015; DeBoever et al., 2015). Because some cryptic 3′ss are too distant from the canonical 3′ss to justify a splicing link between the two, we further examined the 627 cryptic 3′ss that are closer than 100 nt upstream of the canonical 3′ss. Unsupervised clustering of these 3′ss clearly separated SF3B1 mutants from WT SF3B1, suggesting that the different SF3B1 mutations all consistently led to missplicing involving aberrant 3′ss usage (Figure 1B). By setting three stringent threshold parameters (q value < 0.05, closer than 50 nt upstream of the canonical 3′ss, and more than 15 supporting read counts), we obtained 169 high-confidence cryptic 3′ss differentially used by mutant vs. WT SF3B1 (Figure 1C and Table S1).
Figure 1. SF3B1 Mutations Lead to Use of Upstream Cryptic 3′ Splice Sites.

(A) Distribution of cryptic 3′ss around the associated canonical 3′ss. Red and black histograms indicate 1,145 cryptic 3′ss more frequently used (q value < 0.05) in six mutant SF3B1 MDS patient samples and 186 cryptic 3′ss in nine WT SF3B1 samples, respectively.
(B) Hierarchical clustering and heatmap analysis of the 627 cryptic 3′ss differentially used in six mutant vs. nine WT SF3B1 samples (q value < 0.05, and cryptic 3′ss closer than 100 nt upstream of the associated canonical 3′ss). Each row represents one cryptic 3′ss, and each column one MDS patient sample. Z-scores in the matrix represent normalized Percent-Spliced-In values. The color bars above the heatmap indicate the SF3B1 mutations.
(C) Volcano plot representation of genes associated with the 169 cryptic 3′ss differentially used in mutant vs. WT SF3B1 samples (q value < 0.05, closer than 50 nt upstream of the associated canonical 3′ss, and more than 15 supporting reads averaged over mutant SF3B1 samples). The horizontal axis shows the Percent-Spliced-In (PSI) difference between mutant and WT SF3B1 samples, and the vertical axis shows the significance. Genes selected for further experimental validation are highlighted in red.
(D) RT-PCR products of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) of seven select genes (ORAI2, ZNF91, GCC2, KANSL3, MAP3K7, TTI1, and PPP2R5A) in normal control samples (C), WT SF3B1 MDS patient samples (W), and K700E SF3B1 MDS patient samples (K).
(E) HEK293T cells (293T) were transfected with empty vector plasmid (Vec), or expression plasmid for HA-tagged WT (W) or K700E (K) SF3B1, followed by western blotting.
(F) Total RNA was extracted from HEK293T cells as in (E), followed by RT-PCR of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) of the indicated genes.
(G) Each minigene was cotransfected with empty vector plasmid (Vec), or expression plasmid for HA-tagged WT (W) or K700E (K) SF3B1 to HEK293T cells, followed by RT-PCR of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) from the minigenes.
We next examined splicing of seven of the top targets directly using reverse transcription-polymerase chain reaction (RT-PCR). We first verified the mutant SF3B1-specific splicing defects using MDS patient cells and control samples (Figure 1D). To rule out the possibility that the RNA splicing defects were the result of patients’ complex genetic backgrounds in addition to the mutational status of SF3B1, we transiently overexpressed K700E SF3B1 in HEK293T cells and found that the use of upstream cryptic 3′ss was recapitulated (Figures 1E and 1F). Finally, we constructed minigene reporters from four of the target genes with the relevant introns and flanking exons, and used these in cotransfection experiments with K700E or WT SF3B1 expression vector. Again, we observed strong shifts to the upstream cryptic 3′ss (Figure 1G). Together, these results confirm that mutant SF3B1 is by itself sufficient to induce the 3′ss switch.
The K700E Mutation Specifically Impairs Association with SUGP1
We next wished to identify the defect of the mutant SF3B1 spliceosome that is responsible for the RNA missplicing. We hypothesized that SF3B1 mutations disrupt interactions with other spliceosomal protein(s) during assembly of the spliceosome, because all the hotspot mutations are localized in the HEAT domain, which functions as a major scaffold within U2 snRNP that facilitates the early events of spliceosome assembly (Cretu et al., 2016; Gozani et al., 1996; Wahl et al., 2009). To identify proteins differentially associated with mutant and WT SF3B1, we wished to perform affinity purification using cells expressing either K700E or WT SF3B1 with affinity tags. To this end, we engineered K562 myelogenous leukemia cells using CRISPR/Cas9 technology to knock in the hotspot K700E mutation (or synonymous mutations for the WT control), followed by addition of mono-allelic His6 (hexahistidine)-FLAG tandem tags at the N-terminus of the knock-in SF3B1 allele (see STAR METHODS). Using RT-PCR, we confirmed that cells expressing the tagged K700E SF3B1 recapitulated the 3′ss switch (Figure 2A).
Figure 2. The K700E Mutation Specifically Impairs Association with SUGP1.

(A) Total RNA was extracted from K562 parental cells and CRISPR/Cas9 engineered K562 cells expressing mono-allelic His6-FLAG-tagged WT or K700E SF3B1, followed by RT-PCR of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) of two select genes (GCC2 and KANSL3).
(B) SF3B1-associated proteins were purified from CRISPR/Cas9 engineered K562 cells expressing mono-allelic His6-FLAG-tagged WT or K700E SF3B1 using the large-scale affinity purification protocol, followed by SDS-PAGE and staining with QC Colloidal Coomassie Stain (Bio-Rad). M, Precision Plus Protein marker (Bio-Rad).
(C) Mass spectrometry results of three candidate proteins differentially associated with WT (W) and K700E (K) SF3B1, with the second and third columns indicating the numbers of unique peptides and the fourth column the peptide ratio.
(D and E) SF3B1-associated proteins were purified from CRISPR/Cas9 engineered K562 cells expressing mono-allelic His6-FLAG-tagged WT (W) or K700E (K) SF3B1 using the small-scale protocol, and then resolved by SDS-PAGE, followed by silver staining (D) or western blotting (E). M, Precision Plus Protein marker (Bio-Rad).
(F) Mass spectrometry results of SF3b subunits (as well as SUGP1) associated with WT and K700E SF3B1. Note that in the K700E SF3B1 sample there was one unique peptide that belongs to a novel isoform (fragment) of SF3B2, but this single peptide was not included in the total number of unique peptides of SF3B2, which does not affect our conclusion.
We next purified SF3B1 and associated spliceosomal proteins from extracts of the engineered K562 cells using two rounds of affinity purification with anti-DYKDDDDK antibody (for FLAG-tag) and cobalt beads (for His6-tag). After partially resolving the recovered proteins by SDS-PAGE to remove cobalt ions (Figure 2B), we excised the entire gel lanes of both WT and K700E SF3B1 samples for mass spectrometry (MS) to identify all SF3B1-associated proteins. Table S2 shows the identities of the 606 proteins (including 10 protein isoforms or fragments), of which 503 were in WT and 533 in K700E SF3B1 samples. To identify proteins differentially associated with WT and K700E SF3B1, we used stringent filtering to minimize false positives. We selected top candidates from proteins with at least 10 unique peptides recovered in either WT or K700E SF3B1 purification, and found three with a peptide ratio (K700E/WT) of less than or equal to 0.7 (Figure 2C). The most reduced protein in the K700E SF3B1 purification was SUGP1. We confirmed the reduction of SUGP1 levels by silver staining and western blotting of the isolated complexes (Figures S1A and S1B). It is noteworthy that the only protein in the silver-stained gel that appeared reduced in the K700E lane was the one with the predicted size of SUGP1. We further confirmed the loss of SUGP1 in the mutant spliceosome using a small-scale protocol, which we optimized for ease of later experimentation (Figures 2D and 2E). Importantly, all SF3b subunits were recovered at essentially the same levels in both complexes (Figure 2F), consistent with a previous study indicating that the K700E mutation does not affect interactions of SF3B1 with other SF3b subunits (Cretu et al., 2016).
Loss of SUGP1 Recapitulates the Splicing Defects of K700E SF3B1
We reasoned that if weakened association of SUGP1 with SF3B1 in the spliceosome is responsible for the K700E-induced splicing defects, then reducing SUGP1 levels in cells should recapitulate the 3′ss switch. To test this, we knocked down SUGP1 in HEK293T cells using two independent small interfering RNAs (siRNAs). Strikingly, we observed by RT-PCR that SUGP1 KD indeed led to use of upstream cryptic 3′ss in all transcripts tested (Figures 3A and 3B). It is noteworthy that KD of RBM6, which also appeared slightly reduced in the K700E SF3B1 purification based on the MS analysis (Figure 2C), did not induce cryptic 3′ss usage (Figures S2A and S2B). We also depleted 11 other select transcripts (including some encoding SF3a/b subunits, SURP domain and/or G-patch domain containing proteins, and two other control proteins), and in no case did we observe significant 3′ss switching (Figures S2C and S2D), providing evidence that the SUGP1 depletion-induced effect was specific.
Figure 3. Loss of SUGP1 Recapitulates the Splicing Defects of K700E SF3B1.

(A) HEK293T cells were transfected with a negative control siRNA (siC), or one of two independent siRNAs targeting SUGP1, followed by western blotting.
(B) RT-PCR products of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) of the indicated genes in HEK293T cells as in (A).
(C) Each of the indicated WT (shown in green) and mutant (shown in red) minigenes was cotransfected either with expression plasmid for HA-tagged WT (W) or K700E (K) SF3B1 (left panels), or with one of two independent siRNAs targeting SUGP1 (right panels) to HEK293T cells, followed by RT-PCR of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) from the minigenes. As indicated, an empty vector plasmid (Vec) and a negative control siRNA (siC) were also used.
See also Figures S2 and S3.
Two previous studies showed that mutant SF3B1 induces cryptic 3′ss usage by causing the spliceosome to use an upstream BP (Alsafadi et al., 2016; Darman et al., 2015). Using nested RT-PCR followed by sequencing of intron-lariat intermediates, we mapped the BP on the endogenous transcripts of two target genes, ORAI2 and TTI1, in the engineered K562 cells, and indeed found a mutant SF3B1-specific BP at 13- or 11-nt upstream of the corresponding canonical BP, respectively (Figure S3). To examine whether loss of SUGP1 also led to use of upstream BPs, we utilized minigenes from ORAI2 and TTI1 and mutated both constructs by changing the nucleotide corresponding to the upstream BP from A to G, the least likely nucleotide to function as a BP (Mercer et al., 2015). Minigene reporter assays showed that the BP mutations abolished the 3′ss switch by K700E SF3B1, and this effect was recapitulated by SUGP1 KD (Figure 3C). These results provide further evidence that loss of SUGP1 indeed accounts for the splicing defects of the mutant SF3B1 spliceosome.
A Dominant Negative Mutant of SUGP1 Phenocopies Mutant SF3B1
Next, we wished to investigate what specific function of SUGP1 is involved in the splicing misregulation. To this end, we decided to mutate each of the SUGP1 functional domains and examine their effects on splicing. SUGP1, previously known as splicing factor 4 (Sampson and Hewitt, 2003), has two tandem SURP domains and one G-patch domain (Figure S4A), which show high homology to annotated domains in the Conserved Domain Database (Marchler-Bauer et al., 2017). SURP domains are known to interact with the branchsite binding protein SF1, and G-patch domains have been shown to activate RNA helicases for ATP hydrolysis (Crisci et al., 2015; Robert-Paganin et al., 2015). Because both displacement of SF1 and ATP hydrolysis by RNA helicases are required for BP recognition by the SF3B1-containing U2 snRNP (Wahl et al., 2009), the presence of both SURP and G-patch domains in SUGP1 strongly suggests that SUGP1 may be involved in this process. In addition, interaction with U2AF is also involved in BP recognition (Ruskin et al., 1988), leading us to wonder whether SUGP1 interacts with U2AF. We examined the protein sequence of SUGP1 for potential U2AF binding sites, and found a KRKRKSRW sequence that matches the binding site consensus of the RNA recognition motif 3 (RRM3) of U2AF2 (Selenko et al., 2003). To test whether this sequence is in fact able to interact with U2AF2, we expressed a small portion of SUGP1 (containing this region) attached to a GST (glutathione S-transferase) tag in HEK293T cells and performed affinity purification. We found that U2AF2 indeed coimmunoprecipitated with this portion of SUGP1, whereas mutation of the most conserved amino acid residue of this binding site from W to A completely abolished coimmunoprecipitation (Figure S4B). To determine whether the interaction is direct, we purified this portion of SUGP1 and its W-to-A mutant with a GST tag, as well as the RRM3 of U2AF2 with His6-FLAG tags, from E. coli (Figure S4C). In vitro protein-protein interaction assays showed that purified U2AF2 RRM3 directly interacts with this portion of SUGP1 but not with the W-to-A mutant or the GST tag control (Figures S4D and S4E).
To investigate whether any of these functional domains in SUGP1 are essential for the splicing defects, we decided to attempt to disrupt the specific function of each domain to examine possible effects of the mutant proteins on 3′ss usage. Assuming that the most conserved amino acid residue(s) in each domain are important for its function, we mutated each domain of SUGP1 by changing the most conserved residues to alanine based on the sequence alignments of the domains (Aravind and Koonin, 1999; Kuwasako et al., 2006; Selenko et al., 2003). We then expressed these mutant derivatives in HEK293T cells (Figures 4A and 4B). Strikingly, we found that expression of the G-patch domain mutant of SUGP1 robustly switched 3′ss usage, recapitulating the splicing defects of the mutant SF3B1 spliceosome (Figure 4C). In contrast, mutations in either or both of the two SURP domains, or in the U2AF2 motif, did not affect splicing. This result strongly suggests that the G-patch domain of SUGP1 is essential for faithful splicing, likely by activating a currently unknown RNA helicase for ATP hydrolysis required for BP recognition (see DISCUSSION).
Figure 4. A Dominant Negative Mutant of SUGP1 Phenocopies Mutant SF3B1.

(A) Schematic representation of SUGP1 functional domains and mutants.
(B) HEK293T cells were transfected with empty vector plasmid (Vec), or expression plasmid for HA-tagged SUGP1 or one of the SUGP1 mutants (F222A, F297A, F222A-F297A, W387A, and G574A-G582A), followed by western blotting.
(C) RT-PCR products of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) of the indicated genes in HEK293T cells as in (B).
See also Figure S4.
Overexpression of SUGP1 Partially Rescues the Mutant Spliceosome
An important question is whether the function of the mutant SF3B1 spliceosome can be rescued. For example, can overexpression of SUGP1 increase its incorporation into the mutant spliceosome? If so, is this sufficient to restore proper 3′ss selection? To address this, we again used overexpression assays in HEK293T cells. We first tested whether exogenous expression of His6-FLAG-tagged WT or K700E SF3B1 could recapitulate the defective interaction of K700E SF3B1 with endogenous SUGP1. Using the small-scale protocol, we purified the SF3B1-containing complexes from HEK293T cells, and found that SUGP1 associated with exogenously expressed WT SF3B1 but the interaction was defective with the mutant SF3B1 (Figures S5A and S5B). Next, we co-expressed SUGP1 with either WT or K700E SF3B1 and then isolated SF3B1 complexes using affinity tags. We found that overexpression of SUGP1 indeed increased its association with the mutant SF3B1 complexes (Figures 5A and 5B), indicating that the K700E mutation does not entirely preclude SUGP1 association. Importantly, RT-PCR showed that SUGP1 overexpression partially rescued the splicing errors of the K700E SF3B1 spliceosome (Figures 5C–5E). These results suggest that loss of SUGP1 is responsible for, and is the sole determinant of, the splicing errors of the mutant spliceosome. Our findings also show that the mutant spliceosome is “repairable” in principle by restoring SUGP1 assembly.
Figure 5. Overexpression of SUGP1 Partially Rescues the Mutant Spliceosome.

(A and B) The indicated plasmids were cotransfected to HEK293T cells in 10-cm plates, followed by affinity purification using the small-scale protocol. The SF3B1-associated proteins were resolved by SDS-PAGE, followed by silver staining (A) or western blotting (B). Marker, Precision Plus Protein marker (Bio-Rad); Vec, empty vector plasmid; SUGP1, expression plasmid for HA-tagged SUGP1; WT, expression plasmid for His6-FLAG-tagged WT SF3B1; K700E, expression plasmid for His6-FLAG-tagged K700E SF3B1.
(C) The indicated plasmids were cotransfected to HEK293T cells in six-well plates. Total RNA was extracted from the cells, followed by RT-PCR of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) of two select genes (GCC2 and KANSL3). Vec, empty vector plasmid; SUGP1, expression plasmid for HA-tagged SUGP1; WT, expression plasmid for HA-tagged WT SF3B1; K700E, expression plasmid for HA-tagged K700E SF3B1. Note that the PCR products of the cryptic 3′ss of GCC2 and KANSL3 (open arrowheads) in the sixth lane appeared to migrate slightly more slowly than the PCR products in the fifth lane. However, DNA sequencing results indicated that the cryptic 3′ss and canonical 3′ss of GCC2 and KANSL3 in the last two lanes were exactly the same.
(D) Quantification of RT-PCR products in (C). The bar graph was made using Prism (GraphPad). Error bars represent SDs of the means (n = 3, three independent experiments). **p < 0.01 (unpaired, two-tailed, and unequal variance t test, calculated using Microsoft Excel).
(E) Protein extracts from HEK293T cells as in (C) were resolved by SDS-PAGE, followed by western blotting.
See also Figures S5 and S6.
Other SF3B1 Mutations Also Weaken Interaction with SUGP1 in the Spliceosome
In addition to K700E, a number of other SF3B1 mutations are frequently found in MDS and certain cancers (Haferlach et al., 2014; Harbour et al., 2013; Landau et al., 2013; Papaemmanuil et al., 2011, 2013; Yoshida et al., 2011). To examine whether other disease mutations also weaken the interaction of SF3B1 with SUGP1, we introduced each of four common mutations into our SF3B1 expression vector and exogenously expressed the mutant derivatives in HEK293T cells. Affinity purification showed that all the mutations reduced SUGP1 association with SF3B1 (Figures 6A and 6B). As with K700E, these other SF3B1 mutations also led to 3′ss switching (Figures 6C and 6D). These results suggest that loss of SUGP1 during assembly of the mutant spliceosome is a common mechanism by which SF3B1 mutations result in splicing defects in MDS and cancer.
Figure 6. Other SF3B1 Mutations Also Weaken Interaction with SUGP1 in the Spliceosome.

(A and B) HEK293T cells in 10-cm plates were transfected with empty vector plasmid (Vec), or expression plasmid for His6-FLAG-tagged WT, K700E, E622D, R625C, H662Q, or K666N SF3B1, followed by affinity purification using the small-scale protocol. The SF3B1-associated proteins were resolved by SDS-PAGE, followed by silver staining (A) or western blotting (B). Marker, Precision Plus Protein marker (Bio-Rad).
(C and D) HEK293T cells in 6-well plates were transfected with empty vector plasmid (Vec), or expression plasmid for HA-tagged WT, K700E, E622D, R625C, H662Q, or K666N SF3B1, followed by RT-PCR of the cryptic 3′ss (open arrowheads) and canonical 3′ss (solid arrowheads) of GCC2 and KANSL3 (C) or western blotting (D).
DISCUSSION
Since the discovery of high frequencies of splicing factor mutations in MDS and other cancers, considerable progress has been made towards understanding the role of several of these mutant proteins in misregulation of RNA splicing (Alsafadi et al., 2016; Darman et al., 2015; DeBoever et al., 2015; Ilagan et al., 2015; Kim et al., 2015; Komeno et al., 2015; Shiozawa et al., 2018; Shirai et al., 2015; Zhang et al., 2015). However, nothing has been known about how SF3B1 mutations, which are the most common of the splicing factor gene mutations, affect the function of the protein in splicing. Furthermore, in all these cases, progress towards developing effective therapies has been slow.
The fact that splicing gene mutations are consistently mutually exclusive suggests that mutant cells may be more susceptible to additional RNA splicing modulation than WT cells, offering therapeutic opportunities (Lee and Abdel-Wahab, 2016). Indeed, a small-molecule splicing modulator called H3B-8800 showed promising results in selective killing of splicing factor mutant vs. WT cells (Seiler et al., 2018b). However, this splicing modulator, which targets SF3b, binds both WT and mutant complexes similarly. Although this indiscriminate binding is tolerable to the WT cells tested, it may cause side effects in more sensitive WT cells in some parts of the body, e.g., cases of vision blur or loss were reported in clinical trials of another similar splicing modulator, E7107 (Eskens et al., 2013; Hong et al., 2014). Furthermore, cancer cell killing agents often lead to drug resistance after long-term use because of constant selection pressure (Friedman, 2016). Therefore, for mutant SF3B1 cancers, the ideal strategy would be to specifically repair the mutant spliceosome and thereby rescue the diseased cells rather than kill them. Given the complex nature of the spliceosome, the assembly of which requires coordinated interactions among possibly hundreds of proteins, this task has been challenging. Nevertheless, we have elucidated here the defect of the mutant SF3B1 spliceosome. Our data indicate that loss or weakening (to be more precise) of the interaction of SUGP1 with SF3B1 in the spliceosome is solely responsible for the defects in BP recognition and resulting use of cryptic 3′ss typically located 10–30 nt upstream of the canonical 3′ss. Below we present a detailed mechanistic model explaining how SUGP1 loss affects splicing in this way, and also discuss our results in the context of the insights they provide into how restoring SUGP1 assembly into the mutant spliceosome can be developed as a potential cure for mutant SF3B1 cancers.
Based on our data, we propose the following model for SUGP1 function in facilitating BP recognition, and how SF3B1 mutations disrupt this process and force use of a cryptic 3′ss (Figure 7). During the early stages of RNA splicing under normal conditions, SF1 binds to the canonical branchsite with the help of U2AF, which binds the canonical 3′ss and its adjacent polypyrimidine tract (Berglund et al., 1998; Ruskin et al., 1988). U2 snRNP then recruits SUGP1 via the interaction of the SF3B1 HEAT domain with SUGP1, and SUGP1 in turn assists in localizing U2 snRNP to the vicinity of the canonical BP and 3′ss through direct interactions with both SF1 and U2AF2. The SUGP1 G-patch domain then associates with and activates an unknown RNA helicase required for the displacement of SF1 by p14 (Wahl et al., 2009), allowing base pairing between the canonical branchsite and U2 snRNA. As a result, the spliceosome uses the canonical BP and 3′ss for splicing (Figure 7A). When SUGP1 is depleted or mutated in the G-patch domain (Figures 7B and 7C), the coupling between SF1/U2AF2 binding and ATP hydrolysis is absent, such that SF1 is not displaced and thus blocks access to the canonical BP. U2 snRNP is then forced to utilize an unblocked upstream cryptic BP, possibly through scanning of the branchsite’s negatively charged backbone by p14 (Perea et al., 2016). Similarly, in patients carrying SF3B1 HEAT domain mutations, U2 snRNP is unable to recruit SUGP1, the RNA helicase is thus also not recruited and/or activated, SF1 is not displaced, and the mutant spliceosome must again scan for an upstream branchsite and utilize a cryptic 3′ss (Figure 7D).
Figure 7. The Mechanistic Model.

(A) Under normal conditions, the WT SF3B1 spliceosome uses a canonical BP and 3′ss for splicing (see main text for a detailed explanation). The G-patch domain of SUGP1 is shown as “G” in red. Py-tract, polypyrimidine tract.
(B and C) When SUGP1 is depleted (B), or mutated in the G-patch domain (C), the WT SF3B1 spliceosome uses an upstream cryptic BP and cryptic 3′ss for splicing.
(D) Because the interaction between SF3B1 and SUGP1 is disrupted by SF3B1 mutations, the mutant SF3B1 spliceosome uses an upstream BP and cryptic 3′ss for splicing.
An interesting question is why only a relatively small number of introns are affected by SF3B1 mutations, given that SF3B1 is an essential splicing factor. In order for the mutant SF3B1 spliceosome to misregulate RNA splicing, both an upstream BP and a cryptic 3′ss within an appropriate distance of the canonical 3′ss are required (Alsafadi et al., 2016; Darman et al., 2015). These restrictions define a small number of introns in which mutant SF3B1 can induce splicing errors. For the large number of other introns that do not appear misspliced by mutant SF3B1, we envision two different scenarios. In one, where the intron has an upstream cryptic BP but not a cryptic 3′ss, we suggest that the mutant SF3B1 spliceosome proceeds through the normal splicing pathway by utilizing the upstream BP for lariat formation and then uses the canonical 3′ss for exon-exon joining, but at a much reduced rate (Noble et al., 1987; Padgett et al., 1985; Ruskin et al., 1985). In the other scenario, where neither an upstream BP nor a cryptic 3′ss is present in the intron, we suggest that the mutant SF3B1 spliceosome is assembled to the stage of complex B without SUGP1 and then it stalls to wait for SUGP1 incorporation (Utans and Kramer, 1990). Because the mutant SF3B1 still has residual affinity for SUGP1 (likely with a slower on-rate), after a long lag SUGP1 will eventually be incorporated into the stalled complex B, allowing the mutant spliceosome to carry out the subsequent catalytic reactions with the canonical BP and 3′ss (Utans and Kramer, 1990).
The significance of our study is multifaceted. First, we found that the effects of SF3B1 mutations on the function of the mutant spliceosome are subtle. The fact that the WT spliceosome, in the absence of SUGP1 or when SUGP1 is mutated in the G-patch domain, apparently functions identically to the mutant SF3B1 spliceosome indicates that the mutations lead to only a small change in a limited area of the SF3B1 protein, leaving the remaining scaffold intact and functional. This structural change in SF3B1 appears to only impair recruitment of SUGP1 to the U2 snRNP complex during BP recognition, and the subsequent assembly of the active spliceosome as well as the catalytic splicing reactions are not affected. Second, we found that the effects of SF3B1 mutations are reversible. Our data show that the mutant spliceosome is partially rescued by SUGP1 overexpression, indicating that the assembly of SUGP1 into the spliceosome is concentration dependent and that SF3B1 mutations simply reduce affinity for SUGP1. Therefore, a more robust rescue of the mutant spliceosome may in principle be achieved, either by identifying small molecules that stabilize SUGP1 interaction with mutant SF3B1 (Andrei et al., 2017; Doveston et al., 2017), or by complementary mutations (or suppressor mutations) in SUGP1 that fully restore its affinity for mutant SF3B1 (Aakre et al., 2015; Crispino et al., 1999; Li et al., 2018).
Our study also provides insights into how the multiple steps involved in BP recognition are coordinated. Previously, SUGP1 was known to have both SURP and G-patch domains (Sampson and Hewitt, 2003), which likely interact with SF1 and RNA helicase(s), respectively (Crisci et al., 2015; Robert-Paganin et al., 2015). In this study, we show that SUGP1 interacts with U2AF2 and the HEAT domain of SF3B1. The presence of all these functions in the same protein streamlines the BP recognition process, as illustrated in Figure 7A. In keeping with this, our mechanistic model is different from several proposed in previous studies that suggested that SF3B1 mutations might: 1) facilitate selection of cryptic 3′ss by overcoming certain steric hindrance within a region of RNA immediately downstream of a BP (DeBoever et al., 2015); 2) enhance direct interactions of SF3B1 with specific nucleotides flanking the upstream BP (Darman et al., 2015); 3) induce a conformational change in the U2 snRNP complex leading to selection of a stronger upstream BP (Alsafadi et al., 2016); or 4) facilitate the recognition of inaccessible 3′ss buried in RNA secondary structures (Kesarwani et al., 2017). All these models imply that the structural changes to SF3B1 induced by disease mutations are substantial and not readily amenable to rescue by restoring interaction with a partner protein, as argued by our data.
An open question that requires further investigation is the identity of the RNA helicase that the G-patch domain of SUGP1 binds and activates. The human genome encodes 64 distinct RNA helicases (Umate et al., 2011), half of which were identified in the spliceosomal complexes purified in this study (Table S2). A previous systematic analysis of human spliceosomal protein-protein interactions using yeast two-hybrid assays revealed 632 unique pairwise interactions between 196 spliceosomal proteins, among which SUGP1 interacts with the RNA helicase DHX15 (Hegele et al., 2012). However, that study also showed that DHX15 interacts with three other G-patch domain-containing proteins (RBM5, RBM10, and RBM17) in addition to SUGP1. It therefore remains to be determined if it is SUGP1 that DHX15 interacts with in the spatially organized spliceosome. We note that KD of DHX15 did not induce cryptic 3′ss usage (Figures S2C and S2D), and that DHX15 was present almost equally in both WT and K700E SF3B1 purifications (Table S2). If it is indeed DHX15 that the G-patch domain of SUGP1 activates, then DHX15 must be recruited to the mutant spliceosome independent of SUGP1, possibly by the interaction of DHX15 with the SF3b subunit SF3b125 (Hegele et al., 2012). Another helicase candidate might be UAP56, an RNA helicase that can be recruited to the spliceosome via interaction with U2AF2 (Fleckner et al., 1997). UAP56 was shown to be required for U2 snRNP-branchsite interaction, which aligns well with the role of SUGP1. It is thus possible that the G-patch domain of SUGP1 binds UAP56 to activate its ATPase activity required for branchsite recognition. In yeast, Prp5 is an RNA helicase shown to be important for prespliceosome formation and branchsite proofreading in an ATP-dependent manner (Ruby et al., 1993; Xu and Query, 2007). As mentioned in the INTRODUCTION, a previous study showed that SF3B1 mutations affect the interaction of SF3B1 with the N-terminus of Prp5 (Tang et al., 2016). Although the region in Prp5 involved in this interaction is not conserved in the human homolog of Prp5, it remains possible that SUGP1 bridges SF3B1 and the human Prp5 homolog. It is worth mentioning that the G-patch domain mutation is unlikely to affect the interaction of SUGP1 with SF3B1, as the mutant SUGP1 had a dominant-negative effect on splicing, indicating that it is assembled into the spliceosome to induce use of cryptic 3′ss.
Despite frequent SF3B1 mutations in MDS and other cancers, no mutations in SUGP1 have been identified in these diseases. Since loss of SUGP1 recapitulates the splicing errors of the mutant SF3B1 spliceosome, why are there no loss-of-function SUGP1 mutations? To begin to address this and related questions, we examined expression of SUGP1 in a large number of uveal melanoma and breast cancer samples, whose mRNA transcripts were sequenced by The Cancer Genome Atlas (TCGA) and transcript levels estimated by the University of California Santa Cruz (UCSC) Xena platform (Goldman et al., 2018). Interestingly, we found that SUGP1 mRNA levels were elevated by ~60–80% on average in 3′ss-switching mutant SF3B1 vs. WT samples (Figure S6), suggesting that the mutant cells attempt to compensate for the defective SUGP1 interaction by increasing SUGP1 levels. Although the modestly elevated SUGP1 levels are insufficient to rescue mutant SF3B1 splicing, this compensation mechanism may provide an explanation for why there are no loss-of-function SUGP1 mutations in cancer. Specifically, it seems likely that heterozygous loss-of-function mutations in SUGP1 would also induce this feedback mechanism to elevate SUGP1 mRNA levels, which in turn would be sufficient to compensate for SUGP1 haploinsufficiency, thereby preventing disease-driving splicing changes. As a result, SUGP1 loss-of-function mutations would not be selected for during disease progression. Although the rescue mechanism is unknown, it may reflect autoregulation in splicing of SUGP1 precursor mRNAs.
The functional consequences of the splicing errors produced by the mutant SF3B1 spliceosome in pathogenesis remain to be understood. A gene ontology pathway analysis of the 169 misspliced transcripts identified in this study revealed a cluster annotated “innate immune response” that may be of potential interest (Figure S7). Innate immune activation increases the risk of developing MDS, but it is unknown how innate signaling is initiated in the pathogenesis of MDS (Barreyro et al., 2018). Missplicing of transcripts from genes involved in “innate immune response” may contribute to the activation of innate immune signaling in mutant SF3B1 patients. A few prominent splicing targets identified in this study may contribute to other cancer phenotypes. For example, a splicing error in MAP3K7 (mitogen-activated protein kinase kinase kinase 7) transcripts may result in reduced activation of the MAPK pathway required for hematopoiesis (Geest and Coffer, 2009). A splicing error in intron 3 of ZNF91 (zinc finger protein 91) may lead to translation from a start codon in exon 4, producing a truncated ZNF91 protein that lacks the N-terminal KRAB (the Krüppel-associated box) domain. The KRAB domain is important for nuclear localization, and its loss may allow the truncated ZNF91 to localize to the cytoplasm where it may interact with the IκB kinase complex, activating the NF-κB pathway, as demonstrated for another member of this family of zinc-finger proteins (Wang et al., 2012). A splicing error in PPP2R5A (protein phosphatase 2 regulatory subunit B’alpha) transcripts may also contribute to cancer development. PPP2R5A is one of the regulatory subunits of the protein phosphatase 2A and regulates multiple signaling pathways in cancer, such as p53, MAPK, JAK/STAT, c-Myc, and β-Catenin related signaling pathways (Mao et al., 2018). Although these and other potential disease-causing splicing errors may be exploited as targets for therapeutic intervention, a more effective way would be, as alluded to above, to rescue the mutant spliceosome itself to prevent splicing errors altogether.
In summary, we have elucidated the defect of the mutant SF3B1 spliceosome in MDS and cancer, and provided proof-of-principle evidence that the cancer-causing spliceosome is curable. This study not only advances our understanding of how SF3B1 mutations disrupt splicing, but also provides insights into novel strategies for developing treatments for MDS and other cancers caused by SF3B1 mutations.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James L. Manley (jlm2@columbia.edu).
Experimental Model and Subject Details
Patient samples
Bone marrow samples were aspirated from MDS patients. Informed consent was obtained from the human subjects who participated in the study, which was approved by the Institutional Review Board of Columbia University and was carried out in accordance with the Declaration of Helsinki.
Cell lines
HEK293T cells were cultured at 37°C in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a 5% CO2 incubator. CRISPR/Cas9 engineered and parental K562 cells were cultured at 37°C in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FBS in a 5% CO2 incubator.
Method Details
RNA Sequencing of Patient Samples
Mononuclear cells were isolated from bone marrow samples of MDS patients and then lysed in TRIzol reagent (Thermo Fisher Scientific). Total RNA was isolated using the RNeasy kit (QIAGEN), and RNA quality was checked using Agilent’s Bioanalyzer. RNA samples with an RNA integrity number of more than nine were used, and from each RNA sample an Illumina’s TruSeq RNA library was prepared and sequenced using Illumina’s HiSeq2000 sequencer, generating approximately 50–75 million pair-end 101-bp reads. Illumina’s Real-Time Analysis software was used for base calling (BCL), and the resulting BCL file was converted to a FASTQ format using bcl2fastq2 v2.17.
Identification of Novel Cryptic 3′ Splice Sites
We adopted the computational analysis pipeline from DeBoever et al. (2015) for identification and usage calculation of novel splice junctions, as well as identification of canonical 3′ss associated with cryptic 3′ss. Specifically, RNA-sequencing FASTQ files were aligned to the human genome using STAR (Dobin et al., 2013), with a splice junction database that enables highly sensitive detection of novel 3′ss (DeBoever et al., 2015). Counts of junction reads from the STAR output file (SJ.out.tab) were merged into a unique matrix, with each row indicating one splice junction and each column indicating one sample. Splice junctions with fewer than 20 reads (summed up across all samples) were filtered out. The Percent-Spliced-In (PSI) was calculated by transforming raw read counts into a PSI matrix with values between 0 and 1. We used t tests with PSI values (instead of read counts) to identify novel cryptic 3′ss between WT and mutant SF3B1 samples. The resulting p values were further adjusted by Benjamini–Hochberg multiple test correction. Differences in mean PSI values were also calculated as important metrics for differential splicing between WT and mutant samples. A bar plot of log2 distance (nt) was used to illustrate the distribution of distances from cryptic 3′ss to their associated canonical 3′ss.
Heatmap and Hierarchical Clustering
An unsupervised hierarchical clustering of the 627 splice junctions differentially used by mutant vs. WT SF3B1 (q value < 0.05, and cryptic 3′ss closer than 100 nt upstream of the associated canonical 3′ss) was performed using the command “heatmap.2” from the R software package “gplots” (R Core Team, 2013), with (1 − Spearman’s correlation/2) as the clustering distance.
Gene Ontology Analysis and Visualization
A standard gene ontology (GO) analysis was performed to reveal enriched functional pathways affected by RNA missplicing by mutant SF3B1 using DAVID (Huang da et al., 2009a, b). The input was a list of genes associated with the 169 cryptic 3′ss differentially used by mutant vs. WT SF3B1 (q value < 0.05, closer than 50 nt upstream of the associated canonical 3′ss, and more than 15 supporting reads averaged over mutant SF3B1 samples). GO terms with a p value of less than 0.05 were selected, and REVIGO was used to visualize the most informative ontology themes (Supek et al., 2011).
Reverse Transcription-Polymerase Chain Reaction
Total RNA was extracted using TRIzol (Thermo Fisher Scientific), and 2 μg total RNA per sample was reverse transcribed using 0.3 μl Maxima Reverse Transcriptase (Thermo Fisher Scientific) and 50 pmol oligo-dT primer. The synthesized cDNA was diluted 1:10 in H2O, and 1.2 μl was used as template in a 10-μl polymerase chain reaction (PCR) containing 0.6 μCi [α−32P] dCTP. PCR products were resolved in a 6% non-denaturing polyacrylamide gel, and the gel was dried and exposed to a phosphor screen. Radioactive signals were scanned by a Typhoon FLA 7000 imager (GE Healthcare) and quantified using ImageQuant (Molecular Dynamics). Primers used in the PCR reactions were: ORAI2 forward, 5′-GGGGCGAGGGCAGCTC-3′; ORAI2 reverse, 5′-CTCTCCATCCCATCTCCTTG-3′; ZNF91 forward, 5′-CAAGGAAAAGAGCCCTGGA-3′; ZNF91 reverse, 5′-CTCTGCTCTGGCCAAAAGTC-3′; MAP3K7 forward, 5′-TGTCTTGTGATGGAATATGCTG-3′; MAP3K7 reverse, 5′-TCCCTGTGAATTAGCGCTTT-3′; TTI1 forward, 5′-CCACAGCTGAAGACATCGAA-3′; TTI1 reverse, 5′-ACATCTGGACGGGTGTCATT-3′; GCC2 forward, 5′AAGGTGAGCTGGAGGCAAG-3′; GCC2 reverse, 5′-TGCTGTCACTCTCTGCTGGT-3′; KANSL3 forward, 5′-GCAGCGTGATGAATGAGTGT-3′; KANSL3 reverse, 5′-GGGCTTTGAGGATCTTGTTG-3′; PPP2R5A forward, 5′-TTGGCCTCACATACAGTTGG-3′; and PPP2R5A reverse, 5′-TTCCCATAAATTCGGTGCAG-3′.
Expression Plasmid Constructs
The cloning of SF3B1 constructs was not straightforward. We initially tried to clone the human SF3B1 cDNA into plasmid vector p3xFLAG-CMV-7.1 (Sigma), p3xFLAG-CMV-14 (Sigma) both in bacterial SURE strain, or pcDNA3 (Invitrogen) in DH5α strain, but found that the full-length sequence was unclonable. However, we were able to clone the 5′-end portion of the cDNA (nt 1–2331, at the end of which there is a naturally occurring EcoRI site) into p3xFLAG-CMV-7.1, suggesting that there may be an unclonable sequence in the 3′-end portion of the SF3B1 cDNA. To narrow down the unclonable region, we tried to clone ten truncated SF3B1 cDNA fragments into p3xFLAG-CMV-7.1, but found that bacterial transformants of only five of the ten ligation products were able to grow. This result allowed us to determine the boundaries of the unclonable sequence to be nt 2856–2983 (i.e., from the last nt of codon 952 to the first nt of codon 995), which is consistent with a previous study indicating that a fragment of the human SF3B1 cDNA (nt 2797–3042) is toxic to bacteria (Wang et al., 1998). Unclonable sequences are common in the human genome, e.g., there are 127 unclonable regions in euchromatin (Garber et al., 2009). To overcome the difficulty of cloning the SF3B1 cDNA, we introduced extensive synonymous mutations in an extended region covering the unclonable sequence. Specifically, we first cloned the 5′-end portion of the SF3B1 cDNA (nt 1–2331) with an HA (hemagglutinin) tag at the N-terminus into p3xFLAG-CMV-14 using the HindIII and EcoRI sites. Then we used overlapping PCR to make a mosaic sequence, in which nt 2497–3132 of the 3′-end portion of the SF3B1 cDNA was replaced with a sequence harboring extensive synonymous mutations (derived from a DNA synthesized by Life Technologies). The resulting mosaic sequence was cloned into the intermediate construct containing HA-tagged SF3B1 cDNA (nt 1–2331) using the EcoRI and XbaI sites, generating a final construct that encodes the full-length SF3B1 with a portion of the sequence (nt 2497–3132) harboring extensive synonymous mutations. This sequence is 5′-CTGGTGGACACAACAGTCGAACTCGCCAATAAAGTCGGCGCTGCCGAGATCATCAGCAGAATCGTCGACGACCTCAAGGACGAGGCTGAGCAATATCGCAAGATGGTCATGGAAACCATCGAAAAGATCATGGGCAACCTCGGCGCTGCTGACATCGACCACAAGTTGGAGGAGCAGCTCATCGACGGCATCCTGTACGCCTTTCAGGAGCAAACAACCGAAGATAGCGTGATGCTCAATGGATTCGGAACCGTCGTGAACGCCTTGGGAAAGAGGGTGAAGCCCTATCTCCCACAAATTTGCGGCACCGTGCTCTGGAGACTGAACAATAAGAGCGCCAAAGTCAGACAGCAAGCCGCAGATCTCATCTCCCGGACAGCCGTGGTGATGAAAACCTGCCAGGAAGAGAAGCTCATGGGCCATCTGGGCGTGGTGCTGTACGAATACCTCGGAGAGGAATATCCCGAGGTGCTGGGATCCATCTTGGGCGCCTTGAAAGCTATCGTGAACGTGATCGGCATGCACAAAATGACACCTCCCATCAAGGACCTCCTCCCACGGCTGACACCTATTCTGAAAAATCGGCACGAGAAGGTGCAGGAAAACTGCATCGACTTGGTGGGCAGAATCGCCGAT-3′. An N-terminally His6-FLAG-tagged version of SF3B1 construct was generated by replacing the HA-tag with the His6-FLAG tandem tags using overlapping PCR. The K700E, E622D, R625C, H662Q, and K666N mutant constructs were generated by site-directed mutagenesis (Ho et al., 1989). All SF3B1 plasmids were maintained in DH5α cells grown at 30°C.
N-terminally HA-tagged SUGP1 with two stop codons was cloned in p3xFLAG-CMV-14 (Sigma). Because of the two stop codons immediately following the SUGP1 coding sequence, the 3xFLAG tag in the vector was not added to the C-terminus of SUGP1. The F222A, F297A, F222A-F297A, W387A, and G574A-G582A mutant SUGP1 constructs were generated by site-directed mutagenesis (Ho et al., 1989).
Overexpression Experiments
Two rounds of transfections were performed for overexpression experiments in six-well plates. Specifically, HEK293T cells were seeded in a six-well plate with 300,000 cells per well. On the next day, 2 μg of expression plasmid DNA were transfected to each well of cells using Lipofectamine 2000 (Thermo Fisher Scientific). At 24 h post transfection, cells were trypsinized and one third of the cells were re-seeded in a new six-well plate. At 24 h post seeding (i.e., 48 h post initial transfection), cells were transfected a second time with 2 μg of plasmid DNA. At 48 h post second-round transfection (i.e., 96 h post initial transfection), cells were collected for protein isolation and total RNA extraction. For co-expression, a mixture of two plasmids (1 μg each) was used in each round of transfection.
Knockdown Experiments
Small interfering RNA (siRNA) was first mixed with DharmaFECT 1 reagent (Dharmacon), and then this transfection mixture was mixed with 300,000 HEK293T cells, followed by seeding the cells in a six-well plate. For each transfection, a final concentration of 10 nM siRNA was used. At 24 h post transfection, a second-round transfection with 10 nM siRNA was performed. At 24 h post second-round transfection (i.e., 48 h post initial transfection), cells were collected for protein isolation and total RNA extraction. Two independent siRNAs targeting SUGP1 (siSUGP1–1 and siSUGP1–2), two siRNAs targeting RBM6 (siRBM6–1 and siRBM6–2), and one negative control siRNA (siC) were used in this study. The siRNA sequences were: siSUGP1–1 sense strand, 5′-GGAUAACCCAGCAUUUGCAdTdT-3′; siSUGP1–1 antisense strand, 5′-UGCAAAUGCUGGGUUAUCCdTdT-3′; siSUGP1–2 sense strand, 5′-GAAGGUGGCUGAGAUAAGAdTdT-3′; siSUGP1–2 antisense strand, 5′-UCUUAUCUCAGCCACCUUCdTdT-3′; siRBM6–1 sense strand, 5′-GAGUCAUGCUCAAGAGAGAdTdT-3′; siRBM6–1 antisense strand, 5′-UCUCUCUUGAGCAUGACUCdTdT-3′; siRBM6–2 sense strand, 5′-CAAAGACGGAACACAAGUAdTdT-3′; siRBM6–2 antisense strand, 5′-UACUUGUGUUCCGUCUUUGdTdT-3′; siC sense strand, 5′-UUCUCCGAACGUGUCACGUdTdT-3′ (Shanghai GenePharma); and siC antisense strand, 5′-ACGUGACACGUUCGGAGAAdTdT-3′ (Shanghai GenePharma).
Short hairpin RNAs (shRNAs) targeting mRNAs of SF3A1, SF3A2, SF3A3, SF3B4, RBM5, RBM17, CHERP, U2SURP, DHX15, GPATCH11, and CCDC97 were each cloned into pLKO.1-puro (Sigma). The shRNA sequences were: shSF3A1, 5′-CTCCAGACCAAGTCATTGTCCTCTCAACACTGGACAATGACTTGGTCTGGAGTTTTT-3′; shSF3A2, 5′-CAAGGACCCGTACTTCATGCCTCTCAACACTGGCATGAAGTACGGGTCCTTGTTTTT-3′; shSF3A3, 5′-CAGCGACATCTCACTCATGCCTCTCAACACTGGCATGAGTGAGATGTCGCTGTTTTT-3′; shSF3B4, 5′-CCGTCCTATCACCGTATCTCCTCTCAACACTGGAGATACGGTGATAGGACGGTTTTT-3′; shRBM5, 5′-GATACGGTTCCATCATAGACCTCTCAACACTGGTCTATGATGGAACCGTATCTTTTT-3′; shRBM17, 5′-GTGGTCTTACTAAGGAACACCTCTCAACACTGGTGTTCCTTAGTAAGACCACTTTTT-3′; shCHERP, 5′-GAACTGGATGTTCAGCAATCCTCTCAACACTGGATTGCTGAACATCCAGTTCTTTTT-3′; shU2SURP, 5′-GCCATAGTCAAAGTGGTTACCTCTCAACACTGGTAACCACTTTGACTATGGCTTTTT-3′; shDHX15, 5′-GGTCTACAATCCTCGAATCCCTCTCAACACTGGGATTCGAGGATTGTAGACCTTTTT-3′; shGPATCH11, 5′-CAGTTTCGAATGCGACTTACCTCTCAACACTGGTAAGTCGCATTCGAAACTGTTTTT-3′; and shCCDC97, 5′GGAGGAGAGGTACTTTGATCCTCTCAACACTGGATCAAAGTACCTCTCCTCCTTTTT-3′. The pLKO.1-puro non-target shRNA plasmid from Sigma was used as a negative control.
To perform shRNA knockdown, HEK293T cells were seeded in six-well plates, per well with 300,000 cells and 2 ml growth medium (DMEM supplemented with 10% FBS). On the next day, 2 μg of shRNA plasmid DNA were transfected to each well of cells using Lipofectamine 2000 (Thermo Fisher Scientific). At 24 h post transfection, the growth medium was replaced with 2 ml fresh growth medium containing puromycin at a final concentration of 2 μg/ml. After 48 h of puromycin selection (i.e., at 72 h post transfection), cells were collected for total RNA extraction using TRIzol (Thermo Fisher Scientific). Total RNA was then reverse transcribed using Maxima Reverse Transcriptase (Thermo Fisher Scientific), followed by real-time PCR using Maxima SYBR Green Mix (Thermo Fisher Scientific). The 2−ΔΔCt method was used to quantify the relative mRNA levels of each gene, using RPL13A as an internal control. Primers used in the real-time PCRs were: SF3A1 forward, 5′-TTCACGAAGCTAGTGGAACAGT-3′; SF3A1 reverse, 5′-GGTAACACACCTGATCCAAAACT-3′; SF3A2 forward, 5′-GATTGACTACCCTGAGATCGCC-3′; SF3A2 reverse, 5′-CTCCCGGTTCCAGTGTGTC-3′; SF3A3 forward, 5′-TGGTCGTTATCTCGATCTCCAT-3′; SF3A3 reverse, 5′-AGAGGCTTCACTCTATCTGTGT-3′; SF3B4 forward, 5′-CTCCGAGCGGAATCAGGATG-3′; SF3B4 reverse, 5′-GGCATGTGGGTGTTGACTACT-3′; RBM5 forward, 5′-CCATCACAGAGAGCGATATTCG-3′; RBM5 reverse, 5′-CGGCTTACACCTGTTTTCCTC-3′; RBM17 forward, 5′-CTCGCCCCAGTCATTGACC-3′; RBM17 reverse, 5′-TCGTCAGCTAAGGGAATCAGAA-3′; CHERP forward, 5′-ATCCTGGCATCAACGAGCAC-3′; CHERP reverse, 5′-GAAGTAGGGCACATTGGGGAC-3′; U2SURP forward, 5′-TTCAAGAGGAACGTGATGAGAGA-3′; U2SURP reverse, 5′CGTCCATAGAACGACGCTG-3′; DHX15 forward, 5′-GGGGACCGATGGGAAGGAT-3′; DHX15 reverse, 5′-TAGCATTTGTTGAAGCTCGCA-3′; GPATCH11 forward, 5′TGCAGAACAGTTTCGAATGC-3′; GPATCH11 reverse, 5′-CCTCTTCAAGCCTCAACCAG-3′; CCDC97 forward, 5′-ATGCAGCAGTGAGTGCTATG-3′; CCDC97 reverse, 5′CAGTGGCTTCTCGTGGTACAG-3′; RPL13A forward, 5′-GCCATCGTGGCTAAACAGGTA3′; and RPL13A reverse, 5′-GTTGGTGTTCATCCGCTTGC-3′.
Western Blotting
Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad), followed by immunoblotting with primary and secondary antibodies. Primary antibodies were: anti-SF3B1 (Bethyl Laboratories, A300–996A, 1:1,000), anti-ACTIN (Sigma, A2066, 1:2,000), anti-HA rabbit polyclonal (Abm, G166, 1:1,000), anti-HA mouse monoclonal (Sigma, H3663, 1:1,000), anti-DYKDDDDK (GenScript, A00187, 1:1,000), anti-SUGP1 (Bethyl Laboratories, A304–675A-M, 1:1,000), anti-RBM6 (ABclonal, A10391, 1:1,000), anti-U2AF2 (Sigma, U4758, 1;10,000), and anti-GST (Invitrogen, A5800, 1:1,500). Secondary antibodies were: Donkey anti-Rabbit IgG (LI-COR, 926–68073, 1:5,000), and Goat anti-Mouse IgG (LI-COR, 926–32210, 1:5,000). Immunofluorescence was detected using either the Odyssey Infrared Imager (LI-COR) or the ChemiDoc Imaging System (Bio-Rad).
Minigene Assays
Minigenes were cloned into pcDNA3 (Invitrogen), with the ORAI2 minigene containing ORAI2 (NM_001271818.1) exons 1, 2, and 3, and truncated introns 1 and 2; ZNF91 minigene containing ZNF91 (NM_003430.3) exon 3, truncated intron 3, and the 5′-end portion of exon 4; MAP3K7 minigene containing MAP3K7 (NM_145331.2) exons 4, 5, and 6, and truncated introns 4 and 5; and TTI1 minigene containing TTI1 (NM_001303457.2) exons 3, 4, and 5, and truncated introns 3 and 4. The –38 A>G ORAI2 and –35 A>G TTI1 mutant minigenes were generated by site-directed mutagenesis from the ORAI2 and TTI1 minigenes, respectively.
For overexpression minigene experiments, minigene DNA (100 ng) and expression plasmid DNA (1 μg) were cotransfected to HEK293T cells in a six-well plate using Lipofectamine 2000 (Thermo Fisher Scientific). At 48 h post transfection, total RNA was extracted from the transfected HEK293T cells using TRIzol (Thermo Fisher Scientific), followed by treatment with DNase I (New England Biolabs). For knockdown minigene experiments, each siRNA was mixed with DharmaFECT 1 reagent (Dharmacon), and minigene DNA (100 ng) was mixed with Lipofectamine 2000 (Thermo Fisher Scientific). Then these two mixtures were cotransfected to HEK293T cells in a six-well plate. For each transfection, a final concentration of 10 nM siRNA was used. After 24 h, a second-round siRNA transfection was performed using DharmaFECT 1 reagent (Dharmacon). At 24 h post second-round transfection (i.e., at 48 h post initial transfection), total RNA was extracted from the transfected HEK293T cells using TRIzol (Thermo Fisher Scientific), followed by treatment with DNase I (New England Biolabs).
Reverse transcription was carried out with 2 μg DNase-treated total RNA using 0.3 μl Maxima Reverse Transcriptase (Thermo Scientific), 50 pmol oligo-dT primer, and 0.2 pmol vector-specific reverse primer (5′-TAGAAGGCACAGTCGAGG-3′), followed by PCR containing [α−32P] dCTP. PCR products were resolved by 6% non-denaturing PAGE. The gel was dried and exposed to a phosphor screen, which was then scanned by a Typhoon FLA 7000 imager (GE Healthcare). Primers used in the PCR reactions were: vector-specific forward primer, 5′-TAATACGACTCACTATAGGGAG-3′; ORAI2 reverse, 5′-CTCTCCATCCCATCTCCTTG-3′; TTI1 reverse, 5′-ACATCTGGACGGGTGTCATT-3′; ZNF91 reverse, 5′-CTCTGCTCTGGCCAAAAGTC-3′; and MAP3K7 reverse, 5′-TCCCTGTGAATTAGCGCTTT-3′.
CRISPR/Cas9 Genome Editing
The SF3B1 K700E mutation (or synonymous mutations for the WT control) was knocked into K562 cells using CRISPR/Cas9 technology as previously described (Zhang et al., 2015). The human SF3B1 CRISPR guide RNA was 5′-TGGATGAGCAGCAGAAAGTTcgg-3′, and the single-stranded oligodeoxynucleotides (ssODNs, Integrated DNA Technologies) for WT and K700E SF3B1 were 5′-AATGTTGGGGCATAGTTAAAACCTGTGTTTGGTTTTGTAGGTCTTGTGGATGAGCAGCAGAAAGTGCGCACCATCAGTGCTTTGGCCATTGCTGCCTTGGCTGAAGCAGCAACTCCTTATGGTATCGAAT-3′ and 5′-AATGTTGGGGCATAGTTAAAACCTGTGTTTGGTTTTGTAGGTCTTGTGGATGAGCAGCAGGAAGTGCGCACCATCAGTGCTTTGGCCATTGCTGCCTTGGCTGAAGCAGCAACTCCTTATGGTATCGAAT-3′, respectively. Mutation knock-in was confirmed using PCR amplification of genomic DNA (forward primer: 5′-GTTGATATATTGAGAGAATCTGGATG3′; and reverse primer: 5′-AAATCAAAAGGTAATTGGTGGA-3′) and DNA sequencing. The resulting knock-in K562 cell clones were subject to a second-round CRISPR/Cas9 procedure, which introduced the His6-FLAG tandem tags into the N-terminus of the knock-in SF3B1 allele using the CRISPR guide RNA (5′-GTGTGTTCGAGTGGACAAAAtgg-3′) and the ssODN (5′-CTATTTTTCTCCGTGGCGGCGGCGACGAGCGGAAGTTCTTGGGAGCGCCAGTTCCGTCTGTGTGTTCGAGTGGACAAAATGCATCACCATCACCATCACGACTACAAGGACGACGATGACAAGGCGAAGATCGCCAAGACTCACGAAGGTAAGCGGTCTTTCCCTGCTTACGTGTTTTCTTCGTTGCTAGCCTAAT-3′). Confirmation of tag knock-in was performed by PCR of genomic DNA (forward primer: 5′-GATTTCCTGGGTCACCACGC-3′; and reverse primer: 5′-GTGTAAGAGGAGGACGCCATT-3′), followed by DNA sequencing. To select for clones that have the tags and the K700E mutation (or synonymous mutations for the WT control) on the same SF3B1 allele, total RNA was isolated from positive clones with tag knock-in using TRIzol (Thermo Fisher Scientific), and then reverse transcribed using Superscript III (Invitrogen), followed by PCR using Phusion DNA polymerase (New England Biolabs) with the following primer set: 5′-TCACGACTACAAGGACGACG-3′ and 5′-ACCCTTTCCTCTGTGTTGGC-3′. PCR products were sequenced to confirm the presence of the tags and the K700E mutation (or synonymous mutations for the WT control) on the same SF3B1 allele using sequencing primers 5′-CATAATAACCTGTAGAATCGAG-3′ and 5′-GCTGTGTGCAAAAGCAAGAA-3′, respectively.
Large-Scale Affinity Purification of SF3B1-Associated Proteins
A total of 200 million CRISPR/Cas9 engineered K562 cells with His6-FLAG-tagged WT or K700E SF3B1 were harvested by centrifugation at 2,000 × g for 5 min. Cells were lysed with 6.5 ml of a lysis buffer containing 30 mM Tris-Cl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 10 mM Sodium Orthovanadate, 10 mM Sodium Fluoride, protease inhibitor cocktail (Roche), and PhosSTOP (Roche), in the presence of 1,000 μg RNase A. After incubation at 4°C for 20 min and centrifugation at 21,130 × g for 10 min, cell extracts (6 ml of the supernatant) were incubated with 100 μl (0.5 μg/μl) anti-DYKDDDDK monoclonal antibody (GenScript, A00187) at 4 °C for 30 min, followed by precipitation at 4 °C for 3 h with 250 μl Pierce ProteinA/G Magnetic Beads (Thermo Fisher Scientific). After four washes, each with 1 ml Wash Buffer I (30 mM Tris-Cl, pH 7.4, 150 mM NaCl, and 0.5% Triton X-100), proteins were eluted from the magnetic beads using four iterations of elution, each by incubation at 4 °C for 30 min with 300 μl Elution Buffer (30 mM Tris-Cl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 3 mM Imidazole, and 750 ng/μl 3X FLAG peptide). The four eluates were then combined (1.2 ml in total) and subject to a second-round of affinity purification by precipitation at 4 °C for 3 h with 100 μl TALON Superflow Metal Affinity Resin (Clontech). After four washes, each with 1 ml Wash Buffer II (30 mM Tris-Cl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, and 3 mM Imidazole), proteins were eluted from the TALON cobalt beads using SDS loading buffer. A large aliquot of the eluted proteins was partially resolved in an SDS-polyacrylamide gel and stained with QC Colloidal Coomassie Stain (Bio-Rad), followed by mass spectrometry (Taplin Mass Spectrometry Facility at Harvard University). Two smaller aliquots of the eluted proteins were resolved in two SDS-polyacrylamide gels, one for silver staining and the other for western blotting.
Small-Scale Affinity Purification of SF3B1-Associated Proteins
The small-scale protocol was applied to both K562 suspension cells and HEK293T adherent cells. For K562 cells, 20 million CRISPR/Cas9 engineered K562 cells with His6-FLAG-tagged WT or K700E SF3B1 were used to purify SF3B1-associated proteins. For HEK293T cells, 1.5 million cells per plate were first seeded in 10-cm plates. Then on the next day, 4 μg of expression plasmid DNA or, for co-expression, a mixture of two plasmids (2 μg each) were transfected to each plate of cells using Lipofectamine 2000 (Thermo Fisher Scientific). At 24 h post transfection, cells were trypsinized and re-seeded evenly in four 10-cm plates. After 48 h of growth (i.e., at 72 h post initial transfection), cells were harvested for affinity purification of SF3B1-associated proteins.
Cells were lysed with 1.3 ml of a lysis buffer containing 30 mM Tris-Cl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 10 mM Sodium Orthovanadate, 10 mM Sodium Fluoride, protease inhibitor cocktail (Roche), and PhosSTOP (Roche), in the presence of 200 μg RNase A. After incubation at 4°C for 20 min and centrifugation at 21,130 × g for 10 min, cell extracts (1.2 ml of the supernatant) were incubated with 10 μl (0.5 μg/μl) anti-DYKDDDDK monoclonal antibody (GenScript, A00187) at 4 °C for 30 min, followed by precipitation at 4 °C for 3 h with 50 μl Pierce ProteinA/G Magnetic Beads (Thermo Fisher Scientific). After four washes, each with 1 ml Wash Buffer I (30 mM Tris-Cl, pH 7.4, 150 mM NaCl, and 0.5% Triton X-100), proteins were eluted from the magnetic beads using four iterations of elution, each by incubation at 4 °C for 30 min with 300 μl Elution Buffer (30 mM Tris-Cl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 3 mM Imidazole, and 150 ng/μl 3X FLAG peptide). The four eluates were then combined (1.2 ml in total) and subject to a second-round of affinity purification by precipitation at 4 °C for 3 h with 40 μl TALON Superflow Metal Affinity Resin (Clontech). After four washes, each with 1 ml Wash Buffer II (30 mM Tris-Cl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, and 3 mM Imidazole), proteins were eluted from the TALON cobalt beads using SDS loading buffer. Two equal aliquots of the eluted proteins were resolved in two SDS-polyacrylamide gels, one for silver staining and the other for western blotting.
GST Coimmunoprecipitation
GST, and GST-tagged SUGP1 (amino acids 366–401) were cloned into p3xFLAG-CMV-14 (Sigma), and the W387A mutant construct of GST-tagged SUGP1 (amino acids 366–401) was generated by site-directed mutagenesis. HEK293T cells were seeded in 10-cm plates, with 1.5 million cells per plate. On the next day, the cells were transfected with 4 μg plasmid DNA expressing GST, GST-tagged SUGP1 (amino acids 366–401), or the W387A mutant of GST-tagged SUGP1 (amino acids 366–401). At 48 h post transfection, cells were lysed with brief periods of sonication in a lysis buffer containing 50 mM Tris-Cl (pH 7.4), 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, and protease inhibitor cocktail (Roche), in the presence of 100 μg RNase A. After incubation at 4°C for 15 min and centrifugation at 21,130 × g for 15 min, cell extracts were incubated with 20 μl Glutathione Sepharose 4B beads (GE Healthcare) at 4 °C overnight. After four washes with a wash buffer (50 mM Tris-Cl, pH 7.4, 100 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P-40), proteins were eluted from the beads with SDS loading buffer, and then subject to western blotting analysis.
Branch Point Mapping
Total RNA was extracted from CRISPR/Cas9 engineered K562 cells using TRIzol (Thermo Fisher Scientific), and 2 μg total RNA was reverse transcribed using Maxima Reverse Transcriptase (Thermo Scientific) and random hexamer primers. The synthesized cDNA was then used as template in nested PCR reactions (i.e., two successive PCR reactions using outer primers and inner primers). Primers used in the nested PCRs were: ORAI2 outer forward, 5′TCTCACCCCTCCAGTCTCTG-3′; ORAI2 outer reverse, 5′-AGCCTACAGTCGTCCCCTTC-3′; ORAI2 inner forward, 5′-GACCTGCATGCTCAATTCTG-3′; ORAI2 inner reverse, 5′AGCGTCCCCGGTCCAATTCC-3′; TTI1 outer forward, 5′-GCCGTGTTCACACCACTGTA-3′; TTI1 outer reverse, 5′-CCTTTCTGTAATGATGTTTCACCA-3′; TTI1 inner forward, 5′-CAGTCCATTTGTGCTGCTGT-3′; and TTI1 inner reverse, 5′CCAGCTAAGCCAAAATAAACAA-3′. PCR products were resolved in a 1.5% agarose gel, followed by gel extraction and sequencing. The BP was identified as the transition nucleotide from the downstream intronic sequence to the upstream 5′-end of the intron.
Recombinant Protein Purification and In Vitro Protein-Protein Interaction
GST, and GST-tagged SUGP1 (amino acids 366–401) were cloned in pGEX-4T-3 (GE Healthcare), and the W387A mutant construct of GST-tagged SUGP1 (amino acids 366–401) was generated by site-directed mutagenesis. Expression of recombinant proteins were induced in E. coli Rosetta cells at 10°C for 24 h by 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Bacterial cells were lysed using a lysis buffer containing 50 mM Tris-Cl (pH 7.4), 150 mM NaCl, and 0.5% Triton X-100. Recombinant proteins were then purified using Glutathione Sepharose 4B beads (GE Healthcare). Protein concentrations were determined by measuring their optical density absorbance at 280 nm.
His6-FLAG-tagged U2AF2 (amino acids 367–475) was cloned in pET26b (Novagen). Protein expression was induced in E. coli Rosetta cells at 10°C for 24 h by 0.05 mM IPTG. Bacterial cells were lysed using a lysis buffer containing 50 mM Tris-Cl (pH 7.4), 150 mM NaCl, and 0.5% Triton X-100, as well as 10 μg/ml RNase A. The recombinant protein was then purified with nickel affinity chromatography using Ni-NTA agarose beads (QIAGEN). Protein concentration was determined by measuring the optical density absorbance at 280 nm.
To characterize the interaction between SUGP1 and U2AF2, 100 pmol purified His6-FLAG-tagged U2AF2 (amino acids 367–475) was mixed with 100 pmol purified GST, GST-tagged SUGP1 (amino acids 366–401), or the W387A mutant of GST-tagged SUGP1 (amino acids 366–401) in a total volume of 650 μl with a binding buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, and 0.5% Triton X-100). With 50 μl of the mixture saved as an input control, the remaining 600 μl was incubated at 4°C for 30 min, followed by addition of 10 μl (0.5 μg/μl) anti-DYKDDDDK monoclonal antibody (GenScript, A00187) and incubation at 4°C for 30 min. The protein-antibody mixture was then incubated with 50 μl Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific) at 4°C overnight. After four washes with the binding buffer, proteins were eluted from the magnetic beads with 30 μl (1 μg/μl) 3X FLAG peptide. Two equal aliquots of the eluted proteins, as well as the input controls (with a relative amount of 2% by adjusting the loading volume), were resolved in two SDS-polyacrylamide gels, one for silver staining and the other for western blotting.
Quantification and Statistical Analysis
Radioactive signals of RT-PCR products in Figure 5C were quantified using ImageQuant (Molecular Dynamics), from which PSI values of cryptic 3′ss were calculated (Figure 5D). The data represent the mean ± SD of three independent experiments. Unpaired, two-tailed, and unequal variance t tests were performed using Microsoft Excel. Normalized gene expression data in Figure S6 were downloaded from the UCSC Xena platform (https://xenabrowser.net/). Unpaired, two-tailed, and unequal variance t tests were performed using the t.test function in R software (R Core Team, 2013).
Data and Software Availability
RNA-sequencing data reported in this paper were deposited in the Gene Expression Omnibus (GEO) database, and the accession number is GEO: GSE128805.
Supplementary Material
Table S1. Cryptic 3′ Splice Sites Differentially Used by Mutant vs. WT SF3B1, Related to Figure 1
Table S2. Mass Spectrometry Results of Proteins Associated with WT or K700E SF3B1, Related to Figure 2
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-SF3B1 | Bethyl Laboratories | Cat# A300-996A, RRID:AB_805834 |
| anti-ACTIN | Sigma | Cat# A2066, RRID:AB_476693 |
| anti-HA rabbit polyclonal | Abm | G166 |
| anti-HA mouse monoclonal | Sigma | Cat# H3663, RRID:AB_262051 |
| anti-DYKDDDDK | GenScript | Cat# A00187, RRID:AB_1720813 |
| anti-SUGP1 | Bethyl Laboratories | A304-675A-M |
| anti-RBM6 | ABclonal | Cat# A10391, RRID:AB_2757939 |
| anti-U2AF2 | Sigma | Cat# U4758, RRID:AB_262122 |
| anti-GST | Invitrogen | A5800 |
| Donkey anti-Rabbit IgG | LI-COR | Cat# 926-68073, RRID:AB_10954442 |
| Goat anti-Mouse IgG | LI-COR | Cat# 926-32210, RRID:AB_621842 |
| Biological Samples | ||
| Bone marrow samples from MDS patients | This paper | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 2000 | Thermo Fisher Scientific | 11668019 |
| DharmaFECT 1 | Dharmacon | T-2001-03 |
| TRIzol | Thermo Fisher Scientific | 15596018 |
| 3X FLAG peptide | APExBIO | A6001 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 4693159001 |
| PhosSTOP Phosphatase Inhibitor Cocktail | Roche | 4906837001 |
| Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | 88803 |
| TALON Superflow Metal Affinity Resin | Clontech | 635506 |
| Deposited Data | ||
| RNA-sequencing data | This paper | GEO: GSE128805 |
| Experimental Models: Cell Lines | ||
| CRISPR/Cas9 engineered K562 cells | This paper | NA |
| Oligonucleotides | ||
| SF3B1 guide, TGGATGAGCAGCAGAAAGTTcgg | This paper | NA |
| siSUGP1–1 sense, GGAUAACCCAGCAUUUGCAdTdT | This paper | NA |
| siSUGP1–1 antisense, UGCAAAUGCUGGGUUAUCCdTdT | This paper | NA |
| siSUGP1–2 sense, GAAGGUGGCUGAGAUAAGAdTdT | This paper | NA |
| siSUGP1–2 antisense, UCUUAUCUCAGCCACCUUCdTdT | This paper | NA |
| Recombinant DNA | ||
| p3xFLAG-CMV-14-HA-SF3B1 | This paper | NA |
| p3xFLAG-CMV-14-His6-FLAG-SF3B1 | This paper | NA |
| p3xFLAG-CMV-14-HA-SUGP1 | This paper | NA |
| pcDNA3-ORAI2-mini | This paper | NA |
| pcDNA3-ORAI2-mini −38 A>G | This paper | NA |
| pcDNA3-TTI1-mini | This paper | NA |
| pcDNA3-TTI1-mini −35 A>G | This paper | NA |
| Software and Algorithms | ||
| deboever-sf3b1-2015 | DeBoever et al., 2015 | https://github.com/cdeboever3/deboever-sf3b1-2015 |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| R software | R Core Team, 2013 | https://www.R-project.org/ |
| DAVID | Huang da et al., 2009a, b | https://david.ncifcrf.gov/ |
| REVIGO | Supek et al., 2011 | http://revigo.irb.hr/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| ImageQuant | Molecular Dynamics | NA |
| Prism | GraphPad | NA |
| Excel | Microsoft | NA |
| Illustrator | Adobe | NA |
Highlights.
SF3B1 disease mutations disrupt SF3B1 interaction with SUGP1 in the spliceosome
SUGP1 depletion completely recapitulates mutant SF3B1 splicing defects
SUGP1 G-patch domain mutant also reproduces mutant SF3B1 splicing defects
SUGP1 overexpression partially rescues the mutant SF3B1 spliceosome
ACKNOWLEDGEMENTS
We thank Kaori Yamazaki and Ritam Neupane for technical help in this study. This work was supported by NIH grant R35 GM118136 (to J.L.M.), an EvansMDS discovery research grant (to J.L.M.), Partnership for Cures (to A.R.), and NCI grant U54 CA193313 (to R.R.). We also used the resources of the Herbert Irving Comprehensive Cancer Center Flow Cytometry Shared Resources funded in part through Center Grant P30CA013696.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aakre CD, Herrou J, Phung TN, Perchuk BS, Crosson S, and Laub MT (2015). Evolving new protein-protein interaction specificity through promiscuous intermediates. Cell 163, 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. (2016). Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun 7, 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei SA, Sijbesma E, Hann M, Davis J, O’Mahony G, Perry MWD, Karawajczyk A, Eickhoff J, Brunsveld L, Doveston RG, et al. (2017). Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discov 12, 925–940. [DOI] [PubMed] [Google Scholar]
- Aravind L, and Koonin EV (1999). G-patch: a new conserved domain in eukaryotic RNA-processing proteins and type D retroviral polyproteins. Trends Biochem. Sci 24, 342–344. [DOI] [PubMed] [Google Scholar]
- Barreyro L, Chlon TM, and Starczynowski DT (2018). Chronic immune response dysregulation in MDS pathogenesis. Blood 132, 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund JA, Abovich N, and Rosbash M (1998). A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev. 12, 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. (2012). Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu C, Schmitzova J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Luhrmann R, and Pena V (2016). Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 64, 307–319. [DOI] [PubMed] [Google Scholar]
- Crisci A, Raleff F, Bagdiul I, Raabe M, Urlaub H, Rain JC, and Kramer A (2015). Mammalian splicing factor SF1 interacts with SURP domains of U2 snRNP-associated proteins. Nucleic Acids Res. 43, 10456–10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispino JD, Lodish MB, MacKay JP, and Orkin SH (1999). Use of altered specificity mutants to probe a specific protein-protein interaction in differentiation: the GATA-1:FOG complex. Mol. Cell 3, 219–228. [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. (2015). Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 13, 1033–1045. [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, and Frazer KA (2015). Transcriptome sequencing reveals potential mechanism of cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol 11, e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doveston RG, Kuusk A, Andrei SA, Leysen S, Cao Q, Castaldi MP, Hendricks A, Brunsveld L, Chen H, Boyd H, et al. (2017). Small-molecule stabilization of the p53 – 14-3-3 protein-protein interaction. FEBS lett. 591, 2449–2457. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. (2012). Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskens FA, Ramos FJ, Burger H, O’Brien JP, Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ, Baselga J, et al. (2013). Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res 19, 6296–6304. [DOI] [PubMed] [Google Scholar]
- Fleckner J, Zhang M, Valcarcel J, and Green MR (1997). U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 11, 1864–1872. [DOI] [PubMed] [Google Scholar]
- Friedman R (2016). Drug resistance in cancer: molecular evolution and compensatory proliferation. Oncotarget 7, 11746–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K, Masuda A, Matsuura T, and Ohno K (2008). Human branch point consensus sequence is yUnAy. Nucleic Acids Res. 36, 2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Zody MC, Arachchi HM, Berlin A, Gnerre S, Green LM, Lennon N, and Nusbaum C (2009). Closing gaps in the human genome using sequencing by synthesis. Genome Biol. 10, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, and Lander ES (2013). Lessons from the cancer genome. Cell 153, 17–37. [DOI] [PubMed] [Google Scholar]
- Geest CR, and Coffer PJ (2009). MAPK signaling pathways in the regulation of hematopoiesis. J. Leukoc. Biol 86, 237–250. [DOI] [PubMed] [Google Scholar]
- Goldman M, Craft B, Hastie M, Repečka K, Kamath A, McDade F, Rogers D, Brooks AN, Zhu J, and Haussler D (2018). The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv, doi: 10.1101/326470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Feld R, and Reed R (1996). Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Genes Dev. 10, 233–243. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. (2014). Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, and Bowcock AM (2013). Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet 45, 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Luhrmann R, and Stelzl U (2012). Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580. [DOI] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, and Pease LR (1989). Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59. [DOI] [PubMed] [Google Scholar]
- Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O’Brien J, and LoRusso P (2014). A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs 32, 436–444. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, and Bradley RK (2015). U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 25, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesarwani AK, Ramirez O, Gupta AK, Yang X, Murthy T, Minella AC, and Pillai MM (2017). Cancer-associated SF3B1 mutants recognize otherwise inaccessible cryptic 3’ splice sites within RNA secondary structures. Oncogene 36, 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, Chen L, Monterroza DD, Li H, DeKelver RC, et al. (2015). SRSF2 Is Essential for Hematopoiesis, and Its Myelodysplastic Syndrome-Related Mutations Dysregulate Alternative Pre-mRNA Splicing. Mol. Cell. Biol 35, 3071–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwasako K, He F, Inoue M, Tanaka A, Sugano S, Guntert P, Muto Y, and Yokoyama S (2006). Solution structures of the SURP domains and the subunit-assembly mechanism within the splicing factor SF3a complex in 17S U2 snRNP. Structure 14, 1677–1689. [DOI] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. (2013). Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, and Abdel-Wahab O (2016). Therapeutic targeting of splicing in cancer. Nat. Med 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Gumpper RH, Uddin Y, Schmidt-Krey I, and Luo M (2018). Complementary Mutations in the N and L Proteins for Restoration of Viral RNA Synthesis. J. Virol 92, e01417–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Liu C, Lin X, Sun B, and Su C (2018). PPP2R5A: A multirole protein phosphatase subunit in regulating cancer development. Cancer Lett. 414, 222–229. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Clark MB, Andersen SB, Brunck ME, Haerty W, Crawford J, Taft RJ, Nielsen LK, Dinger ME, and Mattick JS (2015). Genome-wide discovery of human splicing branchpoints. Genome Res. 25, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble JC, Pan ZQ, Prives C, and Manley JL (1987). Splicing of SV40 early pre-mRNA to large T and small t mRNAs utilizes different patterns of lariat branch sites. Cell 50, 227–236. [DOI] [PubMed] [Google Scholar]
- Padgett RA, Konarska MM, Aebi M, Hornig H, Weissmann C, and Sharp PA (1985). Nonconsensus branch-site sequences in the in vitro splicing of transcripts of mutant rabbit beta-globin genes. Proc. Natl. Acad. Sci. U.S.A. 82, 8349–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med 365, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. (2013). Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea W, Schroeder KT, Bryant AN, and Greenbaum NL (2016). Interaction between the Spliceosomal Pre-mRNA Branch Site and U2 snRNP Protein p14. Biochemistry 55, 629–632. [DOI] [PubMed] [Google Scholar]
- R Core Team. (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: URL https://www.R-project.org/. [Google Scholar]
- Robert-Paganin J, Rety S, and Leulliot N (2015). Regulation of DEAH/RHA helicases by G-patch proteins. BioMed Res. Int 2015, 931857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby SW, Chang TH, and Abelson J (1993). Four yeast spliceosomal proteins (PRP5, PRP9, PRP11, and PRP21) interact to promote U2 snRNP binding to pre-mRNA. Genes Dev. 7, 1909–1925. [DOI] [PubMed] [Google Scholar]
- Ruskin B, Greene JM, and Green MR (1985). Cryptic branch point activation allows accurate in vitro splicing of human beta-globin intron mutants. Cell 41, 833–844. [DOI] [PubMed] [Google Scholar]
- Ruskin B, Zamore PD, and Green MR (1988). A factor, U2AF, is required for U2 snRNP binding and splicing complex assembly. Cell 52, 207–219. [DOI] [PubMed] [Google Scholar]
- Sampson ND, and Hewitt JE (2003). SF4 and SFRS14, two related putative splicing factors on human chromosome 19p13.11. Gene 305, 91–100. [DOI] [PubMed] [Google Scholar]
- Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, Smith PG, Cancer Genome Atlas Research, N., Buonamici S, and Yu L (2018a). Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 23, 282–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018b). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P, Gregorovic G, Sprangers R, Stier G, Rhani Z, Kramer A, and Sattler M (2003). Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol. Cell 11, 965–976. [DOI] [PubMed] [Google Scholar]
- Shiozawa Y, Malcovati L, Galli A, Sato-Otsubo A, Kataoka K, Sato Y, Watatani Y, Suzuki H, Yoshizato T, Yoshida K, et al. (2018). Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun 9, 3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. (2015). Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 27, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, and Smuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Rodriguez-Santiago S, Wang J, Pu J, Yuste A, Gupta V, Moldon A, Xu YZ, and Query CC (2016). SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev. 30, 2710–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thickman KR, Swenson MC, Kabogo JM, Gryczynski Z, and Kielkopf CL (2006). Multiple U2AF65 binding sites within SF3b155: thermodynamic and spectroscopic characterization of protein-protein interactions among pre-mRNA splicing factors. J. Mol. Biol 356, 664–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umate P, Tuteja N, and Tuteja R (2011). Genome-wide comprehensive analysis of human helicases. Commun. Integr. Biol 4, 118–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utans U, and Kramer A (1990). Splicing factor SF4 is dispensable for the assembly of a functional splicing complex and participates in the subsequent steps of the splicing reaction. EMBO J. 9, 4119–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, and Luhrmann R (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. [DOI] [PubMed] [Google Scholar]
- Wang C, Chua K, Seghezzi W, Lees E, Gozani O, and Reed R (1998). Phosphorylation of spliceosomal protein SAP 155 coupled with splicing catalysis. Genes Dev. 12, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, Hergert S, Yin S, Freeman SS, Levin JZ, et al. (2016). Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 30, 750–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Guo M, Hu L, Cai J, Zeng Y, Luo J, Shu Z, Li W, and Huang Z (2012). The zinc finger protein ZNF268 is overexpressed in human cervical cancer and contributes to tumorigenesis via enhancing NF-kappaB signaling. J. Biol. Chem. 287, 42856–42866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, and Luhrmann R (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol 3, a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, and Query CC (2007). Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol. Cell 28, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, and Ogawa S (2014). Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA 5, 445–459. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, and Manley JL (2015). Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. U.S.A 112, E4726–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Cryptic 3′ Splice Sites Differentially Used by Mutant vs. WT SF3B1, Related to Figure 1
Table S2. Mass Spectrometry Results of Proteins Associated with WT or K700E SF3B1, Related to Figure 2
Data Availability Statement
RNA-sequencing data reported in this paper were deposited in the Gene Expression Omnibus (GEO) database, and the accession number is GEO: GSE128805.