

Abstract
Mitochondrial disorders are the result of nuclear and mitochondrial DNA mutations that affect multiple organs, with the central and peripheral nervous system often affected. Currently, there is no cure for mitochondrial disorders. Currently, gene therapy offers a novel approach for treating monogenetic disorders, including nuclear genes associated with mitochondrial disorders. We utilized a mouse model carrying a knockout of the mitochondrial fusion–fission-related gene solute carrier family 25 member 46 (Slc25a46) and treated them with neurotrophic AAV–PHP.B vector carrying the mouse Slc25a46 coding sequence. Thereafter, we used immunofluorescence staining and western blot to test the transduction efficiency of this vector. Toluidine blue staining and electronic microscopy were utilized to assess the morphology of optic and sciatic nerves following treatment, and the morphology and respiratory chain activity of mitochondria within these tissues were determined as well. The adeno-associated virus (AAV) vector effectively transduced in the cerebrum, cerebellum, heart, liver and sciatic nerves. AAV–Slc25a46 treatment was able to rescue the premature death in the mutant mice (Slc25a46−/−). The treatment-improved electronic conductivity of the peripheral nerves increased mobility and restored mitochondrial complex activities. Most notably, mitochondrial morphology inside the tissues of both the central and peripheral nervous systems was normalized, and the neurodegeneration, chronic neuroinflammation and loss of Purkinje cell dendrites observed within the mutant mice were alleviated. Overall, our study shows that AAV–PHP.B’s neurotrophic properties are plausible for treating conditions where the central nervous system is affected, such as many mitochondrial diseases, and that AAV–Slc25a46 could be a novel approach for treating SLC25A46-related mitochondrial disorders.
Introduction
Mitochondrial defects can affect multiple organ systems, with often irreversible consequences. Unfortunately, there is no cure for most mitochondrial diseases because of their complexity and heterogeneity. Mitochondrial disorders are often hereditary in nature, as mitochondrial structure and function require over 1500 proteins encoded by the nuclear genome (1,2). Since the majority of nuclear genome-related mitochondrial diseases are caused by autosomal recessive mutations in a single gene (1), gene therapy could be a plausible approach to treat this group of disorders.
Adeno-associated virus (AAV) is a 25 nm diameter virus of the Parvoviridae family, comprised of a non-enveloped icosahedral capsid with a linear, single-stranded DNA genome of 4.7 kb (3). AAV has low immunogenicity and is not associated with any human diseases (4). The excellent safety profile and the high transduction efficiency of AAV make it one of the best options for gene therapy if the disease-causing gene is relatively small. In the genome of recombinant AAV vectors used for gene therapy, the two inverted terminal repeats are retained, while the other viral sequences are modified with the desired exogenous transgenic DNA of choice. The capacity of the DNA cargo inside the AAV genome is approximately 5 kb (5), but after the inclusion of promoters, reporters and selection markers, the maximum size available for packaging rescue genes into AAV vectors is generally limited to <2.4 kb.
Solute carrier family 25 member 46 (SLC25A46) is a protein encoded by the nuclear genome that is integrated into the outer membrane of mitochondria. Previously, our own work, as well as the work of other groups, has demonstrated the role of this protein in mitochondrial fusion–fission dynamics (6–8). Loss of function of this protein results in hyperfilamentous mitochondria and mitochondrial hyper-fusion, which impairs mitochondrial function and cellular respiration and eventually leads to ataxia, visual and developmental defects and premature death in both humans and mice (7,9–14). The coding sequence of mouse Slc25a46 is about 1.25 kb, which makes it a suitable size for AAV gene therapy. Because of the global expression of this protein (15), genetic defects of this gene cannot be treated via local administration (16–20). The gene expression vector must be systematically delivered in vivo. AAV–PHP.B, which was developed by Deverman et al. (21) using Cre-recombination-based AAV targeted evolution technology, transfers genes throughout the CNS (central nervous system) with an efficiency 40-fold higher than AAV9. Therefore, it represents an appropriate candidate for non-invasive gene delivery to the central and peripheral nervous systems.
In this report, we systematically delivered an AAV–Slc25a46 rescue vector via face vein injection into Slc25a46−/− mutant pups. As expected, mutants treated with the AAV–Slc25a46 vector showed significant rescue for the majority of phenotypic and physiological abnormalities. These results demonstrate that gene therapy could be a novel method for treating SLC25A46-related mitochondrial genetic disorders in humans.
Results
AAV-PHP.B-eGFP transduces efficiently in multiple organ systems through intravascular injection
To test the transduction efficiency of our AAV-based expression system, we utilized an AAV2–PHP.B vector carrying an integrated enhanced green fluorescent protein (eGFP) reporter. We administered 1 × 1011 genome copies (GC)/g to Slc25a46−/− mutants at postnatal day 2 (P2) and then sacrificed the mice 2 or 4 weeks after injection. The targeted organs brain, heart, liver, sciatic nerve and eye tissues were harvested, and eGFP levels were assessed as a measure of transduction efficiency. After 2 weeks, confocal microscopy revealed widespread eGFP expression in the cerebellum, cerebral cortex, heart, liver and sciatic nerve (Fig. 1A and B) and a weaker signal in the hippocampus (Fig. 1B). Four weeks after injection, we found sparse signal visible in the heart tissue (Fig. 1C) and minimal expression detectable elsewhere. Therefore, in an attempt to create longer-lasting transduction, we injected the second cohort of mice with a higher dose of the vector (2 × 1011 GC/g), which yielded a measurable level of eGFP in the cerebellum, cerebral cortex, heart and sciatic nerve at 4 weeks after injection (Fig. 1D). To further confirm the expression of Slc25a46 protein in the central nervous system after AAV vector administration, we conducted a western blot (WB) analysis on cerebral tissue from treated Slc25a46−/− mutants at 4 and 8 weeks after AAV administration, respectively. WB analysis verified that the Slc25a46 protein had been successfully expressed in the central nervous system of AAV–Slc25a46-treated Slc25a46−/− mutants (Supplementary Material, Fig. S2).
Figure 1.
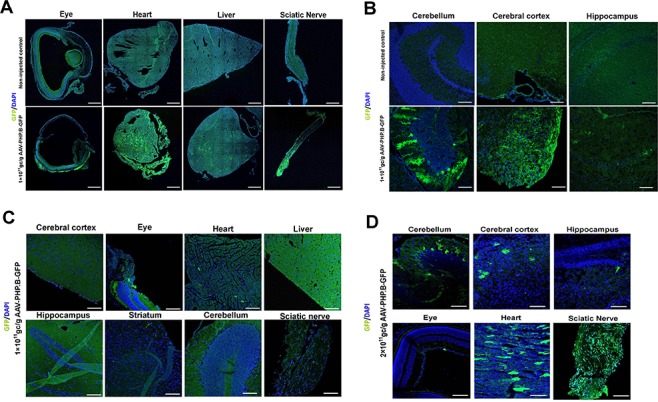
The efficiency of the delivery of AAV–PHP.B-eGFP vector at different dosages and different time points after administration via face vein injection. (A and B) Distribution of the eGFP reporter in peripheral tissues (A) and the central nervous system (B) 2 weeks after face vein injection of the AAV–PHP.B-eGFP vector into neonatal mice. Upper panel, uninjected control; lower panel, mice injected with 1 × 1011 GC/g AAV–PHP.B-eGFP. (C and D) Distribution of eGFP reporter expression in peripheral tissues and the central nervous system 4 weeks after administration of 1 × 1011 GC/g (C) and 2 × 1011 GC/g (D) AAV–PHP.B-eGFP vector via face vein injection.
AAV–Slc25a46 improves lifespan and bodyweight of Slc25a46−/− mice in a dose-dependent manner
Since Slc25a46 mutant mice suffer from severe ataxia (10), they often have difficulty feeding. To avoid this, both injected and uninjected animals were fed with DietGel after weaning age. After 51 days, the Slc25a46−/−mutants that received 1 × 1011 GC/g AAV–Slc25a46 vector had a 45% survival rate (n = 8), while the untreated Slc25a46−/− mutants (n = 16) had only a 22.2% survival rate (Fig. 2A). There was no significant difference in body weight between the groups (Fig. 2B). Mutants that received 2 × 1011 GC/g (n = 15) showed a significantly longer life-span in contrast to untreated Slc25a46−/− mutants (P = 0.0144) (Fig. 2C). The body weights of these ‘double-dose’ treated mutants were also significantly higher than the untreated Slc25a46−/− mutants as well as the mutants treated with low dose 1 × 1011 GC/g (P = 0.0018 and 0.0070, respectively, with n = 6 for each group) (Fig. 2B), suggesting a dosage-dependent effect of AAV–Slc25a46.
Figure 2.
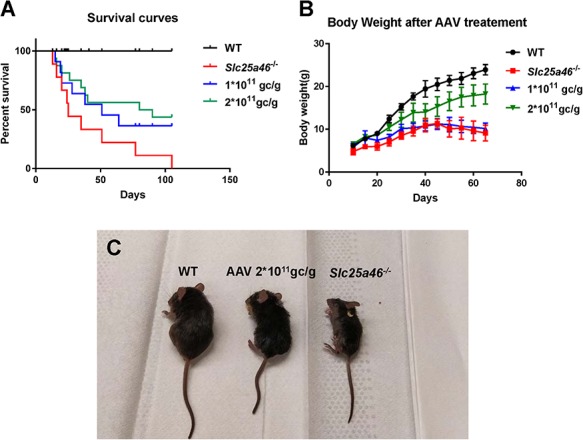
Behavioral manifestations after varying dosages of AAV-Slc25a46 vector treatment in Slc25a46−/− mutants. (A) A Kaplan–Meier survival curve of injected versus uninjected mice shows that the majority of untreated Slc25a46−/− mutants died prematurely. In contrast to untreated mutants, mutants treated with 1 × 1011 GC/g AAV–Slc25a46 vector gained a longer lifespan, but not at a statistically significant level (P = 0.1425). However, the mutants treated with 2 × 1011 GC/g AAV–Slc25a46 vector did show a significantly extended life-span (P = 0.0144) (WT, n = 16; Slc25a46−/− mutants, n = 16; mutants treated with 1 × 1011 GC/g, n = 8; mutants treated with 2 × 1011 GC/g, n = 15.) (B) Bodyweight monitoring starting at P10. The bodyweights of 2 × 1011 GC/g AAV-Slc25a46 vector-treated male mutants were notably and significantly increased relative to untreated Slc25a46−/− male mutants and the male mutants treated with 1 × 1011 GC/g (P = 0.0018 and 0.0070, respectively, with n = 6 for each group). (C) The growth defect was alleviated after treatment with a 2 × 1011 GC/g dose of the AAV–Slc25a46 vector, and the body size of the 2 × 1011 GC/g AAV-Slc25a46 treated mutants was distinctly larger than untreated mutants.
AAV–Slc25a46 vector attenuates central nervous system defects and ataxia in Slc25a46−/− mice
Slc25a46 −/− mutants injected with either dose of AAV–Slc25a46 vector showed better coordination and better hind limb strength than untreated mutants (Supplementary Materials, Movies 1 and 2). The AAV-treated mice were able to maintain an upright posture and did not demonstrate the characteristic ‘flopping’ observed in mutant mice., suggesting that AAV–Slc25a46 is able to rescue movement defects in the mutant mice.
To assess the extent of neuropathy after vector treatment, we performed hexaribonucleotide binding protein-3 (NeuN), glial fibrillary acidic protein (GFAP) and Fluoro-Jade C staining of the cerebellum at P40 from wild-type (WT) mice, untreated Slc25a46−/−mice and Slc25a46−/− mice treated with 2 × 1011 GC/g AAV–Slc25a46 vector. The thickness of granule cells as stained by anti-NeuN was similar across all groups (Fig. 3A). However, the number of astrocytes and microglia stained by GFAP was significantly reduced in the treated as in contrast to the untreated mutants (Fig. 3B and C). Similarly, the number of degenerating neurons (as indicated by Fluro-Jade C staining in the axons, dendrites and somata of Purkinje cells) was notably decreased in the mutants after vector treatment (Fig. 3D). This result demonstrated that AAV–Slc25a46 is able to prevent cerebellar neuropathy in Slc25a46−/−mice.
Figure 3.
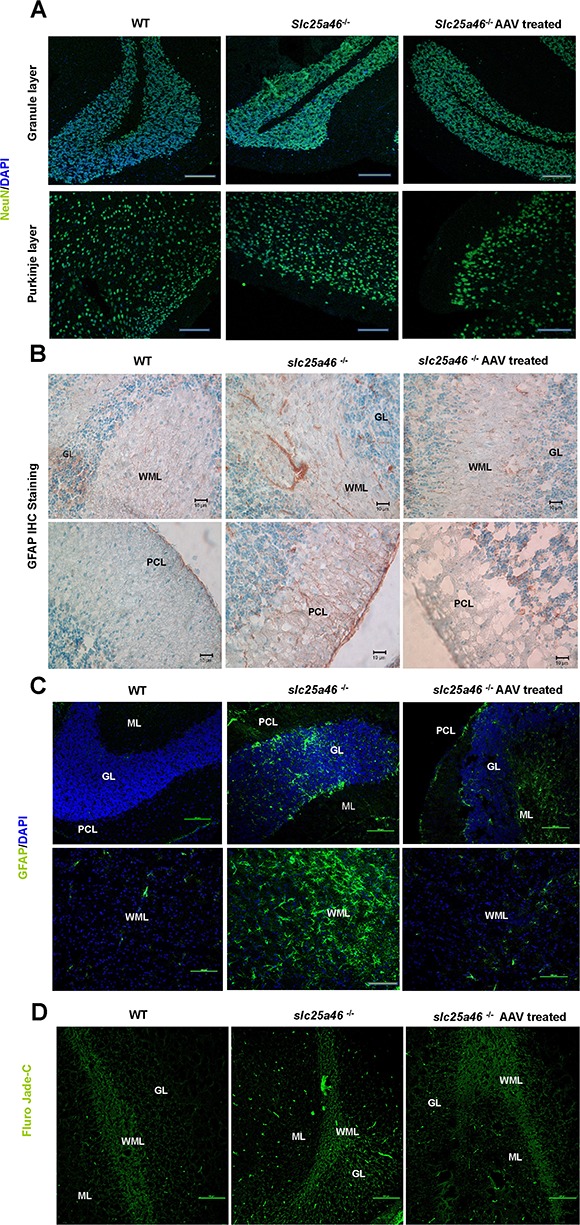
AAV–Slc25a46 treatment relieved chronic neuronal inflammation in the cerebellum of Slc25a46−/− mutants. (A) NeuN staining of the GL in the cerebellum of WT mice, Slc25a46−/− mutants and mutants treated with the AAV–Slc25a46 vector. (B) Immunohistochemical staining of GFAP in the cerebellum of the different mouse groups. (C) IF staining of GFAP in the cerebellum of the different mouse groups. (D) Fluoro-Jade C staining in the cerebellum of the different mouse groups. Green signals indicate neurons are undergoing degeneration. WML, white matter layer; PCL, Purkinje cell layer; GL, granule cell layer. ML, molecular layer. Scale bars: 100 μm.
To determine whether or not Purkinje cell loss is affected by the vector treatment, we used the specific marker calbindin (Swant, Switzerland) to label Purkinje cells in the cerebellum of P40 mice. Golgi staining was also performed to visualize the dendritic spreading of Purkinje cells. The AAV–Slc25a46 vector reduced the Purkinje cell loss in Slc25a46−/− mutants in contrast to untreated mutants (Fig. 4A). Similarly, Golgi staining indicated both wider Purkinje cell distribution and denser Purkinje cell dendrites in the vector-treated group (Fig. 4B and C). To investigate the microstructure of mitochondria inside cerebellar cells, we used electron microscopy (EM) to show that the mitochondrial dysmorphology, enlargement and disorganization characteristic of untreated Slc25a46−/− mutants were not observed in the vector-treated group (Fig. 4D and E), suggesting that AAV–Slc25a46 mitigates the loss of Purkinje cells and dendrites in Slc25a46−/− mice.
Figure 4.
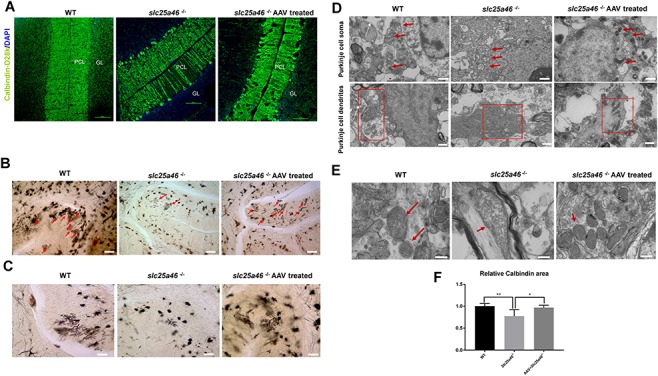
Treatment with AAV–Slc25a46 reduced Purkinje cell loss and enhanced dendrite branching in Slc25a46−/− mutants. (A) IF staining with calbindin in mouse cerebellum. White arrows indicate Purkinje cell deficiency. (B) Golgi staining of the P50 mice cerebellum. Red arrows indicate the distribution of Purkinje dendrites in the cerebellum. Scale bars: 80 μm. (C) Enlarged Golgi staining images show the dendritic branching of Purkinje cells in the cerebellum. Slc25a46−/− mutants demonstrated a remarkable sparseness in the dendrite branching of Purkinje cells, while vector-treated mutants acquired significantly denser dendrites. Scale bars: 40 μm. (D) EMimages of Purkinje cell soma and dendrites. Upper panel: red arrows indicate mitochondria within Purkinje cell soma. Lower panel: red boxes indicate mitochondria inside Purkinje cell dendrites. Note the aggregated and fused mitochondria inside the dendrites of the neurons from untreated mutants. Scale bar: 1 μm. (E) EM images of individual mitochondria inside the mouse cerebellum. Red arrows indicate individual mitochondria. Scale bar: 600 nm. (F) Quantitation for calbindin staining of P50 mice. (*P < 0.05, **P < 0.01, six sections were measured from three mice for each group).
AAV–Slc25a46 attenuates optic atrophy and dysmorphic mitochondria
To investigate whether optic atrophy was reduced in Slc25a46−/− mutants following vector treatment, we performed H&E staining on the retinal ganglion cell (RGC) layer, as well as immunofluorescence (IF) staining using the RGC-specific marker brain-specific homeobox protein 3A (Brn3a). H&E staining indicated that the cell count in the RGC layer of AAV-treated mice was significantly higher than those of the untreated mutants (P = 0.0332) (Fig. 5A and E). IF staining revealed that the number of Brn3a-positive cells in the ganglion cell layer was markedly higher in vector-treated in contrast to untreated mutants (P = 0.0012) (Fig. 5B and F). Meanwhile, toluidine blue staining revealed that the diameter of optic nerve axons from Slc25a46−/− mutants treated with the AAV–Slc25a46 vector was considerably more significant than the diameter observed in untreated mutants (Fig. 5C). After examining the axons at higher magnification under an electron microscope (Fig. 5D), we found that the presence of ring-shaped mitochondria and ‘pushed aside’ cristae was reduced in treated mutants. In summary, these data indicate that AAV–Slc25a46 attenuates optic nerve atrophy in Slc25a46−/− mutant mice.
Figure 5.
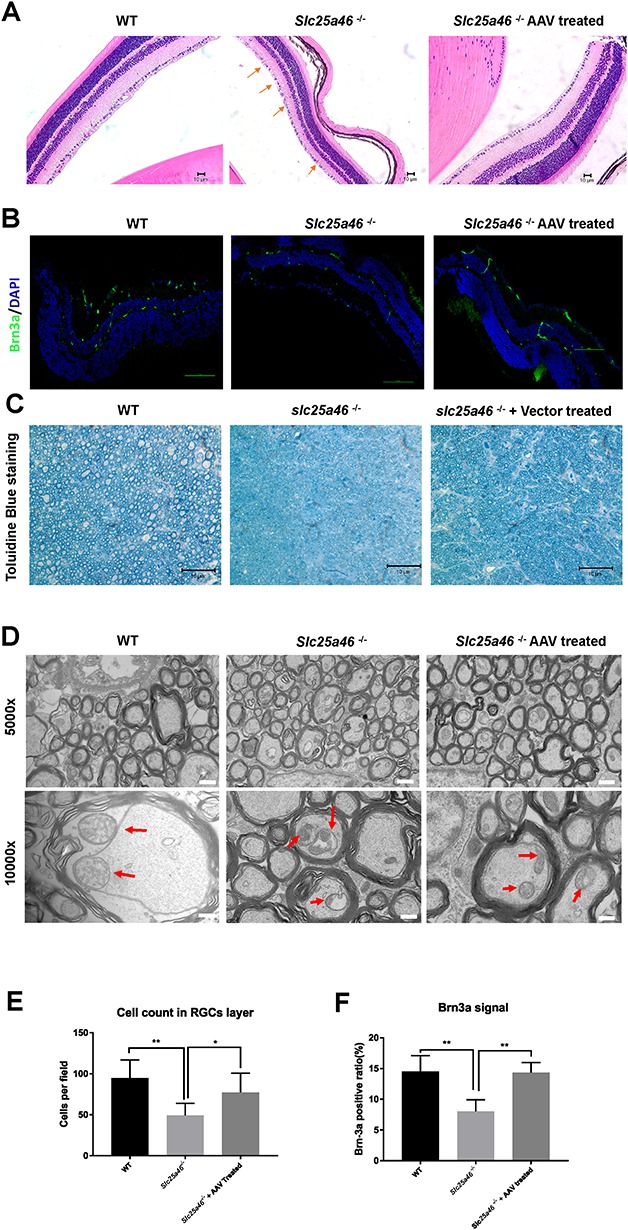
Optic atrophy reduced following AAV–Slc25a46 treatment in Slc25a46−/− mutants. (A) H&E staining of eyes from P50 mouse retinas. Scale bar: 150 μm. Red arrows indicate cell loss in the RGC layer of Slc25a46−/− mutants. (B) Brn3a IF staining of P50 mouse retinas. Scale bar: 100 μm. (C) Toluidine blue staining of semi-thin cross sections of mouse optic nerves. Scale bar: 10 μm. (D) EM images of cross sections of mouse P50 optic nerves. Red arrows specify individual mitochondria inside the axons of optic nerves. Scale bar: upper panel, 1 μm; lower panel, 600 nm. (E) Cell counts in the RGC layer. (WT in contrast to Slc25a46−/− mutant, P = 0.0018; Slc25a46−/− mutant in contrast to AAV–Slc25a46-treated mutant, P = 0.0332.) (F) Quantification of Brn3a–positive RGCs in retinas of P50 mice (WT in contrast to Slc25a46−/− mutant, P = 0.0019; Slc25a46−/− mutant in contrast to AAV–Slc25a46-treated mutant, P = 0.0012). Brn3a–positive RGCs were quantified from six retinal sections of P50 mice. (*P < 0.05, **P < 0.01, six sections were measured from three mice for each group).
AAV–Slc25a46 vector mitigates dysmorphic mitochondria in sciatic nerves
Our next step was to investigate the morphological and physiological state of the sciatic nerve, which plays a vital role in conducting the signals that control hind limb movement. Toluidine blue staining showed no apparent differences in the sciatic nerves of WT, AAV vector-treated and untreated Slc25a46−/− mutant animals (Fig. 6A). However, a more in-depth examination using EM revealed hyper-fused mitochondria with disorganized, vesicular-like cristae in the axons of the mutants, which is consistent with our previous studies (10) (Fig. 6B). Moreover, some axons within the sciatic nerve of the Slc25a46−/− mutant appeared to be undergoing demyelination (Fig. 6B). Vector treatment yielded axonal mitochondria that more closely resembled those of WT mice: they exhibited natural shape, were distributed evenly and showed cristae with normal morphology (Fig. 6B). To further evaluate the neuronal function, we conducted electromyography on P50 mice from the three different groups. Mutants treated with 2 × 1011 GC/g vector showed reduced compound muscle action potential (CMAP) and conduction velocities (CVs) that were significantly different from those of untreated mutants (P = 0.0018 and P < 0.0001, respectively) (Fig. 6C–E).
Figure 6.
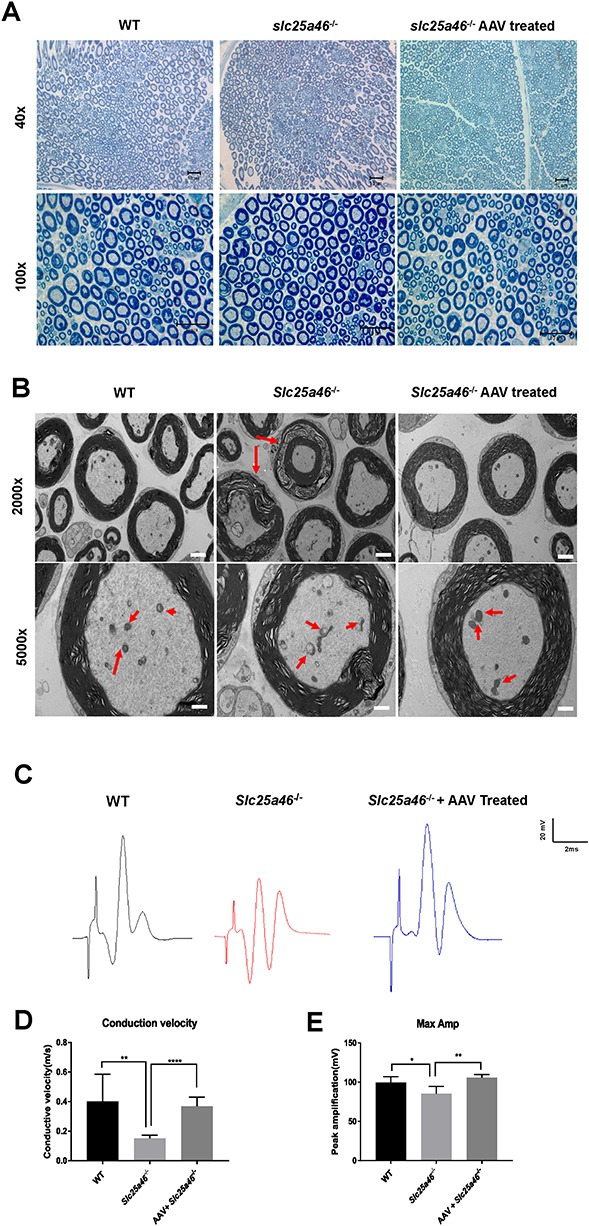
Peripheral neuropathy in Slc25a46−/− mice eased by AAV–Slc25a46 treatment. (A) Toluidine blue staining of semi-thin cross sections of sciatic nerves. Scale bars: upper panel, 10 μm; lower panel, 10 μm. (B) EM images of sciatic nerves. Top panel: red arrows indicate axons that are undergoing demyelination. Bottom panel: red arrows indicate mitochondria inside the sciatic nerves. Scale bar: top panel, 2 μm; bottom panel, 1 μm. (C) Electromyograph wave chart of P55 mice. The unit of scale is volts. (D and E) The CV (D) and CMAP (E) of P55 mice were compared between untreated mutants and mutants treated with AAV–Slc25a46. The mutants treated with AAV–Slc25a46 maintained substantially higher CV as well as CMAP in contrast to untreated mutants (P < 0.0001 and P = 0.0018, respectively). (*P < 0.05, **P < 0.01, ****P < 0.0001, n = 3 for each group).
AAV–Slc25a46 vector partially restores mitochondrial complex activity in various tissues
To test the function of electron transport chain components of WT mice as in contrast to untreated and vector-treated Slc25a46−/− mutants, we isolated mitochondria from tissues with high-energy consumption, including the brain, heart, liver and muscle. These isolated mitochondrial samples were then used to perform activity assays on mitochondrial complexes I through IV. We found that the activities of complex I, II, III and IV were all restored to varying extents (Fig. 7). In contrast to untreated mutants, mutants treated with AAV vector showed significant restoration of complex I activity in heart and muscle tissue (P = 0.0104 and P = 0.0110, respectively). Furthermore, the complex II + III activity levels showed substantial recovery in brain tissues after vector treatment (P = 0.0110). Additionally, the complex IV activity of heart and muscle tissues was restored significantly as well after the AAV vector treatment (P = 0.0163 and P < 0.0001, respectively).
Figure 7.
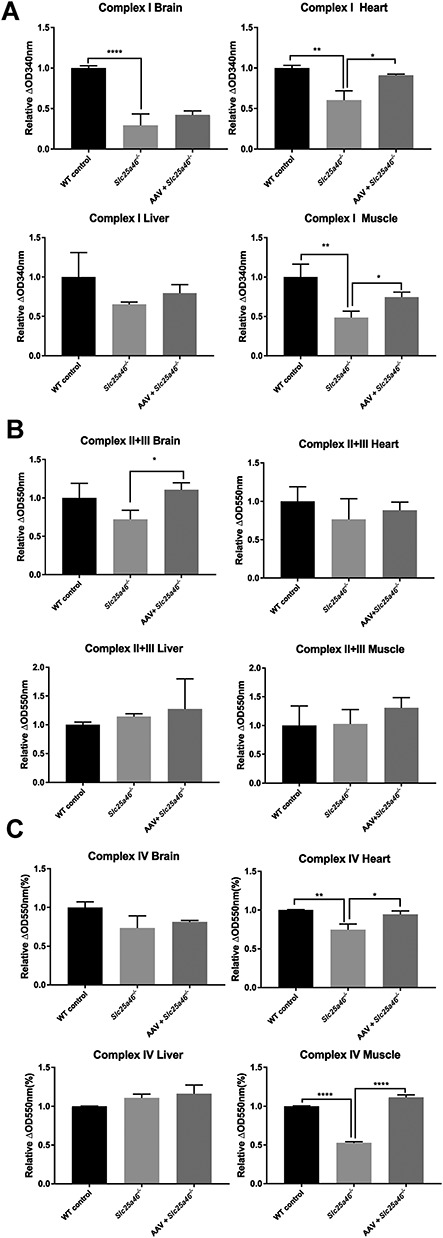
Dysfunction of mitochondrial complexes in Slc25a46−/− mice mitigated by AAV–Slc25a46 vector treatment. (A) Complex I activities in the brain, heart, liver and muscle of P50 mice. AAV–Slc25a46-treated mutants showed a significantly improved function of complex I in heart and muscle tissues (P = 0.0104 and 0.0110, respectively). (B) Complex II + III activities in the brain, heart, liver and muscle of P50 mice. AAV–Slc25a46-treated mutants showed a restored function of complex II + III in brain tissue (P = 0.0110). (C) Complex IVactivities in various tissues. Upper right panel: the activity of complex IV in the heart of AAV-treated mutants was significantly higher than the activity levels observed in untreated mutants (P = 0.016). As expected, WT mice also showed significantly higher activity than untreated mutants (P = 0.0040). Lower right panel: both AAV–Slc25a46-treated mutants and WT mice showed significantly higher complex IV activity in muscle tissue than the activity levels observed in mutants (P < 0.0001 for both comparisons). The dataset of mitochondrial activity assays was generated from three mice of each group. (*P < 0.05, **P < 0.01, ****P < 0.0001).
Discussion
Currently, mitochondrial diseases have no cure, with only treatments that may alleviate the clinical symptoms available (22). The ‘mitochondrial cocktail’ is a combination of vitamins and supplements—often including L-carnitine, coenzyme Q10, riboflavin, thiamine, vitamin C, vitamin E and idebenone—that has been used in mitochondrial disease treatment. These cocktails have shown modest improvements in some clinical trials (23,24), but none has had a decisive effect on improving the most critical clinical outcomes (25). Therefore, gene therapy could offer a more optimistic option for treating mitochondrial disorders caused by mitochondrial-related nuclear gene mutations.
AAV vectors have been widely utilized in the field of gene therapy (26), and many preclinical and clinical trials have shown remarkable success up to this point (27). However, only a few studies have been published for mitochondrial defects treated by AAV (28–30). Meo and colleagues utilized AAV2/AAV9–Hndufs4 by both intravenous and intracerebroventricular delivery, with the latter representing a relatively invasive procedure (28). Therefore, an AAV serotype which is capable of robust transduction in the central nervous system and peripheral organs would be a better option. AAV–PHP.B was created by Deverman and colleagues (31,32) and has proven to have a unique property in contexts requiring penetration of the blood–brain barrier while also maintaining the capacity to spread throughout the whole body. Therefore, we generated an AAV–PHP.B vector carrying the mouse WT Slc25a46 coding sequence and delivered it intravenously to Slc25a46−/− mutants.
In the central nervous system, AAV–PHP.B is reported to transduce the majority of cortical neurons and cerebellar Purkinje cells (33). This neuronal cell tropism was confirmed in our study. eGFP reporter expression in IF images demonstrated a particularly strong tropism for Purkinje cells (Fig. 1). Moreover, the appearance of Slc25a46 protein in the central nervous system has been proven by western blotting (Supplementary Material, Fig. S2A). Given the particularly strong association between Purkinje cell degeneration and ataxia (34) and the fact that the cerebellum is one of the most compromised organs in these mutants (10), neurotropism may explain the relatively stable behavioral rescue by our AAV–Slc25a46 vector.
One crucial issue revealed by this study is the dose-dependency of AAV–Slc25a46 treatment. Our results showed that while an injection titer of 1 × 1011 GC/g produced a modest rescue effect on Slc25a46−/− mutants, this effect was not nearly as robust as a 2 × 1011 GC/g dose (Fig. 2). Accordingly, the low dose did not produce any detectable change in the bodyweight of the mutants, although it did elongate the lifespan. In contrast to other studies involving gene therapy-based rescue (31,35,36), this dose requirement is relatively high, likely because the Slc25a46 gene is globally expressed in vivo (15). As a result, it is expected that different genetic mitochondrial defects will require different doses of rescue vector to show improvement because of the unique expression level of each gene. Nevertheless, as the 2 × 1011 GC/g dose does not exhibit significant rescue of the phenotype of Slc25a46−/− mutants, further exploration of the dosing issue is likely warranted if we intend to translate this approach to a clinical trial in the future. For instance, according to the results from immunoblotting, the expression of our desired Slc25a46 protein after AAV administration was still far from the natural expression levels observed in WT animals. This may be the main reason that the phenotype of the mutant is not fully rescued after AAV vector treatment. However, to achieve an amount equal to WT animals, it might require more than 10 times the presenting dose (Supplementary Material, Fig. S2B) or repeat administrations, which will result in a massive increase in the cost of vector production. This is not a trivial consideration, as the manufacturing of AAV at a clinical scale remains a challenging process (37). An alternative production pathway using insect baculovirus-sf9 cell might be one viable alternative in the future, as this approach was reported to increase production 10–100-fold in contrast to the human embryonic kidney 293 cells (HEK293) cell manufacturing system currently favored by the field (38). On the other hand, it may be worth considering what dose threshold is likely to be effective for the subject undergoing treatment and to use this to establish a dosage that maximizes the relief of symptoms while simultaneously balancing the economic costs of vector production.
Another consideration is how to sustain prolonged expression of the transduced gene. In this study, the signal of the eGFP reporter was significantly reduced after 6 weeks post AAV injection. The diminishing of eGFP in vivo after administration has been previously reported and is believed to be related to neutralizing antibodies generated by the hosts (39). However, the movement ability of treated animals was still noticeably improved. Concurrently, the expression of Slc25a46 also maintained a significant level in the central nervous system. These results might suggest that because of the 2A self-cleaving peptides (P2A) cleavage site, the two transgenes (Slc25a46 and eGFP) can be translated independently (Supplementary Material, Fig. S1). Therefore, as an intact protein, Slc25a46 may not be eliminated by the host. Thus, long-term expression of the therapeutic transgene carried by the AAV vector could be expected. Nonetheless, some reports have claimed that AAV vector-mediated transgene expression can continue for up to 1-year postinjection in mice (40) and perhaps as long as 15 years in the postmortem brain of a cynomolgus macaque (41). However, this point requires further study and corroboration.
Collectively, this study shows that AAV–Slc25a46 is a plausible vector for gene therapy of a mitochondrial disease affecting the central nervous system, as well as multiple other organ systems in our mouse model. Thus, this preclinical study suggests that gene therapy with AAV may represent a formidable approach to the treatment of Slc25a46-related and other mitochondrial disorders.
Materials and Methods
Animal handling and mouse strains
Slc25a46−/− mice were generated on a cross between C57BL/6J (B6) female × DBA/2J (D2) male mice (B6D2) background by the transgenic core of Cincinnati Children’s Hospital Medical Center (CCHMC) and raised in-house, as previously described (10). Animals were housed in a 12 h light/dark cycle inside the animal barrier at CCHMC, and all Slc25a46−/− mice were fed with DietGel® (76A, Clear H2O) mixed with regular chewing food after weaning age. All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee-approved protocol of CCHMC.
AAV vector production
An AAV vector carrying the Slc25a46 coding sequence (Mouse Genome Informatics: 1914703) was designed and shown in (Supplementary Material, Fig. S1). The Slc25a46 coding sequence was obtained from a plasmid carrying the full-length Slc25a46 cDNA (Clone ID: 3990909, purchased from Dharmacon, Lafayette, Colorado, USA). The Slc25a46 coding sequence was then subcloned into the AAV vector with ampicillin selection, cytomegalovirus (CMV) promoter, and FLAG and His tags (Vigene Biosciences, Rockville, Maryland, USA) upstream of an in-frame FLAG tag and under the control of a CMV promoter. The vector also contains an eGFP reporter downstream of the FLAG tag and separated by a P2A self-cleaving peptide. The resulting AAV viral vector containing Slc25a46 cDNA was cross-packaged as serotype PHP.B (i.e. with a serotype PHP.B capsid) to facilitate transduction in the CNS. Packaging, purification and titration of the recombinant viral vector were carried out by Vigene Biosciences (Rockville, Maryland, USA). Purified vectors were stored in 1× PBS (phosphate-buffered saline) containing 350 mm NaCl and 5% D-sorbitol. AAV–PHP.B-eGFP virus was utilized as a negative control for untreated Slc25a46−/− mutants.
AAV vector delivery
To deliver the AAV vector to the mice systemically, aliquots of the vector were injected into the facial vein of pups, based on a previously described protocol (42). First, P1 to P2 mice were immobilized with freezing anesthesia for 30–60 s. An aliquot of the viral vector to be injected was thawed out at the same time, at doses of 1 × 1011 or 2 × 1011 GC/g, diluted in 20 μl PBS. The pups were determined to be under deep anesthesia as soon as they showed a state of decreased activity while still maintaining a smooth rate of breathing. Once under deep anesthesia, the pups were placed under an anatomical microscope. A syringe containing the aliquot of AAV vector was attached to a 31 gauge insulin needle (BD, USA) and inserted obliquely into the temporal vein. The contents of the syringe were injected into the vein slowly, and the lower facial vein was monitored during the process for signs of whitening. After injection, the needle was retracted, and the injection site was pressed with a cotton stick until the bleeding stopped. Pups were warmed with a heat pack to recover from anesthesia and then placed back with the mother.
Golgi staining
Eight-week-old mice were deeply anesthetized by CO2, followed by euthanasia via cervical dislocation as previously described (10). The cerebellum was then removed and prepared for Golgi staining using the FD Rapid Golgi Stain Kit (FD NeuroTechnologies, MD) according to the manufacturer’s protocol (43). After staining, the cerebellum was embedded in 4% agarose and cut into 150 nm sagittal sections by vibratome (LEICA VT1000 S). The sections were then mounted on gelatin-coated slides, dehydrated with gradient ethanol and cleared with xylene and then coverslipped with Permount® (Fisher Scientific, NH). Images were acquired with Zeiss Axioskop.
IF staining
Frozen tissue sections were processed as described previously (44). Briefly, mice were perfused transcardially with PBS followed by fresh 4% paraformaldehyde (PFA) in PBS. After overnight postfixation in 4% PFA at 4°C, tissues were dehydrated in 30% sucrose for at least 24 h. Tissue sections 10 μm in thickness were cut on a cryostat (Leica CM1850) and processed for IF. Sections were labeled with the following primary antibodies: rabbit anti-green fluorescent protein, mouse anti-NeuN (1:2000, Millipore, MA), rabbit anti-calbindin D28k (1:1000, Swant, Switzerland), mouse anti-GFAP (1:1000, Millipore, MA) and mouse anti-Brn3a (1:250, Santa Cruz, TX). Secondary antibodies used were as follows: Alexa Fluor-488 (1:500, Jackson ImmunoResearch). Processed sections were mounted onto SuperFrost microscope slides (Fisher Scientific, NH) using a mounting medium including 4′,6-diamidino-2-phenylindole (Vector, CA) and then imaged with either a Zeiss Axiovert 200M or a Nikon C2 confocal microscope.
Fluoro-Jade C staining
Sections were mounted on gelatin-coated slides, air-dried and subjected to Fluoro-Jade C staining. Slides were first immersed for 5 min in a solution containing 1% NaOH diluted in 80% ethanol, then rinsed in 70% ethanol for 2 min and in distilled water for 2 min and finally incubated for 10 min in a solution of 0.06% potassium permanganate. Following a 2 min rinse in distilled water, slides were transferred to a solution of 0.0001% Fluoro-Jade C stain dissolved in 0.1% acetic acid and incubated for 10 min. After washing three times with water, slides were air-dried on a slide warmer at 50°C for 30 min, then cleared in xylene and coverslipped with DPX mounting medium (Sigma-Aldrich, MO). Signals were visualized using a Zeiss Axiovert 200 M microscope.
EM and toluidine blue staining
P56 mice were anesthetized using CO2 and perfused with 0.9% saline, followed by EM fixative (4% PFA, 2.5% glutaraldehyde, PBS, pH 7.4–7.6). Tissues were postfixed in EM fixative, washed in 0.1 M Na cacodylate buffer (EMS, Hatfield, PA) and finally postfixed in 1% osmium tetroxide (EMS) for 1 h at 4°C. Samples were washed in 0.1 M Na cacodylate buffer and dehydrated through a graded ethanol series. Samples were then embedded in LX-112 (Ladd Research Industries, Williston, VT). Tissue blocks were sectioned to a thickness of 0.5 μm and stained with toluidine blue for examination under a light microscope. For EM, tissue blocks were cut and trimmed with an ultramicrotome (Leica EM UC7, Buffalo Grove, IL) to a thickness of 90 nm. Sections were counterstained with uranyl acetate 2% (EMS) and lead citrate. All images were taken with an 80-kV transmission electron microscope (Hitachi, H-7650, V01.07, Tokyo, Japan).
Western blotting
Cell lysates from mouse tissues were harvested and then homogenized after euthanasia. Briefly, 120 μg of protein from each mouse tissue was mixed with 2× RIPA lysis and extraction buffer (Thermo Fisher Scientific, NH). Then, the mixed samples were heated for 5 min at 95°C for denaturing before separating on a NuPAGE 4–12% Bis–Tris gel (Invitrogen, Carlsbad, CA). The gel was then transferred using an iBlot semidry transferring system onto a polyvinylidene fluoride (PVDF) membrane (Invitrogen, Carlsbad, CA). Afterward, the PVDF membrane was blocked in Intercept™ Blocking Buffer (LI-COR Biosciences, Lincoln, NE) for 45 min and then incubated overnight with primary antibodies diluted in blocking buffer with 0.1% Tween 20 and rabbit anti-SLC25A46 (Abcam, Cambridge, MA) at 1:1000 dilution. After overnight incubation, the membrane was washed three times to removed unbound primary antibody and then incubated for 90 min with IRDye 800CW Goat anti-Rabbit IgG secondary antibodies (LI-COR Biosciences, Lincoln, NE). Bands were visualized using the LI-COR Odyssey CLx Imaging System (LI-COR Biosciences, Lincoln, NE). As a loading control, anti-glyceraldehyde 3-phosphate dehydrogenase (Ambion, Foster City, CA) was used at 1:2000 dilution.
Complex activity assays
Activity levels of respiratory chain complexes were determined as described previously (45). Briefly, complex I activity was assessed by measuring the decrease of NADH absorbance at 340 nm, using decylubiquinone as an electron acceptor. Complex II and III activity levels were determined by the increased absorbance of reduced cytochrome C at 550 nm, with potassium cyanide added into the reaction medium to inhibit subsequent oxidation. Complex IV activity was demonstrated using Complex IV Rodent Enzyme Activity Microplate Assay Kit (Abcam, United Kingdom). The activity of Complex IV is determined colorimetrically by following the oxidation of reduced cytochrome c by the absorbance change at 550 nm. All absorbance values were measured on a Synergy H1 microplate reader (BioTek Instruments, VT). Activity levels were calculated as nmol/min/mg protein and normalized to those of WT mice.
Electromyograph recording
Electromyography was performed as previously described (10). P55 mice were anesthetized with isoflurane and then intraperitoneally injected with 50 mg/kg sodium phenobarbital. The lateral gastrocnemius muscle is then exposed from the knee to approximately 4 mm above the ankle. The sciatic nerve is then exposed near the biceps femoris. A polyester film-coated steel recording line (California thin line) was implanted outside the gastrocnemius muscle, and a reference line was inserted under the skin near the bottom of the tail. A concentric bipolar stimulation electrode was placed on the sciatic nerve for electrical activation.
The composite muscle action potential was amplified, acquired using a Micro 1401 data acquisition unit and analyzed offline using the Spike5 software (Cambridge Electronic Design, Cambridge, UK). The sciatic nerve adjacent to the tibia and fibula was subjected to 2–4 MA electrical stimulation by a stimulation isolator (World Precision Instruments) attached to the micro 1401. After recording, the sciatic nerve axons were removed. The proximal sciatic never was stimulated to ensure that CMAP is produced by direct nerve stimulation. CMAP, CV was calculated from each stimulation pattern. The average stimulation of the sciatic nerve in each mode was obtained and averaged in the animals.
Statistical analysis
P values of the survival curve were generated using the Log-rank (Mantel-Cox) test. The rest of the comparisons between groups were using the student’s t-test. Graphical illustrations and significance were obtained with GraphPad Prism 7 (GraphPad). P < 0.05 was considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Supplementary Material
Acknowledgements
The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Funding
Center for Pediatric Genomics (CpG); Cincinnati Children’s Research Foundation, Hadley Jo Foundation, and National Institutes of Health (1R01EY026609-01 to T.H.); National Institutes of Health (grant R01AR064551-01A1 to M.P.J.); China Scholarship Council (to Y.L.)
Conflict of Interest
The authors declare that they have no competing interests.
References
- 1. Calvo S.E., Clauser K.R. and Mootha V.K. (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res, 44, D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wallace D.C. (2012) Mitochondria and cancer. Nat Rev Cancer, 12, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balakrishnan B. and Jayandharan G.R. (2014) Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr Gene Ther, 14, 86–100. [DOI] [PubMed] [Google Scholar]
- 4. Smith R.H. (2008) Adeno-associated virus integration: virus versus vector. Gene Ther, 15, 817–822. [DOI] [PubMed] [Google Scholar]
- 5. Colella P., Ronzitti G. and Mingozzi F. (2018) Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev, 8, 87–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abrams A.J., Hufnagel R.B., Rebelo A., Zanna C., Patel N., Gonzalez M.A., Campeanu I.J., Griffin L.B., Groenewald S., Strickland A.V. et al. (2015) Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet, 47, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Janer A., Prudent J., Paupe V., Fahiminiya S., Majewski J., Sgarioto N., Des Rosiers C., Forest A., Lin Z.Y., Gingras A.C. et al. (2016) SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol Med, 8, 1019–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steffen J., Vashisht A.A., Wan J., Jen J.C., Claypool S.M., Wohlschlegel J.A. and Koehler C.M. (2017) Rapid degradation of mutant SLC25A46 by the ubiquitin-proteasome system results in MFN1/2-mediated hyperfusion of mitochondria. Mol Biol Cell, 28, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Terzenidou M.E., Segklia A., Kano T., Papastefanaki F., Karakostas A., Charalambous M., Ioakeimidis F., Papadaki M., Kloukina I., Chrysanthou-Piterou M. et al. (2017) Novel insights into SLC25A46-related pathologies in a genetic mouse model. PLoS Genet, 13, e1006656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Z., Peng Y., Hufnagel R.B., Hu Y.C., Zhao C., Queme L.F., Khuchua Z., Driver A.M., Dong F., Lu Q.R. et al. (2017) Loss of SLC25A46 causes neurodegeneration by affecting mitochondrial dynamics and energy production in mice. Hum Mol Genet, 26, 3776–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Charlesworth G., Balint B., Mencacci N.E., Carr L., Wood N.W. and Bhatia K.P. (2016) SLC25A46 mutations underlie progressive myoclonic ataxia with optic atrophy and neuropathy. Mov Disord, 31, 1249–1251. [DOI] [PubMed] [Google Scholar]
- 12. Nguyen M., Boesten I., Hellebrekers D.M., Mulder-den Hartog N.M., Coo I.F., Smeets H.J. and Gerards M. (2017) Novel pathogenic SLC25A46 splice-site mutation causes an optic atrophy spectrum disorder. Clin Genet, 91, 121–125. [DOI] [PubMed] [Google Scholar]
- 13. Wan J., Steffen J., Yourshaw M., Mamsa H., Andersen E., Rudnik-Schoneborn S., Pope K., Howell K.B., McLean C.A., Kornberg A.J. et al. (2016) Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain, 139, 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duchesne A., Vaiman A., Castille J., Beauvallet C., Gaignard P., Floriot S., Rodriguez S., Vilotte M., Boulanger L., Passet B. et al. (2017) Bovine and murine models highlight novel roles for SLC25A46 in mitochondrial dynamics and metabolism, with implications for human and animal health. PLoS Genet, 13, e1006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fagerberg L., Hallstrom B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K. et al. (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics, 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bailey R.M., Armao D., Nagabhushan Kalburgi S. and Gray S.J. (2018) Development of Intrathecal AAV9 gene therapy for Giant axonal neuropathy. Mol Ther Methods Clin Dev, 9, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gong Y., Berenson A., Laheji F., Gao G., Wang D., Ng C., Volak A., Kok R., Kreouzis V., Dijkstra I. et al. (2019) Intrathecal adeno-associated virus vector-mediated gene delivery for Adrenomyeloneuropathy. Hum Gene Ther, 30, 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hammond S.L., Leek A.N., Richman E.H. and Tjalkens R.B. (2017) Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS One, 12, e0188830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meijer D.H., Maguire C.A., LeRoy S.G. and Sena-Esteves M. (2009) Controlling brain tumor growth by intraventricular administration of an AAV vector encoding IFN-beta. Cancer Gene Ther, 16, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fraldi A., Hemsley K., Crawley A., Lombardi A., Lau A., Sutherland L., Auricchio A., Ballabio A. and Hopwood J.J. (2007) Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet, 16, 2693–2702. [DOI] [PubMed] [Google Scholar]
- 21. Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P. et al. (2016) Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol, 34, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slone J., Gui B. and Huang T. (2018) The current landscape for the treatment of mitochondrial disorders. J Genet Genomics, 45, 71–77. [DOI] [PubMed] [Google Scholar]
- 23. Rodriguez M.C., MacDonald J.R., Mahoney D.J., Parise G., Beal M.F. and Tarnopolsky M.A. (2007) Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve, 35, 235–242. [DOI] [PubMed] [Google Scholar]
- 24. Glover E.I., Martin J., Maher A., Thornhill R.E., Moran G.R. and Tarnopolsky M.A. (2010) A randomized trial of coenzyme Q10 in mitochondrial disorders. Muscle Nerve, 42, 739–748. [DOI] [PubMed] [Google Scholar]
- 25. Kerr D.S. (2013) Review of clinical trials for mitochondrial disorders: 1997–2012. Neurotherapeutics, 10, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deverman B.E., Ravina B.M., Bankiewicz K.S., Paul S.M. and Sah D.W.Y. (2018) Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov, 17, 767. [DOI] [PubMed] [Google Scholar]
- 27. Wang D., Tai P.W.L. and Gao G. (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov, 18, 358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di I., Marchet S., Lamperti C., Zeviani M. and Viscomi C. (2017) AAV9-based gene therapy partially ameliorates the clinical phenotype of a mouse model of Leigh syndrome. Gene Ther, 24, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suzuki-Hatano S., Saha M., Rizzo S.A., Witko R.L., Gosiker B.J., Ramanathan M., Soustek M.S., Jones M.D., Kang P.B., Byrne B.J. et al. (2019) AAV-mediated TAZ gene replacement restores mitochondrial and cardioskeletal function in Barth syndrome. Hum Gene Ther, 30, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Torres-Torronteras J., Cabrera-Perez R., Vila-Julia F., Viscomi C., Camara Y., Hirano M., Zeviani M. and Marti R. (2018) Long-term sustained effect of liver-targeted adeno-associated virus gene therapy for mitochondrial neurogastrointestinal encephalomyopathy. Hum Gene Ther, 29, 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morabito G., Giannelli S.G., Ordazzo G., Bido S., Castoldi V., Indrigo M., Cabassi T., Cattaneo S., Luoni M., Cancellieri C. et al. (2017) AAV-PHP.B-mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol Ther, 25, 2727–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hordeaux J., Yuan Y., Clark P.M., Wang Q., Martino R.A., Sims J.J., Bell P., Raymond A., Stanford W.L. and Wilson J.M. (2019) The GPI-linked protein LY6A drives AAV-PHP.B transport across the blood–brain barrier. Mol Ther, 27, 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang Q., Chan K.Y., Tobey I.G., Chan Y.A., Poterba T., Boutros C.L., Balazs A.B., Daneman R., Bloom J.M., Seed C. et al. (2019) Delivering genes across the blood–brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS One, 14, e0225206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoxha E., Balbo I., Miniaci M.C. and Tempia F. (2018) Purkinje cell Signaling deficits in animal models of ataxia. Front Synaptic Neurosci, 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. et al. (2017) Single-dose gene-replacement therapy for spinal muscular atrophy. NEJM, 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- 36. de Moura A.P., Dominguez E., Carcenac R., Duque S., Benkhelifa-Ziyyat S., Astord S., Marais T., Voit T., Barkats M., Chatauret N. et al. (2010) Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet, 20, 681–693. [DOI] [PubMed] [Google Scholar]
- 37. Deverman B.E., Ravina B.M., Bankiewicz K.S., Paul S.M. and Sah D.W.Y. (2018) Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov, 17, 641. [DOI] [PubMed] [Google Scholar]
- 38. Kotin R.M. and Snyder R.O. (2017) Manufacturing clinical grade recombinant adeno-associated virus using invertebrate cell lines. Hum Gene Ther, 28, 350–360. [DOI] [PubMed] [Google Scholar]
- 39. Shinohara Y., Konno A., Nitta K., Matsuzaki Y., Yasui H., Suwa J., Hiromura K. and Hirai H. (2019) Effects of neutralizing antibody production on AAV-PHP.B-mediated transduction of the mouse central nervous system. Mol Neurobiol, 56, 4203–4214. [DOI] [PubMed] [Google Scholar]
- 40. Jiang L., Zhang H., Dizhoor A.M., Boye S.E., Hauswirth W.W., Frederick J.M. and Baehr W. (2011) Long-term RNA interference gene therapy in a dominant retinitis pigmentosa mouse model. PNAS, 108, 18476–18481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshihide S., Ken-ichi F., Kunihiko I., Yuko K., Fumiko O., Naomi T., Mika I., Keiya O. and Shin-ichi M. (2017) Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of Parkinson's disease. Hum Gen Ther Clin Dev, 28, 74–79. [DOI] [PubMed] [Google Scholar]
- 42. Gombash Lampe S.E., Kaspar B.K. and Foust K.D. (2014) Intravenous injections in neonatal mice. J Vis Exp, e52037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ayata P., Badimon A., Strasburger H.J., Duff M.K., Montgomery S.E., Loh Y.E., Ebert A., Pimenova A.A., Ramirez B.R., Chan A.T. et al. (2018) Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci, 21, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Riazifar H., Sun G., Wang X., Rupp A., Vemaraju S., Ross-Cisneros F.N., Lang R.A., Sadun A.A., Hattar S., Guan M.X. et al. (2015) Phenotypic and functional characterization of Bst+/− mouse retina. Dis Model Mech, 8, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simon M., Richard E.M., Wang X., Shahzad M., Huang V.H., Qaiser T.A., Potluri P., Mahl S.E., Davila A., Nazli S. et al. (2015) Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet, 11, e1005097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.