

Abstract
Maturases are prokaryotic enzymes that aid self‐excision of introns in precursor RNAs and have evolutionary ties to the nuclear spliceosome. Both the mitochondria and chloroplast, due to their prokaryotic origin, encode a single intron maturase, MatR for the mitochondria and MatK for the chloroplast. MatK is proposed to aid excision of seven different chloroplast group IIA introns that reside within precursor RNAs for essential elements of chloroplast function. We have developed an in vitro activity assay to test chloroplast group IIA intron excision. Using this assay, we demonstrate self‐excision of the group IIA intron of the second intron of rps12 and the group IIA intron of rpl2. We further show that the addition of heterologously expressed MatK protein increases efficiency of group IIA intron self‐splicing for the second intron of rps12 but not the group IIA intron of rpl2. These data, to our knowledge, provide the first direct evidence of MatK’s maturase activity.
Keywords: chloroplast, group II intron, intron splicing, MatK, maturase
1. INTRODUCTION
Intron removal is an essential step of gene expression, without which aberrant transcripts would be formed leading to nonfunctional RNA and/or protein product. Introns can be excised using one of three mechanisms: self‐excision, maturases, or in the case of the eukaryotic nucleus, the spliceosome, a large multi‐protein and RNA complex capable of binding and excising multiple introns. Maturases are intron‐encoded proteins that serve a catalytic function in intron excision and are found in some bacteria and prokaryotic‐descendent organelles such as the chloroplast and mitochondria (Matsuura, Noah, & Lambowitz, 2001; Sultan et al., 2016; Zoschke et al., 2010). Unlike the nuclear spliceosome, prokaryotic maturases, such as the well‐characterized LtrA maturase of Lactococcus lactis, are highly specific to bind and catalyze self‐excision of a single intron target (Matsuura et al., 1997). Maturases found in plant genomes seem to have diverged from their prokaryotic relatives and tend to bind multiple intron targets (Sultan et al., 2016; Zoschke et al., 2010). Maturase K is one such maturase.
Maturase K (MatK) is a plastid‐encoded group II intron maturase of land plants (Liere & Link, 1995; Mohr, Perlman, & Lambowitz, 1993; Sugita, Shinozaki, & Sugiura, 1985). It is an intron‐encoded protein with a relatively high mutation rate at both nucleotide and subsequent amino acid levels (Hilu, Black, & Oza, 2014; Hilu & Liang, 1997; Young & dePamphilis, 2000). This elevated mutation rate is sufficient for use of the matK gene sequence as a DNA barcoding region and as a molecular marker in plant phylogenetic analyses at both shallow and deep taxonomic levels (e.g., CBOL Plant Working Group1, 2009; Hilu, Alice, & Liang, 1999; Hilu et al., 2003, 2014; Li, Gao, Poudel, Li, & Forrest, 2011; Lutzoni et al., 2018). MatK has been shown to bind to seven different intron targets (Zoschke et al., 2010). There are three classes of group II introns, A, B, and C, divided by sequence and structural characteristics, including how each type recognizes exons for splicing (Costa, Michel, & Westhof, 2000; Lambowitz & Zimmerly, 2011; Martínez‐Abarca, Zekri, & Toro, 1998; Michel, Umesono, & Ozeki, 1989; Toor, Hausner, & Zimmerly, 2001; Toor, Rajashankar, Keating, & Pyle, 2008). All seven intron targets of MatK are classified as group IIA introns (Zoschke et al., 2010). These group IIA introns lie within precursor RNAs for tRNAs (trnK, trnA, trnI, and trnV) and ribosomal proteins (rpl2 and the second intron of rps12 (rps12‐2)) necessary for function of the chloroplast translation machinery, as well as one subunit of ATP synthase (atpF) needed for generation of ATP for photosynthesis (Zoschke et al., 2010). The importance of the tRNAs and protein products from these precursor RNAs for formation and function of the plastid translational machinery implicates MatK as an essential enzyme for chloroplast function, without which no plastid proteins could be made.
Examination of the white barley mutant, albostrians, indirectly supports the essential role of MatK for chloroplast function and intron excision. The albostrians mutant is a chloroplast ribosomal mutant lacking the ability for translation of all chloroplast proteins, including MatK. Nuclear protein translation and import into the chloroplast was not impacted in the albostrians mutant; however, seven precursor RNAs containing group IIA introns lacked or had significantly reduced intron excision in the albostrians mutant compared with wild‐type barley (Hess et al., 1994; Vogel, Börner, & Hess, 1999; Vogel, Hübschmann, Börner, & Hess, 1997). Although a direct knockout of MatK has never been made, the lack of group IIA intron excision in the albostrians mutant suggested that a chloroplast‐encoded factor was required for group IIA intron excision, most likely MatK (Hess et al., 1994; Vogel et al., 1997, 1999). Later studies using RNA immunoprecipitation (RIP) and homoplastomic HA‐tagged MatK protein in tobacco resulted in the pull‐down of the same seven group IIA introns by MatK that were lacking intron excision in the albostrians mutant (Zoschke et al., 2010). In addition, a recent attempt to overexpress MatK in the chloroplast of tobacco led to the reduction in MatK protein in homoplastomic plants, correlating with a variegated phenotype in cotyledon development, and further supporting a link between MatK expression and chloroplast function (Qu et al., 2018). Together, the albostrians mutant and RIP studies highly support the proposed function of MatK as a group IIA intron maturase.
MatK differs from other prokaryotic maturases in the number of targets it binds. This could be due to loss of regions of functional domains meant to restrict intron target association. MatK has lost two of the three main functional domains found in other prokaryotic‐like maturases. The core functional domains of prokaryotic‐like maturases are a reverse transcriptase (RT) domain which comprises the fingers and palm subdomains typical of a DNA polymerase, domain X, which comprises a region analogous to the polymerase thumb domain, and a C‐terminal DNA endonuclease domain (Blocker et al., 2005; Joyce & Steitz, 1994; Mohr et al., 1993; Sultan et al., 2016). MatK contains only four of the seven RT domain sequence motifs and has completely lost the DNA endonuclease domain (Barthet & Hilu, 2008; Mohr et al., 1993). One additional sequence motif of the RT region is RT0, an extended motif that aids RNA binding (Cui, Matsuura, Wang, Ma, & Lambowitz, 2004; Gu et al., 2010). The RT0 and RT sequence motifs 1–4 have been shown to not only contribute to RNA binding but also contribute to target specificity (Gu et al., 2010; Zhao & Pyle, 2016). Although the MatK protein contains part of the RT0 sequence motif, most of this region is missing (Barthet & Hilu, 2008). The lack of part of RT0, as well as the missing elements from the rest of the RT domain, may contribute to the broader range of intron targets for the MatK maturase. MatK retains domain X, the functional domain for maturase (RNA splicing) activity (Cui et al., 2004; Moran et al., 1994). The retention of domain X suggests that this enzyme is capable of facilitating the formation of the catalytic structure necessary for group IIA intron self‐excision, the question remains, however, whether MatK is able to excise group IIA introns alone or requires additional protein factors for activity due to loss of sequence domains found in other related maturases. We have explored self‐excision of MatK‐targeted group IIA introns as well as the proposed enzymatic function of MatK as a group IIA intron maturase using in vitro activity assays.
2. MATERIALS AND METHODS
2.1. Plant material and growth conditions
Oryza sativa japonica (rice) seeds maintained at 4°C were sterilized in 95% ethanol for 1 min, followed by 5 min in a 50% bleach and 0.05% Tween 20 solution. Seeds were then rinsed three times with sterile ultrapure water prior to planting. Seeds were planted in 1/3 AIS (Enriched) Arabidopsis Growth Medium (Lehle Seeds) and 2/3 vermiculite and germinated under 100% humidity with a photoperiod of 16‐hr light/8‐hr dark with ~26.7 µmoles/m2/s of light.
2.2. RNA extraction and RT‐PCR of MatK open reading frame
Approximately 100 mg of leaf tissue from 2‐week‐old O. sativa plants was ground under liquid nitrogen and processed for RNA extraction using the RNAeasy Plant Mini Kit (Qiagen) following the manufacturer's instructions. Extracted RNA was treated with Turbo™ DNase (Ambion) prior to reverse transcription. DNase was removed using the included DNase inactivation solution from the Turbo DNA‐free Kit™ (Ambion). Five micrograms of DNA‐free RNA was converted to cDNA using oligo dT(20) primer with one microliter of SuperScript III Reverse Transcriptase at 200 u/µl (Invitrogen) per a reaction according to the standard two‐step manufacture protocol (Invitrogen). The reaction was terminated by incubation at 70°C for 15 min. Remaining RNA template was removed from the reaction by the addition of two units of Escherichia coli RNase H and incubation at 37°C for 20 min.
2.3. Cloning of the MatK ORF
The O. sativa matK open reading frame (ORF) has an in‐frame upstream initiation codon (R1) that leads to an elongated N‐terminus in comparison with the matK coding region from other related monocots (Figure 1a,b). Translation of matK using the R1 initiation codon in O. sativa produced a full‐length translated protein product of ~553 amino acids (Figure 1b). Translation from the R1 initiation codon in closely related monocots resulted in premature stop codons due to a 1‐bp deletion in the reading frame upstream of the R2 initiation codon relative to the open reading frame in O. sativa (Figure 1a,b). We included the elongated N‐terminal region generated using the R1 initiation codon in our MatK clone in order to ensure the most complete form of the MatK protein was expressed in induced cultures and any lack of function could not be attributed to lack of required protein regions. The full‐length reading frame for matK using the R1 initiation codon from O. sativa japonica was amplified from cDNA using Phusion® DNA polymerase (2,000 units/ml, NEB) with the following primers: 5′ GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGCAACACCCTGTTCTGACCA 3′ (forward primer) and 5′ GGGGACCACTTTGTACAAGAAAGCTGGGTTTTAATTAAGAGGATTCACCAG 3′ (reverse primer) which included attB sites for Gateway cloning (underlined portion of sequences above). Products were amplified using the standard Phusion® PCR protocol (NEB) with a final concentration of 0.6 µM of each primer and 0.5 units of Phusion® polymerase per a reaction. PCR cycling conditions were 98°C for 2 min, followed by 50 cycles of amplification (98°C for 1 min, 52°C for 1 min, 72°C for 3 min), and a final extension of 72°C for 10 min. Amplified product was purified using 30% PEG 8000/30 mM MgCl2 solution and cloned into pDONR™221 entry vector (Invitrogen) followed by subsequent sub‐cloning into pDEST™17 expression vector (Invitrogen) behind a 6X histidine tag. Cloned products were sequenced. The final clone used for MatK expression was found to contain two mutations relative to the genomic DNA sequence for matK based on GenBank accession GU592207. The first mutation at position 605 relative to the initiation codon (G to T transversion) resulted in a silent mutation that did not impact the reading frame or translated amino acid sequence of MatK in anyway. The second mutation was a C to T transition that resulted in a histidine conversion to a tyrosine amino acid (Figure S1). This second mutation is the result of a conserved RNA editing site found previously in other monocots such as barley and maize (Tillich, Schmitz‐Linneweber, Herrmann, & Maier, 2001; Vogel et al., 1997).
Figure 1.

Nucleotide and translated amino acid sequence using the R1 and R2 potential initial codons for the MatK maturase of Oryza sativa as compared to related members of the Poaceae family. (a) Nucleotide sequence and (b) translated amino acid sequence of the matK gene from six related monocots of the Poaceae as described by Soreng et al. (2017). Stop codons are highlighted. Species abbreviations: Oryza sativa (O.s.), Valiha sp. (V.sp.), Brachyelytrum aristosum (B.a.), Lygeum spartum (L.s.), Bromus leptoclados (B.l.), Poa perrieri (P.p.). ATG initiation codons are blocked in red. Dashes in alignment represent gaps. Stop codons (*) in translated amino acid sequence are highlighted. Species chosen for comparison of the matK gene and protein sequence are all from species of the BOP (Bambusoideae, Oryzoideae, and Pooideae) clade of Poaceae and represent the closest relatives of rice outside of the Oryzoideae tribe according to Soreng et al. (2017)
2.4. Heterologous expression of MatK in E. coli
The final expression clone containing the matK reading frame was heat shock transformed into E. coli BL21 DE3 pLysS competent cells (Promega) and protein induced using IPTG following standard transformation and induction protocols using IPTG at a final concentration of 500 µM. Induced cultures were shaken at 210 rpm at 35°C for an additional three hours before pelleting cultures at 10,000 rpm for 10 min for 4°C. Cold temperature spins were used to prevent protein degradation. Induction at 37°C was found to induce proteolysis of MatK protein in E. coli (data not shown); therefore, a lower temperature of 35°C was used for protein induction. Pre‐ and post‐induction samples were assayed for MatK expression by immune detection using one of two different anti‐MatK antibodies, a commercial antibody from Agrisera (AS132720) or an epitope‐derived antibody specifically designed against the MatK protein sequence in rice (Barthet & Hilu, 2007).
2.5. MatK isolation
Following protein induction, cell pellets were resuspended in 3 mls/g of wet weight of 1X binding buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, and 5 mM imidazole), incubated with rLysozyme™ (EMD Millipore) at a final concentration of 45 ku/g of cell paste, and sonicated on ice using a Model 100 Sonic Dismembrator (Fisher Scientific) at 200–300 volts six times with 10‐s cooling between bursts. Benzonase® Nuclease (Merck) 25 µl/ml of lysis (1X binding) buffer was added to protein lysate to ensure removal of nucleic acids. Final lysate was centrifuged at 10,000x g for 30 min at 4°C to remove cell debris. MatK protein was purified using a fused 6X histidine tag using Ni‐NTA His·Bind® SuperFlow™ Resin (EMD Millipore) according to the manufacturer's instructions with the exception of using a 1X Ni‐NTA binding buffer containing 5 mM imidazole. The final eluate (Elute 1) was run through a second column of Ni‐NTA His·Bind® SuperFlow™ Resin (EMD Millipore) to remove any residual contaminating proteins. The flow‐through fraction (fraction 2) generated after running Elute 1 over a second Ni‐NTA column was collected, and proteins from this fraction were resolved by SDS‐PAGE followed by staining to assess the presence of any additional protein bands. Resolved fractions were further transferred to PVDF membrane, and MatK protein was detected using the Agrisera anti‐MatK antibody (AS132720) and/or the anti‐MatK antibody described by Barthet and Hilu (2007) using Pierce™ ECL Western blotting substrate for luminescent detection. Concentration of isolated MatK protein was determined using Coomassie G‐250 (Bio‐Rad) as described by Bradford (1976) with bovine serum albumin (BSA) as the standard. Confirmation that isolated protein fractions contained MatK protein and identification of any background protein contaminants was achieved using electrospray ionization (ESI). To prepare protein for ESI mass spectrometry, protein fractions underwent alkylation, trypsin cleavage, and guanidination using Thermo Scientific™ Pierce™ In‐Solution Tryptic Digestion and Guanidination Kit. Samples were sent to the Mass Spectrometry Center at the University of South Carolina for ESI processing. Proteome Discover™ software using the SEQUEST algorithm (Thermo Scientific™) was used for initial peptide analyses followed by protein identification using the UniProt proteome database (http://www.uniprot.org) and O. sativa as the model organism.
2.6. Preparation of RNA substrates for activity assays
Primers were designed to amplify gene regions that surround postulated MatK‐target group IIA intron substrates (Table S1; Figure 2a). Genomic DNA used as the template for PCR amplification was extracted from 2‐week rice blades using the CTAB method (Doyle & Doyle, 1990) with the modification of adding 0.1% beta‐mercaptoethanol into the CTAB buffer. Amplified products were generated using Phusion® DNA polymerase (NEB), purified using a 30% PEG 8000/30 mM MgCl2 solution, and cloned into pDONR™221 entry vector, followed by sub‐cloning into pDEST™17 using the Gateway™ method (Invitrogen). Recombinant clones were extracted from host cells using the Wizard® Plus SV Minipreps DNA Purification System (Promega) and sequenced. The pDEST™17 expression vector includes a T7 promoter which then was used for in vitro transcription of RNA transcripts using the MEGAscript™ T7 Transcript Kit (Ambion). Rps12 has three exons and two introns. The first intron is trans‐spliced (Hildebrand, Hallick, Passavant, & Bourque, 1988; Tadini et al., 2018; Zaita, Torazawa, Shinozaki, & Sugiura, 1987), and only the second intron has been proposed to require the MatK maturase for self‐excision (Zoschke et al., 2010). Only regions of exons 2 and 3 that surround the second intron of rps12, therefore, were cloned and used to generate RNA substrate for testing MatK activity (Figure 2a).
Figure 2.
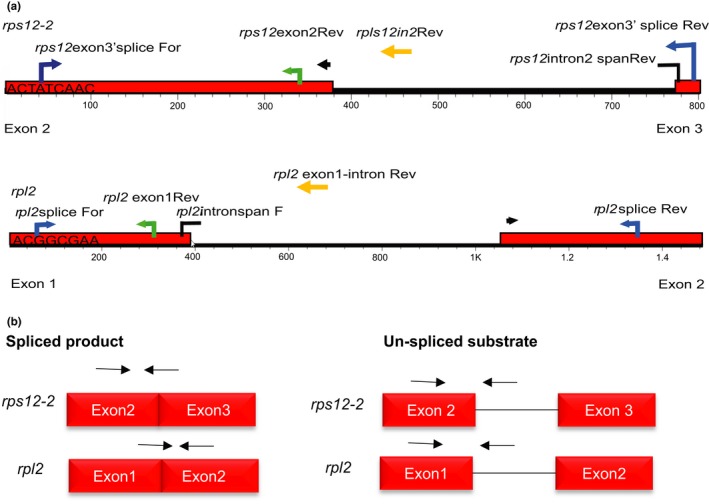
Annotation of primer locations used for cloning and amplification of RNA substrates and qPCR products. (a) Primers mapped onto genomic DNA sequence of rps12 exons 2 to 3 including intron 2 (top) or rpl2 (bottom). Blue arrows = primers used for amplification of RNA substrates; broken black arrows = intron‐spanning primers; yellow arrows = reverse primer used to detect un‐spliced product; green arrows = primers used to detect total RNA. (b) Schematic representation of expected products from qPCR based on spliced and un‐spliced primers annotated in (a). Arrows represent relative position of primers to template needed to amplify spliced products (left) or un‐spliced products (right)
2.7. Group IIA intron self‐excision and maturase activity assay
Twenty nanomolar of group IIA intron‐containing RNA substrate was heat‐denatured at 90°C for 2 min in reaction buffer modified from Holländer and Kück (1999) to contain 5 mM MgCl2, 40 mM Tris‐HCl pH 7.5, and 0.5 M NH4Cl, prior to the addition of MatK protein in order to unfold any previous tertiary structure that may preclude protein binding. Low magnesium (5 mM MgCl2) concentration in buffers for L. lactis was shown to induce a protective tertiary structure preventing self‐excision of the L1.LtrB group IIA intron (Matsuura et al., 2001). Since the main goal of our study was to examine MatK maturase activity, we chose to use a low magnesium (5 mM MgCl2) buffer to prevent or reduce possible self‐excision of targeted group IIA introns. Impact of heat denaturing and low magnesium concentration on group IIA intron self‐excision was tested by incubating RNA in water, in reaction buffer without prior heat denaturing, and after heat denaturing. MatK activity was tested by the addition of 200 nM of isolated Ni‐NTA 6X His‐tagged MatK protein to reaction buffer containing heat‐denatured precursor RNA substrates and the reaction incubated at 26°C for up to 60 min. Aliquots were taken of the initial reaction mixture and assayed for production of spliced product, as well as un‐spliced substrate and total RNA, at zero (immediately after all components of the reaction were mixed together), 15, 30, and 60 min. Several controls were run for each activity assay. Controls of MatK protein alone (just the MatK protein in buffer without any additional components) and mock‐induction alone (E. coli background protein eluted from Ni‐NTA columns in buffer without additional components) were used to check for nucleic acid contamination that could come from the protein isolation itself. Mock induction + RNA was a control for any change in RNA levels due to background protein isolated from Ni‐NTA columns, and RNA alone was a control for RNA self‐splicing activity. Mock‐induction controls included Ni‐NTA isolated protein lysate from E. coli BL21 DE3 pLysS cells lacking MatK‐pDEST™17 plasmids. Briefly, mock‐induction control cells were induced identically to cells that included MatK‐pDEST™17 plasmid with 0.5 mM IPTG at 35°C. Pre‐ and post‐induction samples were taken for protein analyses. Cell pellets were then lysed and protein purified in the exact same manner as was done for induced MatK protein. All reactions were replicated in triplicate. Fifty nanograms of each aliquot was reverse‐transcribed (RT) using SuperScript™ III Reverse Transcriptase according to the manufacturer's instructions (Invitrogen) using a gene‐specific reverse primer for first‐strand synthesis (Table S1). Samples of each activity assay or stock RNA were assayed for genomic DNA contamination by performing an identical reverse transcription reaction but without the SuperScript™ III Reverse Transcriptase (“No RT” controls).
2.8. qPCR
Quantitative PCR was used for the assessment of efficiency of RNA self‐excision with and without the addition of MatK. Although assessment by formaldehyde gel electrophoresis would have been advantageous to directly evaluate splicing activity without concern about RT artifacts, this could not be done due to low concentrations of RNA used in the activity assays (20 nM or 818 ng/100 µl reaction for rps12; 20 nM or 848 ng/100 µl reaction for rpl2) limiting visualization of spliced product by agarose gel electrophoresis. For this reason, qPCR was chosen as the method to quantitatively ascertain splicing efficiency. qPCR has been shown previously to provide accurate quantitative detection of spliced products, even more so than densitometry of products resolved by gel electrophoresis (Yoon et al., 2019). qPCRs were performed using SsoAdvanced™ Universal SYBR® Green Supermix (Bio‐Rad) on the Bio‐Rad CFX 96 Touch™ Real‐time PCR Detection System with at least two technical replicates for each sample and a melting curve used to assess specificity of product formation in the reaction. cDNA was diluted 10‐fold in DEPC‐treated water with 1 µl of each diluted cDNA used for qPCR analyses. Random samples of qPCR products from each qPCR run were resolved on agarose gels to confirm product and ensure qPCR data correlated with formation of resolved product and were not the result of primer dimer or contaminants in the reaction. Products observed from qPCR data and gel electrophoresis were sequenced to confirm identity. Reaction conditions included an initial 95°C denaturing step for 30 s followed by 40 cycles of 95°C for 10 s, annealing temperature (Table S1) for 30 s, and 72°C for 1 min. Plates were read after the extension step for each cycle. The melt curve ran from 65 to 95°C in 0.5°C increments for 0.05 s per a step.
Group IIA self‐excision occurs with two trans‐esterification steps. The first step results in a free 5′ exon and an intron‐3′ exon intermediate. The second step is the nucleophilic attack of the free 3′ OH of the 5′ exon to the 5′ phosphate of the 3′ exon resulting in exon–exon (spliced product) ligation (reviewed in Smathers & Robart, 2019). Intron‐spanning primers were designed that required exons to be ligated together, the final product of intron excision, for at least one of the two primers in the primer pair to bind and allow amplification (Table S1; Figure 2b). Un‐spliced substrate was detected using primers that bound to the 5′ exon and group IIA intron, while total RNA was detected using primers that bound only to the 5′ exon (Table S1; Figure 2b). Both spliced product and un‐spliced substrate would contain the 5′ exon. Some splicing intermediates may not include this exon, but the exon would remain in solution enabling a way to estimate total RNA at each time point.
2.9. Generation of standard curves for qPCR
Standard curves used for absolute quantification of reaction products were generated using 10‐fold dilution series of plasmids that contained either spliced product for each substrate or un‐spliced substrate (exons surrounding the group IIA intron). To clone spliced products, RNA was extracted from approximately 100 mg of 2‐week‐old rice leaf tissue ground under liquid nitrogen and processed for RNA extraction using the RNAeasy Plant Mini Kit (Qiagen). RNA was used as the template to ensure that the group IIA intron was excised from precursor RNA molecules and only spliced product remained. Extracted RNA was treated with Turbo™ DNase (Ambion) prior to reverse transcription. DNase was removed using the included DNase inactivation solution provided in the Turbo DNA‐free Kit™ (Ambion). DNA‐free RNA was converted to cDNA using SuperScript™ III Reverse Transcriptase (Invitrogen) according to the standard two‐step manufacture protocol using an oligo dT(20) primer for first‐strand synthesis followed by RNase H digestion of residual RNA (Invitrogen). Spliced products were then amplified using Phusion® DNA polymerase (NEB) from cDNA template using gene‐specific primers with Gateway adapter sites (Table S1). Amplified product was purified using a 30% PEG 8000/30 mM MgCl2 solution and cloned into pDONR™221 entry vector using BP Clonase™ II (Invitrogen). All plasmids were sequenced to confirm cloning of the correct spliced product. Quantity of plasmid with each cloned spliced substrate was determined by absorbance at 260 nM of light in the Beckman Coulter DU® 730 Life Science UV/Vis Spectrophotometer and then converted based on total size of plasmid to number of target copies per microliter.
Recombinant pDONR™221 clones containing genomic DNA of rps12‐2 and rpl2 used for in vitro transcription of precursor RNA substrates were used to generate the standard curve of un‐spliced substrate. Relative quantity of amplified products (spliced product, un‐spliced substrate, or total RNA, respectively) was determined using the Bio‐Rad CFX Maestro™ software package with the highest product level based on relative fluorescent units set to 1 and all other products shown relative to this.
2.10. Statistical analysis
All statistical comparisons of qPCR data were performed using a standard unpaired t test unless otherwise stated using algorithms provided in the Bio‐Rad CFX Maestro™ software package. Inter‐run calibration of plates in the Bio‐Rad CFX Maestro™ software was used to correlate data from multiple plates and increase number of experimental replicates for self‐splicing assays and number of technical replicates for activity assays when possible to provide more robust statistical analyses. Outliers of technical replicates were removed from statistical analyses.
2.11. Accession numbers
Sequence data from this article can be found in the EMBL/GenBank data libraries under the following accession numbers: O. sativa matK (GU592207), Valiha sp. matK (LN906794), Brachyelytrum aristosum matK (KM974735), Lygeum spartum matK (HE573932), Bromus leptoclados matK (LN906668), and Poa perrieri matK (LN906764.1).
3. RESULTS
3.1. MatK maturase expression
Immune detection of protein fractions after nickel‐NTA purification of induced bacterial lysates that included the pDEST™17 plasmid with the R1 matK coding region resulted in the identification of an ~65 kDa protein product (Figure 3a). An additional ~25 kDa protein band often observed from Western blots using anti‐MatK antibodies was most likely the result of proteolyzed product (Figure 3a). Lower induction temperatures were tested to avoid proteolysis of protein but resulted in no change in amount of product produced after IPTG induction or amount of proteolysis (Figure S2). Additional contaminating protein bands were evident after initial elutes were obtained from the first round of nickel purification. Such protein contaminants are well known to be present from E. coli BL21 DE3 pLysS lysates when using Ni‐NTA columns due to having metal‐binding sites or large clusters of histidine residues on the protein surface (Bolanos‐Garcia & Davies, 2006; Robichon, Luo, Causey, Benner, & Samuelson, 2011). Eluted protein fractions were rerun through a new nickel–histidine column to further remove these contaminants. The subsequent eluted fraction (fraction 2) contained less protein contaminants than the initial elution (Figure 3a). Only the second more “pure” fraction 2 was utilized for activity assays.
Figure 3.
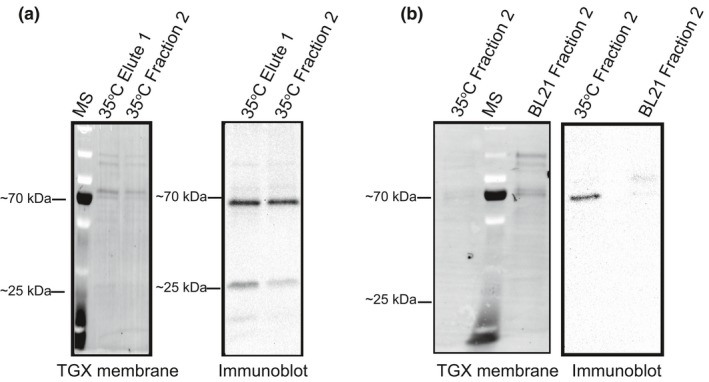
Expressed and purified MatK protein. The Oryza sativa open reading frame for MatK was cloned into pDEST™17 behind a 6X histidine tag and induced for expression in Escherichia coli BL21 DE3 pLysS cells. (a) Fractions from Ni‐NTA isolation of 6X his‐tagged MatK protein induced at 35°C with 0.5 mM IPTG. Resolution of protein bands from fractions transferred to PVDF membrane and detected using TGX stained gel activation (left). Immune detection of proteins using an anti‐MatK antibody (Agrisera) (right). (b) Proteins from fraction 2 (resulting fraction from the second round of purification through Ni‐NTA column) of 6X His‐tagged MatK purification compared with the proteins of the same fraction after Ni‐NTA purification from Escherichia coli Bl21 DE3 pLysS cells lacking the MatK‐pDEST™17 plasmid. Proteins resolved by SDS‐PAGE, transferred to PVDF, and detected using TGX stain activation (left). Immune detection of proteins using anti‐MatK antibody (Agrisera) (right). MS = PageRuler™ Plus prestained protein ladder (Thermo Fisher). Position of the ~70 kDa and ~25 kDa protein standards are shown for mass comparison of detected protein bands
The presence of residual E. coli protein contaminants in MatK Ni‐NTA fractions was not unexpected. Secondary affinity tags are often employed to enable better target protein isolation. These secondary tags, however, may result in proteolysis or aggregation of target protein, as well as decreased concentration of isolated protein due to additional purification steps (Robichon et al., 2011). An engineered BL21 (DE3) strain has come out in recent years that includes a chitin‐binding domain (CBD) on some of the major E. coli protein contaminants of Ni‐NTA purification (Robichon et al., 2011). We attempted to utilize this newer expression strain (NiCo21(DE3) NEB) for expressing and isolating MatK but found MatK expression greatly decreased in this strain compared with the pLysS strain and Ni‐NTA purification still resulted with an abundance of contaminating E. coli proteins (data not shown). We, therefore, chose to continue our work using the BL21 DE3 pLysS strain without the additional CBD‐tagged E. coli proteins and rely on two rounds of isolation of MatK using Ni‐NTA columns.
Confirmation of MatK as the primary protein in fraction 2 was done using electrospray ionization mass spectrometry. The MatK maturase had the highest identity match based on epitope matches using the UniProt (http://www.uniprot.org) database with O. sativa (rice) as the model organism (Table 1). An anti‐MatK reactive protein band was strongly detected only from fractions of MatK‐induced cultures with very limited amounts of cross‐reactivity to bands from mock‐induction fractions further supporting the expression and isolation of the MatK maturase used in activity assays (Figure 3b). Based on these results, we believe that the ~65 kDa protein observed from SDS‐PAGE gels and Western blots of resolved protein after Ni‐NTA purification from BL21 DE3 pLysS cells that included the matK recombinant expression clone was the MatK maturase.
Table 1.
Top four proteins identified through electrospray ionization mass spectrometry and UniProt (https://www.uniprot.org/) peptide analysis from Ni‐NTA isolated protein fractions after two rounds of purification
| Model species | Identity score | Protein |
|---|---|---|
| Oryza sativa L. | 51 | Maturase K |
| 50 | Cytokinin dehydrogenase 4 | |
| 41 | 30S ribosomal protein S12 | |
| 39 | Parkeol synthase | |
| Escherichia coli (Migula 1895) | 3,238 | 60 kDa chaperonin |
| 1,220 | Elongation factor Tu | |
| 1,136 | Triosephosphate isomerase | |
| 980 | Bifunctional polymyxin resistance protein ArnA |
Size discrepancy of the MatK maturase expressed in the current study (~65 kDa) with previous assessments of MatK protein from rice extract (~55 kDa; Barthet & Hilu, 2007) was attributed to use of an upstream initiation codon for heterologous expression (Figure 1a) as well as the addition of the 6X histidine tag. The upstream initiation codon added 32 amino acids to the N‐terminus of our heterologous‐expressed MatK protein. The ~65 kDa mass matched the predicted mass for MatK based on amino acid sequence when expressed using the R1 initiation codon.
3.2. Self‐excision of the group IIA introns of rps12‐2 and rpl2 without additional protein factors
We chose to begin our investigation into determining the role of MatK as a group IIA intron maturase using the group IIA introns of and rps12 and rpl2 as representative models of proposed protein‐coding MatK‐precursor RNA target transcripts. The protein products of rps12 and rpl2 are required for formation of the plastid translation machinery implicating a vital importance for their protein products in plastid function. We tested self‐excision of in vitro transcribed rps12 and rpl2 precursor RNAs that included the group IIA intron under three different buffer conditions, water (RNA + water), low magnesium (5mM MgCl2) buffer (no heat + buffer), and after a 2 min 90°C heat denaturing in the same low magnesium buffer (plus heat + buffer). Even without the addition of protein, spliced product indicative of self‐splicing was observed using intron‐spanning primers for rps12‐2 and the group IIA intron of rpl2 under all three conditions (Figure 4a,c). Spliced products from RT‐qPCR of rps12‐2 excision were resolved on agarose gels and revealed the amplification of a ~200‐bp product (Figure 4b). RT‐PCR using 2 µl of undiluted cDNA for rpl2 was used to confirm RT‐qPCR data and resulted in amplification of a ~300‐bp product in all three conditions tested (Figure 4d). Product sizes amplified by either RT‐qPCR or RT‐PCR are close to those estimated for spliced product for each of these transcripts (190 bp for rps12‐2 and 240 bp for rpl2, respectively). Sequencing confirmed identity of amplified products as rps12 (exons 2–3 ligated) or rpl2, respectively (data not shown). For rps12‐2, self‐splicing was significantly higher when precursor substrate was added to water than in low magnesium buffer after the addition of heat (p = .01 for rps12‐2). Similar results were obtained for rpl2 in which the addition of rpl2 precursor substrate to water resulted in a significant increase in spliced product compared with both no heat + buffer and plus heat + buffer trial (p = .0004; .0002, respectively; Figure 4c). Levels of self‐excision for rps12‐2 and rpl2 in buffer after the initial heating period stayed relatively constant with no significant change in relative quantity of spliced product produced through self‐excision even after 60 min of incubation (Figures 5a and 6a). All reactions were assayed immediately after the addition of RNA to each buffer/water or after incubation at 90°C for 2 min for the “plus heat + buffer” trials. qPCR of negative controls (water alone and buffer alone) and No RT reactions confirmed that product was not due to background contamination of solutions or RNA stocks (relative quantity = 0, 0, and 0.00062, respectively). For simplicity, only results of water controls are shown (Figure 4a,c). All treatments produced a significant amount of spliced product compared with water controls (p < .01).
Figure 4.
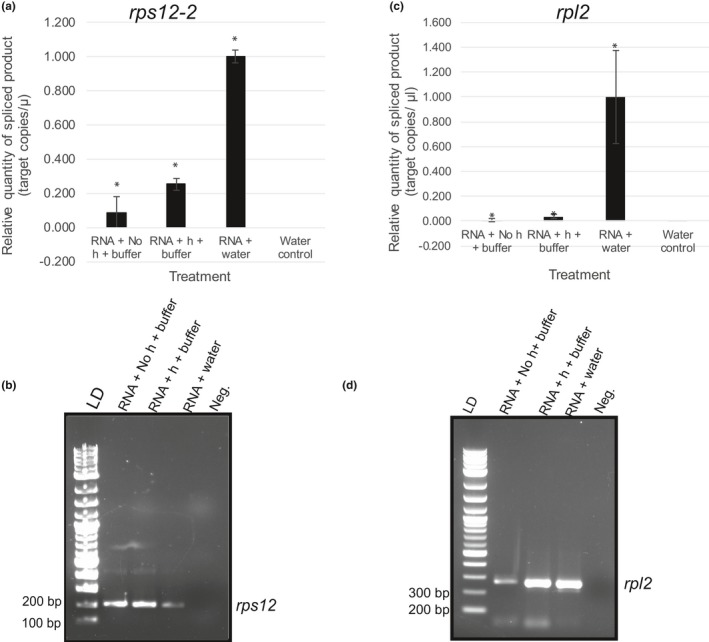
Self‐splicing of the rps12‐2 and rpl2 group IIA introns under three treatments: without heat (RNA + No heat + buffer), after an initial denaturing period of 90°C for 2 min to remove secondary structure (RNA + heat +buffer), or in DEPC‐treated dH20 (RNA + water). Activity assays were performed by adding 20 nM of rps12 (exons 2–3 including intron 2) and rpl2 in vitro transcribed precursor RNA to buffer modified from Holländer and Kück (1999) or water. Buffer (not shown) and water controls lacking RNA also were run to ensure results were not from contamination. Amount of self‐splicing was determined by assaying spliced product using RT‐qPCR. In brief, for each treatment, 50 ng, based on initial mass added of precursor RNA, was reverse‐transcribed using SuperScript™ III Reverse Transcriptase with subsequent cDNA diluted 1:10 in nuclease‐free water. One microliter of 1:10 diluted cDNA was used for qPCR of spliced product using SsoAdvanced™ Universal SYBR® Green Supermix (Bio‐Rad) on the Bio‐Rad CFX 96 Touch™ Real‐time PCR Detection System. Amount of spliced product was determined using absolute quantification based on a standard curve diluted 10‐fold. Standards were generated by cloning spliced product for rps12‐2 or rpl2, respectively, in pDONR™221. Concentration of plasmid was used to determine number of single‐stranded target copies/µl. ΔCq was normalized to zero. Relative quantity is shown with the highest level of expression for each substrate set to 1.00 and all other quantities shown relative to this. (a) Relative quantity of spliced product of rps12‐2. (b) Gel analysis of rps12‐2 RT‐qPCR products. LD = 2‐log ladder (NEB). Critical sizes of standard needed to determine product size indicated to the left. Negative control (Neg.) for the rps12 qPCR. (c) Relative quantity of rpl2 spliced product. (d) RT‐PCR analysis of rpl2 spliced product. Products were sequenced to confirm identity (data not shown). LD = 2‐log ladder (NEB). Negative control (Neg.) for the rpl2 RT‐PCR. Critical sizes of standard needed to determine product size indicated to the left. All qPCRs were run with at least two technical replicates for each sample. Error bars = ± corrected SEM as determined using the Bio‐Rad CFX Maestro™ Software (n = 3 experimental replicates for all assays except rpl2 RNA + water where n = 6). (*) represent statistical significance (p < .005) of treatment versus water control
Figure 5.
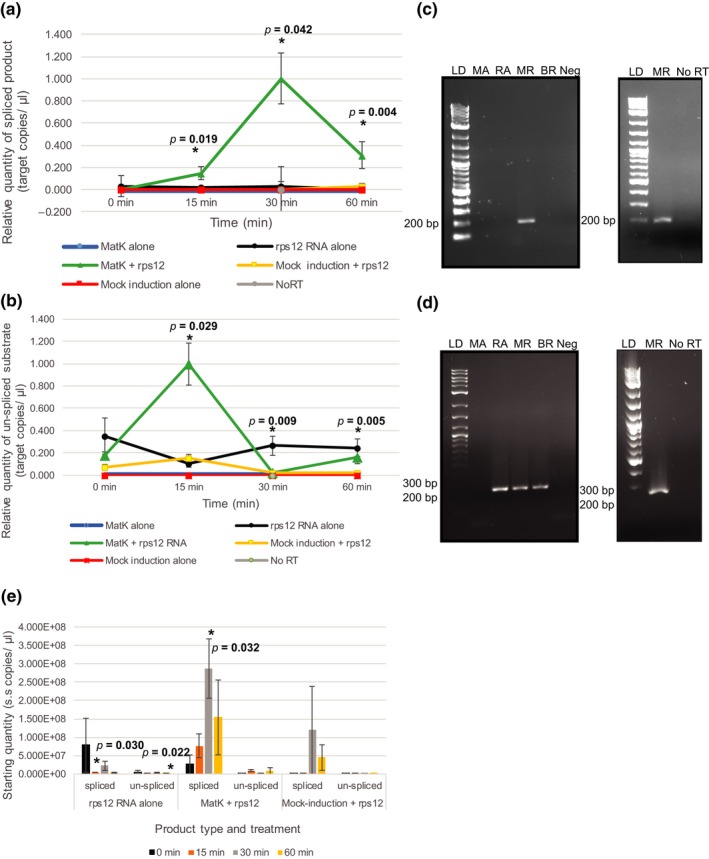
Efficiency of rps12‐2 intron excision produced with and without addition of the MatK maturase. MatK protein (200 nM) or control mock‐induced protein (200 nM) was incubated with 20 nM rps12‐2 precursor RNA up to 60 min in reaction buffer at 26°C to determine MatK activity. Controls of rps12‐2 (rps12) RNA alone, MatK protein alone, or mock‐induced protein alone in buffer also were assayed under the same conditions. Amount of spliced product/residual substrate was determined using RT‐qPCR. In brief, 50 ng based on initial mass added of precursor RNA of each RNA sample was reverse‐transcribed using SuperScript™ III Reverse Transcriptase with subsequent cDNA diluted 1:10 in nuclease‐free water. One microliter of 1:10 diluted cDNA was used for qPCR of product using SsoAdvanced™ Universal SYBR® Green Supermix (Bio‐Rad) on the Bio‐Rad CFX 96 Touch™ Real‐time PCR Detection System. Relative quantity of (a) spliced product or (b) un‐spliced substrate was determined based on a standard curve using number of target copies/µl of cloned spliced product, or un‐spliced precursor substrate, respectively, and ΔCq normalized to zero. The 0‐min time point reflects sample collection immediately after addition of protein to the reaction. Prior to the addition of protein, all RNA samples were heat‐denatured for 2 min at 90°C. All qPCR reactions included at least two technical replicates. Error bars = ±corrected SEM as determined using the Bio‐Rad CFX Maestro™ Software (n = 3 experimental replicates). *indicates statistical significance when comparing relative quantity of spliced product formed at each time point for MatK + rps12 to rps12 RNA in buffer alone or rps12 RNA in buffer alone to mock induction +rps12. Significant p values are given. (c, d) Left: RT‐PCR products using the same 1:10 cDNA templates as used for qPCR assessment of (c) spliced rps12‐2 and (d) un‐spliced rps12‐2 precursor substrate resolved by gel electrophoresis. Right: Amplified products of both RT reaction and No RT‐PCR that included MatK + rps12‐2 precursor RNA taken after 30 min of incubation at 26°C in activity buffer. LD: 2‐log ladder (NEB), MA: MatK protein + buffer negative control, RA: rps12‐2 precursor RNA + buffer, MR: MatK protein + rps12‐2 precursor RNA, BR: mock‐induction protein + rps12‐2 precursor RNA, BA: mock‐induction protein + buffer negative control. (e) Starting quantity of rps12‐2 spliced product and un‐spliced substrate determined by absolute quantification based on standard curve of diluted plasmid DNA calculated in single‐stranded copies/µl. Significant p values are given. Error bars = ±SEM
Figure 6.
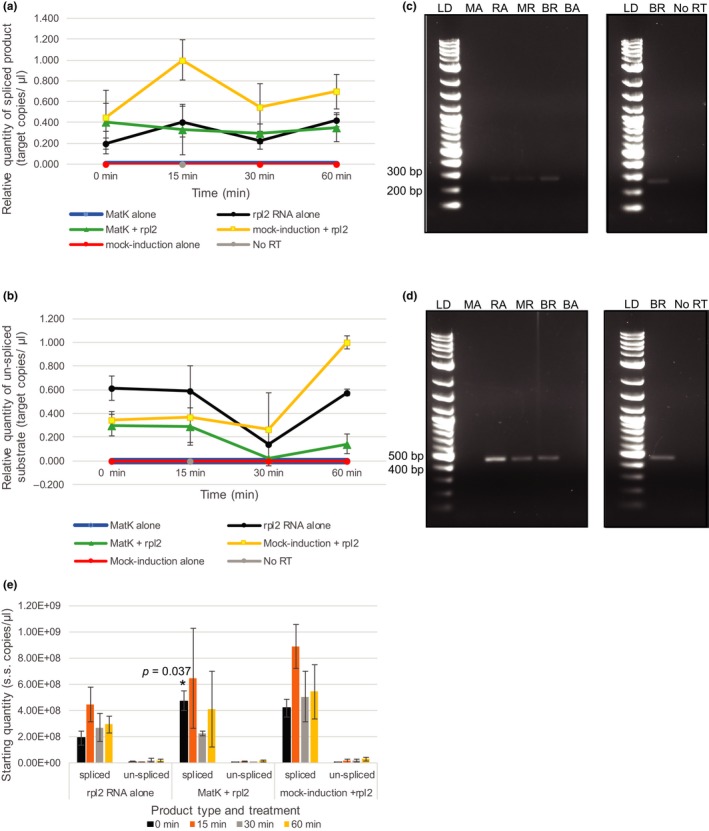
Efficiency of rpl2 intron excision produced with and without addition of the MatK maturase. MatK protein (200 nM) or control mock‐induced protein (200 nM) was incubated for up to 60 min at 26°C with 20 nM rpl2 precursor RNA to determine MatK activity. Controls of rpl2 RNA alone, MatK protein alone, or mock‐induced protein alone in buffer also were assayed under the same conditions. Amount of spliced product/residual substrate was determined using RT‐qPCR. In brief, 50 ng based on initial mass added of precursor RNA of each RNA sample was reverse‐transcribed using SuperScript™ III Reverse Transcriptase with subsequent cDNA diluted 1:10 in nuclease‐free water. One microliter of 1:10 diluted cDNA was used for qPCR of product using SsoAdvanced™ Universal SYBR® Green Supermix (Bio‐Rad) on the Bio‐Rad CFX 96 Touch™ Real‐time PCR Detection System. Relative quantity of (a) spliced product or (b) un‐spliced substrate was determined based on a standard curve using number of target copies/µl of cloned spliced product, or un‐spliced precursor substrate, respectively, and ΔCq normalized to zero. The 0‐min time point reflects sample collection immediately after addition of protein to the reaction. Prior to the addition of protein, all RNA samples were heat‐denatured for 2 min at 90°C. All qPCR reactions included at least two technical replicates. Error bars = ±corrected SEM as determined using the Bio‐Rad CFX Maestro™ Software (n = 3 experimental replicates). No significant levels of spliced product or un‐spliced substrate were detected. All comparisons resulted in p > .05. (c, d) Left: RT‐PCR products using the same 1:10 cDNA templates as used for qPCR assessment of (c) spliced rpl2 and (d) un‐spliced rpl2 precursor substrate resolved by gel electrophoresis. Right: Amplified products of both RT reaction and No RT‐PCR that included mock‐induced protein + rpl2 precursor RNA taken after 15 min of incubation at 26°C in activity buffer. LD: 2‐log ladder (NEB), MA: MatK protein + buffer negative control, RA: rpl2 precursor RNA + buffer, MR: MatK protein + rpl2‐2 precursor RNA, BR: mock‐induction protein + rpl2 precursor RNA, BA: mock‐induction protein + buffer negative control. (e) Starting quantity of rpl2 spliced product and un‐spliced substrate determined by absolute quantification based on standard curve of diluted plasmid DNA calculated in single‐stranded copies/µl. Significant p values are given. Error bars = ±SEM
RT reactions are known to produce artifacts including false ligation of exons regions from noncanonical splice sites (Houseley & Tollervey, 2010). We ran products directly from self‐splicing assays undiluted on formaldehyde gels stained with SYBR gold to analyze the legitimacy of splicing activity directly from the self‐splicing assays. However, concentration of RNA used in the assays was extremely low and precluded visualization of product on gels (data not shown). If self‐splicing products were the result of RT artifacts, it would be assumed equal amounts of product would be observed under all three conditions tested. This was not the case for either substrate tested in the current study in which self‐splicing differed among conditions tested with a much higher quantity of product observed when RNA was incubated in water compared with the addition of RNA in buffer (Figure 4a,c).
3.3. MatK maturase activity
MatK was found to significantly increase spliced product formation for rps12‐2 but not rpl2. MatK protein from fraction 2 after nickel‐NTA purification (Figure 3a) was added to in vitro transcribed rps12 (exons 2–3) or rpl2 precursor RNA containing the group IIA intron predicted to require MatK for intron excision (Hess et al., 1994; Zoschke et al., 2010). Although minimum levels of self‐excision without addition of the MatK maturase were evident in low magnesium buffer, the addition of 200 nM of MatK maturase to 20 nM rps12 RNA increased production of spliced product relative to rps12 RNA alone controls over 60 min, with the highest amount of spliced product (30‐fold higher than self‐splicing controls, p = .042, as determined using a standard t test in Bio‐Rad CFX Maestro™ Software) evident after 30 min of incubation in reaction buffer (Figure 5a). No significant difference was observed between spliced product levels of rps12‐2 RNA alone and with the addition of mock‐induced E. coli protein at any time point supporting that increased splicing resulting from MatK was not the result of any residual background contaminating proteins from Ni‐NTA purification. RT‐PCRs using the same 1:10 dilution of cDNA as used for qPCR of activity assays produced a band of ~200 bp using intron‐spanning primers only for activity assays that included both rps12 (exons 2–3) precursor RNA and the MatK maturase (Figure 5c) supporting qPCR data. No RT controls of the same RNA from activity assays with both rps12 precursor RNA and the MatK maturase after 30 min of incubation in reaction buffer produced no product after PCR with the same intron‐spanning primers supporting that RT‐PCR results were not due to contamination (Figure 5a,c).
A complimentary pattern of un‐spliced precursor rps12 (exons 2–3) substrate was found to that of spliced product (Figure 5b). The lowest levels of un‐spliced precursor RNA were detected after 30 min of incubation when MatK was added to the reaction complimenting the time point in which the highest levels of spliced product were observed (Figure 5a). The highest levels of precursor substrate were found after 15 min of incubation when MatK was added to the reaction (an almost 10‐fold greater amount of un‐spliced substrate compared with rps12 RNA alone controls, p = .029). The change in un‐spliced substrate levels for MatK + rps12 RNA dropped 40‐fold from 15 to 30 min and then rose again from 30 to 60 min with a 6.7‐fold increase, though neither change was determined to be statistically significant (p = .063 and .437, respectively). A reduction in un‐spliced substrate also was observed with the mock‐induced protein + rps12 RNA treatment from 15 to 30 min (7.6‐fold reduction of un‐spliced substrate, p = .016) but not with RNA alone, which had significantly more un‐spliced substrate at 30 and 60 min compared with mock‐induction + RNA controls (p = .009 and .005, respectively). Gel analysis of RT‐PCR products using the same 1:10 dilution of cDNA resolved a single band of the expected size (277 bp) for all three RNA treatments at 30 min (Figure 5d). No products (spliced or un‐spliced) were detected from MatK protein + buffer alone or mock‐induced protein + buffer alone controls using either RT‐qPCR or RT‐PCR (Figure 5a–d) supporting that results were not due to background RNA or other contaminants of protein purification. The amplification of product for all three RNA treatments (rps12 RNA alone, MatK + rps12, mock induction + rps12) after 30 min of incubation supported qPCR data and demonstrated that un‐spliced rps12‐2 RNA was present at this time point for all three treatments compared with spliced product which was only formed when MatK was added to the reaction (Figure 5a,c). Overall, rps12 (exons 2–3) precursor RNA levels remained unchanged during the 60‐min incubation period when incubated alone and only changed when MatK or mock‐induced protein was added to the reaction.
Similar results were obtained when comparing starting quantity of treatments at each time point with a significant difference in spliced product observed when MatK was added to the reaction after 30 min of incubation (p = .032 as determined using Student's t test with equal variance) compared with rps12‐2 RNA alone self‐splicing controls (Figure 5e). Mock induction + rps12‐2 RNA had significantly lower levels of spliced product compared with rps12‐2 RNA alone at 15 min (p = .030). Comparison of starting quantities determined from standard curves of un‐spliced precursor levels revealed no significant difference in un‐spliced substrate levels for MatK + rps12‐2 versus rps12‐2 RNA self‐splicing controls at any time point (p > .05), but just as seen with relative quantity comparisons, significantly more un‐spliced substrate was observed for rps12‐2 RNA alone compared with mock induction + rps12‐2 RNA at 60 minutest (p = .022, Figure 5e).
Unlike rps12‐2, MatK activity on the group IIA intron of rpl2 was less effective. The addition of the MatK maturase to rpl2 precursor RNA resulted in the same levels and efficiency of spliced product formation as produced by the catalysis of the rpl2 group IIA intron self‐excision alone (Figure 6a). Activity assays that included rpl2 precursor RNA with mock‐induced protein revealed slightly higher levels of rpl2 spliced product as that from self‐splicing of the RNA alone although not at a significant level at any time point (p = .07, .06, .39, and .29 at 0, 15, 30, and 60 min, respectively, of incubation compared with rpl2 self‐splicing controls; Figure 6a). Control assays with MatK alone or mock‐induced protein alone both showed no exterior contamination of RNA products supporting the spliced product was the result of addition of RNA to assays not contaminants in the buffers or protein fractions (Figure 6a,c). No RT controls taken from mock‐induced protein plus rpl2 RNA trials after 15 min of incubation in buffer did not show product supporting results are not due to nucleic acid contamination (Figure 6a,c). Un‐spliced precursor rpl2 RNA also was assayed for comparison to spliced product levels. Although a reduction in un‐spliced rpl2 substrate was observed based on qPCR at 30 min for all three treatments that included rpl2, none of these were considered significant based on treatment or time point using a standard unpaired t test (all p > .05; Figure 6b,d). Assessment of starting quantity based on standard curves for rpl2 spliced product and un‐spliced precursor substrate revealed a similar pattern to relative quantity assessment (Figure 6e). However, the immediate addition of MatK to heat‐denatured rpl2 RNA (time 0 min) was determined to result in a significant increase in spliced product based on starting quantity (p = .037 as determined using Student's t test and assuming equal variance, Figure 6e). It is to be noted that the total amount of possible rpl2 spliced product based on the mass of substrate added to the reaction and spliced product size was 9.79E + 08 single‐stranded copies/µl. The addition of mock‐induced protein to rpl2 RNA produced almost this maximum amount of spliced product after 15 min of incubation (Figure 6e).
The decrease in spliced product for both rps12‐2 and rpl2 from the maximum splicing time point (30 or 15 min, respectively) to 60 min (Figures 5a and 6a) suggests possible RNA degradation. We attempted to examine total cDNA levels as a reflection of total RNA from each of these maximum high points to 60 min using primers that only bound to the exon (Table S1; Figure 2a) in order to assess RNA degradation. The same cDNA stocks were used to assess total cDNA levels as used for assessment of rps12‐2 and rpl2 activity assays (Figures 5 and 6). No significant change in levels of total cDNA based on relative quantity of cDNA was evident from these early time points to the end of the assay time period for rpl2 (Figure S3). Total RNA was not able to be assessed for rps12. The qPCR for rps12 using primers targeted against exon 2 resulted in poor amplification (data not shown).
4. DISCUSSION
Since the discovery of an open reading frame coded within the group IIA intron of trnK, this reading frame has been predicted to contain an enzyme (MatK) that aids in RNA maturation by folding the intron into the catalytically active structure needed for excision (e.g., Barthet & Hilu, 2008; Hess et al., 1994; Neuhaus & Link, 1987; Sugita et al., 1985; Vogel et al., 1999; Vogel et al., 1997; Zoschke et al., 2010). Although indirect studies of chloroplast function and RNA‐binding studies have supported the presumed function of MatK (Hess et al., 1994; Liere & Link, 1995; Vogel et al., 1997, 1999; Zoschke et al., 2010), none of these studies directly show MatK’s maturase activity. We have pursued the question of MatK’s exact role in chloroplast group IIA intron excision by developing an in vitro activity assay for this maturase. Our work has revealed that the group IIA introns of both rps12‐2 and rpl2 are able to self‐splice in vitro and that efficiency of rps12‐2 excision is increased when MatK is added to the reaction. MatK, however, had little if any effect on excision of the group IIA intron of rpl2.
4.1. Excision of chloroplast group IIA introns
Group IIA introns are known to self‐splice in vitro under certain conditions without the need for any protein factors (Hebbar, Belcher, & Perlman, 1992; Matsuura et al., 2001). Specific buffer conditions, such as the amount of Mg+2, may inhibit or enhance self‐excision of group II introns in vitro. Self‐excision of L1.LtrB group IIA intron from L. lactis was shown to require levels above 5 mM MgCl2 for efficient excision (Matsuura et al., 2001; Noah & Lambowitz, 2003). Contrary to this, 5 mM MgCl2 was shown by Jarrell, Peebles, Dietrich, Romit, and Perlman (1988) to be sufficient for in vitro autocatalysis of group II introns. Interestingly, our data indicate that the group IIA introns of rps12‐2 and rpl2 self‐excise even in DEPC‐treated sterile water (Figure 4a–d). This would suggest that the group IIA introns of rps12‐2 and rpl2 do not require magnesium for catalysis of self‐excision. Although this is possible, it is more likely that self‐splicing observed in water was due to small amounts of magnesium that remained with template RNA as residual contaminants of the original in vitro transcription reaction even after subsequent RNA cleanup steps. This minimal amount of magnesium would then be sufficient for formation of the catalytically active structure needed for self‐excision of these two group IIA introns. Functional variants of the L1.LtrB group II intron have been generated that excise in extremely low levels of Mg2+ (1.5 mM; Truong, Sidote, Russell, & Lambowitz, 2013). It is possible that the plastid group IIA introns of rps12‐2 and rpl2 have evolved in a similar fashion to that of the Truong et al. (2013) mutants and only require extremely minimal levels of Mg+2 for self‐excision. It is also possible that observed product from self‐splicing reactions is due to artifacts of the reverse transcription reaction itself and is not the result of actual self‐excision of the RNA intron. RT reactions are known to result in trans‐splicing events that can lead to misinterpretation of true product from splicing reactions (Houseley & Tollervey, 2010). Trans‐splicing refers to the joining of exons on two independently transcribed RNAs. The splicing from the in vitro assays in our study is the result of cis‐splicing events. Further, spliced product reflective of artifact from the RT reactions should have resulted in relatively equal amounts of product from all three conditions tested in our study. This was not the case for either the rps12‐2 or rpl2 group IIA introns, both of which had a much higher level of self‐excision in water compared with treatment in low magnesium buffer (Figure 4a,c). Levels of spliced product, however, were extremely low for self‐excision of both substrates suggestive of very minimal or contaminant levels of template. It is possible that self‐excision observed for rps12‐2 in buffer was only the result of background contamination as the observed extremely low product levels did not change even after 60 min of incubation and only was detected by amplification with qPCR reagents that are highly sensitive (Figure 5a,c). Self‐splicing product levels of rps12‐2, however, were significantly higher than buffer alone controls at 15, 30, and 60 min of incubation (p = 3.00E‐06, 2.00E‐03, and 2.00E‐04, respectively). Further, this would not explain the significantly higher level of rps12‐2 spliced product evident in water to buffer. Rpl2 also was shown to self‐splice more in water than in buffer and had variable levels of spliced product over time (Figures 4c and 6a), suggestive of active self‐excision.
4.2. Impact of the MatK maturase on group IIA intron self‐excision
The almost nonexistent amount of spliced product formed by self‐excision of rps12‐2 in buffer suggests that another protein factor must be involved in catalysis of intron excision from this precursor RNA substrate. Studies of the white barley mutant albostrians were the first studies to imply that the MatK maturase catalyzes excision of the rps12‐2 intron (Hübschmann, Hess, & Börner, 1996). These original studies in barley were then supported by several independent studies ranging from evolutionary analyses showing co‐evolutionary reduction in MatK with its target intron substrates from the plastid genome (McNeal, Kuehl, Borre, Leebens‐Mach, & dePamphilis, 2009) to RNA‐binding studies (Zoschke et al., 2010) that show a more direct ability for MatK to bind to its proposed intron targets including rps12‐2. We added heterologously expressed MatK protein to rps12 precursor RNA that spanned from exon 2 to exon 3 and included the second intron of this RNA. The in vitro activity assay demonstrated that levels of spliced product were greatly increased upon addition of the MatK maturase after 30 min of incubation with a corresponding decrease in un‐spliced substrate (Figure 5a,b). These results were not achieved when background protein from E. coli (mock‐induced protein) was added to the reactions (Figure 5a,b) confirming that the increase in splicing activity of rps12‐2 was the result of the addition of the MatK maturase and not contaminants of the Ni‐NTA fractions or reaction buffer. This is the first direct evidence of MatK’s functional role as a group IIA intron maturase.
In addition to supporting the long‐proposed catalytic role of MatK as a chloroplast group IIA maturase, we have also demonstrated with our activity assays that MatK alone is sufficient for catalyzing group IIA intron excision, at least for rps12‐2. This was not the case for rpl2. The addition of the MatK maturase to rpl2 precursor RNA did not have any effect on the amount or timing to produce spliced product compared with self‐splicing of the RNA without additional protein factors (Figure 6a), implying the role of MatK in catalyzing intron self‐excision may vary for each intron substrate.
The lack of impact of MatK enzymatic activity on formation of rpl2‐spliced product was intriguing. Studies have shown the requirement for several nuclear‐encoded protein factors to facilitate rpl2 group IIA intron excision. These factors include nuclear‐encoded SOT5 and WTF1 (Huang et al., 2018; Kroeger, Watkins, Friso, Wijk, & Barkan, 2009). Importantly, although these same studies indicated additional proteins were involved in splicing of rps12‐2, our findings imply that MatK is sufficient for rps12‐2 excision but not sufficient for rpl2. Instead, additional protein factors likely are required for efficient rpl2 group IIA intron removal. Supporting this proposition is our finding of slightly elevated levels of rpl2 spliced product using protein from mock‐induction trials (Figure 6a,c). These results for rpl2 were likely due to contaminants isolated through Ni‐NTA column purification from the E. coli BL21 DE3 pLysS strain.
Several E. coli proteins are consistently isolated through immobilized metal affinity chromatography and almost impossible to exclude from any isolation. Native E. coli proteins that tend to co‐purify include SlyD, EF‐Tu, and Hsp60 (Bolanos‐Garcia & Davies, 2006; Robichon et al., 2011). Of particular concern for our study was Hsp60, a chaperone implicated in aiding excision of the group II intron of rpl2 in the mitochondria (Hsu, Juan, Wang, & Jauh, 2019) and was found as a contaminant in our MatK protein isolations using Ni‐NTA (Table 1). It is highly probable that E. coli Hsp60 was responsible for the slightly elevated levels of rpl2 spliced product compared with self‐excision controls in our study (Figure 6a). Based on the work of Zoschke et al. (2010) and earlier studies by Hess et al. (1994) that indicated MatK binds to the rpl2 group IIA intron and a plastid‐encoded factor is required for rpl2 group IIA intron excision, it is likely that MatK is required for increasing efficiency of rpl2 group IIA intron removal. However, additional proteins may be required to work with MatK to enable efficient intron removal from rpl2, possibly in a manner akin to the nuclear spliceosome where many protein components are required to facilitate intron removal.
A pertinent finding for both rps12‐2 and rpl2 intron excision was that the amount of spliced product for both substrates decreased after hitting maximum product levels (30 min for rps12‐2 (MatK + rps12) and 15 min for rpl2 (mock induction + rpl2); Figures 5a and 6a). This was surprising since it would be assumed that the amount of product would not decrease but either plateau or increase over time. Several factors may contribute to this decline. One very likely possibility is that RNA is degraded during the assay resulting in the observed decline in spliced product. Although possible, the relative quantity of un‐spliced substrate for rps12 was approximately the same at 0 and 60 min for rps12 RNA alone and MatK + rps12 (Figure 5b). Further, total cDNA levels for rpl2 remained relatively unchanged from 15 to 60 min for all three treatments that included the RNA substrate (Figure S3). We propose based on our data that the reduced level of spliced product observed in our assays after the initial increase in splicing activity is due to alteration between the forward and reverse splicing pathways. The complimentary increase in un‐spliced rps12 substrate from 30 to 60 min (MatK + rps2) relative to the decrease in spliced product at these same points (Figure 5a,b) supports this proposition. Group II introns have been shown to reverse self‐splice in vitro, a process that enables the excised intron to reinsert itself within ligated exons (Augustin, Müller, & Schweyen, 1990; Roitzsch & Pyle, 2009). Reverse splicing of rps12‐2 and the group IIA intron of rpl2 may serve as a form of autocatalytic regulation for amount of mature transcript for use as a template for protein translation. Further work needs to be done to confirm the hypothesis presented here regarding reverse splicing of rps12‐2 and rpl2.
5. CONCLUSIONS AND FUTURE PERSPECTIVES
We have provided here the first direct evidence of MatK splicing activity using an in vitro activity assay. The fact that MatK can increase splicing efficiency of at least one of its proposed substrates without additional protein components suggests that domain X of the MatK maturase is sufficient for catalyzing group IIA intron self‐excision in vitro. The difference in MatK activity between the two substrates tested here implies that the importance of MatK for catalysis of the structure necessary for group IIA intron self‐excision varies with each substrate. Future studies need to be done to ascertain the differences in binding affinities or requirements for additional catalytic components for rps12‐2 and rpl2 intron excision. In addition, the experiments performed in the current study utilized an upstream initiation codon for MatK expression. The existence of two different potential initiation codons for MatK in rice may have structural, as well as functional, implications for this maturase. Experiments need to be performed using MatK expressed from the R2 initiation codon to determine the significance of the upstream region used in the current study in MatK activity. Although much work as yet is required to completely define MatK’s maturase activity and the importance of this enzyme in plant physiology, the current work provides an in vitro method for testing MatK activity and the first direct evidence of MatK’s functional role as a group IIA intron maturase.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
M.M.B., C.L.P., and E‐K.T. conceptualized the study; involved in methodology; validated the study; investigated the data; curated the data; and wrote, reviewed, and edited the manuscript. M.M.B. involved in formal analysis; provided resources; wrote the original manuscript and prepared the draft; visualized the data; supervised the study; administered the project; and acquired funding.
Supporting information
Fig S1
Fig S2
Fig S1‐S3
Table S1
Supplementary Material
ACKNOWLEDGMENTS
This publication was supported in part by grant P20GM103499 (SC INBRE) from the National Institute of General Medical Sciences, National Institutes of Health, the Coastal Carolina University Research Enhancement Grant, the Department of Biology at Coastal Carolina University, and the Gupta College of Science at Coastal Carolina University. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The authors thank Alexandra Margets, Lauren Angello, Ymani Wright, Austen Lu Kin, Alexandra Agee, Alanna Swain‐Jarrett, and James Hatton for their assistance with growth and collection of tissue samples as well as experimental assistance. Special thanks to Michael Walla and Bill Cotham at the University of South Carolina for aid in mass spectrometry analysis of purified MatK protein samples. The authors also thank Scott Parker and Khidir Hilu for statistical and editorial advice.
Barthet MM, Pierpont CL, Tavernier E‐K. Unraveling the role of the enigmatic MatK maturase in chloroplast group IIA intron excision. Plant Direct. 2020;4:1–17. 10.1002/pld3.208
Funding information
M.M.B. was supported in part by grant P20GM103499 (SC INBRE) from the National Institute of General Medical Sciences, National Institutes of Health, the Coastal Carolina University Research Enhancement Grant, the Department of Biology at Coastal Carolina University, and the Gupta College of Science at Coastal Carolina University.
REFERENCES
- Augustin, S. , Müller, M. W. , & Schweyen, R. J. (1990). Reverse self‐splicing of group II intron RNAs in vitro. Nature, 343(6256), 383–386. 10.1038/343383a0 [DOI] [PubMed] [Google Scholar]
- Barthet, M. M. , & Hilu, K. W. (2007). Expression of matK: Functional and evolutionary implications. American Journal of Botany, 94, 1402–1412. 10.3732/ajb.94.8.1402 [DOI] [PubMed] [Google Scholar]
- Barthet, M. M. , & Hilu, K. W. (2008). Evaluating evolutionary constraint on the rapidly evolving gene matK using protein composition. Journal of Molecular Evolution, 66, 85–97. 10.1007/s00239-007-9060-6 [DOI] [PubMed] [Google Scholar]
- Blocker, F. J. H. , Mohr, G. , Conlan, L. H. , Qi, L. , Belfort, M. , & Lambowitz, A. M. (2005). Domain structure and three‐dimensional model of a group II intron‐encoded reverse transcriptase. RNA, 11, 14–28. 10.1261/rna.7181105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos‐Garcia, V. M. , & Davies, O. R. (2006). Structural analysis and classification of native proteins from E. coli commonly co‐purified by immobilized metal affinity chromatography. Biochimica et Biophysica Acta, 1760, 1304–1313. 10.1016/j.bbagen.2006.03.027 [DOI] [PubMed] [Google Scholar]
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Analytical Biochemistry, 72, 248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- CBOL Plant Working Group1 , Hollingsworth, P. M. , Forrest, L. L. , Spouge, J. L. , Hajibabae, M. , Ratnasingham, S. , … Little, D. P. (2009). A DNA barcode for land plants. Proceedings of the National Academy of Sciences of the United States of America, 106, 12794–12797. 10.1073/pnas.0905845106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, M. , Michel, F. , & Westhof, E. (2000). A three‐dimensional perspective on exon binding by a group II self‐splicing intron. The EMBO Journal, 19, 5007–5018. 10.1093/emboj/19.18.5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, X. , Matsuura, M. , Wang, Q. , Ma, H. , & Lambowitz, A. M. (2004). A group II intron‐encoded maturase functions preferentially in cis and requires both the reverse transcriptase and X domains to promote RNA splicing. Journal of Molecular Biology, 340, 211–231. 10.1016/j.jmb.2004.05.004 [DOI] [PubMed] [Google Scholar]
- Doyle, J. J. , & Doyle, J. L. (1990). Isolation of plant DNA from fresh tissue. Focus, 12, 13–25. [Google Scholar]
- Gu, S.‐Q. , Cui, X. , Mou, S. , Mohr, S. , Yao, J. , & Lambowitz, A. M. (2010). Genetic identification of potential RNA‐binding regions in a group II intron‐encoded reverse transcriptase. RNA, 16, 732–747. 10.1261/rna.2007310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbar, S. K. , Belcher, S. M. , & Perlman, P. S. (1992). A maturase‐encoding group IIA intron of yeast mitochondria self‐splices in vitro . Nucleic Acids Research, 20, 1747–1754. 10.1093/nar/20.7.1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, W. R. , Hoch, B. , Zelts, P. , Hübschmann, T. , Kössel, H. , & Börner, T. (1994). Inefficient rpl2 splicing in barley mutants with ribosome‐deficient plastids. The Plant Cell, 6, 1455–1465. 10.1105/tpc.6.10.1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand, M. , Hallick, R. B. , Passavant, C. W. , & Bourque, D. P. (1988). Trans‐splicing in chloroplasts: the rps 12 loci of Nicotiana tabacum . Proceedings of the National Academy of Sciences of the United States of America, 85(2), 372–376. 10.1073/pnas.85.2.372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilu, K. W. , Alice, L. A. , & Liang, H. (1999). Phylogeny of poaceae inferred from matK sequences. Annals of the Missouri Botanical Garden, 86(4), 835–851. 10.2307/2666171 [DOI] [Google Scholar]
- Hilu, K. W. , Black, C. M. , & Oza, D. (2014). Impact of gene molecular evolution on phylogenetic reconstruction: A case study in the rosids (superorder Rosanae, angiosperms). PLoS ONE, 9, e99725 10.1371/journal.pone.0099725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilu, K. W. , Borsch, T. , Muller, K. , Soltis, D. E. , Soltis, P. S. , Savolainen, V. , … Chatrou, L. W. (2003). Angiosperm phylogeny based on matK sequence information. American Journal of Botany, 90, 1758–1776. 10.3732/ajb.90.12.1758 [DOI] [PubMed] [Google Scholar]
- Hilu, K. W. , & Liang, H. (1997). The matK gene: Sequence variation and application in plant systematics. American Journal of Botany, 84, 830–839. 10.2307/2445819 [DOI] [PubMed] [Google Scholar]
- Holländer, V. , & Kück, U. (1999). Group II intron splicing in chloroplasts: Identification of mutations determining intron stability and fate of exon RNA. Nucleic Acids Research, 27, 2345–2353. 10.1093/nar/27.11.2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley, J. , & Tollervey, D. (2010). Apparent non‐canonical trans‐splicing is generated by reverse transcriptase in vitro . PLoS ONE, 5, e12271 10.1371/journal.pone.0012271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, Y. W. , Juan, C. T. , Wang, C. M. , & Jauh, G. Y. (2019). Mitochondrial Heat Shock Protein 60s interact with What’s This Factor 9 to regulate RNA splicing of ccmFC and rpl2 . Plant and Cell Physiology, 60, 116–125. 10.1093/pcp/pcy199 [DOI] [PubMed] [Google Scholar]
- Huang, W. , Zhu, Y. , Wu, W. , Li, X. , Zhang, D. , Yin, P. , & Huang, J. (2018). The pentatricopeptide repeat protein SOT6/EMB2279 is required for plastid rpl2 and trnK intron splicing. Plant Physiology, 177, 684–697. 10.1104/pp.18.00406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübschmann, T. , Hess, W. R. , & Börner, T. (1996). Impaired splicing of the rps12 transcript in ribosome‐deficient plastids. Plant Molecular Biology, 30, 109–123. 10.1007/bf00017806 [DOI] [PubMed] [Google Scholar]
- Jarrell, K. A. , Peebles, C. L. , Dietrich, R. C. , Romit, S. L. , & Perlman, P. S. (1988). Group II intron self‐splicing. Alternative reaction conditions yield novel products. The Journal of Biological Chemistry, 263, 3432–3439. [PubMed] [Google Scholar]
- Joyce, C. M. , & Steitz, T. A. (1994). Function and structure relationships in DNA polymerases. Annual Review of Biochemistry, 63, 777–822. 10.1146/annurev.bi.63.070194.004021 [DOI] [PubMed] [Google Scholar]
- Kroeger, T. S. , Watkins, K. P. , Friso, G. , van Wijk, K. J. & Barkan, A. (2009). A plant‐specific RNA‐binding domain revealed through analysis of chloroplast group II intron splicing. Proceedings of the National Academy of Sciences of the United States of America, 106(11), 4537–4542. 10.1073/pnas.0812503106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz, A. M. , & Zimmerly, S. (2011). Group II introns: Mobile ribozymes that invade DNA. Cold Spring Harbor Perspectives in Biology, 3, a003616 10.1101/cshperspect.a003616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Gao, L.‐M. , Poudel, R. C. , Li, D.‐Z. , & Forrest, A. (2011). High universality of matK primers for barcoding gymnosperms. Journal of Systematics and Evolution, 49, 169–175. 10.1111/j.1759-6831.2011.00128.x [DOI] [Google Scholar]
- Liere, K. , & Link, G. (1995). RNA‐binding activity of the matK protein encoded by the chloroplast trnK intron from mustard (Sinapis alba L.). Nucleic Acids Research, 23, 917–921. 10.1093/nar/23.6.917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzoni, F. , Nowak, M. D. , Alfaro, M. E. , Reeb, V. , Miadlikowska, J. , Krug, M. , … Magallón, S. (2018). Contemporaneous radiations of fungi and plants linked to symbiosis. Nature Communications, 9, 5451 10.1038/s41467-018-07849-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Abarca, F. , Zekri, S. , & Toro, N. (1998). Characterization and splicing in vivo of a Sinorhizobium meliloti group II intron associated with particular insertion sequences of the IS630‐Tc1/IS3 retrotransposon superfamily. Molecular Microbiology, 28, 1295–1306. 10.1046/j.1365-2958.1998.00894.x [DOI] [PubMed] [Google Scholar]
- Matsuura, M. , Noah, J. W. , & Lambowitz, A. M. (2001). Mechanism of maturase‐promoted group II intron splicing. EMBO Journal, 20, 7259–7270. 10.1093/emboj/20.24.7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura, M. , Saldanha, R. , Ma, H. , Wank, H. , Yang, J. , Mohr, G. , … Labmowitz, A. M. (1997). A bacterial group II intron encoding reverse transcriptase, maturase, and DNA endonuclease act: Biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes and Dev., 11, 2910–2924. 10.1101/gad.11.21.2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeal, J. R. , Kuehl, J. V. , Borre, J. L. , Leebens‐Mach, J. , & dePamphilis, C. W. (2009). Parallel loss of plastid introns and their maturase in the genus, Cuscuta . PLoS ONE, 4, e5982 10.1371/journal.pone.0005982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, F. , Umesono, K. , & Ozeki, H. (1989). Comparative and functional anatomy of group II catalytic introns – A review*. Gene, 82, 5–30. 10.1016/0378-1119(89)90026-7 [DOI] [PubMed] [Google Scholar]
- Mohr, G. , Perlman, P. S. , & Lambowitz, A. M. (1993). Evolutionary relationships among group II intron‐encoded proteins and identification of a conserved domain that may be related to maturase function. Nucleic Acids Research, 21, 4991–4997. 10.1093/nar/21.22.4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, J. V. , Mecklenburg, K. L. , Sass, P. , Belcher, S. M. , Mahnke, D. , Lewin, A. , & Perlman, P. (1994). Splicing defective mutants of the COXI gene of yeast mitochondrial DNA: Initial definition of the maturase domain of the group II intron aI2. Nucleic Acids Research, 22, 2057–2064. 10.1093/nar/22.11.2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus, H. , & Link, G. (1987). The chloroplast tRNALys (UUU) gene from mustard (Sinapsis alba) contains a class II intron potentially coding for a maturase‐related polypeptide. Current Genetics, 11, 251–257. 10.1007/bf00355398 [DOI] [PubMed] [Google Scholar]
- Noah, J. W. , & Lambowitz, A. M. (2003). Effects of maturase binding and Mg2+ concentration on group II intron RNA folding investigated by UV cross‐linking. Biochemistry, 42, 12466–12480. 10.1021/bi035339n [DOI] [PubMed] [Google Scholar]
- Qu, Y. , Legen, J. , Arndt, J. , Henkel, S. , Hoppe, G. , Thieme, C. , … Schmitz‐Linneweber, C. (2018). Ectopic transplastomic expression of a synthetic MatK gene leads to cotyledon‐specific variegation. Frontiers in Plant Science, 9, 1453 10.3389/fpls.2018.01453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichon, C. , Luo, J. , Causey, T. B. , Benner, J. S. , & Samuelson, J. C. (2011). Engineering Escherichia coli BL21 (DE3) derivative strains to minimize E. coli protein contamination after purification by immobilized metal affinity chromatography. Applied and Environmental Microbiology, 77, 4634–4646. 10.1128/AEM.00119-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitzsch, M. , & Pyle, A. M. (2009). The linear form of a group II intron catalyzes efficient autocatalytic reverse splicing, establishing a potential for mobility. RNA, 15, 473–482. 10.1261/rna.1392009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smathers, C. M. , & Robart, A. R. (2019). The mechanism of splicing as told by group II introns: Ancestors of the spliceosome. Biochimica et Biophysica Acta, 1862, 194390 10.1016/j.bbagrm.2019.06.001 [DOI] [PubMed] [Google Scholar]
- Soreng, R. J. , Peterson, P. M. , Romaschenko, K. , Davidse, G. , Teisher, J. K. , Clark, L. G. , … Zuloaga, F. O. (2017). A worldwide phylogenetic classification of the Poaceae (Gramineae) II: An update and a comparison of two 2015 classifications. Journal of Systematics and Evolution, 55, 259–290. 10.1111/jse.12262 [DOI] [Google Scholar]
- Sugita, M. , Shinozaki, K. , & Sugiura, M. (1985). Tobacco chloroplast tRNA (UUU) gene contains a 2.5‐kilobase‐pair intron: an open reading frame and a conserved boundary sequence in the intron. Proceedings of the National Academy of Sciences of the United States of America, 82, 3557–3561. 10.1073/pnas.82.11.3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan, L. D. , Mileshina, D. , Grewe, F. , Rolle, K. , Abudraham, S. , Głodowicz, P. , … Ostersetzer‐Biran, O. (2016). The reverse transcriptase/ RNA maturase protein MatR is required for the splicing of various group II introns in Brassicaceae Mitochondria. The Plant Cell, 28, 2805–2829. 10.1105/tpc.16.00398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadini, L. , Ferrari, R. , Lehniger, M.‐K. , Mizzotti, C. , Moratti, F. , Resentini, F. , … Pesaresi, P. (2018). Trans‐splicing of plastid rps12 transcripts, mediated by AtPPR4, is essential for embryo patterning in Arabidopsis thaliana . Planta, 248, 257–265. 10.1007/s00425-018-2896-8 [DOI] [PubMed] [Google Scholar]
- Tillich, M. , Schmitz‐Linneweber, C. , Herrmann, R. G. , & Maier, R. M. (2001). The plastid chromosome of maize (Zea mays): Update of the complete sequence of the transcript editing sites. Maize Genetics Cooperation Newsletter, 75, 42–44. [Google Scholar]
- Toor, N. , Hausner, G. , & Zimmerly, S. (2001). Coevolution of group II intron RNA structures with their intron‐encoded reverse transcriptases. RNA, 7, 1142–1152. https://doi.org/10.1017.S1355838201010251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor, N. , Rajashankar, K. , Keating, K. S. , & Pyle, A. M. (2008). Structural basis for exon recognition by a group II intron. Nature Structural & Molecular Biology, 15, 1221–1221. 10.1038/nsmb.1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong, D. M. , Sidote, D. J. , Russell, R. , & Lambowitz, A. M. (2013). Enhanced group II intron retrohoming in magnesium‐deficient Escherichia coli via selection of mutations in the ribozyme core. Proceedings of the National Academy of Sciences of the United States of America, 110(40), E3800–E3809. 10.1073/pnas.1315742110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, J. , Börner, T. , & Hess, W. (1999). Comparative analysis of splicing of the complete set of chloroplast group II introns in three higher plant mutants. Nucleic Acids Research, 27, 3866–3874. 10.1093/nar/27.19.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, J. , Hübschmann, T. , Börner, T. , & Hess, W. R. (1997). Splicing and intron‐internal RNA editing of trnK‐matK transcripts in barley plastids: Support for MatK as an essential splicing factor. Journal of Molecular Biology, 270, 179–187. 10.1006/jmbi.1997.1115 [DOI] [PubMed] [Google Scholar]
- Yoon, S.‐B. , Park, Y.‐H. , Choi, S.‐A. , Yang, H.‐J. , Jeong, P.‐S. , Cha, J.‐J. , … Kim, S.‐U. (2019). Real‐time PCR quantification of spliced X‐box binding protein 1 (XBP1) using a universal primer method. PLoS ONE, 14, e0219978 10.1371/journal.pone.0219978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, N. D. , & dePamphilis, C. W. (2000). Purifying selection detected in the plastid gene matK and flanking ribozyme regions within a group II intron of nonphotosynthetic plants. Molecular Biology and Evolution, 17, 1933–1941. 10.1093/oxfordjournals.molbev.a026295 [DOI] [PubMed] [Google Scholar]
- Zaita, N. , Torazawa, K. , Shinozaki, K. , & Sugiura, M. (1987). Trans splicing in vivo: Joining of transcripts from the 'divided' gene for ribosomal protein S12 in the chloroplasts of tobacco. FEBS Letters, 210, 153–156. 10.1016/0014-5793(87)81326-1 [DOI] [Google Scholar]
- Zhao, C. , & Pyle, A. M. (2016). Crystal structures of group II intron maturase reveal a missing link in spliceosome evolution. Nature Structural & Molecular Biology, 23, 558–565. 10.1038/nsmb.3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoschke, R. , Nakamura, M. , Liere, K. , Sugiura, M. , Börner, T. , & Schmitz‐Linneweber, C. (2010). An organellar maturase associates with multiple group II introns. Proceedings of the National Academy of Sciences of the United States of America, 107, 3245–3250. 10.1073/pnas.0909400107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S1‐S3
Table S1
Supplementary Material