

Abstract
Reaction of FeIII(O2•−)(TPP) with 2,3-dimethylindole at −40 °C gives the ring-opened, dioxygenated N-(2-acetyl-phenyl)-acetamide product. The reaction was monitored in situ by low-temperature UV-Vis and 1H NMR spectroscopies. This work demonstrates that a discrete iron(III)(superoxo) porphyrin is competent to carry out indole oxidation, as proposed for the tryptophan and indoleamine 2,3-dioxygenases.
Graphical Abstract
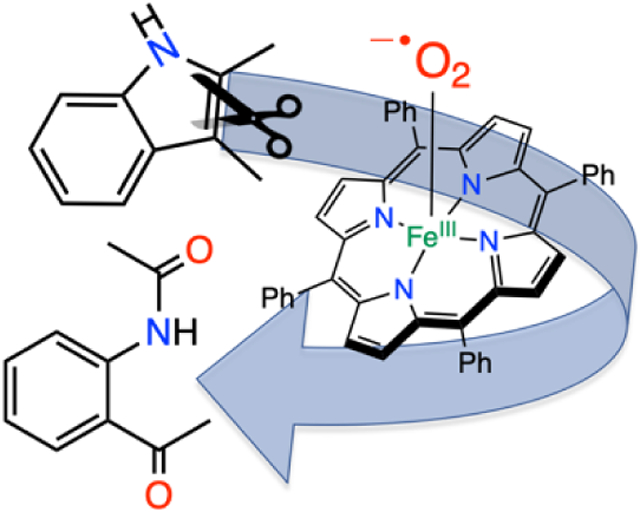
Reaction of FeIII(O2•−)(TPP) with 2,3-dimethylindole at low temperature leads to the ring-cleaved, dioxygenated product, N-(2-acetyl-phenyl)-acetamide, analogous to TDO/IDO enzymes.
Transition metal dioxygen/superoxide adducts (Mn+(O2)/ Mn+1(O2•−)) (e.g. M = Fe, Cu, Ni) are postulated as key intermediates in oxygenases1 and other metalloenzymes2 that utilize O2 to carry out the oxidation of organic compounds. Iron superoxide (FeIII(O2•−)) species are proposed to initiate the attack on the substrate in both heme3 and nonheme4 iron environments. However, direct spectroscopic evidence for the formation of an FeIII(O2•−) intermediate is often difficult to obtain. It can also be challenging to demonstrate that such an intermediate is capable of oxidizing the substrate.
The heme enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO) catalyse the oxidation of tryptophan to N-formylkynurenine (NFK). This O2-dependent reaction involves the cleavage of a C–C double bond and the incorporation of two atoms of oxygen into the substrate (Scheme 1). The catalytic activity of TDO/IDO has been known for over 50 years, but the mechanisms of these enzymes are still poorly understood and remain a subject of intense debate.3d, 5 Recent efforts aimed at delineating the mechanisms of TDO/IDO are partly motivated by therapeutic strategies that target these enzymes to treat a variety of diseases, from neurological disorders to cancer.5a, 5b, 6
Scheme 1.
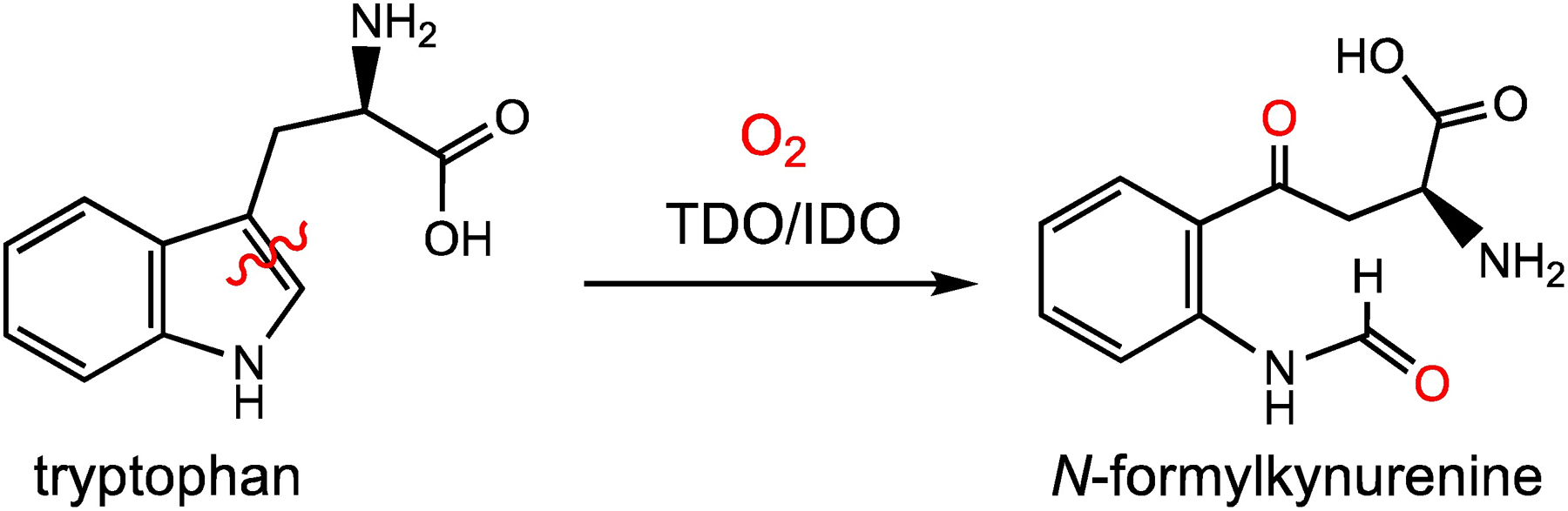
Oxidation of tryptophan to N-formylkynurenine catalyzed by TDO/IDO
One common feature of the proposed mechanisms for TDO/IDO involves the initial attack of an FeII(O2)/FeIII(O2•−) adduct on the indole substrate. In support of this hypothesis, an Fe(O2) adduct was characterized by resonance Raman spectroscopy in human IDO as a ternary complex with L-Trp substrate, and the O–O stretch was consistent with superoxide character.5d A crystal structure of a ternary complex, [TDO-O2-Trp], was recently obtained via diffusion of O2 into solid-state crystals of human TDO (hTDO) loaded with L-Trp.5b The positioning of the Fe(O2) moiety in the crystal structure is in line with the hypothesis that the dioxygenation reaction begins with attack of the O2-derived ligand on the C2 atom of the indole ring. A stopped-flow UV-vis study of hTDO provided evidence for an Fe(O2) adduct, and the data suggested that the decay of the ternary complex [hTDO-O2-Trp] was the rate-determining step, in contrast to IDO.7 Stopped-flow UV-vis spectroscopy was also employed to examine the kinetics of Pseudomonas TDO (PaTDO) reconstituted with different heme derivatives. This study suggested that an FeII(O2), as opposed to an FeIII(O2•−) species, was responsible for L-Trp oxidation, favouring an electrophilic over radical addition mechanism.5e
There are a few limited reports on the catalytic oxidation of indole derivatives with synthetic metalloporphyrins and other transition metal complexes.8 Reaction of iron(III) 5,10,15,20-tetraphenylporphyrin (TPP) with 3-methylindole, OH−, and excess O2 in CH2Cl2/MeOH led to an oxidized product analogous to NFK, and X-band EPR spectroscopy suggested that a ternary complex [FeIII–O2–indole] may be an intermediate in this reaction.8a Given that ferric porphyrin should be inert to O2, it was later suggested9 that the deprotonated 3-methylindole anion may serve as a reductant to give the O2-active FeII(TPP), although no spectroscopic evidence for either the reduced FeII(TPP) or the hypothesized FeIII(O2•−)(TPP) intermediate was presented.
Demonstrating that a well-defined Fe(O2•−)(porphyrin) is capable of initiating the oxidation of an indole substrate would provide good evidence for the hypothesized first step in TDO/IDO. To our knowledge, no direct reaction between a synthetic Fe(O2) adduct, porphyrin or non-porphyrin, and an indole substrate is known.10 Herein we show that the iron superoxide complex FeIII(O2•−)(TPP), stabilized in solution at low temperature, reacts with 2,3-dimethylindole (2,3-DMI) to form the ring-opened, dioxygenated product N-(2-acetyl-phenyl)-acetamide in good yield, analogous to NFK. Product analysis and in situ spectroscopic measurements show that reaction of the iron superoxide complex with 2,3-DMI results in dioxygenation of the indole substrate, as proposed for the heme dioxygenases TDO/IDO.
The synthesis of the ferrous porphyrin starting material, FeII(TPP), and subsequent generation of the superoxide complex FeIII(O2•−)(TPP), followed the recent methods of Mayer et al.11 The specific protocols used in our laboratory, including characterization data, are given below. The starting ferrous porphyrin was synthesized from metalation of TPP with FeBr2 and LiHMDS (HMDS = hexamethyldisilazide) in tetrahydrofuran under strict anaerobic conditions. The metallated product was crystallized from pentane/toluene or THF/heptane solutions. Crystalline FeII(TPP) was dissolved in DMF to give a violet solution with λmax (ε × 104 M−1 cm−1) 538(sh) (7.8), 563 (10.4), and 606 (5.1) nm in the Q-band region, matching literature values (Figure 1).11, 12 The crystalline FeII starting material is a 6-coordinate bis-THF adduct as described in the literature,13 and as reproduced in our laboratory. Upon dissolution in DMF, it is likely that at least one or both axial THF ligands are replaced by the DMF and are involved in rapid binding equilibria. For simplicity, we refer to the FeII starting material as FeII(TPP), without specifying the exact nature of the axial ligands, following Mayer.
Fig. 1.
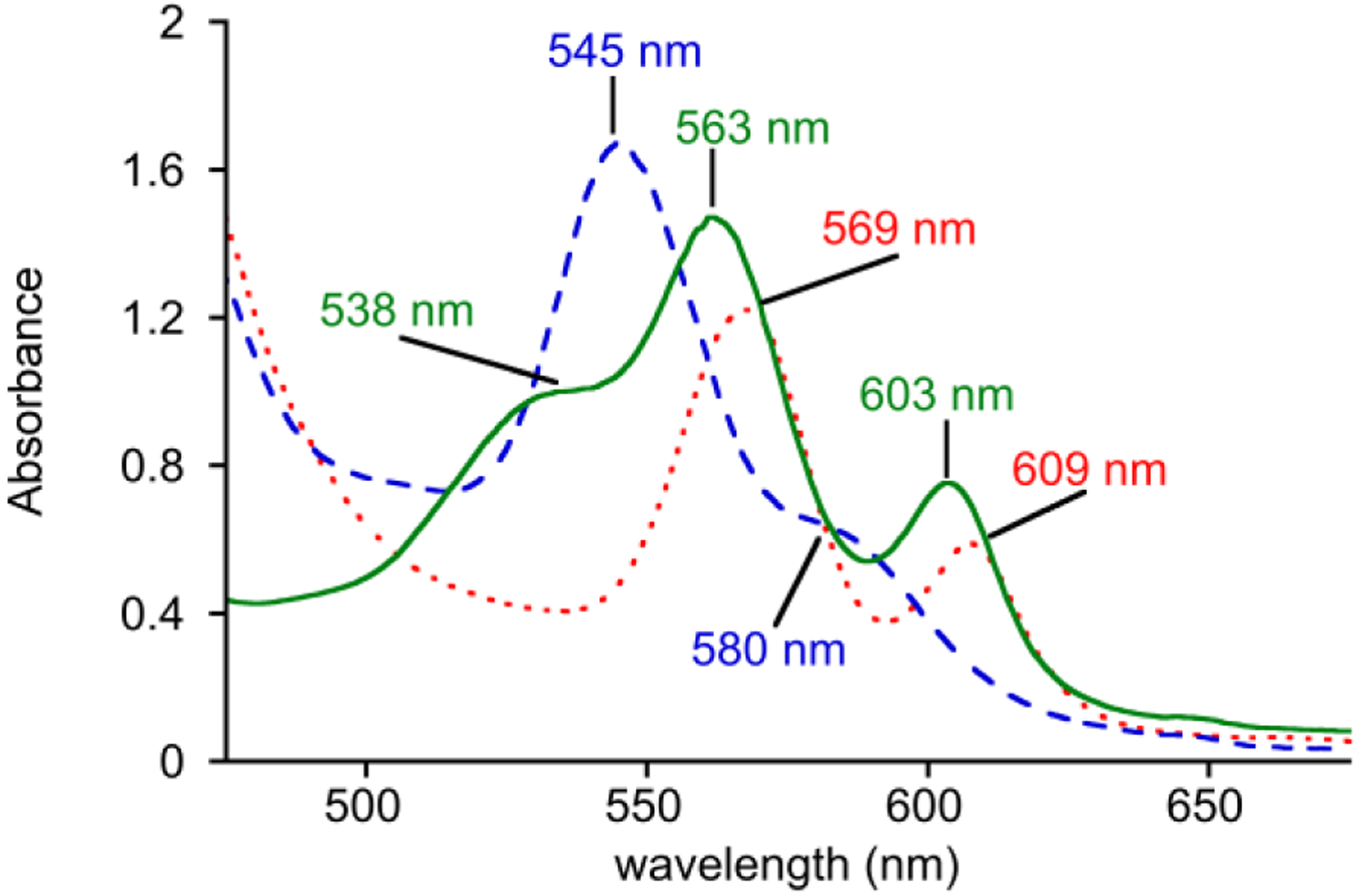
UV-vis spectra for the conversion of FeII(TPP) (0.6 mM) (green solid line) to FeIII(O2•−)(TPP) (blue dashed line), followed by decay to (FeIII(TPP))2(μ-O) (red dotted line) in DMF at −40 °C.
Lowering the temperature of FeII(TPP) in DMF to −40 °C, followed by addition of excess O2, leads to a colour change from violet to bright red and the appearance of a new spectrum with λmax (ε × 104 M−1 cm−1) 545 (13.6), 580(sh) (5.3) nm (Figure 1). This new spectrum is characteristic of the class of ferric tetraarylporphyrin superoxide complexes, including the TPP derivative.14 Purging with inert gas or application of vacuum causes the FeII(TPP) spectrum to reappear, consistent with reversible O2 binding. The stability of the superoxide complex at −40 °C was monitored by observing the change of the spectrum for the superoxo complex to the final spectrum in red in Figure 1, which corresponds to the well-known (FeIII(TPP))2(μ-O) complex and has peaks at λmax (ε × 104 M−1 cm−1) 569 (9.1), 609 (4.9) nm.14b, 15 This conversion, which is accompanied by a change from red to green-brown, is complete within 120 min at a concentration of 0.6 mM and −40 °C.
This window of stability of FeIII(O2•−)(TPP) at −40 °C allowed for the examination of reactivity with indole derivatives (indole, 2-methylindole, 3-methylindole, 2,3-dimethylindole). The iron superoxide complex was generated in DMF at −40 °C, and the indole derivatives were added separately from DMF stock solutions. Addition of excess 2,3-DMI (73 equiv) leads to a significant enhancement in the decay rate of the superoxide complex, showing conversion of (FeIII(O2•−)(TPP) to the μ-oxo dimer in approximately 40 min, compared to decay over 120 min in the absence of substrate at the same FeII(TPP) concentration (Figure 2). However, the other indole substrates did not show the same increase in the rate of decay. The spectral changes in Fig. 2 do not show overall isosbestic behaviour, consistent with conversion to one or more intermediate iron species prior to formation of the final (FeIII(TPP))2(μ-O) product.
Fig. 2.
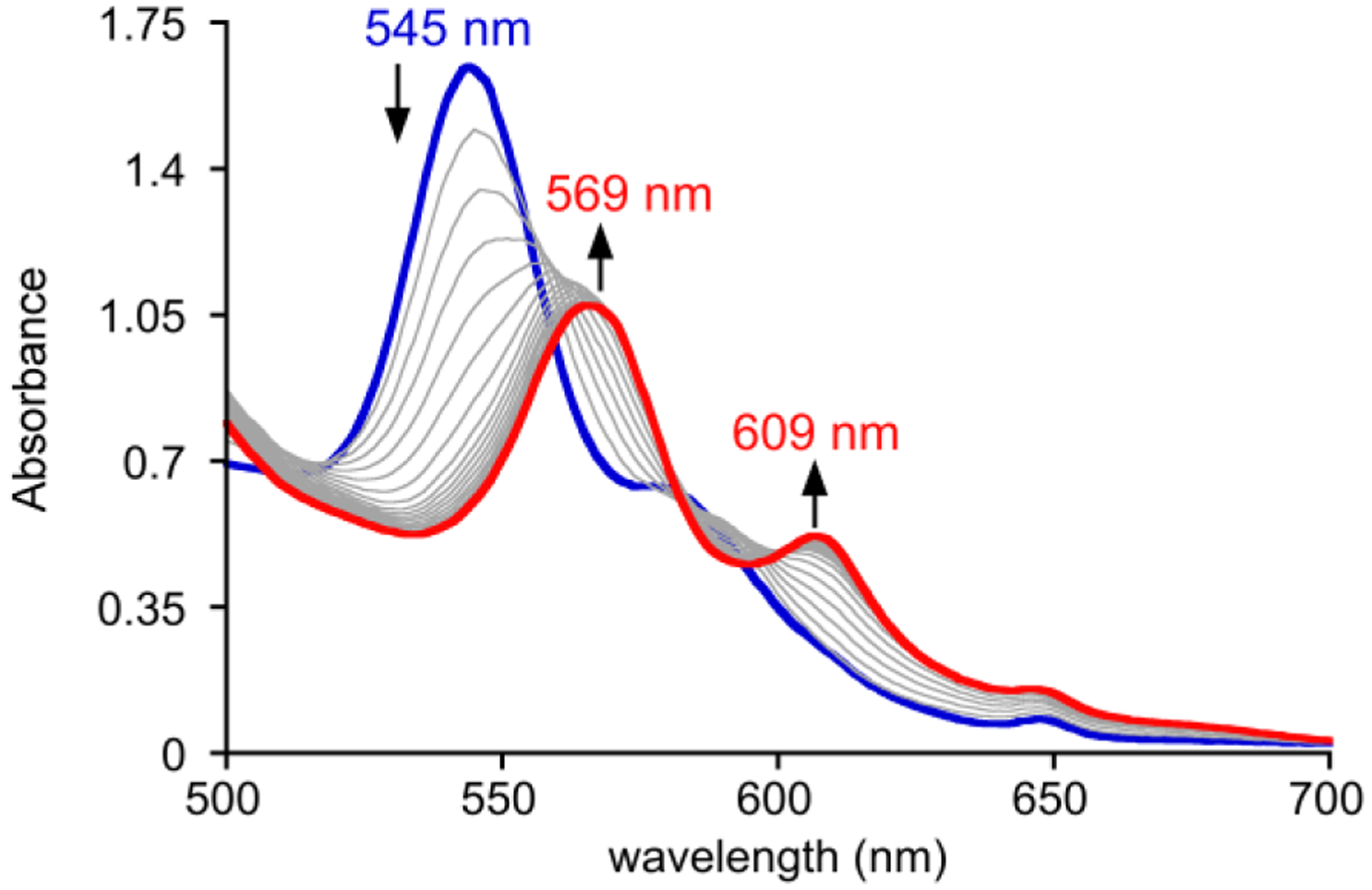
Time-resolved UV-Vis spectra (0 – 40 min) for the reaction between FeIII(O2•−)(TPP) (0.6 mM) and 2,3-dimethylindole (0.044 M) in DMF at −40 °C.
Encouraged by the observation of a reaction with the 2,3-DMI derivative by UV-vis, we increased the scale of this reaction in order to isolate and characterize the organic product(s). Generation of FeIII(O2•−)(TPP) (12 mM) at −40°C, followed by addition of 2,3-DMI (45 equiv) led to a color change from bright red to green. The reaction mixture was then warmed to 25 °C, and TLC analysis revealed one major product. Separation of the organic components by aqueous/ethyl acetate extraction followed by chromatography on neutral alumina led to isolation of the product as a yellow solid. Analysis by 1H NMR indicated that this product was the ring-opened, dioxygenated N-(2-acetyl-phenyl)-acetamide. This assignment was confirmed by comparison with an authentic standard (Figure S5–6). The overall reaction is shown in Scheme 2.
Scheme 2.
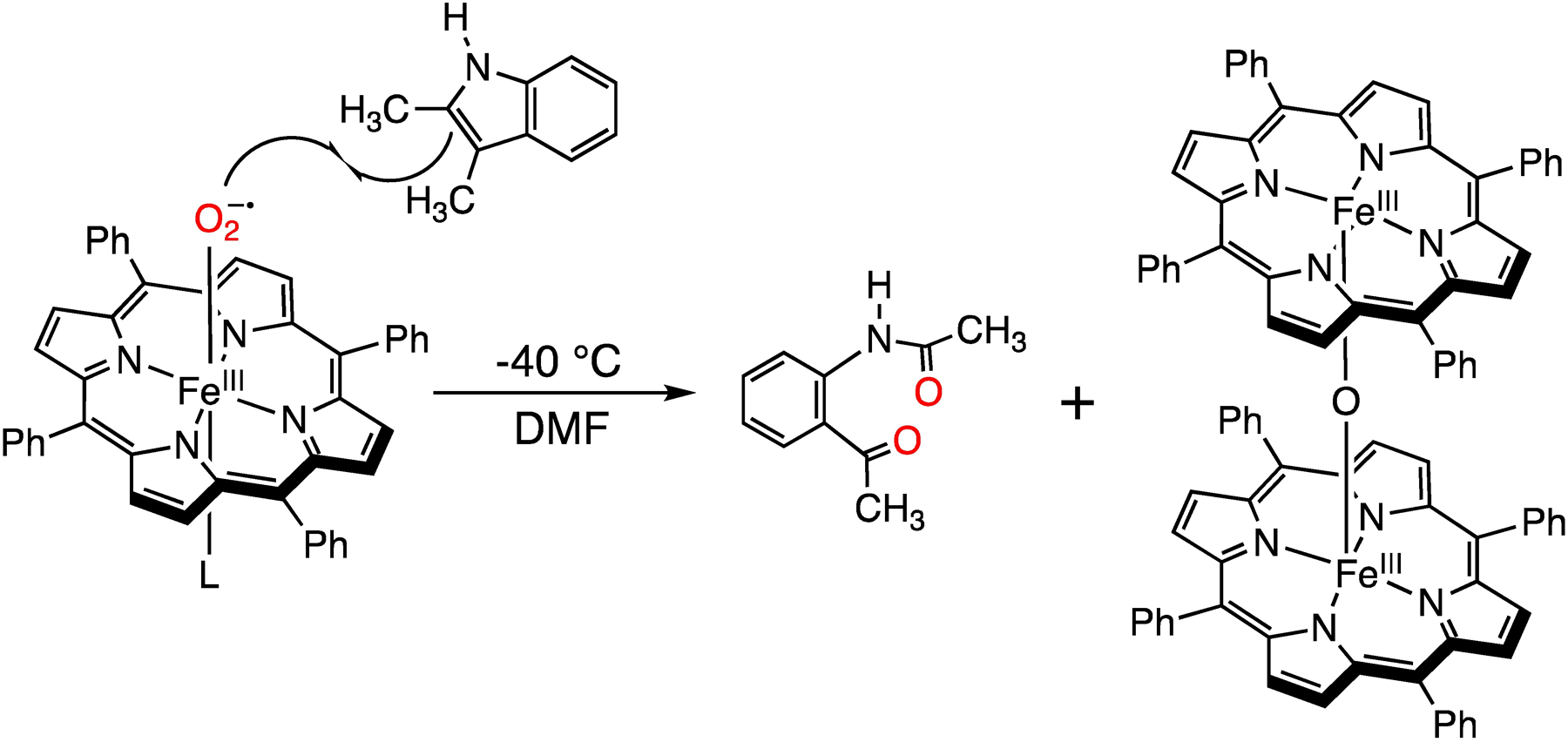
Reaction of FeIII(O2•−)(TPP) with 2,3-dimethylindole in DMF at −40 °C.
The same reaction was monitored in situ by low-temperature 1H NMR spectroscopy. The FeII(TPP) gives a paramagnetically shifted peak at 67 ppm in DMF-d7, which is consistent with a 5- coordinate high-spin FeII porphyrin with a single axial ligand.16 This peak disappears upon exposure to O2 and is replaced by a diamagnetic spectrum with two new peaks at 9.1, 9.8 ppm. The diamagnetic nature of the spectrum is consistent with an FeIII(O2•−) species with a spin ground state of S = 0 that arises from antiferromagnetic coupling between the iron and the superoxide ligand.17 Addition of 2,3-DMI leads to the growth of a peak at 13.1 ppm over 30 min, and loss of the peaks at 9.1, 9.8 ppm corresponding to the superoxide complex. The peak at 13.1 ppm comes from the (FeIII(TPP))2(μ-O) complex, which is the expected final decay product for the iron porphyrin species (Fig. 3). Qualitative observations of the concentrated NMR samples showed that the change from the red color of FeIII(O2•−)(TPP) to the green of (FeIII(TPP))2(μ-O) for the reaction with 2,3-DMI was significantly faster than control samples without the 2,3-DMI substrate, in good agreement with the UV-vis data.
Fig. 3.
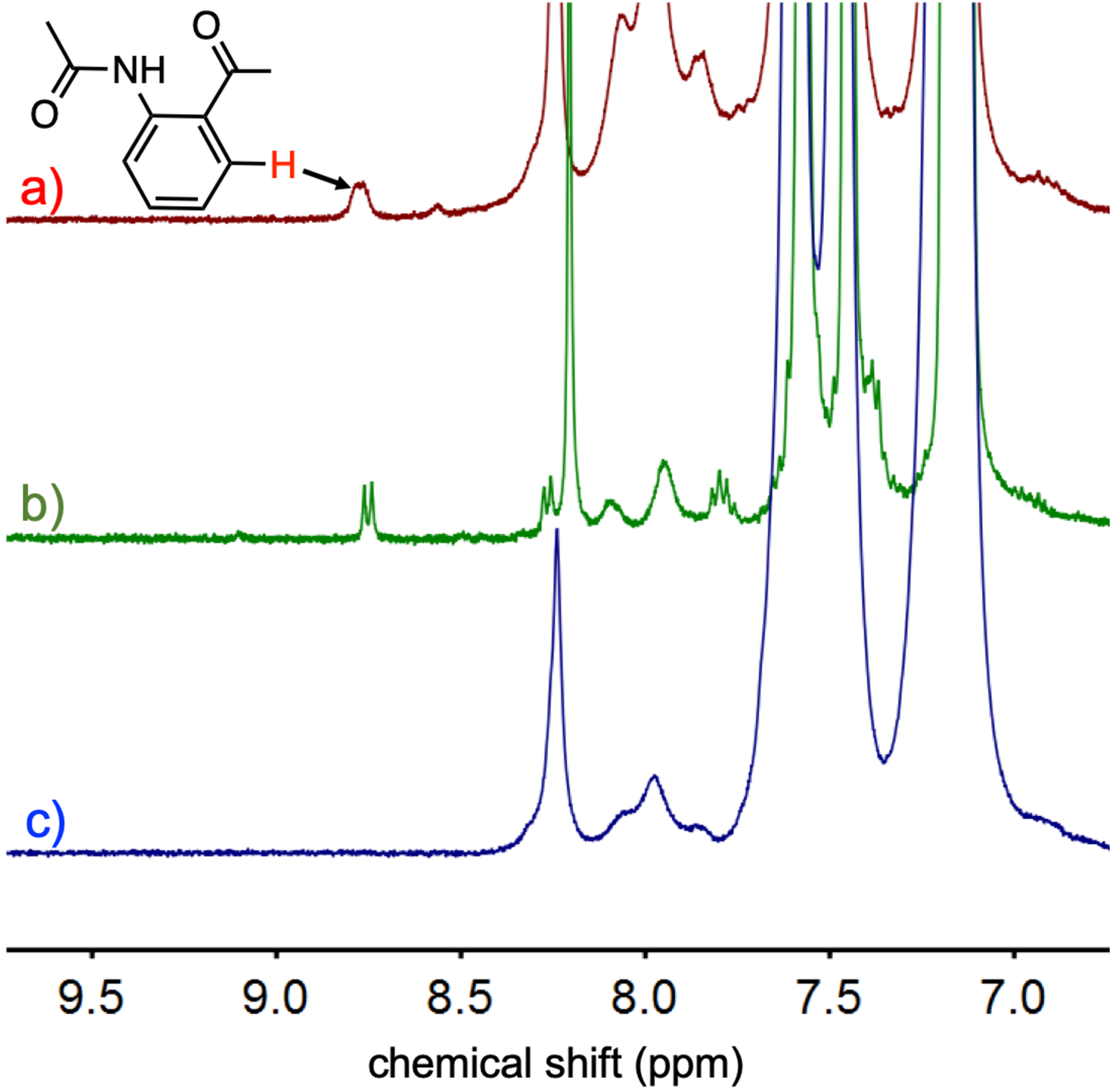
1H NMR spectra of a) FeII(TPP) (4 mM), b) FeIII(O2•−)(TPP) and c) reaction of FeIII(O2•−)(TPP) with 2,3-dimethylindole (30 equiv) in DMF-d7 at −40 °C.
Examination of the diamagnetic region of the low-temperature 1H NMR spectra for the in situ reaction between FeIII(O2•−)(TPP) and 2,3-DMI was also informative. The initial appearance of a peak at 8.8 ppm was seen within 10 min, and can be assigned to the aromatic proton ortho to the acetyl group of the ring-opened, dioxygenated product (Figure 4a). Integration of this peak and comparison to an internal standard at the end of the reaction at −40 °C gave a yield of 49 ± 8% (average of 3 runs). Thermal equilibration of the reaction mixture up to 25 °C and recollection of the NMR data leads to a yield of 61 ± 9% (average of 3 runs), which is within error of the yields observed at low-temperature. Direct addition of O2 to the same FeII(TPP)/2,3-DMI solution at 23 °C gives the N-(2-acetyl-phenyl)-acetamide product in similar yield, suggesting the transient formation of the superoxide complex is followed by the same reaction with 2,3-DMI. To rule out participation of the thermally stable, oxo-bridged (FeIII(TPP))2(μ-O) in the oxidation of the indole substrate, a solution of independently synthesized (FeIII(TPP)2)(μ-O) (4 mM) was combined with excess 2,3-DMI at −40 °C and monitored by 1H NMR. No new peaks are observed, indicating that there is no reaction between (FeIII(TPP)2)(μ-O) and 2,3-DMI (Figure 4c).
Fig. 4.
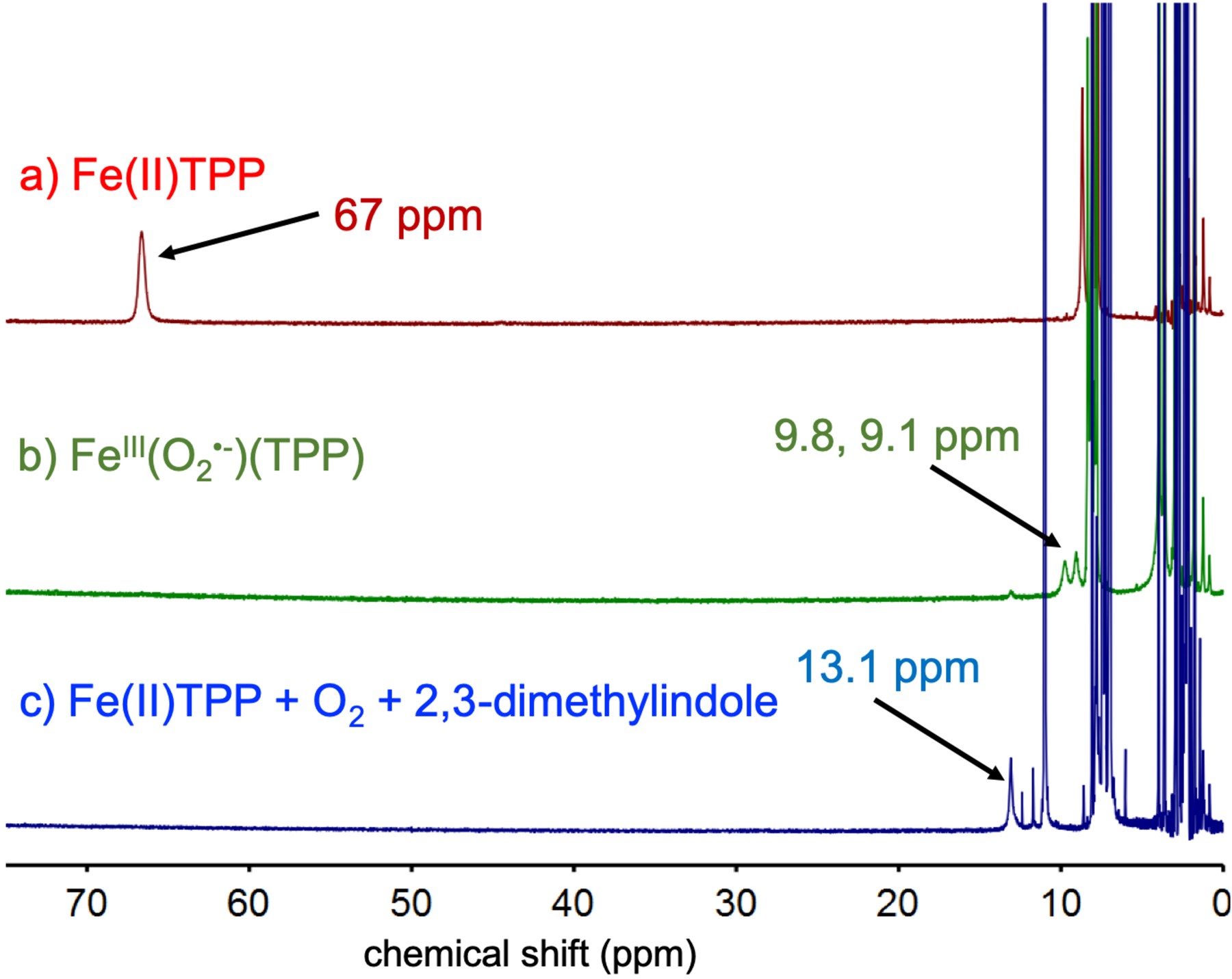
1H NMR spectra in DMF-d7 of the aromatic region of a) the reaction of FeIII(O2•−)(TPP) and 2,3-dimethylindole at −40 °C, b) after warm-up to 25 °C, and c) a mixture of independently synthesized (FeIIITPP)2(μ-O) and 2,3-dimethylindole at −40 °C.
The UV-vis and NMR data show that the iron(III) superoxide complex is competent to oxidize 2,3-DMI and give the ring- opened, dioxygenated N-(2-acetyl-phenyl)-acetamide product in reasonable yield at low temperature. The data also show that the overall rate of conversion of the starting FeIII(O2•−) species to the final μ-oxo product is enhanced by the presence of the substrate. However, the changes over time seen in the UV-vis data are non-isosbestic and suggest the presence of one or more intermediates over the course of the reaction with 2,3-DMI. These intermediates may involve the FeIII-O-O-FeIII and FeIV(O) porphyrin species that have been reported to form under certain conditions involving FeII(TPP) and O2.10,18 Although the overall reactivity and influence of 2,3-DMI on the rate of superoxo conversion to μ-oxo dimer suggests that the superoxo species is likely the initial oxidant for 2,3-DMI, the participation of the other O2-derived intermediates is possible, and will need further mechanistic analyses to sort out. In fact, proposed mechanisms for TDO/IDO invoke the participation of FeIII(O2•−), and further downstream FeIV(O) species, as active oxidants in the dioxygenation of substrate.5d, 7a
The results described in this Communication show that an iron(III) superoxide porphyrin is capable of oxidizing an indole substrate, leading to C2–C3 bond cleavage and dioxygenation as seen in TDO/IDO. This work also shows that the optimized environment of an enzyme active site is not required for this reaction to occur. There is current debate on whether the first step in TDO/IDO is an electrophilic attack on substrate by an FeII(O2) species, or a radical attack by an FeIII(O2•−) species (Scheme 3). Given the likely superoxide character of FeIII(O2•−)(TPP),17b, 17c, 19 we favour a radical-based mechanism, although an electrophilic mechanism remains a possibility for both the synthetic system and the enzymes. A more detailed investigation of the mechanism, including further kinetic and spectroscopic analyses, is warranted to gain further insight into the dioxygenation of indole substrates. This work also adds to the small but growing list of oxidations that can be initiated by a metal superoxide.
Scheme 3.
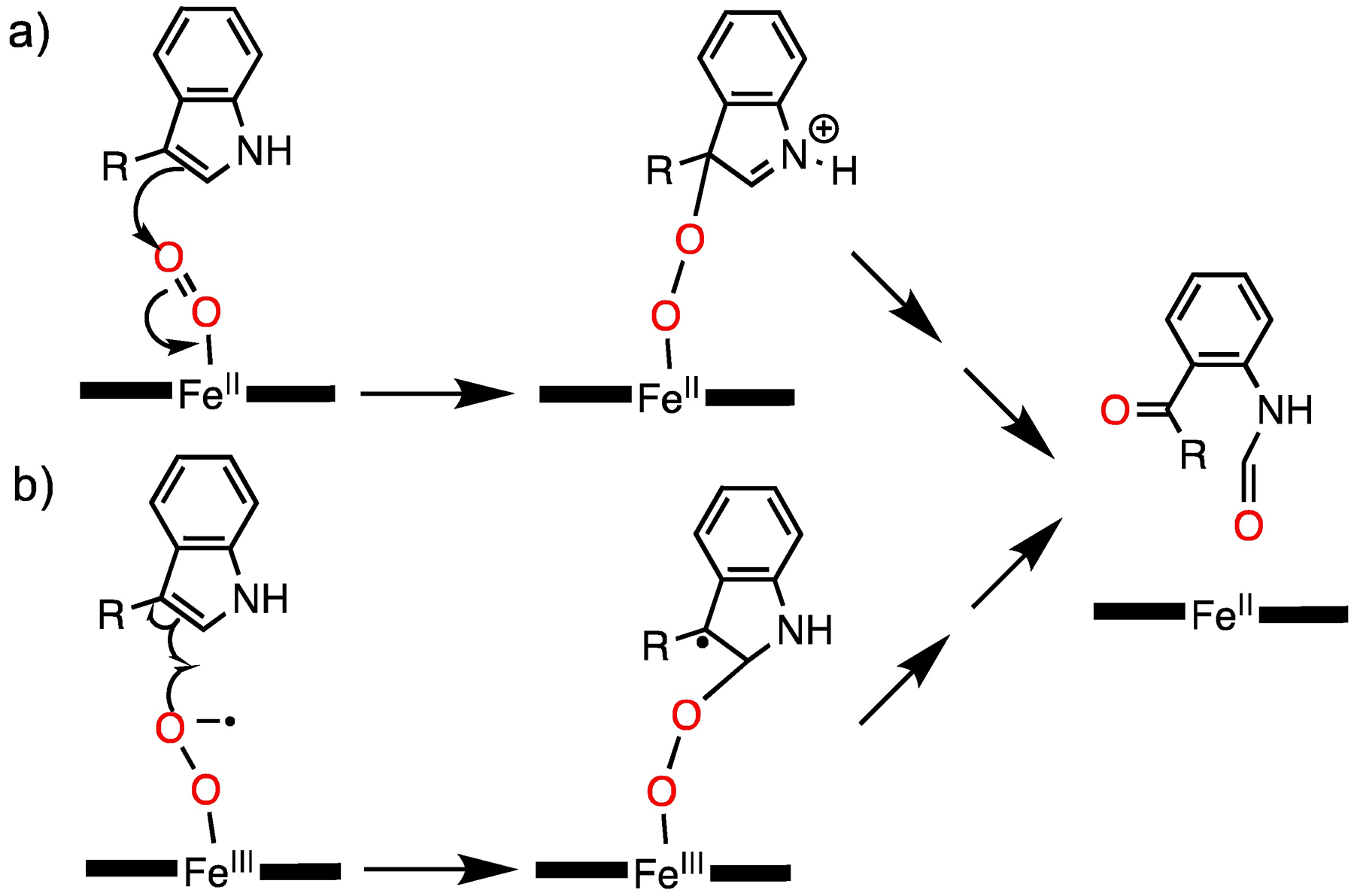
a) electrophilic and b) radical addition mechanisms for the reaction between an Fe(O2)(porphyrin) adduct and Trp substrate proposed for TDO/IDO.
Supplementary Material
Acknowledgments
The authors acknowledge research support from the NIH (GM101153 to D. P. G.).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Huang X and Groves JT, Chem. Rev, 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Sahu S and Goldberg DP, J. Am. Chem. Soc, 2016, 138, 11410–11428; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fiedler AT and Fischer AA, J. Biol. Inorg. Chem, 2017, 22, 407–424. [DOI] [PubMed] [Google Scholar]
- 3.a) Gardner PR, J. Inorg. Biochem, 2005, 99, 247–266; [DOI] [PubMed] [Google Scholar]; b) Huang H, Hah J-M and Silverman RB, J. Am. Chem. Soc, 2001, 123, 2674–2676; [DOI] [PubMed] [Google Scholar]; c) Zaragoza JP and Goldberg D, in Dioxygen-dependent Heme Enzymes, eds. Ikeda-Saito M and Raven EL, Royal Society of Chemistry, Cambridge, UK, 2019, pp. 3–30; [Google Scholar]; d) Efimov I, Basran J, Thackray SJ, Handa S, Mowat CG and Raven EL, Biochemistry, 2011, 50, 2717–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Tchesnokov EP, Faponle AS, Davies CG, Quesne MG, Turner R, Fellner M, Souness RJ, Wilbanks SM, de Visser SP and Jameson GN, Chem. Commun, 2016, 52, 8814–8817; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tamanaha E, Zhang B, Guo Y, Chang WC, Barr EW, Xing G, Clair J. St, Ye S, Neese F, Bollinger JM Jr. and Krebs C, J. Am. Chem. Soc, 2016, 138, 8862–8874; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Faponle AS, Seebeck FP and de Visser SP, J. Am. Chem. Soc, 2017, 139, 9259–9270; [DOI] [PubMed] [Google Scholar]; d) Mukherjee A, Cranswick MA, Chakrabarti M, Paine TK, Fujisawa K, Munck E and Que L Jr., Inorg Chem, 2010, 49, 3618–3628; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Noh H and Cho J, Coord. Chem. Rev, 2019, 382, 126–144; [Google Scholar]; f) Hong S, Lee Y-M, Ray K and Nam W, Coord. Chem. Rev, 2017, 334, 25–42; [Google Scholar]; g) Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Munck E and Lipscomb JD, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 16788–16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Shin I, Ambler BR, Wherritt D, Griffith WP, Maldonado AC, Altman RA and Liu A, J. Am. Chem. Soc, 2018, 140, 4372–4379; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lewis-Ballester A, Forouhar F, Kim S-M, Lew S, Wang Y, Karkashon S, Seetharaman J, Batabyal D, Chiang B-Y, Hussain M, Correia MA, Yeh S-R and Tong L, Nature, 2016, 6, 35169; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Davydov RM, Chauhan N, thackray SJ, Anderson JLR, Papadopoulou ND, Mowat CG, Chapman SK, Raven EL and Hoffman BM, J. Am. Chem. Soc, 2010, 132, 5494–5500; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lewis-Ballester A, Batabyal D, Egawa T, Lu C, Lin Y, Marti MA, Capece L, Estrin DA and Yeh SR, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 17371–17376; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Makino R, Obayashi E, Hori H, Iizuka T, Mashima K, Shiro Y and Ishimura Y, Biochemistry, 2015, 54, 3604–3616. [DOI] [PubMed] [Google Scholar]
- 6.Cervenka I, Agudelo LZ and Ruas JL, Science, 2017, 357. [DOI] [PubMed] [Google Scholar]
- 7.a) Basran J, Booth ES, Lee M, Handa S and Raven EL, Biochemistry, 2016, 55, 6743–6750; [DOI] [PubMed] [Google Scholar]; b) Booth ES, Basran J, Lee M, Handa S and Raven EL, J. Biol. Chem, 2015, 290, 30924–30930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Tajima K, Yoshino M, Mikami K, Edo T and Ishizu K, Inorg. Chim. Acta, 1990, 172, 83–91; [Google Scholar]; b) Ohkubo K, Sagawa T and Ishida H, Inorg. Chem, 1992, 31, 2682–2688; [Google Scholar]; c) Dufour MN, Crumbliss AL, Johnston G and Gaudemer A, J. Mol. Cat, 1980, 7, 277–287; [Google Scholar]; d) Tsuji J, Kezuka H, Takayanagi H and Yamamoto K, Bull. Chem. Soc. Jpn, 1981, 54, 2369–2373. [Google Scholar]
- 9.Oka S, Tajima K and Sakurai H, Chem. Pharm. Bull, 1998, 46, 377–383. [Google Scholar]
- 10.While this paper was under revision, a related work appeared, Mondal P, Wijeratne GB, J. Am. Chem. Soc 2020, 142, 1846–1856. [DOI] [PubMed] [Google Scholar]; In this study, the authors observed the reactivity of three Fe(O2•−)(porphyrin) derivatives (including TPP) with indoles to give the dioxygenated products, under different conditions than reported here. These results are consistent with our conclusion that Fe(O2•−)(porph) is competent to oxidize indole substrates. Spectroscopic data in this study showed that other species, including FeIV(O), could be involved in the oxidation.
- 11.Pegis ML, Martin DJ, Wise CF, Brezny AC, Johnson SI, Johnson LE, Kumar N, Raugei S and Mayer JM, J. Am. Chem. Soc, 2019, 141, 8315–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lexa D, Momenteau M and Mispelter J, Biochim. Biophys. Acta, 1974, 338, 151–163. [DOI] [PubMed] [Google Scholar]
- 13.Reed CA, Mashiko T, Scheidt WR, Spartalian K and Lang G, J. Am. Chem. Soc, 1980, 102, 2302–2306. [Google Scholar]
- 14.a) Anderson DL, Weschler CJ and Basolo F, J. Am. Chem. Soc, 1974, 96, 5599–5600; [DOI] [PubMed] [Google Scholar]; b) McCandlish E, Miksztal AR, Nappa M, Sprenger AQ, Valentine JS, Stong JD and Spiro TG, J. Am. Chem. Soc, 1980, 102, 4268–4271. [Google Scholar]
- 15.Boersma AD and Goff HM, Inorg. Chem, 1982, 21, 581–586. [Google Scholar]
- 16.Walker FA, in The Porphyrin Handbook, eds. Kadish K, Smith K and Guilard R, Academic Press, 1999, vol. 5, ch. 36, pp. 81–175. [Google Scholar]
- 17.a) Weiss JJ, Nature, 1964, 202, 83–84; [DOI] [PubMed] [Google Scholar]; b) Bren KL, Eisenberg R and Gray HB, Proc Natl Acad Sci U S A, 2015, 112, 13123–13127; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Phung QM and Pierloot K, Phys. Chem. Chem. Phys, 2018, 20, 17009–17019. [DOI] [PubMed] [Google Scholar]
- 18.Chin D-H, La Mar GN and Balch AL, J. Am. Chem. Soc, 1980, 102, 4344–4350. [Google Scholar]
- 19.a) Wagner WD, Paeng IR and Nakamoto K, J. Am. Chem. Soc, 1988, 110, 5565–5567; [Google Scholar]; b) Watanabe T, Ama T and Nakamoto K, J. Phys. Chem, 1984, 88, 440–445. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.