

Abstract
Pineoblastoma is a rare embryonal tumor of childhood that is conventionally treated with high-dose craniospinal irradiation (CSI). Multi-dimensional molecular evaluation of pineoblastoma and associated intertumoral heterogeneity is lacking. Herein, we report outcomes and molecular features of children with pineoblastoma from two multi-center, risk-adapted trials (SJMB03 for patients ≥3 years; SJYC07 for patients <3 years) complemented by a non-protocol institutional cohort. The clinical cohort consisted of 58 patients with histologically diagnosed pineoblastoma (SJMB03=30; SJYC07=12; non-protocol=16, including 12 managed with SJMB03-like therapy). The SJMB03 protocol comprised risk-adapted CSI (average-risk=23.4 Gy, high-risk=36 Gy) with radiation boost to the primary site and adjuvant chemotherapy. The SJYC07 protocol consisted of induction chemotherapy, consolidation with focal radiation (intermediate-risk) or chemotherapy (high-risk), and metronomic maintenance therapy. The molecular cohort comprised 43 pineal parenchymal tumors profiled by DNA methylation array (n=43), whole-exome sequencing (n=26), and RNA-sequencing (n=16). Respective 5-year progression-free survival rates for patients with average-risk or high-risk disease on SJMB03 or SJMB03-like therapy were 100% and 56.5±10.3% (P=0.007); respective 2-year progression-free survival rates for those with intermediate-risk or high-risk disease on SJYC07 were 14.3±13.2% and 0% (P=0.375). Of patients with average-risk disease treated with SJMB03/SJMB03-like therapy, 17/18 survived without progression. DNA-methylation analysis revealed four clinically relevant pineoblastoma subgroups: PB-A, PB-B, PB-B–like, and PB-FOXR2. Pineoblastoma subgroups differed in age at diagnosis, propensity for metastasis, cytogenetics, and clinical outcomes. Alterations in the miRNA-processing pathway genes DICER1, DROSHA, and DGCR8 were recurrent and mutually exclusive in PB-B and PB-B–like subgroups; PB-FOXR2 samples universally overexpressed the FOXR2 proto-oncogene. Our findings suggest superior outcome amongst older children with average-risk pineoblastoma treated with reduced-dose CSI. The identification of biologically and clinically distinct pineoblastoma subgroups warrants consideration of future molecularly-driven treatment protocols for this rare pediatric brain tumor entity.
Keywords: Pineoblastoma, clinical trial, molecular subgroups, DICER1, microRNA processing, FOXR2
INTRODUCTION
Pineoblastoma (PB) is a malignant embryonal neoplasm arising from the pineal gland, with predilection for onset during the first two decades of life [31,20]. Given the rarity of these tumors, literature on PB is scarce and mostly limited to retrospective cohorts in which patients were collectively managed with other supratentorial primitive neuroectodermal tumors (sPNETs) [11,10,23,8,14,26,28,12,4]. Most children with PB beyond infancy receive high-dose (36 Gy) craniospinal irradiation (CSI) with multi-modal CNS embryonal tumor-type chemotherapy, regardless of disease extent [28,26,4,10,14]. Older children with PB have a more favorable prognosis than do their younger counterparts, but the impact of clinical characteristics beyond age at diagnosis remains unclear [11,10,23,8,14,26,28,12].
Recent application of genome-wide DNA methylation profiling has augmented CNS tumor classification and histologic diagnosis [37,15,39,2,33,20]. PB is epigenetically distinct from other embryonal CNS tumors, with its molecular identity further substantiated by the discovery of presumed driver alterations [16,30,7,34,2]. Nevertheless, intertumoral heterogeneity among PBs has not been comprehensively evaluated through multi-omic studies, nor have genetic events been overlaid with clinical and demographic features. St. Jude Children’s Research Hospital recently led two prospective, multi-institutional brain tumor trials that enrolled patients with PB. SJMB03 was a phase III trial of patients aged ≥3 years with risk-adapted CSI, wherein reduced-dose CSI (23.4 Gy) was administered to treat PB with average-risk (AR) features. In parallel, SJYC07 was a phase II, risk-adapted trial for children <3 years [29,41]. Herein, we summarize the molecular features and clinical outcomes of patients from these trials and a complementary institutional cohort.
PATIENTS AND METHODS
Design and Study Cohort
SJMB03 was a multi-institutional, phase III study (NCT00085202) accepting patients aged 3–21 years with newly diagnosed CNS embryonal tumors that was conducted between June 24, 2003, and March 7, 2013. SJYC07 was a multi-institutional Phase II, risk-adapted study (NCT00602667) that enrolled children younger than 3 years with newly diagnosed brain tumors between November 9, 2007, and April 19, 2017. Histologically diagnosed PB patients treated on these trials are included in the current report (Fig. 1a). The database of St. Jude Children’s Research Hospital was then reviewed for patients with pineal parenchymal tumors managed off-protocol between January 1, 2003, and June 30, 2018, constituting our non-protocol cohort. SJMB03 and SJYC07 were approved by the Institutional Review Board with written informed consent obtained from patients and families; the review of non-protocol patients was approved with the need for consent waived.
Fig. 1.
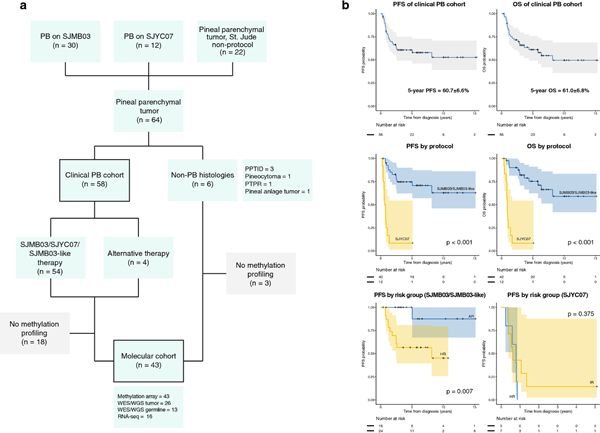
Cohort compositions and key clinical outcomes
(a) Composition of the clinical and molecular cohorts. Patients with histologically diagnosed pineoblastoma (PB) comprised the clinical cohort; patients whose tumors were profiled on methylation array (PB=40, pineal parenchymal tumor of intermediate differentiation [PPTID]=1, pinealocytoma=1, pineal anlage tumor=1) comprised the molecular cohort.
(b) Progression-free survival (PFS) and overall survival (OS) of the entire clinical PB cohort (top) or stratified by treatment protocol (middle) and PFS according to risk strata for patients on SJMB03/SJMB03-like therapy and SJYC07 (bottom).
PTPR, papillary tumor of pineal region; WES, whole exome sequencing; WGS, whole genome sequencing
Treatment Strategies for SJMB03 and SJYC07
Patients were enrolled on SJMB03 and SJYC07 after maximal safe surgical resection (Supplementary Fig. S1, online resource). Baseline evaluations with MRI of the brain and spine as well as cerebrospinal fluid cytology were obtained. Extent of resection and residual disease, if any, was determined by early post-operative imaging. SJMB03 patients were stratified into AR or high-risk (HR) arms, with HR criteria including presence of metastasis (M+) and/or bulky residual disease >1.5 cm2 at the primary site (R+). Risk-adapted CSI (AR=23.4 Gy; HR=36 Gy) and focal boost to the primary site were delivered, followed by four sequential cycles of high-dose chemotherapy (cisplatin/cyclophosphamide/vincristine) with autologous stem cell rescue. The study approach for SJYC07 has been reported [29,41]. SJYC07 patients with localized disease were enrolled on the intermediate-risk (IR) arm, and M+ patients were enrolled on the HR arm. Four cycles of induction chemotherapy (high-dose methotrexate/cisplatin/cyclophosphamide/vincristine; vinblastine added for HR disease) were given, followed by consolidation with focal radiation (IR) or two cycles of cyclophosphamide and topotecan (HR), and metronomic maintenance therapy. IR patients younger than 12 months at the time of consolidation received low-risk arm consolidation to defer radiation. Second-look surgery before the start of consolidation was permitted, and patients were allowed to remain on protocol in case of progression during the induction phase. In both trials, serial hearing evaluations were performed to monitor for ototoxicity, and other adverse events were graded according to Common Terminology Criteria for Adverse Events v3.0, capturing events Grade 3 or above [40]. Adverse effects in patients with PB were similar to those from larger published medulloblastoma and ependymoma cohorts and are not detailed here [29,41,6].
Treatment for non-protocol cohort
Among the non-protocol cohort with pineal parenchymal tumors, 16/22 patients had histologically diagnosed PB (Fig. 1a). Eleven patients with PB were treated when the corresponding studies were not actively recruiting (eight patients >3 years; three patients <3 years); four patients older than three years received chemotherapy before CSI to facilitate second-look surgery and one received CSI only due to significant neurological deficits. Patients were considered to have received an “SJMB03-like” non-protocol regimen (n=12) if they were risk-stratified according to the SJMB03 protocol and treated by CSI followed by cisplatin/cyclophosphamide/vincristine. Chemotherapy (carboplatin/cyclophosphamide/etoposide or cisplatin/cyclophosphamide/vincristine for two cycles) before CSI to facilitate staged resection were allowed in this designation. Non-SJMB03-like regimens (n=4) included CSI only (n=2), and young-children type induction regimen (cisplatin/cyclophosphamide/vincristine/etoposide with intrathecal mafosfamide) followed by focal or CSI (n=2). Patients with non-PB diagnoses (n=6) received individualized treatment and are reported separately (Supplementary Table S1, online resource).
Genome-wide DNA methylation profiling and next-generation sequencing
DNA and RNA were extracted from formalin-fixed paraffin-embedded or fresh-frozen tumor samples (Supplementary Table S3, online resource). Genome-wide DNA methylation profiling was performed on 43 tumor samples (PB, n=40; non-PB histology, n=3) using the Infinium Methylation EPIC BeadChip array as described previously, comprising our molecular cohort [41,29]. Epigenomic classification was conducted on the molecular cohort, together with reference profiles obtained from a published brain tumor dataset, and using the MolecularNeuropathology (MNP) brain tumor classifier (www.molecularneuropathology.org) [2]. Whole-exome sequencing (WES, n=24), whole-genome sequencing (WGS, n=2), and RNA-sequencing (RNA-seq, n=16) data were generated for samples with adequate tumor DNA/RNA. Germline (blood) DNA from 13 patients was available for paired sequencing. Details on the experimental pipelines conducted above are included in Supplementary Methods (Supplementary Tables S4 and S5, S5 online resource).
Survival Analyses
The date of diagnosis was defined as the date of first biopsy or resection. Overall survival (OS) was defined as the duration between the date of diagnosis and date of either death from any cause or last follow-up; progression-free survival (PFS) was defined as the duration between the date of diagnosis and date of either progression, relapse, death from any cause, or last follow-up. Survival comparisons were performed via log-rank testing. Fisher’s exact test was used to compare categorical variables. Statistical analyses were performed using R v3.6.0 (www.R-project.org).
RESULTS
Demographics, clinical characteristics, and risk-stratification
Fifty-eight patients with histologically diagnosed PB were included in our study (median age at diagnosis, 6.2 years): 30 from SJMB03, 12 from SJYC07, and 16 from the non-protocol cohort (including 12 treated with SJMB03-like regimens, Fig. 1a). Baseline risk features and stratification are detailed in Table 1. Twenty-six (45%) patients had M+ disease at diagnosis. Gross-total resection (GTR) was achieved in 22 (38%) patients, subtotal resection (STR) in 16 (28%), and biopsy-only in 20 (34%). Twelve patients (21%) underwent staged resection facilitated by chemotherapy. Of patients who received SJMB03/SJMB03-like therapy, 22/42 (52%) had R+ disease. Patients with non-metastatic (M0) disease were more likely to have received GTR (20/32) than were those with M+ disease (2/26, P<0.001). Forty-six patients (70%) required CSF diversion. Three patients had DICER1 syndrome (molecular cohort=1, clinical cohort=2); and one patient (clinical cohort) had DiGeorge syndrome diagnosed by 22q11.2 fluorescence in situ hybridization.
Table 1.
Demographic, clinical, and treatment characteristics of the clinical pineoblastoma cohort
| Entire Clinical Cohort | SJMB03 | SJYC07 | Non-protocol SJMB03-like | Non-protocol Others | ||
|---|---|---|---|---|---|---|
| N | % | N | N | N | N | |
| Total | 58 | 100 | 30 | 12 | 12 | 4 |
| Sex | ||||||
| Male | 31 | 53 | 14 | 8 | 7 | 2 |
| Female | 27 | 47 | 16 | 4 | 5 | 2 |
| Age (mean in years [range]) | 6.2 (0.4– 21.6) | - | 8.48 (3.1–20.4) | 1.2 (0.4–2.6) | 7.4 (2.9–17.2) | 3.6 (1.9–21.6) |
| Risk group | ||||||
| AR/IR | 26 | 45 | 11 | 7 | 7 | 1 |
| HR | 32 | 55 | 19 | 5 | 5 | 3 |
| Metastasis | ||||||
| No (M0) | 32 | 55 | 16 | 7 | 8 | 1 |
| Yes (M+) | 26 | 45 | 14 | 5 | 4 | 3 |
| Residual tumor (SJMB03/SJMB0 3-like only) | ||||||
| No (R0) | 22 | 52 | 13 | - | 9 | - |
| Yes (R+) | 20 | 48 | 17 | - | 3 | - |
| Radiotherapy | ||||||
| CSI | 45 | 78 | 30 | 0 | 12 | 3 |
| Focal RT | 7 | 12 | 0 | 6 | 0 | 1 |
| No RT | 6 | 10 | 0 | 6 | 0 | 0 |
AR, average-risk (SJMB03/SJMB03-like: M0 and R0 disease); Bx, biopsy; CSI, craniospinal irradiation; GTR, gross-total resection; HR, high-risk (SJMB03/SJMB03-like: M+ or R+ disease, SJYC07: M+ disease); IR, intermediate-risk (SJYC07: M0 disease); M0, non-metastatic; M+, metastatic; N, number; R0, residual disease ≤1.5cm2; R+, residual disease >1.5cm2; RT, irradiation; STR, subtotal resection
Treatment outcome and prognostic factors
Median follow-up duration for the entire clinical cohort was 3.6 years (range: 0.4–15.3); for patients surviving without disease, median follow-up was 5.8 years (range: 0.4–15.3). Progression occurred in 22 (38%) patients (distant failure=19, distant and local failure=3), who died of disease; two patients without evidence of disease died of treatment-related complications (sepsis in one patient, and pulmonary fibrosis, potentially the result of cyclophosphamide treatment, in the second patient). Respective 5-year PFS and OS rates of the entire cohort were 60.7±6.6% and 61.0±6.8% (Fig. 1b). PFS and OS for patients on SJMB03 and patients treated off-protocol with SJMB03-like therapy did not differ significantly (P=0.435 and P=0.619, respectively; Fig. S2a, online resource), prompting us to combine these cohorts for subsequent analyses.
The 5-year PFS and OS for patients with AR disease on SJMB03/SJMB03-like therapy were both 100%, and for patients with HR disease, 56.5±10.3% and 60.3±10.3% respectively (PFS, P=0.007, OS, P=0.014; Fig. 1b, Supplementary Table S2, online resource). The respective 2-year PFS/OS for patients with IR or HR disease on SJYC07 were 14.3±13.2%/14.3±13.2% and 0%/0% (PFS, P=0.375, OS P=0.040). Presence of M+ disease was associated with inferior outcome in patients receiving SJMB03/SJBM03-like (PFS, P<0.001, OS, P=0.001) and SJYC07 therapy (OS, P=0.040) (Supplementary Fig. S2, online resource). GTR was significantly associated with superior outcome for patients on SJMB03/SJMB03-like therapy (PFS, P=0.005, OS, P=0.008). Seventeen of 18 (94%) patients with AR disease who received 23.4 Gy CSI on SJMB03/SJMB03-like therapy survived without disease progression (median follow-up for survivors=4.1 years). Two patients treated on the HR arm of SJYC07 died of disease despite receiving CSI as relapse treatment.
Novel PB subgroups
Unsupervised clustering of tumor samples based on DNA methylation profiles suggested considerable heterogeneity within our molecular cohort (Supplementary Fig. S3a, online resource). Integrating results from the MNP classifier and unsupervised clustering with published CNS tumor profiles revealed four PB subgroups (Fig. 2a): PB-A, PB-B, PB-B-like, and PB-FOXR2. Other samples clustered with non-PB entities or control tissues (Fig. 2a, Supplementary Fig. S7–8, online resource).
Fig. 2.
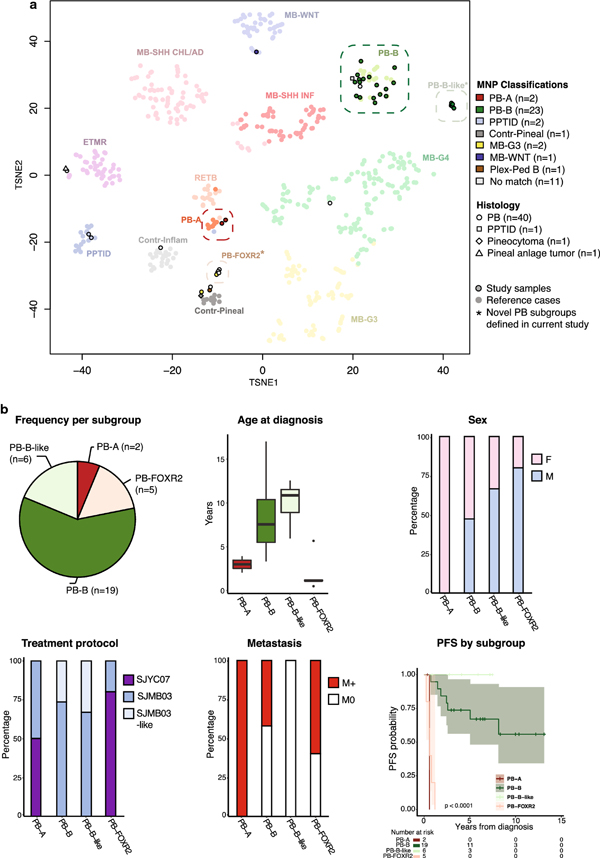
Novel molecular pineoblastoma (PB) subgroups defined by DNA-methylation profiling
(a) t-stochastic neighbor embedding (t-SNE) analysis of pineal parenchymal tumors (histological PB, n= 40; PPTID, n = 1; pineal anlage tumor, n = 1; pineocytoma, n = 1) and relevant reference classes from a published brain tumor dataset by Capper et al.[14] Four main PB subgroups, PB-A, PB-B, PB-B–like, and PB-FOXR2, are apparent, with further heterogeneity observed in the outliers. Study samples are colored by their methylation class assignment by the MolecularNeuropathology (MNP) brain tumor classifier, revealing two previously undescribed subgroups (PB-B–like and PB-FOXR2).
(b) Summary demographic and clinical features of the newly defined PB subgroups. Differences in age at diagnosis, sex, treatment, propensity for metastasis, and progression-free survival are depicted.
Contr-Pineal, control pineal tissue; Contr-Inflamm, control inflammatory tumor; ETMR, embryonal tumor with multilayered rosettes; F, female; M, male; M0, non-metastatic; M+, metastatic; MB-G3, medulloblastoma group 3; MB-G4, medulloblastoma group 4; MB-SHH CHL/AD, medulloblastoma SHH-activated (children and adult); MB-SHH INF, medulloblastoma SHH-activated (infant); MB-WNT, medulloblastoma WNT-activated; PB-A, pineoblastoma group A; PB-B, pineoblastoma group B; PB-B–like, pineoblastoma group B–like; PB-FOXR2, pineoblastoma FOXR2-overexpressed; Plex-Ped B, plexus tumor subclass pediatric B; PPT, pineal parenchymal tumor of intermediate differentiation (MNP classifier); PPTID, pineal parenchymal tumor of intermediate differentiation (histology); RETB, retinoblastoma
Clinical, genomic, and transcriptomic characterization of novel PB subgroups
PFS was significantly different among molecular subgroups of PB (p<0.0001) (Fig. 2b). Tumors assigned to PB-A were from young patients (2.1 and 3.9 years) with M+ disease (Fig. 2b). Chromosome 1q gain and 16q loss were identified in both tumors (Fig. 3a, Supplementary Fig. S3b, online resource). Neither patient had a history of retinoblastoma, and sequencing of one tumor showed no RB1 mutation. Patients with PB-B tumors (n=19) had a median age at diagnosis of 7.6 years (range: 3.3–17.0); eight (42%) had M+ disease, and six experienced disease progression (5-year PFS=73.7±10.1%). Frequent arm-level gains of chromosomes 12 (63%), 7 (47%), and 17 (47%) and losses of 3p (37%) and 16 (37%) were present; focal losses of 5p (16%) and 22q (11%), respectively involving the DROSHA and DGCR8 loci, were also observed. Patients from the PB-B–like subgroup were the oldest in our series (median age=10.9 years, range: 5.9–12.6); all had localized disease without progression (median duration of follow-up=5.0 years). PB-B–like tumors exhibited chromosomal losses involving DICER1 (5/6; 14q) or DROSHA (1/6; focal 5p). Mutually exclusive alterations in microRNA-processing pathway genes DICER1, DROSHA, and DGCR8 were common but restricted to the PB-B and PB-B–like tumors (Fig. 3). Protein-truncating mutations and/or focal deletions of DICER1 (n=5), DROSHA (n=4), and DGCR8 (n=2) occurred in the majority (11/13, 85%) of PB-B and PB-B–like tumors for which NGS data was available. Among PB-B tumors lacking NGS data, two samples had focal 5p deletion involving DROSHA, and another tumor had focal 22q deletion involving DGCR8 (Fig. 3c). No putative driver mutations were identified in samples (5/5 sequenced) from the PB-FOXR2 subgroup.
Fig. 3.
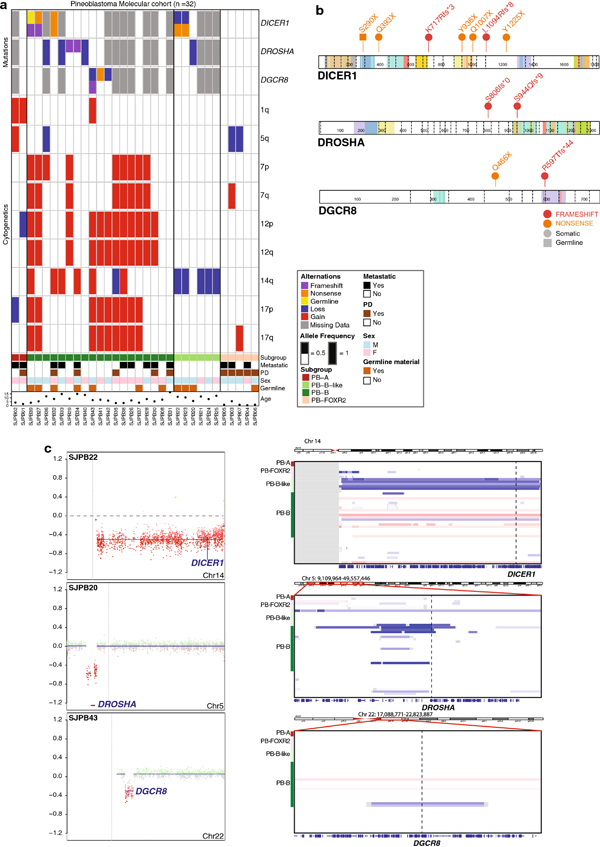
Genomic alterations in molecularly defined pineoblastomas
(a) Oncoprint depicting loss-of-function mutations and/or allelic deletions in miRNA pathway genes DICER1, DROSHA, and DGCR8 in addition to subgroup-specific chromosomal arm–level copy-number alterations, patient demographics, and outcome.
(b) Lollipop plots depicting deleterious somatic and germline mutations in miRNA pathway genes DICER1, DROSHA, and DGCR8.
chr, chromosome; fs, frameshift; X, stop codon.
(c) Three exemplary copy-number plots highlighting 14q loss involving the DICER1 locus, focal 5p loss involving the DROSHA locus, and focal 22q loss involving the DGCR8 locus. Corresponding chromosomal regions of all samples from the molecular cohort, showcasing the restriction of these cytogenetic events to PB-B and PB-B–like subgroups.
Chr, chromosome; F, female; M, male; PD, progressive disease
At the transcriptomic level, unsupervised clustering of PB tumors (PB-B=4, PB-B–like=3, PB-FOXR2=4) yielded subgroups concordant with those determined by methylation profiling (Fig. 4, Supplementary Fig. S5, online resource). Differential expression analysis revealed highly significant FOXR2 overexpression that defined PB-FOXR2 (Fig. 4a–b). Based on DNA methylation profiling, PB-FOXR2 subgroup tumors were distinct from CNS neuroblastoma with FOXR2 activation (CNS NB-FOXR2; Supplementary Fig. S4, online resource). High expression of GABRA5 (gamma-aminobutyric acid A receptor alpha 5 subunit, also highly expressed in MYC-driven medulloblastoma), GNAT1 (rod alpha-transducin), and photoreceptor-specific transcription factors CRX and NRL, together with enrichment in “phototransduction” and “detection of light stimulus” gene sets were also observed in PB-FOXR2 (Supplementary Fig. S6, online resource) [25,5]. PB-B tumors had the highest expression levels of ASMT (acetylserotonin O-methyltransferase) and ADRB1 (beta-1 adrenergic receptor), key markers of pinealocytes, whereas PB-B–like tumors had high expression of NPS (neuropeptide S), a neuropeptide implicated in arousal and anxiolysis [21,45].
Fig. 4.
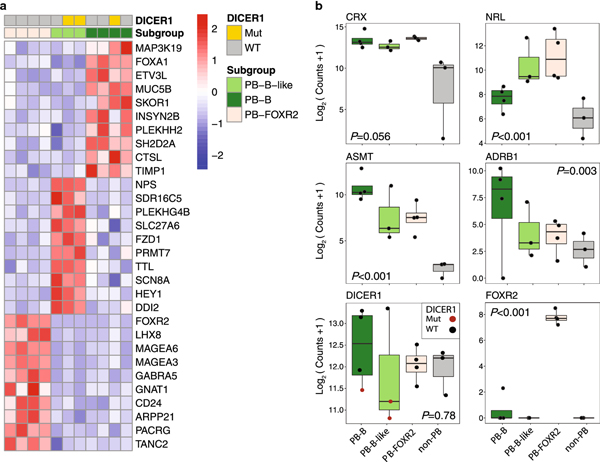
Subgroup-specific expression signature and pathway enrichment in pineoblastoma (PB)
(a) Heatmap of top differentially expressed genes by subgroup. PB-A was excluded here and from subsequent panels because only one expression profile was available.
(b) Expression levels of pinealocyte markers (CRX, NRL, ASMT, ADRB1), DICER1, and FOXR2 among molecularly defined PB subgroups (PB-B, PB-B-like, PB-FOXR2) and outliers (non-PB). P-values were derived from the likelihood ratio test.
Mut, mutant; PB-A, pineoblastoma group A; PB-B, pineoblastoma group B; PB-B–like, pineoblastoma group B–like; PB-FOXR2, pineoblastoma FOXR2-overexpressed; WT, wild-type
Additional heterogeneity in pineal region tumors of childhood
Nine histologically defined PBs from our molecular cohort were not molecularly assigned to the subgroups described above. These tumors either clustered with existing non-PB reference CNS tumor classes or were more closely associated with control pineal tissue. Clustering of histologic PB tumors with methylation class PPTID (n=2) and histological PPTID with the molecular PB-B class (n=1) highlights the potential discrepancy between histology and molecular classification (Fig. 2a). Two tumors from our molecular cohort (histologically PB=1, pineal anlage tumor=1, no matching class for either tumor based on the MNP classifier) were associated with embryonal tumors with multilayered rosettes (ETMR) on t-SNE (Fig. 2a, Supplementary Fig. S7, online resource). Instead of ETMR-defining chromosome 19 miRNA cluster amplification (C19MC), both tumors had concurrent nonsense and missense DICER1 mutations [35]. Further clustering analysis using 10 ETMR tumors with typical histological and molecular features and two reported “ETMR-like infantile cerebellar embryonal tumors” demonstrated similarities between our tumors and the latter [42]. Additionally, two PB tumors, one clustering with WNT subgroup medulloblastoma (MB-WNT) and one clustering close to control pineal tissue, carried missense mutations in exon 3 of CTNNB1, a defining feature of WNT-MB (Supplementary Fig. S8, online resource) [9,38]. Review of imaging confirmed no posterior fossa involvement in these patients, and neither tumor exhibited monosomy 6, which characterizes >85% of MB-WNT [38]. These results exemplify additional molecular heterogeneity among embryonal tumors of the pineal region.
DISCUSSION
We report the outcome of a multi-institutional pediatric PB cohort systematically treated with an age-stratified, risk-adapted approach. Radiation with chemotherapy resulted in 75% 5-year PFS for patients older than 3 years, and 100% 5-year PFS for those with AR disease, despite the reduction of CSI dosage. This outcome compares favorably with those in the published literature [11,10,23,8,14,26,28,12,4]. The randomized Children’s Oncology Group ACNS0332 trial (NCT00392327) for patients older than 3 years treated patients with PB or sPNET regardless of metastatic status with 36 Gy CSI, concurrent vincristine, followed by cisplatin, cyclophosphamide, and vincristine (with or without carboplatin or isotretinoin), reporting a 5-year EFS of 62.8% [14]. A pooled outcome analysis of pediatric PB from 11 national or cooperative group studies using heterogeneous approaches described respective 5-year PFS rates of 63% and 11% for patients older or younger than four years [23]. Our data strongly support the use of reduced-dose CSI (23.4 Gy) as the new standard-of-care for children older than three years with AR PB, potentially mitigating treatment-related neurocognitive sequalae [22]. In contrast, most young children experienced progression on the SJYC07 regimens, echoing the abysmal outcome for young children in previous reports [23,8,10,12]. As all patients progressed on SJYC07 with metastatic failure, delayed CSI might prove beneficial in a subset of young patients.
DNA methylation profiling demonstrated molecular heterogeneity among PBs, defining four molecular subgroups. The two pre-existing PB subgroups detailed on the MNP classifier include PB-A, which encompasses young patients characterized by germline RB1 mutation, as well as sporadic PB cases akin to our two PB-A patients, and PB-B, which includes older pediatric patients and adults, with previously undefined genetic drivers [2]. Our study implicates two novel molecular subgroups, PB-B–like and PB-FOXR2, offering insights into subgroup-specific genomic alterations and expanding the spectrum of genetic events affecting known drivers such as DICER1 and DROSHA (Fig. 5).
Fig. 5.
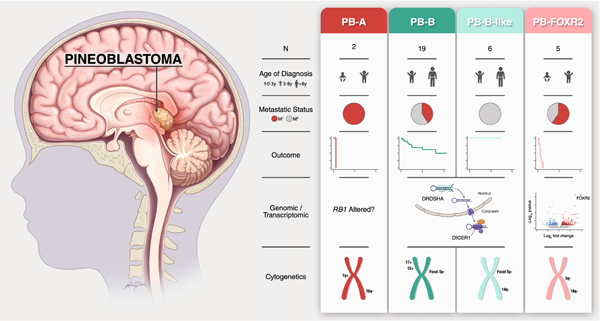
Summary of key clinical, genomic, and cytogenetic findings according to molecularly defined pineoblastoma subgroups based on the current dataset
Progression-free survival (PFS) for each subgroup is shown. Note that RB1 alteration underlying the PB-A subgroup has not been detected in our study but is included for completeness.
M0, non-metastatic; M+, metastatic; N, number; PB-A, pineoblastoma group A; PB-B, pineoblastoma group B; PB-B-like, pineoblastoma group B–like; PB-FOXR2, pineoblastoma FOXR2-overexpressed; y, years
miRNA biogenesis plays an important role in posttranscriptional gene regulation, and altered miRNA profiles have been extensively described across a spectrum of different cancers[19]. The canonical miRNA processing pathway requires the nuclear ribonuclease III enzyme DROSHA, the RNA-binding partner DGCR8, and the cytoplasmic ribonuclease III DICER1 [13]. Association between PB and altered miRNA processing, as seen in the PB-B and PB-B–like subgroups, was first established in phenotypic studies of patients with germline DICER1 loss-of-function mutations [30,7]. Unlike typical DICER1 syndrome tumors where “hotspot” missense mutation in the DICER1 ribonuclease IIIb domain represents the “second hit”, prior studies in PB suggest loss-of-heterozygosity at the DICER1 locus as the disease-specific somatic event [32]. Among the five DICER1-mutated tumors in our cohort, three exhibited nonsense mutations coupled with loss of the wild-type allele, and two had concurrent nonsense and frameshift mutations, consistent with biallelic loss-of-function in tumor cells. All but one of these mutations were previously undescribed in PB. Homozygous DROSHA deletion was recently found in PBs without DICER1 mutations [34]. In addition to focal 5p deletions involving DROSHA in four PB-B/PB-B–like study samples, we observed frameshift mutations in DROSHA upstream of the ribonuclease III domains in two tumors. Furthermore, DGCR8 alterations, seen in high-risk nephroblastomas, were previously unreported in PBs [44]. Involvement of DGCR8 (DiGeorge syndrome critical region 8) in our molecular cohort consolidates the oncogenic role of disrupted miRNA processing and provides an explanation for the rare association between DiGeorge syndrome and PB [24,36]. Despite the incomplete evaluation of cancer predisposition in this cohort, the presence of germline DICER1 mutation or 22q11.2 deletion in at least 9% of our patients older than three years highlights the importance of genetic assessment beyond germline RB1 mutations in patients with PB.
Our findings suggest FOXR2 overexpression characterizes an aggressive subgroup of PB in young children. As a proto-oncogene, FOXR2 physically interacts with and promotes the transcriptional activities of MYC, enhancing cellular proliferation and oncogenic transformation [18]. FOXR2 is overexpressed in numerous adult-onset epithelial cancers, a subset of SHH subgroup medulloblastomas, and pediatric glioblastoma; its activation through gene fusion and chromosomal rearrangements is the driver event in CNS NB-FOXR2 [18,37,17,43]. Unlike CNS NB-FOXR2, we did not identify any fusion events or structural rearrangements to substantiate the mechanism of FOXR2 overexpression seen in PB-FOXR2. PB-FOXR2 also clustered separately from CNS NB-FOXR2 in t-SNE analysis and appeared to be more clinically aggressive than CNS NB, suggesting that there are relevant context-specific differences influencing the molecular and biological features of these entities. Nonetheless, in view of the poor outcome of young patients with PB even with more intensive high-dose chemotherapy, targeting the FOXR2-MYC axis may represent a rational therapeutic approach for this lethal subgroup [3,1].
There are some limitations to our study. Despite consolidating multiple prospective trial cohorts and a non-protocol cohort, the number of patients within each molecular subgroup remains modest. We were thus underpowered to provide robust claims pertinent to subgroup-specific patient outcome in the context of the described treatment strategies. This is in part explained by the rarity of PB, as well as the scarce amount of tissue typically retrieved from surgeries for tumors of the pineal region. Future collaborative meta-analyses will be essential to confirm our findings in sufficiently powered cohorts before adopting these subgroup designations clinically. Moreover, differences in outcome between subgroups may be confounded by age-stratified therapy, as the CSI-containing regimen can only be offered to older children, such as those with PB-B and PB-B-like tumors. Nevertheless, the molecular alterations characteristic to each subgroup support divergent oncogenic processes underlying histologically diagnosed PB. Our findings may therefore inform biomarker discovery and clinical trial design to enhance patient survival and reduce treatment-related toxicities.
In conclusion, we demonstrate biological and clinical heterogeneity among children with PB and provide motivation for a risk-adapted approach in future clinical trials. Integration of methylation profiling and targeted sequencing will provide additional diagnostic detail of prognostic significance. Further research to decipher the link between observed molecular alterations and tumorigenesis could enable new avenues to target this rare, aggressive neoplasm.
Supplementary Material
Supplementary Fig. S1 Schematic summary of treatment approach from (a) SJMB03 and (b) SJYC07
Supplementary Fig. S2 Survival analysis of patients from the clinical pineoblastoma study according to study protocol and clinical characteristics Progression-free survival (PFS) and overall-survival (OS) for (a) patients treated on SJMB03 and SJMB03-like non-protocol therapy; (b) patients on SJYC07 with or without radiation (RT), patients on SJMB03/SJMB03-like or SJYC07 therapy according to (c-d) metastatic status; (e-f) extent of resection (gross-total resection [GTR] vs. subtotal resection [STR] or biopsy [Bx]). (g) PFS and OS for patients on SJMB03/SJMB03-like therapy according to presence/absence of bulky residual disease. (h) OS by risk group for patients on SJMB03/SJMB03-like therapy and SJYC07
Supplementary Fig. S3 Methylation profiling reveals heterogeneity among samples from the study’s molecular cohort (n=43). Sample names are highlighted according to their respective pineoblastoma (PB) subgroups. (a) Heatmap and dendrogram from hierarchical clustering using the top 10,000 most variable probes. (b) Genome-wide chromosome copy-number alterations for each sample. PB-A, pineoblastoma group A; PB-B, pineoblastoma group B; PB-B–like, pineoblastoma group B–like; PB-FOXR2, pineoblastoma FOXR2-overexpressed
Supplementary Fig. S4 t-stochastic neighbor embedding (t-SNE) analysis of samples from the study’s molecular cohort (n=43, open circles) with 2,801 published reference samples from 91 methylation classes by Capper et al. [14] Relevant reference classes were selected for a focused representation in Fig. 2a
Supplementary Fig. S5 Hierarchical clustering of transcriptomic profiles (PB-B=4, PBB–like=3, PB-FOXR2=4) using the top 200 most variably expressed genes revealed results supporting subgrouping by DNA-methylation profiling. Mut, mutated; PB-B, pineoblastoma group B; PB-B–like, pineoblastoma group B–like; PB-FOXR2, pineoblastoma FOXR2-overexpressed; WT, wild-type
Supplementary Fig. S6 ClueGO plots showing enriched pathways in molecularly defined pineoblastoma subgroups. The top 20 terms are shown for PB-B.
Supplementary Fig. S7 Clinical, radiographic, and molecular features of two embryonal tumor with multilayered rosette (ETMR)-like samples harboring nonsense and missense DICER1 mutations (SJPB15 [histologically pineoblastoma] and SJPB16 [pineal anlage tumor]) (Fig. 2a). (a) In both cases, MRI showed a pineal region mass, and copy number plots showed the absence of chromosome 19 miRNA cluster (C19MC) amplification. (b) t-stochastic neighbor embedding (t-SNE) analysis with methylation profiles derived from 10 in-house typical ETMRs and two recently published “ETMR-like infantile cerebellar embryonal tumors” (which also lacked C19MC amplification and carried the same pattern of DICER1 mutations) showed clustering of our samples with the latter [42]. CSI, craniospinal irradiation; DOD, died of disease; Dx, diagnosis; M0, non-metastatic; PD, progressive disease; TSNE, t-stochastic neighbor embedding; WES, whole-exome sequencing
Supplementary Fig. S8 Clinical, radiographic, and molecular features of two samples from the molecular cohort (SJPB17, clustering with WNT-activated medulloblastoma [MB-WNT], and SJPB19, clustering close to control pineal tissue; Fig. 2a) carrying a missense mutation in exon 3 of CTNNB1. In both cases, MRI showed a pineal region mass without posterior fossa involvement, and copy number plots showed the absence of monosomy 6. CTNNB1 mutations detected are highlighted in lollipop plots. CDDP, cisplatin; CPM, cyclophosphamide; CSI, craniospinal irradiation; Dx, diagnosis; IT, intrathecal; M0, non-metastatic; MB-G3, medulloblastoma group 3; MNP, MolecularNeuropathology classifier; t-SNE, t-stochastic neighbor embedding; VCR, vincristine; VP16, etoposide; WES, whole-exome sequencing
Supplementary Methods: Supplementary methods and references on genome-wide DNA methylation profiling and next-generation sequencing pipelines
Supplementary Table S1 Clinical characteristics, treatment, outcome, and molecular features of patients with non-pineoblastoma histologies
Supplementary Table S2 Progression-free survival (PFS) and overall survival (OS) of study patients
Supplementary Table S3 Metadata for the study cohort
Supplementary Table S4 Filtered list of variants called from whole-exome sequencing
Supplementary Table S5 Differential expression and gene set enrichment analysis in molecular pineoblastoma subgroups
ACKNOWLEDGEMENTS
We thank all patients and families; physicians; and nursing, research, and administrative staff from all participating institutions. We appreciate the following core facilities at St Jude Children’s Research Hospital: the Biorepository, for providing and processing study tissues; the Diagnostic Biomarkers Shared Resource, for nucleic acid extractions; and the Hartwell Center, for conducting DNA-methylation profiling and next-generation sequencing. We thank Brandon Stelter for assistance with figure preparation and artwork and Cherise Guess, PhD, ELS, for editing the manuscript.
This work was sponsored by the American Lebanese Syrian Associated Charities, the National Cancer Institute (grant CA21765), and Musicians Against Childhood Cancer. P.A.N. is a Pew-Stewart Scholar for Cancer Research (Margaret and Alexander Stewart Trust) and recipient of The Sontag Foundation Distinguished Scientist Award. P.A.N. was also supported by the National Cancer Institute (R01), American Association for Cancer Research (NextGen Grant for Transformative Cancer Research), The Brain Tumour Charity (Quest for Cures), the American Lebanese Syrian Associated Charities (ALSAC), and St. Jude.
Footnotes
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The authors disclose no conflict of interest.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCES
- 1.AbdelBaki M, Abu Arja M, Funk Z, Stanek J, Davidson T, Fangusaro J, Dunkel I, Dhall G, Gardner S, Finlay J (2018) PDCT-13. PINEOBLASTOMA IN CHILDREN: THE HEAD START EXPERIENCE. Neuro-oncol 20:vi203-vi203. doi: 10.1093/neuonc/noy148.842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capper D, Jones DT, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, Liu H, Qing G (2018) Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduction and Targeted Therapy 3:5. doi: 10.1038/s41392-018-0008-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chintagumpala M, Hassall T, Palmer S, Ashley D, Wallace D, Kasow K, Merchant TE, Krasin MJ, Dauser R, Boop F, Krance R, Woo S, Cheuk R, Lau C, Gilbertson R, Gajjar A (2009) A pilot study of risk-adapted radiotherapy and chemotherapy in patients with supratentorial PNET. Neuro-oncol 11:33–40. doi: 10.1215/15228517-2008-079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho Y-J, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, Lau CC, Olson JM, Gilbertson RJ, Gajjar A, Delattre O, Kool M, Ligon K, Meyerson M, Mesirov JP, Pomeroy SL (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430. doi: 10.1200/JCO.2010.28.5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ClinicalTrials.gov (2019) Treatment of Patients With Newly Diagnosed Medulloblastoma, Supratentorial Primitive Neuroectodermal Tumor, or Atypical Teratoid Rhabdoid Tumor. https://clinicaltrials.gov/ct2/show/NCT00085202. Accessed June 10 2019
- 7.de Kock L, Sabbaghian N, Druker H, Weber E, Hamel N, Miller S, Choong CS, Gottardo NG, Kees UR, Rednam SP, van Hest LP, Jongmans MC, Jhangiani S, Lupski JR, Zacharin M, Bouron-Dal Soglio D, Huang A, Priest JR, Perry A, Mueller S, Albrecht S, Malkin D, Grundy RG, Foulkes WD (2014) Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 128:583–595. doi: 10.1007/s00401-014-1318-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng (2018) Prognosis of Pediatric Patients with Pineoblastoma: A SEER Analysis 1990–2013. World Neurosurgery 118:e871–e879 [DOI] [PubMed] [Google Scholar]
- 9.Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, Pearson AD, Clifford SC (2005) beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol 23:7951–7957. doi: 10.1200/jco.2005.01.5479 [DOI] [PubMed] [Google Scholar]
- 10.Friedrich C, von Bueren AO, von Hoff K, Gerber NU, Ottensmeier H, Deinlein F, Benesch M, Kwiecien R, Pietsch T, Warmuth-Metz M, Faldum A, Kuehl J, Kortmann RD, Rutkowski S (2012) Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro-oncol 15:224–234. doi: 10.1093/neuonc/nos292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilheeney SW, Saad A, Chi S, Turner C, Ullrich NJ, Goumnerova L, Scott RM, Marcus K, Lehman L, De Girolami U, Kieran MW (2008) Outcome of pediatric pineoblastoma after surgery, radiation and chemotherapy. J Neurooncol 89:89–95. doi: 10.1007/s11060-008-9589-2 [DOI] [PubMed] [Google Scholar]
- 12.Gururangan S, McLaughlin C, Quinn J, Rich J, Reardon D, Halperin EC, Herndon J 2nd, Fuchs H, George T, Provenzale J, Watral M, McLendon RE, Friedman A, Friedman HS, Kurtzberg J, Vredenbergh J, Martin PL (2003) High-dose chemotherapy with autologous stem-cell rescue in children and adults with newly diagnosed pineoblastomas. J Clin Oncol 21:2187–2191. doi: 10.1200/jco.2003.10.096 [DOI] [PubMed] [Google Scholar]
- 13.Han J, Lee Y, Yeom K-H, Kim Y-K, Jin H, Kim VN (2004) The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 18:3016–3027. doi: 10.1101/gad.1262504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang EI, Kool M, Burger PC, Capper D, Chavez L, Brabetz S, Williams-Hughes C, Billups C, Heier L, Jaju A, Michalski J, Li Y, Leary S, Zhou T, Deimling Av, Jones DTW, Fouladi M, Pollack IF, Gajjar A, Packer RJ, Pfister SM, Olson JM (2018) Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children’s Oncology Group Randomized ACNS0332 Trial. J Clin Oncol 36:3388–3395. doi: 10.1200/jco.2017.76.4720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DTW, Sturm D, Hermann C, Segura Wang M, Korshunov A, Rhyzova M, Gröbner S, Brabetz S, Chavez L, Bens S, Gröschel S, Kratochwil F, Wittmann A, Sieber L, Geörg C, Wolf S, Beck K, Oyen F, Capper D, van Sluis P, Volckmann R, Koster J, Versteeg R, von Deimling A, Milde T, Witt O, Kulozik AE, Ebinger M, Shalaby T, Grotzer M, Sumerauer D, Zamecnik J, Mora J, Jabado N, Taylor MD, Huang A, Aronica E, Bertoni A, Radlwimmer B, Pietsch T, Schüller U, Schneppenheim R, Northcott PA, Korbel JO, Siebert R, Frühwald MC, Lichter P, Eils R, Gajjar A, Hasselblatt M, Pfister SM, Kool M (2016) Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29:379–393. doi: 10.1016/j.ccell.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 16.Kivela T (1999) Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17:1829–1837. doi: 10.1200/jco.1999.17.6.1829 [DOI] [PubMed] [Google Scholar]
- 17.Koso H, Tsuhako A, Lyons E, Ward JM, Rust AG, Adams DJ, Jenkins NA, Copeland NG, Watanabe S (2014) Identification of FoxR2 as an oncogene in medulloblastoma. Cancer Res 74:2351–2361. doi: 10.1158/0008-5472.Can-13-1523 [DOI] [PubMed] [Google Scholar]
- 18.Li X, Wang W, Xi Y, Gao M, Tran M, Aziz KE, Qin J, Li W, Chen J (2016) FOXR2 interacts with MYC to promote its transcriptional activities and tumorigenesis. Cell reports 16:487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin S, Gregory RI (2015) MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 15:321–333. doi: 10.1038/nrc3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (Berl) 131:803–820 [DOI] [PubMed] [Google Scholar]
- 21.Mays JC, Kelly MC, Coon SL, Holtzclaw L, Rath MF, Kelley MW, Klein DC (2018) Single-cell RNA sequencing of the mammalian pineal gland identifies two pinealocyte subtypes and cell typespecific daily patterns of gene expression. PLoS ONE 13:e0205883. doi: 10.1371/journal.pone.0205883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulhern RK, Palmer SL, Merchant TE, Wallace D, Kocak M, Brouwers P, Krull K, Chintagumpala M, Stargatt R, Ashley DM (2005) Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. J Clin Oncol 23:5511–5519 [DOI] [PubMed] [Google Scholar]
- 23.Mynarek M, Pizer B, Dufour C, van Vuurden D, Garami M, Massimino M, Fangusaro J, Davidson T, Gil-da-Costa MJ, Sterba J, Benesch M, Gerber N, Juhnke BO, Kwiecien R, Pietsch T, Kool M, Clifford S, Ellison DW, Giangaspero F, Wesseling P, Gilles F, Gottardo N, Finlay JL, Rutkowski S, von Hoff K (2017) Evaluation of age-dependent treatment strategies for children and young adults with pineoblastoma: analysis of pooled European Society for Paediatric Oncology (SIOP-E) and US Head Start data. Neuro Oncol 19:576–585. doi: 10.1093/neuonc/now234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen L, Crawford JR (2018) Pineoblastoma in a child with 22q11.2 deletion syndrome. BMJ Case Reports 2018:bcr-2018–226434. doi: 10.1136/bcr-2018-226434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414. doi: 10.1200/JCO.2009.27.4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pizer BL, Weston CL, Robinson KJ, Ellison DW, Ironside J, Saran F, Lashford LS, Tait D, Lucraft H, Walker DA, Bailey CC, Taylor RE (2006) Analysis of patients with supratentorial primitive neuroectodermal tumours entered into the SIOP/UKCCSG PNET 3 study. Eur J Cancer 42:1120–1128. doi: 10.1016/j.ejca.2006.01.039 [DOI] [PubMed] [Google Scholar]
- 27.Poh B, Koso H, Momota H, Komori T, Suzuki Y, Yoshida N, Ino Y, Todo T, Watanabe S (2019) Foxr2 promotes formation of CNS-embryonal tumors in a Trp53-deficient background. Neuro-oncol. doi: 10.1093/neuonc/noz067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy AT, Janss AJ, Phillips PC, Weiss HL, Packer RJ (2000) Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer 88:2189–2193. doi: [DOI] [PubMed] [Google Scholar]
- 29.Robinson GW, Rudneva VA, Buchhalter I, Billups CA, Waszak SM, Smith KS, Bowers DC, Bendel A, Fisher PG, Partap S (2018) Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19:768–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabbaghian N, Hamel N, Srivastava A, Albrecht S, Priest JR, Foulkes WD (2012) Germline DICER1 mutation and associated loss of heterozygosity in a pineoblastoma. J Med Genet 49:417–419 [DOI] [PubMed] [Google Scholar]
- 31.Schild SE, Scheithauer BW, Schomberg PJ, Hook CC, Kelly PJ, Frick L, Robinow JS, Buskirk SJ (1993) Pineal parenchymal tumors: clinical, pathologic, and therapeutic aspects. Cancer 72:870–880 [DOI] [PubMed] [Google Scholar]
- 32.Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, Turner J, Shah R, Schneider K, Schneider KW (2018) DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res 24:2251–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma T, Schwalbe EC, Williamson D, Sill M, Hovestadt V, Mynarek M, Rutkowski S, Robinson GW, Gajjar A, Cavalli F, Ramaswamy V, Taylor MD, Lindsey JC, Hill RM, Jager N, Korshunov A, Hicks D, Bailey S, Kool M, Chavez L, Northcott PA, Pfister SM, Clifford SC (2019) Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol. doi: 10.1007/s00401-019-02020-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snuderl M, Kannan K, Pfaff E, Wang S, Stafford JM, Serrano J, Heguy A, Ray K, Faustin A, Aminova O, Dolgalev I, Stapleton SL, Zagzag D, Chiriboga L, Gardner SL, Wisoff JH, Golfinos JG, Capper D, Hovestadt V, Rosenblum MK, Placantonakis DG, LeBoeuf SE, Papagiannakopoulos TY, Chavez L, Ahsan S, Eberhart CG, Pfister SM, Jones DTW, Karajannis MA (2018) Recurrent homozygous deletion of DROSHA and microduplication of PDE4DIP in pineoblastoma. Nat Commun 9:2868. doi: 10.1038/s41467-018-05029-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spence T, Sin-Chan P, Picard D, Barszczyk M, Hoss K, Lu M, Kim S-K, Ra Y-S, Nakamura H, Fangusaro J (2014) CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol (Berl) 128:291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens T, ten Bosch JvdW, De Rademaeker M, Van Den Bogaert A, van den Akker M (2017) Risk of malignancy in 22q11. 2 deletion syndrome. Clinical case reports 5:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I, Ryzhova M, Koelsche C, Pfaff E, Allen SJ, Balasubramanian G, Worst BC, Pajtler KW, Brabetz S, Johann PD, Sahm F, Reimand J, Mackay A, Carvalho DM, Remke M, Phillips JJ, Perry A, Cowdrey C, Drissi R, Fouladi M, Giangaspero F, Lastowska M, Grajkowska W, Scheurlen W, Pietsch T, Hagel C, Gojo J, Lotsch D, Berger W, Slavc I, Haberler C, Jouvet A, Holm S, Hofer S, Prinz M, Keohane C, Fried I, Mawrin C, Scheie D, Mobley BC, Schniederjan MJ, Santi M, Buccoliero AM, Dahiya S, Kramm CM, von Bueren AO, von Hoff K, Rutkowski S, Herold-Mende C, Fruhwald MC, Milde T, Hasselblatt M, Wesseling P, Rossler J, Schuller U, Ebinger M, Schittenhelm J, Frank S, Grobholz R, Vajtai I, Hans V, Schneppenheim R, Zitterbart K, Collins VP, Aronica E, Varlet P, Puget S, Dufour C, Grill J, Figarella-Branger D, Wolter M, Schuhmann MU, Shalaby T, Grotzer M, van Meter T, Monoranu CM, Felsberg J, Reifenberger G, Snuderl M, Forrester LA, Koster J, Versteeg R, Volckmann R, van Sluis P, Wolf S, Mikkelsen T, Gajjar A, Aldape K, Moore AS, Taylor MD, Jones C, Jabado N, Karajannis MA, Eils R, Schlesner M, Lichter P, von Deimling A, Pfister SM, Ellison DW, Korshunov A, Kool M (2016) New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164:1060–1072. doi: 10.1016/j.cell.2016.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931. doi: 10.1200/jco.2005.04.4974 [DOI] [PubMed] [Google Scholar]
- 39.Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan P, Vasiljevic A, Norman JD, Guilhamon P, Garzia L, Agamez NR, Lu M, Chan TS, Picard D, de Antonellis P, Khuong-Quang D-A, Planello AC, Zeller C, Barsyte-Lovejoy D, Lafay-Cousin L, Letourneau L, Bourgey M, Yu M, Gendoo DMA, Dzamba M, Barszczyk M, Medina T, Riemenschneider AN, Morrissy AS, Ra Y-S, Ramaswamy V, Remke M, Dunham CP, Yip S, Ng H-K, Lu J-Q, Mehta V, Albrecht S, Pimentel J, Chan JA, Somers GR, Faria CC, Roque L, Fouladi M, Hoffman LM, Moore AS, Wang Y, Choi SA, Hansford JR, Catchpoole D, Birks DK, Foreman NK, Strother D, Klekner A, Bognár L, Garami M, Hauser P, Hortobágyi T, Wilson B, Hukin J, Carret A- S, Van Meter TE, Hwang EI, Gajjar A, Chiou S- H, Nakamura H, Toledano H, Fried I, Fults D, Wataya T, Fryer C, Eisenstat DD, Scheinemann K, Fleming AJ, Johnston DL, Michaud J, Zelcer S, Hammond R, Afzal S, Ramsay DA, Sirachainan N, Hongeng S, Larbcharoensub N, Grundy RG, Lulla RR, Fangusaro JR, Druker H, Bartels U, Grant R, Malkin D, McGlade CJ, Nicolaides T, Tihan T, Phillips J, Majewski J, Montpetit A, Bourque G, Bader GD, Reddy AT, Gillespie GY, Warmuth-Metz M, Rutkowski S, Tabori U, Lupien M, Brudno M, Schüller U, Pietsch T, Judkins AR, Hawkins CE, Bouffet E, Kim S- K, Dirks PB, Taylor MD, Erdreich-Epstein A, Arrowsmith CH, De Carvalho DD, Rutka JT, Jabado N, Huang A (2016) Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 30:891–908. doi: 10.1016/j.ccell.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, Langer C, Murphy B, Cumberlin R, Coleman CN CTCAE v3. 0: development of a comprehensive grading system for the adverse effects of cancer treatment. In: Semin Radiat Oncol, 2003. vol 3 Elsevier, pp 176–181 [DOI] [PubMed] [Google Scholar]
- 41.Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Billups CA, Bowers DC, Bendel AE, Hassall T, Crawford JR, Partap S (2019) Molecular Grouping and Outcomes of Young Children with Newly Diagnosed Ependymoma Treated on the Multi-Institutional SJYC07 Trial. Neuro-oncol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uro-Coste E, Masliah-Planchon J, Siegfried A, Blanluet M, Lambo S, Kool M, Roujeau T, Boetto S, Palenzuela G, Bertozzi AI, Gambart M, Coupier I, Oliver-Petit I, Golmard L, Julia S, Savagner F, Mohand-Oumoussa B, Tauziede-Espariat A, Delisle MB, Figarella-Branger D, Bourdeaut F, Rigau V (2019) ETMR-like infantile cerebellar embryonal tumors in the extended morphologic spectrum of DICER1-related tumors. Acta Neuropathol 137:175–177. doi: 10.1007/s00401-0181935-7 [DOI] [PubMed] [Google Scholar]
- 43.van Rijn S, Vouri M, Pfister SM, Kawauchi D, Kool M (2018) EMBR-04. WHAT THE FOX SAY? A MOLECULAR ANALYSIS OF FOXR2 IN PEDIATRIC BRAIN TUMORS. Neuro-oncol 20:i69-i69. doi: 10.1093/neuonc/noy059.189 [DOI] [Google Scholar]
- 44.Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM, Meerzaman D, Chen QR, Hsu CH, Yan C, Nguyen C, Hu Y, Bowlby R, Brooks D, Ma Y, Mungall AJ, Moore RA, Schein J, Marra MA, Huff V, Dome JS, Chi YY, Mullighan CG, Ma J, Wheeler DA, Hampton OA, Jafari N, Ross N, Gastier-Foster JM, Perlman EJ (2015) Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27:286–297. doi: 10.1016/j.ccell.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O (2004) Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron 43:487–497. doi: 10.1016/j.neuron.2004.08.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1 Schematic summary of treatment approach from (a) SJMB03 and (b) SJYC07
Supplementary Fig. S2 Survival analysis of patients from the clinical pineoblastoma study according to study protocol and clinical characteristics Progression-free survival (PFS) and overall-survival (OS) for (a) patients treated on SJMB03 and SJMB03-like non-protocol therapy; (b) patients on SJYC07 with or without radiation (RT), patients on SJMB03/SJMB03-like or SJYC07 therapy according to (c-d) metastatic status; (e-f) extent of resection (gross-total resection [GTR] vs. subtotal resection [STR] or biopsy [Bx]). (g) PFS and OS for patients on SJMB03/SJMB03-like therapy according to presence/absence of bulky residual disease. (h) OS by risk group for patients on SJMB03/SJMB03-like therapy and SJYC07
Supplementary Fig. S3 Methylation profiling reveals heterogeneity among samples from the study’s molecular cohort (n=43). Sample names are highlighted according to their respective pineoblastoma (PB) subgroups. (a) Heatmap and dendrogram from hierarchical clustering using the top 10,000 most variable probes. (b) Genome-wide chromosome copy-number alterations for each sample. PB-A, pineoblastoma group A; PB-B, pineoblastoma group B; PB-B–like, pineoblastoma group B–like; PB-FOXR2, pineoblastoma FOXR2-overexpressed
Supplementary Fig. S4 t-stochastic neighbor embedding (t-SNE) analysis of samples from the study’s molecular cohort (n=43, open circles) with 2,801 published reference samples from 91 methylation classes by Capper et al. [14] Relevant reference classes were selected for a focused representation in Fig. 2a
Supplementary Fig. S5 Hierarchical clustering of transcriptomic profiles (PB-B=4, PBB–like=3, PB-FOXR2=4) using the top 200 most variably expressed genes revealed results supporting subgrouping by DNA-methylation profiling. Mut, mutated; PB-B, pineoblastoma group B; PB-B–like, pineoblastoma group B–like; PB-FOXR2, pineoblastoma FOXR2-overexpressed; WT, wild-type
Supplementary Fig. S6 ClueGO plots showing enriched pathways in molecularly defined pineoblastoma subgroups. The top 20 terms are shown for PB-B.
Supplementary Fig. S7 Clinical, radiographic, and molecular features of two embryonal tumor with multilayered rosette (ETMR)-like samples harboring nonsense and missense DICER1 mutations (SJPB15 [histologically pineoblastoma] and SJPB16 [pineal anlage tumor]) (Fig. 2a). (a) In both cases, MRI showed a pineal region mass, and copy number plots showed the absence of chromosome 19 miRNA cluster (C19MC) amplification. (b) t-stochastic neighbor embedding (t-SNE) analysis with methylation profiles derived from 10 in-house typical ETMRs and two recently published “ETMR-like infantile cerebellar embryonal tumors” (which also lacked C19MC amplification and carried the same pattern of DICER1 mutations) showed clustering of our samples with the latter [42]. CSI, craniospinal irradiation; DOD, died of disease; Dx, diagnosis; M0, non-metastatic; PD, progressive disease; TSNE, t-stochastic neighbor embedding; WES, whole-exome sequencing
Supplementary Fig. S8 Clinical, radiographic, and molecular features of two samples from the molecular cohort (SJPB17, clustering with WNT-activated medulloblastoma [MB-WNT], and SJPB19, clustering close to control pineal tissue; Fig. 2a) carrying a missense mutation in exon 3 of CTNNB1. In both cases, MRI showed a pineal region mass without posterior fossa involvement, and copy number plots showed the absence of monosomy 6. CTNNB1 mutations detected are highlighted in lollipop plots. CDDP, cisplatin; CPM, cyclophosphamide; CSI, craniospinal irradiation; Dx, diagnosis; IT, intrathecal; M0, non-metastatic; MB-G3, medulloblastoma group 3; MNP, MolecularNeuropathology classifier; t-SNE, t-stochastic neighbor embedding; VCR, vincristine; VP16, etoposide; WES, whole-exome sequencing
Supplementary Methods: Supplementary methods and references on genome-wide DNA methylation profiling and next-generation sequencing pipelines
Supplementary Table S1 Clinical characteristics, treatment, outcome, and molecular features of patients with non-pineoblastoma histologies
Supplementary Table S2 Progression-free survival (PFS) and overall survival (OS) of study patients
Supplementary Table S3 Metadata for the study cohort
Supplementary Table S4 Filtered list of variants called from whole-exome sequencing
Supplementary Table S5 Differential expression and gene set enrichment analysis in molecular pineoblastoma subgroups