

Abstract
Heritable mitochondrial DNA (mtDNA) mutations are common, yet only a few recurring pathogenic mtDNA variants account for the majority of known familial cases in humans. Purifying selection in the female germline is thought to be responsible for the elimination of most harmful mtDNA mutations during oogenesis. Here we show that deleterious mtDNA mutations are abundant in ovulated mature mouse oocytes and preimplantation embryos recovered from PolG mutator females but not in their live offspring. This implies that purifying selection acts not in the maternal germline per se, but during post-implantation development. We further show that oocyte mtDNA mutations can be captured and stably maintained in embryonic stem cells and then reintroduced into chimeras, thereby allowing examination of the effects of specific mutations on fetal and postnatal development.
Keywords: oocyte, mitochondria, mtDNA
Our studies show that high heteroplasmy deleterious mtDNA mutations presenting in mature mouse oocytes are eliminated during post-implantation development. The occurrence of purifying selection against deleterious mtDNA mutations during fetal development plays an important role in preventing the accumulation of pathogenic mutations that would cause consequences to species survival.
Introduction
The mammalian mitochondrial DNA (mtDNA) genome is composed of 37 genes encoding 13 protein coding genes (i.e., 11 subunits of the electron transport chain and 2 subunits of ATP synthase, 22 tRNAs and 2 rRNAs (16S RNA and 12S RNA) critical for the production of energy via oxidative phosphorylation (OXPHOS). Typically, each mitochondrion contains multiple copies of the mitochondrial genome and each cell contains thousands of mitochondria [1]. Mutation rates in mtDNA are considerably higher than in the nuclear genome [2, 3], possibly due to the lack of protective histones and less active mtDNA repair mechanisms [4]. Pathogenic mtDNA mutations have been implicated in a variety of human diseases affecting multiple organs and tissues. Co-existence of mutant and wildtype mtDNA in the same cell is often observed, a state known as heteroplasmy. The incidence of somatic mtDNA mutations is particularly high, occurring randomly across cell, tissue, and organ types and accumulating during development and further in aging [5, 6]. In addition, heritable mtDNA mutations cause maternally transmitted diseases in offspring [7–9]. Although inherited mtDNA diseases are common and reported at a frequency of 1 in 5000, only a few recurring mtDNA variants account for the majority of known familial cases [7, 10, 11].
Naturally occurring animal models of mtDNA disease are rare. The mtDNA mutator mouse (PolGmut/mut), expressing a deficient proof-reading 3–5’exonuclease in the catalytic subunit of polymerase gamma, is an experimental knock-in model. Deficient polymerase allows replicative errors leading to the accumulation of mtDNA mutations [12, 13], the frequency of which increases with age, causing premature aging phenotypes including weight and hair loss, reduced fertility, heart enlargement, anemia, thin skin, and osteoporosis [12]. Although the occurrence of somatic mtDNA mutations in PolGmut/mut mutator mice is high, the transmission of deleterious germline mutations to live offspring is relatively rare [7, 8, 14]. These observations imply that most deleterious mtDNA mutations are selectively purged during intergenerational transmission [8]. While the exact timing or developmental stage at which such purifying selection operates is unknown [15], it has been suggested that most mtDNA mutations may adversely affect oocyte development and maturation in ovaries leading to selective elimination of most deleterious mutations in mature oocytes [8, 9, 16, 17]. We reasoned that many deleterious mtDNA mutations may actually escape negative selection during oogenesis owing to low energy dependence of female germ cells on OXPHOS [18]. However, these variants are negatively selected after fertilization, during embryonic and fetal development when the emergence of the somatic lineage and energy demands on OXPHOS are activated.
To test this hypothesis, we directly examined the spectrum of mtDNA mutations in ovulated MII oocytes and wildtype PolGwt/wt blastocysts, embryonic stem cells (ESCs), fetuses, and full-term offspring from heterozygous PolGmut/wt females. Similar to that found in mother’s somatic tissues, high heteroplasmy, deleterious mtDNA mutations were found in mature oocytes, blastocysts, and ESCs but were largely absent in viable PolGwt/wt fetuses and live offspring. It is important to note that oocytes, blastocysts, and ESCs represent various developmental phases of the female germline. We also demonstrate that most mtDNA mutations can be captured and stably maintained in individual PolGwt/wt ESCs supporting the generation of stem cell libraries carrying defined mtDNA defects. Moreover, reintroduction of mutant ESCs into chimeras provides an approach to study effects of specific mtDNA variants on fetal and postnatal development.
Materials and methods
Animals
All mice were maintained under specific-pathogen-free conditions in the Small Lab Animal Unit at Oregon Health & Science University (OHSU). Mice were housed under controlled lighting conditions (daily light period, 6 AM–6 PM). All animal experiments were approved by the Institutional Animal Care and Use Committee at OHSU. All guidelines of the approved protocols were fully followed in the current study. Chimeric animals were weaned at 21 days. To produce wildtype PolGwt/wt offspring, heterozygous PolGmut/wt females (C57BL/6N-Polgmut/N; kindly provided by Dr. Nils-Göran Larsson) [12] were crossed with wildtype PolGwt/wt males (C57BL/6N; Charles River laboratory).
Collection of MII oocytes, blastocysts, fetuses, and tissues
Heterozygous PolGmut/wt females were generated by pairing wildtype PolGwt/wt (C57BL/6N) females with heterozygous PolGmut/wt male and wildtype PolGwt/wt (C57BL/6N) females and wildtype PolGwt/wt F1 offspring from heterozygous PolGmut/wt females were superovulated with 5 international units pregnant mare’s serum gonadotropin and human chorionic gonadotropin. MII oocytes and blastocysts were individually collected. Skin samples from the tail were biopsied from PolGmut/wt mothers and their offspring after weaning. Fetuses were collected from euthanized pregnant PolGmut/wt mothers. Various tissues (brain, eye, heart, kidney, liver, skin, spleen, ovary, testis, and tail) were collected from euthanized chimeric animals. Primary skin fibroblast (SF) cell lines were derived from tail tip skin as described before [5]. Single SF colonies were seeded on P-100 plates and subcloned before extracting mtDNA for sequencing.
DNA extraction
MII oocytes, blastocysts, fetuses, and single SF colonies were individually collected and transferred into a 6 μl of DNA lysis buffer, and DNA was extracted with a Pico Pure DNA extraction kit (Applied Biosystems). Chimera animal skin DNA was extracted using a Purogen DNA purification kit (Qiagen) according to the manufacturer’s instructions.
Whole mtDNA sequencing
Mitochondrial DNA was amplified from whole DNA by single or double fragment(s) PCR reaction as previously described [5, 19, 20] using the following primers designed with Primer-BLAST tool (NCBI) that specifically recognize genuine mtDNA but not nuclear mitochondrial pseudo genes.
Single Fw-1546: TCTGGAACGGAAAAAACCTTTAATAGTG.
Single Rv-1545: AGAGCTGTCCCTCTTTTGGCTATAATTTA.
Double Fw-3222: GGATCCTACTCTCTACAAAC.
Double Rv-11423: TAGTTTGCCGCGTTGGGTGG.
Double Fw-11271: CTACCCCCTTCAATCAATCT.
Double Rv-3335: CCGGTTTGTTTCTGCTAGGG.
Single PCR conditions were: 94°C for 1 min then 98°C for 10 s, 64.2°C for 30 s and 68°C for 16 min, 30 cycles and then 72°C for 10 min. Double PCR conditions were: 95°C for 5 min then 94°C for 30 s, 56°C for 30 s and 68°C for 10 min, 30 cycles and then 68°C for 20 min (TAKARA LA Taq, Cat No. RR02AG). The concentration of PCR products was measured by Qubit 2.0 Fluorometer (Invitrogen). PCR products were used for library preparation following Nextera XT DNA kit (Illumina). Sequencing was performed on the Illumina MiSeq platform at Molecular & Cellular Biology Support Core at Oregon National Primate Research Center.
MiSeq data analysis
MiSeq sequencing data were analyzed using NextGENe software [19]. In brief, sequence reads ranging from 100 to 200 bps were quality filtered and processed using NextGENe software and an algorithm similar to BLAT. Sequence error correction feature (condensation) was performed to reduce false positive variants and produce sample consensus sequence and variant calls. Alignment without sequence condensation was used to calculate the percentage of the mitochondrial genome with depth of coverage of 1000. Starting from quality FASTQ reads, the reads were quality filtered and converted to FASTA format. Filtered reads were then aligned to the Mus musculus strain C57BL/6 J mitochondrion sequence reference (GenBank: AY172335.1) followed by variant calling. Variant heteroplasmy was calculated by NextGENe software as follows; Base heteroplasmy (Mutant Allele Frequency %) = mutant allele (forward + reverse)/total coverage of all alleles C, G, T, A (Forward + Reverse) *100. The clinical significance of the mtDNA variants was analyzed by homology search to human database in MitoMaster (http://www.mitomap.org/MITOMASTER/WebHome).
Validation of mtDNA mutations by Sanger sequencing
Some mtDNA mutations with > 15% heteroplasmy were confirmed by Sanger sequencing. The PCR products were purified, sequenced, and analyzed by Sequencher v. 5.0 (GeneCodes).
Derivation and culture of ESCs
Superovulated PolGwt/wt and PolGwut/wt females were paired with PolGwt/wt or C57BL/6JUbc-GFP (Jackson laboratory) males. Embryos (morula-blastocyst stage) were flushed and collected from the uteri of E2.5 mated females. Embryos were then cultured in KSOM medium for 24h. The zona pellucida was removed with acidic Tyrode’s solution (Sigma-Aldrich) and blastocysts were individually transferred into mitomycin C (Sigma-Aldrich)-treated mouse embryonic fibroblast (mEF) feeder layers in ESC derivation medium: KODMEM (Invitrogen) containing 1 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, 100 μM β-mercaptoethanol (Sigma), 100 μM nonessential amino acids (Invitrogen), 1000 units/ml LIF (Millipore), 10% FBS, and 10% KOSR (Invitrogen).
After ICM outgrowth, the colony was picked up manually and dispersed into single cells using 0.15% trypsin solution (Sigma-Aldrich) before seeding onto mEF in ESC medium containing 1 μM PD0325901 (Axon) and 3 μM CHIR99021 (Axon).
Chimera generation from mutant ESCs
The method of chimera injection, embryo transfer, and delivery of offspring has been described elsewhere [21, 22]. In brief, approximately 10 ESCs were injected into 4–8-cell stage host embryos from ICR strain. Injected embryos were cultured 24h in AA-KSOM (Millipore) at 37°C under 5% CO2 in air.
The presence of GFP (+) cells in embryos was examined by fluorescence. Chimeric blastocysts were transferred into the uteri of pseudo-pregnant ICR females. Cesarean sections were performed on embryonic day 20.5 if animals did not deliver spontaneously. Implantation sites were examined at weaning on day 21 or at caesarian section.
Fasting glucose and glucose tolerance test
Fasting blood glucose levels in mice were determined from tail vein blood drops after 12h of fasting using One Touch Ultra kit (Johnson and Johnson).
For glucose tolerance test, 1 g/kg glucose was injected into the peritoneum after 12h fasting and blood glucose levels were determined from tail vein blood drops collected before injection and after injection in 15 and 30 min and 1 and 2h intervals using One Touch Ultra kit.
Echocardiography
Under light anesthesia (0.5–1.5% isoflurane), cardiac structure and function were evaluated in vivo by high-frequency ultrasound (Vevo2100 with MS-400 prove, FUJIFILM VisualSonics). Ventricular size was quantified in the parasternal long-axis. Left ventricular (LV) ejection fraction (%) was calculated as [(LVEDV−LVESV)/LVEDV] X 100, where LVEDV is LV end-diastolic volume and LVESV is LV end-systolic volume [23].
Optokinetic response test and optical coherence tomography
Optokinetic response test (OKR) thresholds were measured using a virtual optomotor system (VOS; CerebralMechanics) that allows evaluation of the left and right eyes independently. Thresholds were evaluated at every third month, using methods described elsewhere [24–27]. Thresholds were evaluated by one primary operator with periodic confirmation from a second operator. Mice were imaged when OKR showed a blind pattern. Before imaging, pupils were dilated with 1% tropicamide and 2.5% phenylephrine. Artificial tears were used throughout the procedure to maintain corneal clarity. Sedation was induced by ketamine (80 mg/kg)/xylazine (5 mg/kg). SD-optical coherence tomography (OCT) images were obtained using the Envisu R2200-HR SD-OCT device (Bioptigen, Durham, NC) with the reference arm placed at approximately 1187 mm. Horizontal and vertical linear scans (2 mm width, 1500 a-scans/b-scan · 20 frames/b-scan) were obtained first while centered on the optic nerve, then with the nerve displaced either temporally/nasally or superiorly/inferiorly [28].
Auditory brainstem response test
Auditory brainstem response (ABR) thresholds were obtained in mice in response to tone bursts at 4, 8, 16, and 32 kHz. Waveforms were generated from a Tektronix AWG 2020 using a 256 kHz sampling rate with 12-bit amplitude resolution. The speaker (Realistic, SuperTweeter, Cat. No. 40-1310B) delivered sound via a tightly fit tube into the animal’s ear canal. Stainless-steel needle electrodes were placed on the animal’s vertex (noninverting), below the ipsilateral ear (inverting) and on the front leg (ground). Biological signals were bandpass filtered (high pass at 300 Hz and low pass at 3000 Hz) and amplified (100 000) with a differential amplifier (Grass P511). Responses to 300 stimulus repetitions were averaged using a digital storage oscilloscope (Tektronix, TDS 420) with a sampling rate of 25 kS/s. Thresholds were scored as the lowest intensity at which a response could be visually detected above noise in at least two of the five waves of the ABR [29].
Histological analyses
Mice were euthanized by exposure to CO2 followed by cervical dislocation. The euthanasia method followed the OHSU policy on Carbon Dioxide Euthanasia of Rodents, and is consistent with the American Veterinary Medical Association guidelines for the Euthanasia of Animals. Tissues were extracted and fixed in 10% neutral buffered formalin overnight, embedded in paraffin, and processed by the pathology service unit of the Oregon National Primate Research Center.
Mitochondrial respiratory chain enzyme assays
Respiratory chain complex I and IV activities were assayed in each ESC line and muscle tissue homogenates by spectrophotometric methods as described previously [30]. A VersaMax microplate reader system (Molecular Devices) in combination with SoftMax Pro software was used for activity measurements and calculations. Enzyme activities were expressed as a percentage relative to rotenone inhibition in complex I and as absolute value (nmol/min/mg protein) in complex IV.
Statistical analyses
Fisher’s protected least significant difference test (Fisher’s test) was used for the comparisons in Table 1 and Figure 3B, D and E. Student’s t-test was used for the remaining data, which were presented as the mean ± SEM. Significance was set at P < 0.05.
Table 1.
Pregnancy outcomes of ESC chimeric embryos carrying mtDNA mutations.
| Cell line (sex) | Pathogenic mutations (heteroplasmy %) | Type of negative selection for mutations | No. of ET | No. of implantation (%/ET) | No. of total term pups (%/ET) | No. of live pups (%/ET) | No. of chimeras | No. of high chimerism chimerasb (sex) |
|---|---|---|---|---|---|---|---|---|
| ND6-100 (female) | 6106A > T COXI (94%) 13026 T > C ND5 (95%) 13967C > T ND6 (100%) | High heteroplasmy | 533 | 195 (36.6%) | 90 (16.9%) | 76 (14.3%) | 35 | 4 (female) |
| ND2-50 (female) | 4871A > T ND2 (50%) | High heteroplasmy | 588 | 227 (38.6%) | 74 (12.6%) | 64 (10.9%) | 34 | 2 (female) 1 (male) |
| ATP8-62 (male) | 7769C > G ATP8 (62%) | High heteroplasmy | 394 | 139 (35.3%) | 47 (11.9%) | 39 (9.9%) | 25 | 7 (male) |
| COXIII-81 (female) | 6726A > G COXI (63%) 9138G > A/9278A > T COXIII (81%/81%) 12853C > T/13441C > T ND5 (52%/91%) | Selected gene with high heteroplasmy | 278 | 93 (33.5%) | 23 (8.3%)a | 19 (6.8%)a | 12 | 1 (male) |
| Control-1 (female) | None | None | 239 | 92 (38.4%) | 40 (16.7%)a | 35 (14.6%)a | 17 | 3 (female) 1 (male) |
aThe rate of term pups in COXIII-81 chimeras was significantly lower than that in control-1 chimeras (P < 0.01).
bHigh chimerism chimera was defined as ≥50% chimerisms determined by coat colors and whole mtDNA sequencing for C4891T SNP between B6N ESC (C) and host ICR (T).
Figure 3.
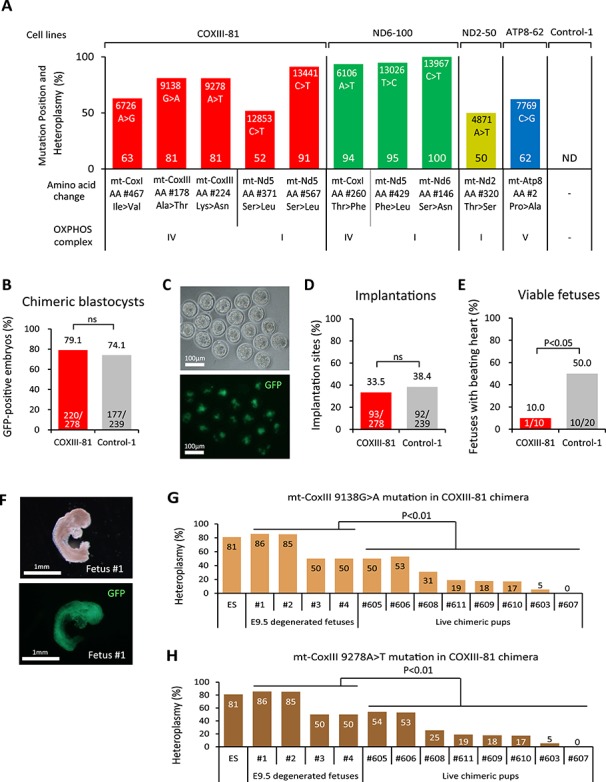
Fetal lethality of COXIII-81 chimeras. (A) Heteroplasmy levels (%) and locations of non-synonymous protein-coding mutations in COXIII-81, ND6-100, ND2-50, and ATP8-62 ESC lines. ND indicates that mtDNA mutations were not detected in Control-1 ESC line. (B) Proportion of GFP-positive ESCs in COXIII-81 and chimeric blastocysts (E3.5). ns denotes P ≥ 0.05. (C) Detection of COXIII-81 ESC contribution by GFP in chimeric blastocysts. The upper panel is a bright field image and the lower panel is a fluorescent image. Scale bar = 100 μm. (D) Implantation rate of COXIII-81 and Control-1 chimeric embryos. The implantation rate was calculated as the number of implantation site/number of embryos transferred. ns denotes P ≥ 0.05. (E) Percent of viable E9.5 fetuses in COXIII-81 and Control-1 chimeras. The viable fetus rate was estimated as the number of viable fetuses/number of implantation sites. (F) Morphology of degenerated E9.5 fetus (Fetus #1) from COXIII-81 chimeras. The upper panel shows a bright field image and the lower panel shows a fluorescent image. Scale bar = 1 mm. (G) Heteroplasmy levels of 9138 G > A mutation in mt-CoxIII in degenerated E9.5 fetuses and in live chimeric pups. (H) Heteroplasmy level of 9278 A > T mutation in mt-CoxIII in degenerated E9.5 fetuses and in live chimeric pups.
Data availability
Sequencing data have been uploaded to the sequence read archive under accession number SRP131410. All other data are available from the authors upon reasonable request.
Results
MtDNA mutations in oocytes and offspring
To investigate the exact timing during which the negative selection for mtDNA mutations operates, we initially examined the entire spectrum of somatic mtDNA mutations in SF of the PolGmut/wt females by whole mtDNA sequencing and compared those to the germline mutations measured directly in individual MII oocytes and live PolGwt/wt F1 offspring generated by intercrossing with PolGwt/wt males (Figure 1A). Heterozygous PolGmut/wt F1 offspring were omitted from analyses due to masking of the initial germline mtDNA mutations with additional de novo mutations generated by deficient polymerase gamma. Whole mtDNA sequencing was performed using Illumina MiSeq-based platform and single-nucleotide variants with heteroplasmy levels ≥2% were included in analyses [31]. This cut-off level was set based on a previous report determining that heteroplasmy levels less than 2% may also represent technical error of the method [32].
Figure 1.
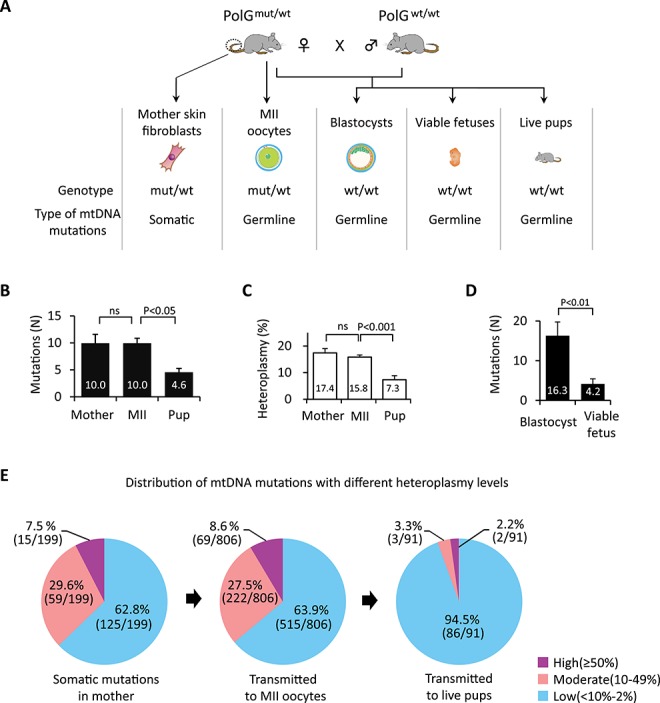
Spectrum of mtDNA mutations in somatic cells of PolGmut/wt females and their germline transmission. (A) Schematic illustration of experimental design. To investigate the timing when purifying selection for mtDNA mutations operates, somatic mutations in the SF of PolGmut/wt mothers (Mother) and germline transmitted variants in MII oocytes (MII), PolGwt/wt blastocysts, PolGwt/wt viable fetuses at E9.5, and PolGwt/wt live offspring SF (Pup) were examined by mtDNA whole genome sequencing (MiSeq). (B) Quantification of mtDNA mutations in PolGmut/wt female SFs (Mother, n = 20) and their MII oocytes (MII, n = 81), and PolGwt/wt pup SFs (Pup, n = 20). (C) Quantification of heteroplasmy levels of mtDNA variants in PolGmut/wt female SFs (n = 20) and their MII oocytes (n = 81) and PolGwt/wt pup SFs (n = 20). (D) Quantification of mtDNA mutations in PolGwt/wt blastocysts at E3.5 (n = 20) and PolGwt/wt viable fetuses at E9.5 (n = 20). (E) Effect of heteroplasmy levels on germline transmission of mtDNA mutations in PolGmut/wt females SFs (n = 20) and their MII oocytes (n = 81) and PolGwt/wt pup SFs (n = 20). Purple, pink, and blue colors represent mtDNA variants with high (≥50%), moderate (10–49%), and low (<10–2%) heteroplasmy levels, respectively. In B–D, error bars are mean ± SEM; ns denotes P ≥ 0.05.
Sequencing in individual SF (n = 20) from tails of four young PolGmut/wt females revealed that each cell carried multiple mtDNA variants (Table S1) with a mean number of 10.0 ± 1.7 and mean heteroplasmy level of 17.4 ± 1.6% (Figure 1B and C; Supplemental Table S1). Remarkably, individual mature MII oocytes (n = 81) collected from the same females also showed a comparable number of mtDNA variants with a mean number of 10.0 ± 0.9 and mean heteroplasmy level of 15.8 ± 0.8% (P > 0.05). As expected, individual SF from PolGwt/wt live offspring contained significantly lower number of mutations with a mean number of 4.6 ± 0.7 and mean heteroplasmy of 7.3 ± 1.5% (P < 0.05). Since more than half mtDNA variants present in oocytes of PolGmut/wt females disappear in live born offspring, these results suggest that the purifying selection likely acts after fertilization.
To further dissect the developmental stage of purifying selection, we examined mtDNA mutations in PolGwt/wt blastocysts (E3.5) and viable fetuses (E9.5; with beating hearts) recovered from PolGmut/wt females. PolGwt/wt blastocysts (n = 20) carried, on average, 16.3 ± 3.4 variants while viable fetuses (n = 20) showed significantly fewer mutations at 4.2 ± 1.0 (Figure 1D and Supplemental Table S1). These results imply that the purifying selection process is operative during post-implantation development.
To evaluate the role of heteroplasmy levels in the transmission of mtDNA mutations from mothers to their oocytes and offspring, mtDNA variants were divided into high (≥50%), moderate (11–49%), and low (2–10%) heteroplasmy subgroups for comparison. The number of mutations with various heteroplasmy levels in mother’s somatic cells was comparable to those in MII oocytes, while most variants in live pups were at low heteroplasmy (Figure 1E and Supplemental Figure S1A). These results support the conclusion that most mtDNA mutations, including those with high and moderate heteroplasmy levels are tolerated in mature oocytes (i.e., female germline) but not in live offspring.
Gene-specific negative selection of mtDNA variants
The majority of mtDNA variants detected in mothers’ somatic cells, MII oocytes, and transmitted to live pups resided in protein-coding genes (76.9–84.7%), while frequencies of rRNA (6–15.4%), tRNA (5.5–7.7%) variants, and the variants in the non-coding control region (CR) (0–2%) were relatively lower (Supplemental Figure S1B).
To determine whether selective elimination of mutations is gene-specific, we examined germline transmission of non-synonymous, protein-coding variants in OXPHOS enzyme complexes. In MII oocytes, 45.8% (367/800) of mtDNA variants were non-synonymous in protein-coding genes (Supplemental Table S2). The mean number of variants in complexes III and V was comparable between mother’s SFs, MII oocytes, and live born PolGwt/wt pups (Figure 2A). The mean numbers of variants found in complexes I and IV, including mt-Nd4l, mt-Nd5, mt-CoxI, mt-CoxII, mt-CoxIII, and mt-Atp6 were similar between mother’s SFs and MII oocytes but significantly reduced in PolGwt/wt pups (P < 0.05; Figure 2A and B). In contrast, the mean number of mutations in mt-Nd1, mt-Nd2, mt-Nd3, mt-Nd4, mt-Nd6, mt-Cytb, and mt-Atp8 genes was comparable between the groups (Supplemental Figure S2A).
Figure 2.
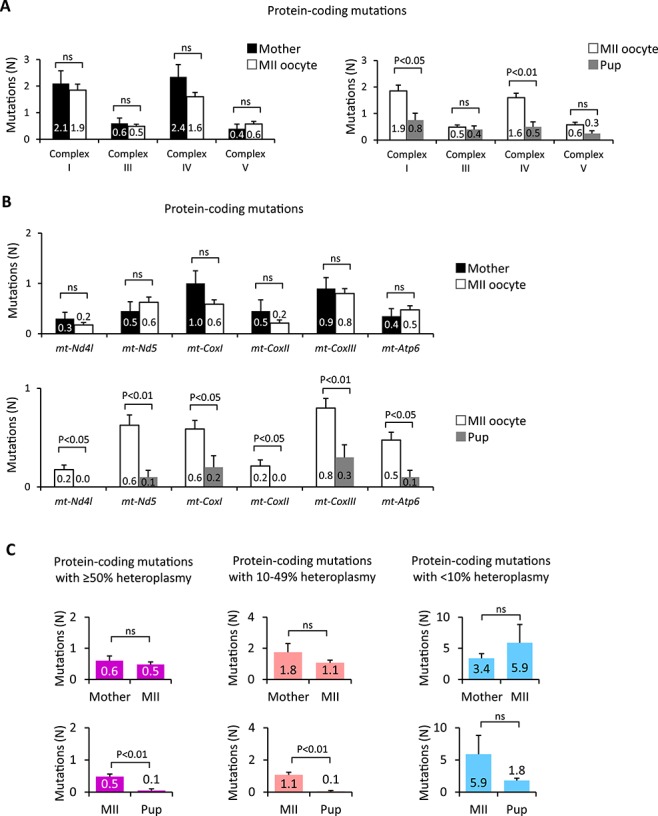
Gene-specific negative selection of mtDNA mutations. (A) Quantification of non-synonymous mtDNA mutations in genes encoding OXPHOS complexes I, III, IV, and V in PolGmut/wt female SFs (Mother, n = 20) and their MII oocytes (MII, n = 81) and PolGwt/wt pup SFs (Pup, n = 20). (B) Germline transmission of non-synonymous mtDNA mutations in mt-Nd4l, mt-Nd5, mt-CoxI, mt-CoxII, mt-CoxIII, and mt-Atp6 measured in PolGmut/wt female SFs (Mother, n = 20) and their MII oocytes (MII, n = 81) and PolGwt/wt pup SFs (Pup, n = 20). (C) Quantification of non-synonymous protein-coding mutations with different heteroplasmy levels in PolGmut/wt female SFs (Mother, n = 20) and their MII oocytes (MII, n = 81) and PolGwt/wt pup SFs (Pup, n = 20). Heteroplasmy levels were grouped into high (≥50%), moderate (10-49%), and low (≤10%) groups. In A–C, error bars are mean ± SEM; ns denotes P ≥ 0.05.
To further dissect the impact of heteroplasmy on germline transmission of mtDNA variants, non-synonymous protein-coding variants were grouped into high, moderate, and low heteroplasmy levels. The mean number of high and moderate heteroplasmy protein coding variants transmitted to PolGwt/wt pups was significantly lower compared to mother’s somatic cells or MII oocytes (P < 0.05; Figure 2C). In contrast, the mean numbers of low heteroplasmy mutations in mother, MII oocytes, and PolGwt/wt pups were comparable. These results suggest that the purifying selection varies for specific mtDNA genes and their heteroplasmy levels.
Purifying selection for RNA-coding variants was also examined. The mean numbers of variants with high and moderate heteroplasmy levels found in tRNAs (0.3 ± 0.07) and rRNAs (0.3 ± 0.06) in oocytes were similar to that in mother’s SF (tRNA0.3 ± 0.09; rRNA0.3 ± 0.09) (Supplemental Figure S2B and C and Supplemental Table S2). The mean numbers of variants in tRNAs and rRNAs transmitted to pups (tRNA0.1 ± 0.05; rRNA0.1 ± 0.07) were reduced compared to those in oocytes, but the difference was not significant (P > 0.05; Supplemental Figure S2B and C and Supplemental Table S2), likely indicating that most RNA-coding mutations are inconsequential. The mean number of mutations observed in the CR in mother’s SF was at 0.1 ± 0.05, which was comparable to the mean number of 0.2 ± 0.06 in MII oocytes and no mutations were found in the CR in PolGwt/wt pups (Supplemental Figure S2D). In addition, no mutations were found in the conserved sequence block of the CR implicated in replication or transcription [33, 34] (Supplemental Table S2).
In summary, these results suggest that most deleterious mtDNA variants with high heteroplasmy levels are tolerated by mature, ovulated oocytes but negatively selected in live offspring.
Generation of ESCs with specific mtDNA variants
We assumed that most mtDNA variants found in MII oocytes are pathogenic based on the observation that they are negatively selected in live offspring. Here, we developed an assay to directly test the developmental consequences of selected oocyte mtDNA mutations. Since the majority of oocyte mtDNA variants are also present in preimplantation blastocysts, we asked if we could capture these variants in PolGwt/wt ESCs. To this end, we established a total of 28 stable ESC lines from 37 expanded blastocysts produced from crossing PolGmut/wt females with PolGwt/wt males. Genotyping revealed that 8 ESC lines were PolGwt/wt and each carried between 1 and 5 high heteroplasmy (≥50%) non-synonymous variants in protein-coding genes that were stably maintained during extended culture (Supplemental Figure S3A and Supplemental Table S3). We further characterized four individual ESC lines designated ND6-100, ND2-50, ATP8-62, and COMIII-81, each carrying between 1 and 5 mtDNA variants (Figure 3A). An unaffected ESC line (Control-1) derived from PolGwt/wt was used as a control. To predict the possible phenotype of these variants, mouse ESC mutations were first compared to a human mtDNA disease database (MITOMAP: http://www.mitomap.org/MITOMAP). We noted that all non-synonymous variants are either reported as coding polymorphisms or not described in the human database, and therefore, their function was unclear (Supplemental Table S3).
We next measured OXPHOS complex activities in undifferentiated mutant ESCs. COXIII-81 and ND6-100 cells displayed significantly reduced complex I and IV activities while ND2-50 line presented reduced complex I activity (Supplemental Figure S3B and C). ATP8-62 cell line carrying variants in complex V displayed normal complex I and IV activities. These outcomes demonstrate the feasibility of creating a library of ESCs carrying specific mtDNA variants (Figure 3A and Supplemental Figure S3B and C).
Impact of mtDNA variants on fetal development in chimeras
Since most deleterious mtDNA mutations cause early fetal demise, we reasoned that complementation of the mutant ESCs with normal extra-embryonic tissues of host embryos in chimeras could allow extension of phenotypic studies in fetal and postnatal stages [35, 36]. ESCs were injected into host 4-cell ICR embryos, and chimeric blastocysts were transferred to recipients. The degree of ESC contribution in chimeric embryos and fetuses was determined by GFP expression or by coat color pigmentation in live offspring. In addition, we used ESC mtDNA mutations and neutral polymorphism at position (4891C > T) between ESCs and ICR strains to aid in chimerism detection (Supplemental Table S4).
In blastocysts, a contribution of GFP positive COXIII-81 ESCs was seen in 79.1% (220/278) of embryos with an implantation rate of 33.5% (93/278), which was comparable to controls (74.1%, 177/239 and 38.4%, 92/239, respectively) (Figure 3B–D). However, the number of viable E9.5 fetuses with beating hearts detected in COXIII-81 chimeras (10.0%, 1/10) was significantly reduced over controls (50.0%, 10/20) (Figure 3E). In addition, degenerated fetuses were observed in COXIII-81 chimeras but not in controls (Figure 3F). Similarly, development to full-term (8.3%, 23/278) and to live pups (6.8%, 19/278) was also significantly reduced over controls (16.7% 40/239 and 14.6% 35/239) (Table 1). Only one surviving (chimera #605 in Supplemental Figure S4) out of 12 born pups from COMIII-81 ESCs carried a high stem cell contribution (Table 1 and Supplemental Figure S4A). Genotyping of four degenerated E9.5 fetuses revealed that affected chimeras had high ESC contributions (Supplemental Figure S4A). In addition, heteroplasmy levels of 9138G > A and 9278A > T mutations in mt-CoxIII gene carried in degenerated fetuses were significantly higher compared to live born chimeras (Figure 3G and H). Similar patterns were observed for three other mutations in this cell line including 6726A > G in mt-CoxI and 12853C > T and 13441 C > T in mt-Nd5 (Supplemental Figure S4B–D). These results indicate that these mtDNA variants adversely affect early fetal development, particularly before E9.5, corroborating pathogenicity in these variants (Figure 2B). It should be noted that chimeras with low ESC contribution may avoid this negative selection and develop to full-term.
In contrast to COXIII-81 ESCs, implantation, full-term and live pup development rates for ND6-100, ND2-50, and ATP8-62 high ESC-chimeras were comparable to controls (Table 1). These results suggest that these mutations can be rescued in chimeras allowing assessment of these specific mtDNA defects on postnatal development.
Postnatal phenotypes of mtDNA variants
Postnatal survival to weaning at day 21 in ESC chimeras was monitored. Most live born chimeric offspring including COXIII-81, ND2-50, and ND6-100 survived to weaning with an exception of ATP8-62 pups (Figure 4A). Since some mtDNA mutations are associated with diabetes mellitus in humans [37, 38], we performed glucose tolerance tests on several high contribution (≥50%) 3–6 month old ND6-100, ND2-50, and ATP8-62 chimeras and compared those to controls. For each animal, two times of measurements were performed. Mean 2h blood glucose levels in ND6-100 (145.8 ± 9.0 mg/dl), ND2-50 (172.3 ± 18.3 mg/dl), and ATP8-62 (145.5 ± 13.5 mg/dl) chimeras were all significantly elevated compared to controls (105.5 ± 11.4 mg/dl) (Figure 4B). Mean fasting blood glucose levels in ND6-100 (151.5 ± 9.4 mg/dl), ND2-50 (125.8 ± 3.4 mg/dl), and ATP8-62 (129.7 ± 10.3 mg/dl) chimeras was also significantly higher than in controls (92.7 ± 8.2 mg/dl) (Supplemental Figure S5A). In addition, intractable cutaneous ulcers that are associated with severe diabetes [39] were observed in ND6-100, ND2-50, and ATP8-62 chimeras (Supplemental Figure S5B), but not in controls. COXIII-81 chimera (n = 1) also displayed elevated fasting blood glucose (117.5 ± 33.5 mg/dl in Supplemental Figure S5A) and 2h glucose levels (154.5 ± 45.5 mg/dl in Figure 4B); however, the differences did not reach significance perhaps reflecting the small animal number.
Figure 4.
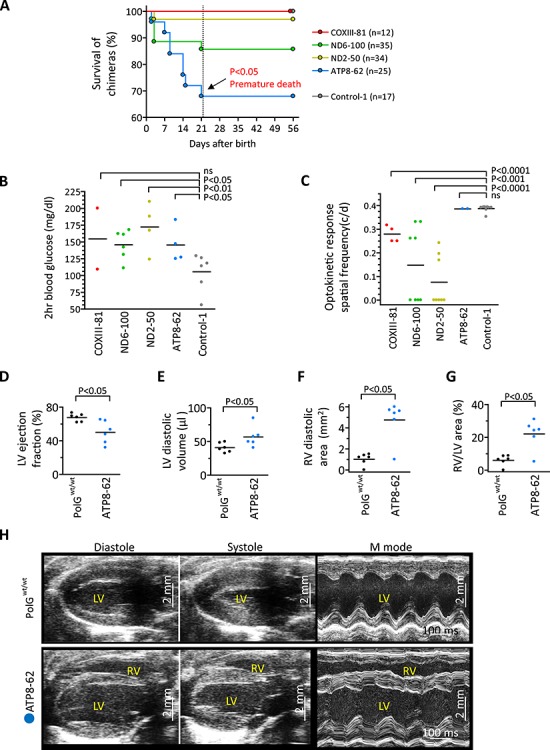
Phenotypes of chimeras carrying deleterious mtDNA mutations. (A) Survival rate of chimeras with COXIII-81 (n = 12), ND6-100 (n = 35), ND2-50 (n = 34), ATP8-62 (n = 25), and control (n = 17) ESC lines at 21 days (dotted line). Premature death was observed in ATP8-62 chimeras, resulting in a significantly lower survival rate compared to controls. (B) Two hour blood glucose levels (mg/dl) in high contribution chimeras with COXIII-81 (n = 1), ND6-100 (n = 3), ND2-50 (n = 2), ATP8-62 (n = 2), and Control-1(n = 3) ESCs after 1 mg/g glucose injection. For each animal, two measurements were performed. Each dot represents biological replicates, ns denotes P ≥ 0.05. Reduced glucose tolerances were observed in ND6-100, ND2-50, and ATP8-62 chimeras compared to Control-1 animals. (C) Optokinetic response spatial frequency (c/d) of pigmented eye chimeras from COXIII-81 (n = 2), ND6-100 (n = 4), ND2-50 (n = 4), ATP8-62 (n = 1), and Control-1(n = 4) ESCs. Measurement was performed on both eyes of animals. Each dot represents biological replicates, ns denotes P ≥ 0.05. Optic neuropathy was observed as low optokinetic response in COXIII-81, ND6-100, and ND2-50 chimeras compared to Control-1 animals. (D–G) Mean cardiac ultrasound values documenting reduced ejection fraction (D) and increased left ventricular (LV) volume (E) in ATP8-62 chimeras (n = 6) compared with age- and sex-matched control PolGwt/wt (n = 6). In tandem, right ventricles (RV) were significantly enlarged in ATP8-62 chimeric hearts (F–G). (H) Echocardiography of ATP8-62 chimeras showing biventricular chamber dilation and reduced pump function in hearts as dilated cardiomyopathy. LV/RV, left/right ventricle. Vertical scale bars = 2 mm.
Since mutations in MT-COXIII, MT-ND2, and MT-ND6 genes have been implicated in vision impairment in humans [40], eye response tests were performed in 4 month old chimeras. Chimeric mice with pigmented eyes (ESC phenotype) were selected from COXIII-81 (n = 2), ND6-100 (n = 4), ND2-50 (n = 4), and ATP8-62 (n = 1) groups and compared to controls (n = 4). The mean optokinetic response spatial frequencies (c/d) in COXIII-81 (0.279 ± 0.02), ND6-100 (0.148 ± 0.06), and ND2-50 (0.076 ± 0.04) chimeras were all significantly lower than in controls (0.388 ± 0.005) (Figure 4C). However, the mean optokinetic response spatial frequency (c/d) in ATP8-62 (0.386 ± 0.02) was comparable to controls (Figure 4C). These results imply that mt-CoxIII, mt-Nd6, and mt-Nd2 but not mt-Atp8 variants are implicated in eye-related impairments.
We also examined hearing impairment by ABR and found that all chimeras were unaffected (Supplemental Figure S5C). We next sacrificed one high contribution chimera from each ND6-100, ND2-50, and ATP8-62 groups and performed histological examinations of all major organs. Histological staining revealed no significant findings except for extramedullary hematopoiesis in livers (Supplemental Figure S5D), suggestive of abnormal blood system. We also measured mitochondrial OXPHOS complex I and IV activities in muscles of ND6-100 (n = 3), ND2-50 (n = 3), and ATP8-62 (n = 1) high contribution chimeras and compared those to controls (n = 3). Tissues were not available from the high contribution COXIII-81 chimeras. ND6-100 chimeras showed significantly reduced complex I and IV activities while ND2-50 chimeras displayed only reduced complex I activity compared to controls. As expected, ATP8-62 chimera carrying mutations in the complex V showed normal complex I and IV activities (Supplemental Figure S5E and F).
These outcomes demonstrate that chimeras carrying specific gene mutations in mitochondrial complex I and IV may develop metabolic deficiencies. Particularly, high heteroplasmy mutations in mt-Nd6, mt-Nd2, and mt-Atp8 genes are associated with various phenotypes in chimeras including premature death and impairment of glucose metabolism and vision.
Mutations in MT-ATP6 and MT-ATP8 genes affecting mitochondrial respiratory chain complex V deficiency cause infantile cardiomyopathy in humans, often resulting in life-threatening heart dysfunction [41]. Here, ATP8-62 chimeras carried a high heteroplasmy mutation in the mt-Atp8 gene (7769C > G) that results in proline to alanine at position 2 of ATPase 8. We examined heart function using echocardiography in high contribution chimeric males (n = 6) at 4–5 months of age and compared to age-matched controls (n = 6). The mean ejection fraction of ATP8-62 chimeras (50.1 ± 5.5%) was significantly lower than in controls (67.6 ± 1.9%) (Figure 4D), and the mean diastolic left ventricle volume (56.8 ± 6.2 μl) was higher than that of controls (40.8 ± 2.9 μl) (Figure 4E). In addition, right ventricular dilatation was observed in ATP8-62 chimeras relative to controls (Figure 4F and G) and 5 of 6 chimeras showed evidence of bi-ventricular failure (Figure 4H). Thus, ATP8-62 chimeras demonstrate reduced pumping function with chamber enlargement, salient features of human cardiomyopathy.
Discussion
Only a few mutations, among numerous potential mtDNA variants, are implicated in inherited mitochondrial disorders in children [42]. The prevailing concept proposes that the vast majority of deleterious mtDNA mutations are negatively selected in the female germline implying that such purifying selection eliminates mature oocytes with mutations. We set out to investigate this assumption directly in ovulated oocytes from heterozygous PolG mutator model [8, 43]. Using whole mtDNA genome sequencing at the specific developmental stages of the mouse germline, we show that mature MII oocytes, PolGwt/wt blastocysts, and ESCs carry mtDNA variants similar to those found in somatic cells of PolGmut/wt females. These results contrast with assumptions that severe mtDNA mutations may adversely affect oogenesis [9, 44] but are consistent with conclusions that many pathogenic mtDNA variants are present in mature oocytes [45]. However, our results could not rule out the possibility that oocytes carrying specific deleterious mutations were eliminated during oogenesis.
We also show that many deleterious mtDNA variants do not affect normal fertilization and preimplantation development, and can be captured in stable ESC lines. However when re-introduced into host embryos, such mutant ESC lines demonstrate metabolic deficiencies resulting in fetal demise in chimeras. Although less information is available concerning how specific mtDNA defects contribute to early development, evidence indicates that cardiac development occurring between embryonic days 9.5 and 13.5 is highly dependent on mitochondrial oxidative metabolism [46, 47]. It is likely that high heteroplasmy mtDNA mutations that change amino acids in mtDNA-encoded proteins and thus cause severe biochemical defects in OXPHOS are strongly selected against due to their fetal lethality. The occurrence of purifying selection against deleterious mtDNA mutations during fetal development plays an important role in preventing the accumulation of pathogenic mutations that would cause damage to mitochondrial OXPHOS with possible consequences to species survival.
Pregnancy loss in humans occurs frequently in couples but the underlying cause is unknown. Recent studies show possible associations between repeated pregnancy loss and mtDNA mutations in the D-loop region [48] or in MT-ND1 [49] and suggest that mitochondrial mutations in oocytes could be a critical determinant of the developmental competence of an early human embryo. We show that some mtDNA variants, particularly in OXPHOS protein-coding genes are associated with fetal demise in mice before E9.5. These results support conclusions from recent studies that spontaneous mtDNA mutations in oocytes can cause fetal loss [48, 49].
In contrast to strong purifying selection against protein-coding mutations, our study shows that selection against tRNA and rRNA gene mutations was less distinct, possibly due to mild pathogenicity or low heteroplasmy levels [45, 50]. Interestingly, tRNA genes comprise less than 10% of the mtDNA genome but account for almost 60% of known germline mutations in humans. Strong purifying selection was reported for m.3875delC tRNAmet mutation [45]. Pathogenic tRNA mutations often must reach high heteroplasmy thresholds before they induce clinical effects [50]. For example, over 85% tRNALys mutant molecule in skeletal muscle of patients is a prerequisite for the phenotypic onset of myoclonic epilepsy and ragged-red fibers [51].
Since most mtDNA mutations cause early fetal demise, it is difficult to perform phenotypic studies on the impact of pathogenic mtDNA mutations on somatic tissues during postnatal development. Indeed, we show that selected pathogenic mtDNA mutations in ESCs cause fetal loss even in chimeras. For example, high incidences of fetal demise were seen in COXIII-81 chimeras. However, other chimeras with mutant ESCs (ND6-100, ND2-50, ATP8-62) developed to full-term and provided an opportunity to study their phenotypic expression in postnatal mice. Observations in these animals detected premature death, impairment of glucose metabolism and vision, or cardiomyopathy. All these conditions are typically associated with mitochondrial disease in humans [52–55]. The limitation of this approach is that specific mtDNA mutations cannot be generated at will but rather must be passively selected from the available pool of mutant ESC lines. Nevertheless, this technology provides a critical tool to study mtDNA mutations in vivo.
Supplementary Material
Acknowledgments
The authors acknowledge the Small Lab Animal Unit at Oregon National Primate Research Center (ONPRC) and the Center for Health and Healing at OHSU for providing expertise and services. We thank Drs. Nils-Göran Larsson and James Bruce Stewart at Max Planck Institute for providing C57BL/6N-Polgmut/N mice and helpful discussions. We thank Dr. Don P. Wolf at OHSU for providing helpful advice in manuscript preparation. We also thank Barbra Mason in the Histopathology- Morphology Core of ONPRC and Yibing Jia in the Molecular Core of ONPRC for their expertise and services. We are also grateful to Rebecca Tippner-Hedges, Chunlong Xu, and Midori Hayama for their excellent technical support. The study was supported by grants from the National Institutes of Health R56-AG045137, R01-EY021214, and P51-OD011092, grants from Burroughs Wellcome Fund and Leducq Foundation, Marriott Family Foundation, OHSU institutional funds, and Global Research Development Center Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (NRF-2015K1A4A3046807).
Conflict of interest
The authors declare no conflict of interest.
Author contributions
T.H., H.M., and S.M. conceived and planned the study. T.H., C.V.D., Y.L., and R.A. performed oocyte and embryo collections and cell culture. C.V.D. and Y.L. performed animal management. T.H., H.M., Y.M.L., C.V.D., Y.L., A.K., M.N., H.D., D.L., and T.G. performed DNA studies, whole genome sequencing, and mitochondrial complex activity assays. T.H. carried out micromanipulation and embryo transfer with help from E.K., N.M.G., Y.M.L., C.V.D., and Y.L. T.J.M and M.A. performed vision tests. S.Y. and A.T. performed cardiac function test. J.B. and B.K. performed hearing tests. H.M., T.H., P.A., and S.M. analyzed data and wrote the manuscript.
REFERENCES
- 1. Hagstrom E, Freyer C, Battersby BJ, Stewart JB, Larsson NG. No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res 2014; 42:1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khrapko K, Coller HA, Andre PC, Li XC, Hanekamp JS, Thilly WG. Mitochondrial mutational spectra in human cells and tissues. Proc Natl Acad Sci U S A 1997; 94:13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wallace DC, Ye JH, Neckelmann SN, Singh G, Webster KA, Greenberg BD. Sequence analysis of cDNAs for the human and bovine ATP synthase beta subunit: Mitochondrial DNA genes sustain seventeen times more mutations. Curr Genet 1987; 12:81–90. [DOI] [PubMed] [Google Scholar]
- 4. Kazak L, Reyes A, Holt IJ. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 2012; 13:659–671. [DOI] [PubMed] [Google Scholar]
- 5. Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes Clifford DL, Gutierrez Nuria M, Lee Y, Van Dyken C, Ahmed R, Li Y, Koski A, Hayama T et al. Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell 2016; 18:625–636. [DOI] [PubMed] [Google Scholar]
- 6. Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 1999; 286:774–779. [DOI] [PubMed] [Google Scholar]
- 7. Rebolledo-Jaramillo B, Su MS, Stoler N, McElhoe JA, Dickins B, Blankenberg D, Korneliussen TS, Chiaromonte F, Nielsen R, Holland MM, Paul IM, Nekrutenko A et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A 2014; 111:15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson NG. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol 2008; 6:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, Macgregor GR, Wallace DC. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 2008; 319:958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders--past, present and future. Biochim Biophys Acta 2004; 1659:115–120. [DOI] [PubMed] [Google Scholar]
- 11. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev Genet 2005; 39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004; 429:417–423. [DOI] [PubMed] [Google Scholar]
- 13. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005; 309:481–484. [DOI] [PubMed] [Google Scholar]
- 14. Ross JM, Stewart JB, Hagstrom E, Brene S, Mourier A, Coppotelli G, Freyer C, Lagouge M, Hoffer BJ, Olson L, Larsson NG. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013; 501:412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shoubridge EA, Wai T. Mitochondrial DNA and the mammalian oocyte. Curr Top Dev Biol 2007; 77:87–111. [DOI] [PubMed] [Google Scholar]
- 16. Cummins J. Mitochondrial DNA in mammalian reproduction. Rev Reprod 1998; 3:172–182. [DOI] [PubMed] [Google Scholar]
- 17. Floros VI, Pyle A, Dietmann S, Wei W, Tang WCW, Irie N, Payne B, Capalbo A, Noli L, Coxhead J, Hudson G, Crosier M et al. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat Cell Biol 2018; 20:144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramalho-Santos J, Varum S, Amaral S, Mota PC, Sousa AP, Amaral A. Mitochondrial functionality in reproduction: From gonads and gametes to embryos and embryonic stem cells. Hum Reprod Update 2009; 15:553–572. [DOI] [PubMed] [Google Scholar]
- 19. Ma H, Folmes CDL, Wu J, Morey R, Mora-Castilla S, Ocampo A, Ma L, Poulton J, Wang X, Ahmed R, Kang E, Lee Y et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 2015; 524:234–238. [DOI] [PubMed] [Google Scholar]
- 20. Salomonis N, Dexheimer PJ, Omberg L, Schroll R, Bush S, Huo J, Schriml L, Ho Sui S, Keddache M, Mayhew C, Shanmukhappa SK, Wells J et al. Integrated genomic analysis of diverse induced pluripotent stem cells from the progenitor cell biology consortium. Stem Cell Rep 2016; 7:110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang E, Wu G, Ma H, Li Y, Tippner-Hedges R, Tachibana M, Sparman M, Wolf DP, Scholer HR, Mitalipov S. Nuclear reprogramming by interphase cytoplasm of two-cell mouse embryos. Nature 2014; 509:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kobayashi T, Yamaguchi T, Hamanaka S, Kato-Itoh M, Yamazaki Y, Ibata M, Sato H, Lee YS, Usui J, Knisely AS, Hirabayashi M, Nakauchi H. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 2010; 142:787–799. [DOI] [PubMed] [Google Scholar]
- 23. Yamada S, Arrell DK, Martinez-Fernandez A, Behfar A, Kane GC, Perez-Terzic CM, Crespo-Diaz RJ, McDonald RJ, Wyles SP, Zlatkovic-Lindor J, Nelson TJ, Terzic A. Regenerative therapy prevents heart failure progression in dyssynchronous nonischemic narrow QRS cardiomyopathy. J Am Heart Assoc 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci 2004; 45:4611–4616. [DOI] [PubMed] [Google Scholar]
- 25. Douglas RM, Alam NM, Silver BD, McGill TJ, Tschetter WW, Prusky GT. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis Neurosci 2005; 22:677–684. [DOI] [PubMed] [Google Scholar]
- 26. McGill TJ, Lund RD, Douglas RM, Wang S, Lu B, Silver BD, Secretan MR, Arthur JN, Prusky GT. Syngeneic Schwann cell transplantation preserves vision in RCS rat without immunosuppression. Invest Ophthalmol Vis Sci 2007; 48:1906–1912. [DOI] [PubMed] [Google Scholar]
- 27. McGill TJ, Cottam B, Lu B, Wang S, Girman S, Tian C, Huhn SL, Lund RD, Capela A. Transplantation of human central nervous system stem cells - neuroprotection in retinal degeneration. Eur J Neurosci 2012; 35:468–477. [DOI] [PubMed] [Google Scholar]
- 28. Pennesi ME, Michaels KV, Magee SS, Maricle A, Davin SP, Garg AK, Gale MJ, Tu DC, Wen Y, Erker LR, Francis PJ. Long-term characterization of retinal degeneration in rd1 and rd10 mice using spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci 2012; 53:4644–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitchell C, Kempton JB, Creedon T, Trune D. Rapid acquisition of auditory brainstem responses with multiple frequency and intensity tone-bursts. Hear Res 1996; 99:38–46. [DOI] [PubMed] [Google Scholar]
- 30. Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 2012; 7:1235–1246. [DOI] [PubMed] [Google Scholar]
- 31. Ma H, Lee Y, Hayama T, Van Dyken C, Marti-Gutierrez N, Li Y, Ahmed R, Koski A, Kang E, Darby H, Gonmanee T, Park Y et al. Germline and somatic mtDNA mutations in mouse aging. PLoS One 2018; 13:e0201304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010; 464:610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Agaronyan K, Morozov YI, Anikin M, Temiakov D. Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 2015; 347:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kang E, Wu J, Gutierrez NM, Koski A, Tippner-Hedges R, Agaronyan K, Platero-Luengo A, Martinez-Redondo P, Ma H, Lee Y, Hayama T, Van Dyken C et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016; advance online publication. [DOI] [PubMed] [Google Scholar]
- 35. Sligh JE, Levy SE, Waymire KG, Allard P, Dillehay DL, Nusinowitz S, Heckenlively JR, MacGregor GR, Wallace DC. Maternal germ-line transmission of mutant mtDNAs from embryonic stem cell-derived chimeric mice. Proc Natl Acad Sci U S A 2000; 97:14461–14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marchington DR, Barlow D, Poulton J. Transmitochondrial mice carrying resistance to chloramphenicol on mitochondrial DNA: Developing the first mouse model of mitochondrial DNA disease. Nat Med 1999; 5:957–960. [DOI] [PubMed] [Google Scholar]
- 37. van den JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, van de Kamp JJ, Maassen JA. Mutation in mitochondrial tRNA (Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet 1992; 1:368–371. [DOI] [PubMed] [Google Scholar]
- 38. Perucca-Lostanlen D, Narbonne H, Hernandez JB, Staccini P, Saunieres A, Paquis-Flucklinger V, Vialettes B, Desnuelle C. Mitochondrial DNA variations in patients with maternally inherited diabetes and deafness syndrome. Biochem Biophys Res Commun 2000; 277:771–775. [DOI] [PubMed] [Google Scholar]
- 39. Nakada K, Hayashi J. Transmitochondrial mice as models for mitochondrial DNA-based diseases. Exp Anim 2011; 60:421–431. [DOI] [PubMed] [Google Scholar]
- 40. Meyerson C, Van Stavern G, McClelland C. Leber hereditary optic neuropathy: Current perspectives. Clin Ophthalmol 2015; 9:1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Imai A, Fujita S, Kishita Y, Kohda M, Tokuzawa Y, Hirata T, Mizuno Y, Harashima H, Nakaya A, Sakata Y, Takeda A, Mori M et al. Rapidly progressive infantile cardiomyopathy with mitochondrial respiratory chain complex V deficiency due to loss of ATPase 6 and 8 protein. Int J Cardiol 2016; 207:203–205. [DOI] [PubMed] [Google Scholar]
- 42. Chinnery PF, Thorburn DR, Samuels DC, White SL, Dahl HM, Turnbull DM, Lightowlers RN, Howell N. The inheritance of mitochondrial DNA heteroplasmy: Random drift, selection or both? Trends Genet 2000; 16:500–505. [DOI] [PubMed] [Google Scholar]
- 43. Stewart JB, Freyer C, Elson JL, Larsson NG. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat Rev Genet 2008; 9:657–662. [DOI] [PubMed] [Google Scholar]
- 44. Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 1996; 14:146–151. [DOI] [PubMed] [Google Scholar]
- 45. Freyer C, Cree LM, Mourier A, Stewart JB, Koolmeister C, Milenkovic D, Wai T, Floros VI, Hagstrom E, Chatzidaki EE, Wiesner RJ, Samuels DC et al. Variation in germline mtDNA heteroplasmy is determined prenatally but modified during subsequent transmission. Nat Genet 2012; 44:1282–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. St John JC, Facucho-Oliveira J, Jiang Y, Kelly R, Salah R. Mitochondrial DNA transmission, replication and inheritance: A journey from the gamete through the embryo and into offspring and embryonic stem cells. Hum Reprod Update 2010; 16:488–509. [DOI] [PubMed] [Google Scholar]
- 47. Folmes CD, Terzic A. Metabolic determinants of embryonic development and stem cell fate. Reprod Fertil Dev 2014; 27:82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seyedhassani SM, Houshmand M, Kalantar SM, Modabber G, Aflatoonian A. No mitochondrial DNA deletions but more D-loop point mutations in repeated pregnancy loss. J Assist Reprod Genet 2010; 27:641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Colagar AH, Mosaieby E, Seyedhassani SM, Mohajerani M, Arasteh A, Kamalidehghan B, Houshmand M. T4216C mutation in NADH dehydrogenase I gene is associated with recurrent pregnancy loss. Mitochondrial DNA 2013; 24:610–612. [DOI] [PubMed] [Google Scholar]
- 50. Shimizu A, Mito T, Hayashi C, Ogasawara E, Koba R, Negishi I, Takenaga K, Nakada K, Hayashi J. Transmitochondrial mice as models for primary prevention of diseases caused by mutation in the tRNA (Lys) gene. Proc Natl Acad Sci U S A 2014; 111:3104–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boulet L, Karpati G, Shoubridge EA. Distribution and threshold expression of the tRNA (Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers MERRF. Am J Hum Genet 1992; 51:1187–1200. [PMC free article] [PubMed] [Google Scholar]
- 52. Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol 2013; 5:a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chinnery PF, Turnbull DM. Mitochondrial DNA mutations in the pathogenesis of human disease. Mol Med Today 2000; 6:425–432. [DOI] [PubMed] [Google Scholar]
- 54. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005; 6:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN, Turnbull DM. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet 2002; 30:145–146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been uploaded to the sequence read archive under accession number SRP131410. All other data are available from the authors upon reasonable request.