

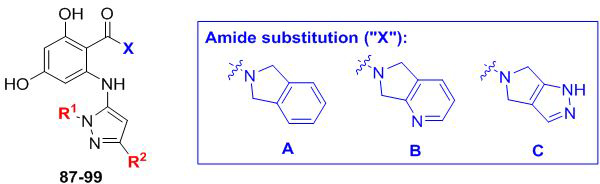
Abstract
The molecular chaperone Hsp90, essential in all eukaryotes, plays a multifaceted role in promoting survival, virulence and drug resistance across diverse pathogenic fungal species. The chaperone is also critically important, however, to the pathogen’s human host, preventing the use of known clinical Hsp90 inhibitors in antifungal applications due to concomitant host toxicity issues. With the goal of developing Hsp90 inhibitors with acceptable therapeutic indices for the treatment of invasive fungal infections, we initiated a program to design and synthesize potent inhibitors with selective activity against fungal Hsp90 isoforms over their human counterparts. Building on our previously-reported derivatization of resorcylate natural products to produce fungal-selective compounds, we have developed a series of synthetic aminopyrazole-substituted resorcylate amides with broad, potent, and fungal-selective Hsp90 inhibitory activity. Herein we describe the synthesis of this series, as well as biochemical structure-activity relationships driving selectivity for the Hsp90 isoforms expressed by Cryptococcus neoformans and Candida albicans, two pathogenic fungi of major clinical importance.
Graphical Abstract
INTRODUCTION
The morbidity and mortality caused by fungal infections cripple human health across the globe. Over a billion people are affected by superficial infections, such as ringworm and athlete’s foot. Adding to these numbers are the burden of oral and other mucosal infections. Of most concern is the increasing number of invasive systemic infections, which leads to over one million deaths each year.1 People with compromised immune function, such as patients receiving cancer chemotherapies, organ transplant recipients and those infected with HIV, are most vulnerable to invasive fungal infections. The pathogens responsible for > 90% of invasive mycoses are Candida albicans, Aspergillus fumigatus and Cryptococcus neoformans. Once diagnosed, treatment options are limited to only three major classes of antifungal drugs, notoriously hampered by problems with host toxicity, the emergence of resistance, or limited spectrum of activity.2 In fact, the only new class of antifungals to reach the clinic in decades has no efficacy against C. neoformans and related species.3
Selective targeting of fungal stress responses provides a promising therapeutic strategy to mitigate resistance and more effectively combat invasive mycoses. The essential molecular chaperone Hsp90 has been extensively validated as a regulator of virulence and antifungal drug resistance in Candida and Aspergillus species.4, 5 For instance, in C. albicans, genetic depletion or pharmacological inhibition of Hsp90 increases the efficacy of current antifungal drugs, reduces acquired antifungal resistance in clinical isolates, and improves clearance in a mouse model of disseminated candidiasis.6 Recent studies have demonstrated the critical importance of Hsp90 for C. neoformans thermotolerance and shown that Hsp90 inhibition alters capsule assembly and sensitivity to antifungals, influencing virulence of the pathogen.7,8 While targeting Hsp90 offers a promising but relatively unexplored strategy for antifungal drug development, the chaperone has been intensively explored as a target in oncology. A structurally diverse array of drugs targeting the ATP-binding pocket of human Hsp90 continue to be evaluated for anticancer activity in patients. In contrast, allosteric approaches to targeting the function of Hsp90 at sites other than its N-terminal ATPase have only been explored in preclinical studies,9 the exception being a putative C-terminal inhibitor (RTA901) which has recently completed Phase I testing in humans (NCT0266693).
Unfortunately, dose-limiting toxicities coupled with relatively limited therapeutic efficacy have so far precluded FDA approval of any N-terminal Hsp90 inhibitor either alone or in combination with other therapeutic agents. In the course of these anticancer drug development and testing campaigns, no effort has been devoted to the pursuit of fungal selectivity and an Hsp90 inhibitor with the properties required for use as an antifungal has yet to be reported.
Fungal selectivity is a crucial feature for an Hsp90 inhibitor to be developed as an antifungal given that Hsp90 is essential in all eukaryotes. Its function supports protein quality control mechanisms, productive folding and the stability of conformationally labile proteins, many involved in key signaling cascades.10 The chaperoning by Hsp90 of its so-called client proteins is ATP-dependent and coordinated by a suite of co-chaperones and accessory factors that impart client selectivity and help regulate progression through the chaperoning cycle. Although Hsp90 is highly conserved across phylogenetic kingdoms, species-specific variations are observed at the level of conformational flexibility, intrinsic ATPase activity, chaperoning dynamics, and the involvement of specific co-chaperone/accessory proteins.11 Therefore, despite a very high degree of conservation at the primary sequence level, these important functional differences provide hope that species-selectivity can be achieved, either at the classical N-terminal ATP-binding pocket or alternatively via allosteric inhibitors acting at other sites.12
While efforts to achieve species-selectivity are just beginning, the pursuit of human paralog-specific Hsp90 inhibitors has already achieved considerable success. These efforts have been focused on achieving selectivity at the N-terminal nucleotide-binding domain (NBD) across the four family members expressed in humans: Hsp90α, Hsp90β, Trap1 and Grp94.13, 14 For example, Blagg and coworkers have described successful efforts to modify the resorcylate scaffold to confer selectivity towards specific human paralogs, including selective Grp94 inhibitors with applications in oncology and glaucoma,15–19and more recently, the first Hsp90β-selective inhibitor with applications in cancer.20 In addition, isoform-selective purine mimetics, such as Hsp90α/β-specific inhibitor TAS-11621 and modified analogs of BIIB021 selectively targeting Trap114 have been described. Modified benzamides resembling SNX-2112 have also been diverted to both Hsp90α/β-specific22 and Trap1-specific23 activities for neurological applications.
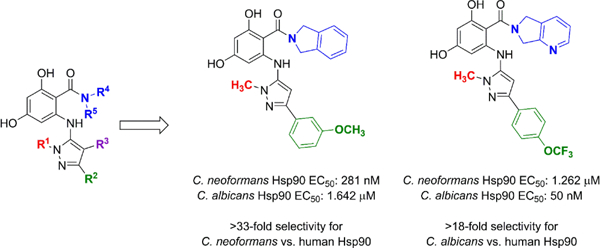
We recently disclosed the discovery of the first fungal-selective Hsp90 inhibitors,11 with activity against the C. albicans Hsp90 isoform, based on semi-synthetic oxime-derivatization of the resorcylate macrocycle natural products radicicol (1) and monocillin I (2). For therapeutic applications, fungal-selectivity is critical as current inhibitors targeting host Hsp90 have deleterious effects that preclude their use in the context of systemic infection. Our most promising lead from this series, monocillin-derived oxime CMLD013075 (3) (Figure 1A), has >25-fold binding selectivity for the C. albicans Hsp90 NBD compared to the human ortholog, limits fungal proliferation in whole cell assays, and is less toxic to human cells compared to the non-selective compound radicicol. Importantly, the co-crystal structure of C. albicans Hsp90 NBD with CMLD013075 displayed unique structural rearrangements, including remodeling of the ATP-binding site, N-terminus, and lid region of the fungal chaperone. Aided by structural insights, key residues were identified as critical for the fungal selectivity of this derivative. Encouraged by these findings and using 3 as a point of departure, we now report the structure activity relationship (SAR)-guided efforts to develop fully synthetic, non-macrocyclic resorcylate inhibitor chemotypes, focusing on selectivity toward both C. neoformans and C. albicans Hsp90.
Figure 1.

Design of aminopyrazole resorcylate-type inhibitor chemotype 8 based on precedented fungal-selective natural-product-derived inhibitors (A) and truncated resorcylates under clinical evaluation in oncology (B).
Replacement of the macrolactone of radicicol with acyclic isosteres including amides (Onalespib (4)24–27), oxazoles (Luminespib (5)28–33), triazolones (Ganetespib (6)34–42), and ketones (KW-2478 (7)43–46) has been a widely successful strategy for the development of multiple classes of synthetic Hsp90 inhibitors currently in clinical evaluation (Figure 1B). Using our macrocyclic oxime CMLD013075 as a lead template, our initial efforts focused on the replacement of the selectivity-imparting oxime with a suitable heterocyclic isostere, with the parallel goals of removing the isomerizable oxime (which we postulated could obfuscate selectivity analysis), and reducing rotational degrees of freedom to enhance binding affinity. After evaluating various heterocyclic options for similarity and synthetic tractability, we selected the aminopyrazole of general type 8 (Figure 1C) for initial development. We hypothesized that a pendant aminopyrazole could project substituents (R1/R2/R3) in orientations similar to that of the CMLD013075 oxime, to impart fungal selectivity in the binding of Hsp90. In addition to the attractiveness of the pyrazole from the standpoint of developability,47 we also postulated that structure-activity relationships at three points of diversity (R1/R2/R3) could be easily elaborated through the coupling of aryl bromide 9 with a combination of commercial and synthetic aminopyrazoles (10).
RESULTS AND DISCUSSION
Synthesis of resorcylate aminopyrazole analogs
Our initial synthesis of aminopyrazole resorcylates began with 1-bromo-3,5-dimethoxybenzene 11 (Scheme 1). Formylation, de-methylation, MOM protection, and Pinnick oxidation afforded carboxylic acid 12, which was then subjected to HATU-mediated amidation with isoindoline to produce amide 13. We initially selected the isoindoline amide as it is conserved across multiple classes of acyclic resorcylate heat shock protein inhibitors, 48–53 providing a simple, precedented model scaffold on which our selectivity-inducing strategy could be evaluated. We next installed the aminopyrazole using Pd-mediated coupling; after a brief exploration of coupling conditions54 we ultimately settled on Pd2(dba)3/Xantphos/NaOPh in dioxane under microwave irradiation55 as the optimal conditions across a wide scope of substrates. Following amination, acid-mediated MOM deprotection produced the desired aminopyrazole-substituted resorcylates.
Scheme 1.

First-generation synthetic route to aminopyrazole/isoindoline resorcylate amides Conditions: a) POCl3, DMF, 100 °C; b) BBr3, CH2Cl2, −78 °C to RT; c) MOMCl, DIPEA, DMF; d) NaOCl2, NaH2PO4•H2O, 2-methyl-2-butene, THF/tBuOH/H2O; e) isoindoline•HCl, HATU, Et3N, THF/CH2Cl2; f) tBuXphos Pd G1 (10 mol%), tBuXPhos (10 mol%), NaOtBu, tBuOH, or Pd2(dba)3 (4 mol%), Xantphos (8 mol%), NaOPh, dioxane, 60 °C to 120 °C, or Pd2(dba)3 (10 mol%), Xantphos (10 mol%), NaOPh, dioxane, 170 °C, microwave; g) HCl, methanol, 50 °C.
We also applied this first-generation synthetic sequence to explore replacement of the isoindoline amide for several early compounds (Scheme 2). During the course of analog synthesis, however, we found that reversing the order of coupling/amidation resulted in a more efficient procedure with improved yields and product purities; the resultant second-generation route is depicted in Scheme 3. Following esterification of carboxylic acid 12, the resulting ester 17 was subjected to Pd-mediated coupling with 10 to afford intermediate 18. Following ester hydrolysis, carboxylic acid 19 was subsequently amidated, which was initially performed using the HATU-mediated conditions, and later optimized to employ polymer-supported carbonyldiimidazole (PS-CDI) as a coupling reagent for improved parallel processing. Finally global MOM-deprotection provided the desired products for testing. All tested compounds were purified by mass-targeted HPLC.
Scheme 2.

Early exploration of amide SAR using first-generation synthetic route Conditions: a) HNR4R5, HATU, Et3N, THF/CH2Cl2; b) 10a, Pd2(dba)3 (4 mol%), Xantphos (10 mol%), NaOPh, dioxane, 170 °C, microwave; c) HCl, methanol, 50 °C.
Scheme 3.

Second-generation synthetic route to aminopyrazole resorcylate amides Conditions: a) CH3I, K2CO3, DMF, 80 °C; b) 10, Pd2(dba)3 (4 mol%), Xantphos (10 mol%), NaOPh; c) KOH, EtOH, 95 °C; d) HNR4R5, HATU, Et3N, CH2Cl2/THF, RT or HNR4R5, PS-CDI, HOBt•xH2O, Et3N, THF/CH2Cl2; e) HCl, methanol, 50 °C.
Measurement of fungal Hsp90 binding affinity and selectivity
All analogs were assessed for Hsp90 binding affinity using a fluorescence polarization (FP)-based equilibrium competition assay in fungal and human whole cell lysates. Notably, this approach allows for the assessment of compound binding while the target protein is in native complexes with co-chaperones; and, in the case of human cell lysate, in a biologically relevant mix of Hsp90 paralogs. Using lysates, we were able to measure the relative potency and selectivity for fungal Hsp90 versus the entire ensemble of human Hsp90 isoforms in microplate format using small amounts of test materials. To confirm target engagement with an alternative biochemical approach, the most selective analogs were also assessed by protein thermal shift assays using purified recombinant Hsp90 nucleotide binding domains (NBD) of the relevant fungal species. Thermal shift assays were performed under saturating ligand conditions, i.e. equimolar concentrations (10 μM) of protein and ligand. As a result, they provided qualitative evidence of target binding, but not a quantitative measurement of ligand affinity. For quantitation, a ligand dissociation constant (Ki) for key compounds was also determined using purified NBDs in FP assays and KD measurements were made by surface plasmon resonance (SPR) using a Biacore instrument. Finally, all analogs were assessed for whole cell antifungal activity against the pathogens C. albicans and C. neoformans. Quantitative dose-response assays were performed for all compounds found to inhibit growth at a concentration ≤ 50 μM.
Structure activity relationships for resorcylate aminopyrazoles
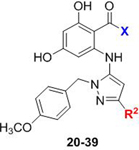
We first examined N-(para)-methoxybenzyl substituted aminopyrazoles, designed to mimic our parent Candida-selective inhibitor CMLD013075. We began by making systematic alterations to the resorcylate amide, with R2 substitution limited to methyl and phenyl. Our initial amide diversification utilized several pyrrolidine/isoindoline-based heterocycles, which are prevalent among resorcylate amide Hsp90 inhibitors reported by Astex and Pfizer (20–21, 26-29, 32–36, 38),49, 50 as well as new isoindoline isosteres (pyrido- and pyrazolopyrrolidines 22-25). We also pursued a small series of acyclic mono- and disubstituted amides, both new (30-31) and precedented (37, 39).56 From this initial set, we were pleased to find a number of compounds had < 200 nM EC50 values against one or both fungal species (Table 1). Consistent with published inhibitors in this space, larger, substituted isoindoline-type moieties (32–36) generally exhibited excellent potency, but with no apparent selectivity for the fungal Hsp90 isoforms. In contrast, we found that the pairing of smaller heterobicylic amides with a phenyl group at the R2 position (compounds 21, 23, 25, 27, and 29) afforded modestly fungal-selective compounds; as a general trend, their R2 = CH3 analogs (20, 22, 24, 26 and 28) were more potent but nonselective. Activity was mainly relegated to the heterobicyclic amides; our limited set of acyclic and monocyclic amides (30–31, 37–39) were for the most part less active and also nonselective, with the interesting exception of low potency cryptococcal-selective compound 30. Based on these results, and given our early hypothesis that the installation of functionality at the aminopyrazole would be the key driver in imparting selectivity, we opted to progress forward with the lower-molecular weight isoindoline, pyridopyrrolidine, and pyrazolopyrrolidine amides, selected to represent both precedented and novel resorcylate amide substitutions with varying basicities.
Table 1.
Structure-activity relationships for N-(4-methoxybenzyl)-substituted aminopyrazoles, exploring variation of the resorcylate amide with methyl- and phenyl-substitution at R2. Fold-selectivity > 5 for any compound is highlighted in red.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | X | R2 |
C. neoformans
EC50a (μM) |
C. neoformans fold- selectivityb |
C. albicans EC50c (μM) |
C. albicans fold- selectivityb |
| 1 | 20 |  |
CH3 | 0.040 | 0.8 | 0.011 | 0.9 |
| 2 | 21 | Ph | 0.877 | 2.5 | 0.511 | 2.2 | |
| 3 | 22 |  |
CH3 | 0.087 | 1.2 | 0.184 | 0.4 |
| 4 | 23 | Ph | 0.142 | 4.0 | 0.068 | 6.2 | |
| 5 | 24 | 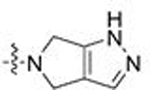 |
CH3 | 0.063 | 1.7 | 0.157 | 0.5 |
| 6 | 25 | Ph | 0.121 | 2.7 | 0.063 | 3.9 | |
| 7 | 26 | 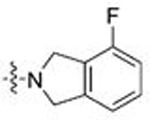 |
CH3 | 0.109 | 0.6 | 0.117 | 0.4 |
| 8 | 27 | Ph | 0.787 | 2.2 | 1.089 | 1.2 | |
| 9 | 28 | 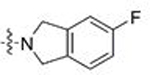 |
CH3 | 0.592 | 0.1 | 0.054 | 0.5 |
| 10 | 29 | Ph | 0.705 | 2.7 | 1.043 | 1.3 | |
| 11 | 30 | 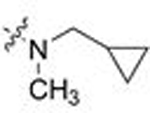 |
CH3 | 1.330 | 5.8 | > 9 | - |
| 12 | 31 | Ph | > 9 | - | > 9 | - | |
| 13 | 32 | 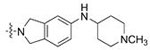 |
CH3 | 0.096 | 1.0 | 0.171 | 0.4 |
| 14 | 33 | Ph | 0.146 | 0.8 | 0.023 | 1.8 | |
| 15 | 34 | CH3 | 0.086 | 1.2 | 0.115 | 0.7 | |
| 16 | 35 | Ph | 0.091 | 0.8 | 0.014 | 1.7 | |
| 17 | 36 | 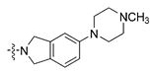 |
CH3 | 0.115 | 0.9 | 0.143 | 0.6 |
| 18 | 37 | 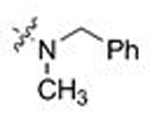 |
CH3 | 4.814 | 1.6 | > 6 | 0.0 |
| 19 | 38 | 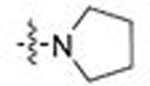 |
Ph | 0.464 | 2.1 | 0.282 | 1.2 |
| 20 | 39 |  |
Ph | >10 | - | >10 | - |
EC50 values were determined by FP-based equilibrium competition assay performed in 384-well format using whole cell lysates prepared from C. neoformans (a) and C. albicans (c) and serial compound dilutions. All determinations were performed in duplicate. To calculate fold-selectivity (b), the EC50 value determined in human HepG2 cell lysate was divided by the EC50 value determined in fungal cell lysate. The resulting ratio was then normalized to values determined in the same assay for the non-selective inhibitor geldanamycin using lysate of each cell type. Results for key selective compounds were confirmed by repeat assay (see Supplemental Table 1).
Our next series of analogs explored additional R2/R3 substitutions on the aminopyrazole, again keeping the R1 para-methoxybenzyl group intact (Table 2). For the R2 unsubstituted pyrazoles (40–44), we found that substitution at R3 was tolerated, but with decreasing potency as steric bulk increased. Several of these compounds also exhibited modest undesired selectivity toward the human isoform. Based on these results, we opted not to pursue this substitution pattern further. In contrast, and similar to our initial cohort, we identified wider tolerance for substitution at the R2 position with several acyclic (45–48) and cyclic (50–53) aliphatic groups, as well as furan (54–55) substitution. A drop in potency was limited to the bulkier R2 = tBu analog 49. Disappointingly, however, none of the inhibitors exhibited the modest fungal selectivity that had been observed in their R2 = Ph substituted counterparts 21, 23 and 25 (Table 1).
Table 2.
Exploration of SAR at R2/R3 for R1 = p-methoxybenzyl substituted aminopyrazoles
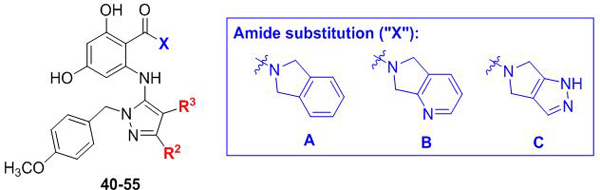 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound | X | R2 | R3 |
C. neoformans EC50a (μM) |
C. neoformans fold- selectivityb |
C. albicans EC50c (μM) |
C. albicans fold- selectivityb |
| 1 | 40 | A | H | H | 0.072 | 0.8 | 0.041 | 1.0 |
| 2 | 41 | A | H | CH3 | 0.094 | 0.3 | 0.022 | 0.7 |
| 3 | 42 | A | H | iPr | 0.396 | 0.4 | 0.147 | 0.5 |
| 4 | 43 | A | H | Ph | 1.623 | 1.6 | 0.615 | 2.1 |
| 5 | 44 | A | H | Bn | 1.756 | 0.5 | 0.465 | 1.1 |
| 6 | 45 | B | Et | H | 0.022 | 0.9 | 0.014 | 0.5 |
| 7 | 46 | C | 0.025 | 0.7 | 0.012 | 0.6 | ||
| 8 | 47 | B | iPr | H | 0.025 | 1.0 | 0.013 | 0.7 |
| 9 | 48 | C | 0.023 | 0.8 | 0.009 | 0.8 | ||
| 10 | 49 | A | tBu | H | 1.026 | 1.4 | 0.816 | 1.0 |
| 11 | 50 | B |  |
H | 0.026 | 0.9 | 0.014 | 0.6 |
| 12 | 51 | C | 0.023 | 0.9 | 0.014 | 0.5 | ||
| 13 | 52 | B |  |
H | 0.021 | 0.7 | 0.006 | 1.0 |
| 14 | 53 | C | 0.041 | 0.6 | 0.009 | 1.0 | ||
| 15 | 54 | B | 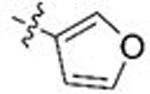 |
H | 0.039 | 1.3 | 0.022 | 0.9 |
| 16 | 55 | C | 0.040 | 0.9 | 0.018 | 0.7 | ||
EC50 and selectivity values were determined as described for Table 1.
We next assessed replacement of the p-methoxybenzyl group at R1. Initially, this group had been chosen based on analogy to our Candida-selective inhibitor CMLD013075. Our X-ray crystallographic analysis11 (PDB ID: 6CJP) indicates that the aryl ring participates in a key binding interaction following a major structural rearrangement of the Candida Hsp90 lid region, serving as a donor in an N-H…π interaction with C. albicans Asn40. However, given the limited scope of radicicol- and monocillin-derived analogs that we previously explored, coupled with a current lack of structural information about the cryptococcal isoform, it is not clear that this group represents an “ideal” binding moiety for either fungal species. As an initial probe, we focused on varying solely the R1 group across the isoindoline, tetrahydropyrrolopyridine and tetrahydropyrrolopyrazole amides, leaving the R2 and R3 sites unsubstituted. The results for this series are summarized in Table 3. We once again identified a wide array (aliphatic, aromatic, heteroaromatic) of aminopyrazole substitutions that afforded in most cases sub-125 nM potencies for both fungal species (56, 59, 62–77 and 80–86), but all broadly nonselective with the exception of isoindoline 83. This compound was exemplary as the first compound in our aminopyrazole series to exhibit sub-100 nM EC50 with greater than 10-fold selectivity. Interestingly, however, in isolated cases the tetrahydropyrrolopyridine and tetrahydropyrrolopyrazole amides diverged from their isoindoline counterparts with a slight decrease in potency (compounds 78–79), which was in some cases coupled with a slight increase in cryptococcal selectivity (57–58 and 60–61). These compounds, bearing aliphatic N-substitutions of varying size, showed 2- to 5-fold selectivity toward C. neoformans, with no apparent selectivity toward C. albicans.
Table 3.
Exploring alternative R1 substituents on R2/R3-unsubstituted aminopyrazoles. Fold selectivity >5 for any compound is highlighted in red.
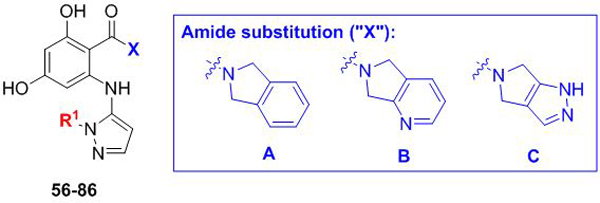 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | R1 | X |
C. neoformans EC50a (μM) |
C. neoformans fold- selectivityb |
C. albicans EC50c (μM) |
C. albicans fold- selectivityb |
| 1 | 56 | CH3 | A | 0.088 | 1.5 | 0.111 | 0.4 |
| 2 | 57 | B | 0.286 | 4.4 | 0.624 | 0.7 | |
| 3 | 58 | C | 0.142 | 5.1 | 0.377 | 0.7 | |
| 4 | 59 | iPr | A | 0.125 | 1.3 | 0.076 | 0.7 |
| 5 | 60 | B | 0.250 | 2.6 | 0.309 | 0.7 | |
| 6 | 61 | C | 0.097 | 3.6 | 0.161 | 0.7 | |
| 7 | 62 | iBu | A | 0.062 | 1.1 | 0.035 | 0.7 |
| 8 | 63 | B | 0.103 | 1.7 | 0.117 | 0.6 | |
| 9 | 64 | C | 0.041 | 2.3 | 0.057 | 0.6 | |
| 10 | 65 |  |
A | 0.051 | 0.7 | 0.009 | 1.2 |
| 11 | 66 | B | 0.026 | 1.7 | 0.015 | 0.9 | |
| 12 | 67 | C | 0.018 | 1.3 | 0.008 | 0.8 | |
| 13 | 68 | Ph | A | 0.061 | 0.5 | 0.014 | 0.8 |
| 14 | 69 | B | 0.044 | 0.9 | 0.021 | 0.7 | |
| 15 | 70 | C | 0.037 | 0.8 | 0.015 | 0.8 | |
| 16 | 71 | Cy | A | 0.087 | 0.4 | 0.013 | 0.9 |
| 17 | 72 | B | 0.045 | 1.0 | 0.016 | 1.0 | |
| 18 | 73 | C | 0.035 | 1.0 | 0.014 | 1.0 | |
| 19 | 74 | Bn | A | 0.052 | 0.8 | 0.015 | 0.8 |
| 20 | 75 | B | 0.043 | 1.1 | 0.018 | 0.8 | |
| 21 | 76 | C | 0.036 | 1.1 | 0.015 | 0.8 | |
| 22 | 77 |  |
A | 0.058 | 1.7 | 0.041 | 0.7 |
| 23 | 78 | B | 0.207 | 1.6 | 0.143 | 0.7 | |
| 24 | 79 | C | 0.199 | 1.8 | 0.164 | 0.7 | |
| 25 | 80 | 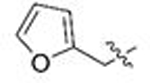 |
A | 0.074 | 0.9 | 0.033 | 0.7 |
| 26 | 81 | B | 0.106 | 1.2 | 0.080 | 0.6 | |
| 27 | 82 | C | 0.058 | 1.4 | 0.041 | 0.8 | |
| 28 | 83 | 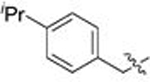 |
A | 0.044 | 12.8 | 0.366 | 1.1 |
| 29 | 84 | B | 0.040 | 0.8 | 0.012 | 0.9 | |
| 30 | 85 | C | 0.036 | 0.8 | 0.012 | 0.8 | |
| 31 | 86 |  |
B | 0.063 | 0.9 | 0.025 | 0.8 |
| 32 | 87 | C | 0.048 | 1.1 | 0.021 | 0.9 | |
EC50 and selectivity values were determined as described for Table 1.
We next progressed to examining the combined modifications of the R1 N-substitution with additional groups at R2 (Table 4). Again mindful of keeping physicochemical properties such as molecular weight and lipophilicity within an acceptable “druglike” range, we imposed a limitation for this series that each pyrazole should contain a maximum of one aryl ring at either R1 or R2, but not at both.57 This series produced a number of analogs with more modest sub-micromolar potency and cryptococcal selectivity greater than 4-fold (91–95). Of these, compounds 94 and 95 also exhibited modest selectivity for C. albicans Hsp90 over human Hsp90 paralogs, which was consistent with their early near neighbor analogs 21, 23 and 25 (Table 1).
Table 4.
Examining varied parings of R1/R2 substitutions on the aminopyrazole ring. Fold selectivity >5 for any compound is highlighted in red.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound | X | R1 | R2 |
C. neoformans EC50a (μM) |
C. neoformans fold- selectivityb |
C. albicans EC50c (μM) |
C. albicans fold- selectivityb |
| 1 | 88 | A | 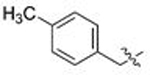 |
CH3 | 0.252 | 0.2 | 0.121 | 0.3 |
| 2 | 89 | A | 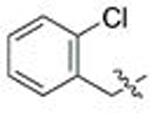 |
CH3 | 0.560 | 0.2 | 0.116 | 0.4 |
| 3 | 90 | A | 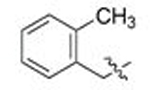 |
CH3 | 0.700 | 0.2 | 0.134 | 0.4 |
| 4 | 91 | A | CH3 | Ph | 0.078 | 9.2 | 0.328 | 0.4 |
| 5 | 92 | B | 0.127 | 8.2 | 0.395 | 0.8 | ||
| 6 | 93 | C | 0.066 | 6.7 | 0.213 | 0.6 | ||
| 7 | 94 | B | tBu | Ph | 0.517 | 9.7 | 0.379 | 3.9 |
| 8 | 95 | C | 0.379 | 6.7 | 0.182 | 4.1 | ||
| 9 | 96 | B | 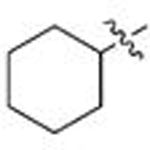 |
Ph | 0.615 | 0.4 | 0.059 | 1.7 |
| 10 | 97 | C | 0.419 | 0.3 | 0.034 | 1.4 | ||
| 11 | 98 | B | iBu | Ph | 0.315 | 1.7 | 0.100 | 1.6 |
| 12 | 99 | C | 0.147 | 1.8 | 0.070 | 1.4 | ||
EC50 and selectivity values were determined as described for Table 1.
Among our initial fungal-selective leads from this effort, compounds 91–93 stood out as having high cryptococcal selectivity without a concomitant loss in cryptococcal potency as seen in earlier analogs. To follow up, we designed a final array of analogs N-methylated at R1, probing more diverse aliphatic and aryl substituents at R2 (Table 5).
Table 5.
Variation of R2 substituent for N-methylated aminopyrazoles yields C. neoformans- and C. albicans-selective Hsp90 inhibitors with diverging isoform selectivities. Fold selectivities > 5 are highlighted in red.
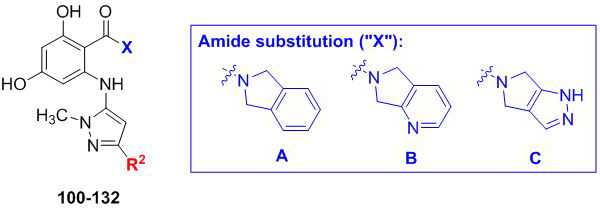 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | R2 | X |
C. neoformans EC50a (μM) |
C. neoformans fold- selectivityb |
C. albicans EC50c (μM) |
C. albicans fold- selectivityb |
| 1 | 100 | iPr | A | 0.087 | 0.9 | 0.048 | 0.6 |
| 2 | 101 | B | 0.062 | 1.6 | 0.156 | 0.2 | |
| 3 | 102 | C | 0.040 | 3.0 | 0.094 | 0.5 | |
| 4 | 103 | Cy | A | 0.156 | 0.7 | 0.033 | 1.4 |
| 5 | 104 | B | 0.110 | 6.5 | 0.670 | 0.4 | |
| 6 | 105 | C | 0.037 | 3.7 | 0.096 | 0.6 | |
| 7 | 106 | 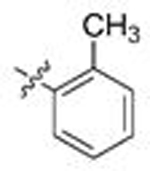 |
A | 0.065 | 14.1 | 0.599 | 0.7 |
| 8 | 107 | B | 0.139 | 12.9 | 0.795 | 0.8 | |
| 9 | 108 | C | 0.084 | 12.4 | 0.398 | 0.9 | |
| 10 | 109 | 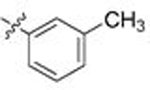 |
A | 0.267 | 16.3 | 0.573 | 3.0 |
| 11 | 110 | B | 0.421 | 15.1 | 1.030 | 2.4 | |
| 12 | 111 | C | 0.176 | 14.6 | 0.396 | 2.5 | |
| 13 | 112 | 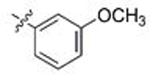 |
A | 0.281 | 33.3 | 1.642 | 2.0 |
| 14 | 113 | B | 0.852 | 27.6 | 5.000 | 1.7 | |
| 15 | 114 | C | 0.244 | 26.5 | 1.881 | 1.3 | |
| 16 | 115 | 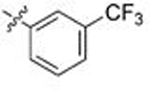 |
A | 4.601 | 3.2 | 2.236 | 2.1 |
| 17 | 116 | B | 1.523 | 5.5 | 0.594 | 4.4 | |
| 18 | 117 | C | 0.434 | 9.8 | 0.294 | 4.6 | |
| 19 | 118 | 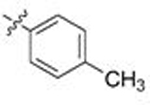 |
A | 1.318 | 6.3 | 1.067 | 2.6 |
| 20 | 119 | B | 1.781 | 9.5 | 1.779 | 4.0 | |
| 21 | 120 | C | 0.424 | 14.9 | 0.636 | 3.4 | |
| 22 | 121 | 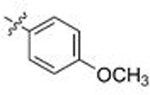 |
A | 0.630 | 4.4 | 0.186 | 5.8 |
| 23 | 122 | B | 1.289 | 6.9 | 1.139 | 3.1 | |
| 24 | 123 | C | 0.489 | 6.8 | 0.376 | 3.4 | |
| 25 | 124 | 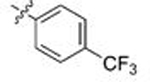 |
A | 9.530 | 1.0 | 1.084 | 3.1 |
| 26 | 125 | B | 5.815 | 0.8 | 0.319 | 4.8 | |
| 27 | 126 | C | 1.385 | 1.1 | 0.103 | 4.9 | |
| 28 | 127 | 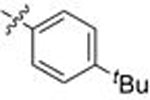 |
A | >10 | - | 1.197 | 2.1 |
| 29 | 128 | B | 8.337 | 0.4 | 0.237 | 4.5 | |
| 30 | 129 | C | 2.262 | 0.5 | 0.071 | 5.0 | |
| 31 | 130 | 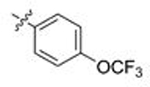 |
A | 5.402 | 1.0 | 0.134 | 15.3 |
| 32 | 131 | B | 1.262 | 1.0 | 0.050 | 18.2 | |
| 33 | 132 | C | 0.514 | 1.3 | 0.016 | 15.9 | |
EC50 and selectivity values were determined as described for Table 1.
Gratifyingly, this series produced highly selective inhibitors for both the C. neoformans and C. albicans isoforms of Hsp90. While our exploration of aliphatic substitution was limited, high cryptococcal potency (EC50 < 160 nM), and in some cases modestly Cryptococcus-selective compounds (3- to 6.5-fold) were observed with isopropyl (100–102) and cyclohexyl (103–105) substitution at R2. The most highly selective compounds, however, were observed among the R2 arylated analogs, with diverging species-selectivity based on the nature and position of the aryl ring substituent. The ortho-methylated analogs 106–108 displayed slightly enhanced cryptococcal selectivity and similar cryptococcal potency (<150 nM) as compared to their unsubstituted congeners 91–93 (Table 4), with no apparent selectivity and significantly lower potencies (≥ 400 nM) in lysate of C. albicans. Movement of the methyl substituent from ortho- to meta- (compounds 109–111) afforded similarly Cryptococcus-selective compounds, albeit with lower potencies. Interestingly, the meta-methoxy substituted 112–114 exhibited a significant improvement in cryptococcal selectivity (27- to 33-fold) despite only modest cryptococcal potency (EC50s all >250 nM). Trifluoromethylation at the same meta- position (compounds 115–117), resulted in a dramatic reduction in both cryptococcal selectivity and activity.
Moving from testing in C. neoformans lysate to C. albicans lysate, the aforementioned meta-substituted compounds 109–117 also exhibited modest selectivity, with the best Candida-selectivity observed m-trifluoromethylated analogs 116 and 117 (4.4- and 4.6-fold, respectively). The meta-substituted series also exhibited consistently poor C. albicans potencies, with EC50 values ranging from ~300 nM to 5 μM. In contrast, improved C. albicans selectivities and potencies were observed among the analogs that were para-substituted on the R2 phenyl ring. para-Methylated (119–120) and para-methoxy substituted (121–123) aminopyrazoles exhibited moderate selectivities and, in most cases, equivalently low potencies against both fungal species, with EC50 values generally ranging from 0.5–2 μM. Incorporation of larger lipophilic substituents at the para-position such as trifluoromethyl (124–126) and tert-butyl (127–129) further depressed cryptococcal potency, with EC50s ranging from 2 to >10 μM and no apparent selectivity. In contrast these compounds (124–129) maintained improved potencies and similar selectivities against Candida Hsp90 to their para-methyl- and para-methoxy- counterparts 124–127. This series also highlights what we have observed to be an occasional sensitivity to the nature of the amide/aminopyrazole pairing; for example in direct contrast to the cryptococcal potency trends observed with unsubstituted analogs 56–58, pairing of the pyrido- and pyrazolopyrrolidine with the bulkier 3-CF3-Ph (116–117), 4-CF3-Ph (125–126) and 4-tBu-Ph (128–129) substituents at R2 improved C. albicans potency and selectivity relative to their isoindoline counterparts 115, 124 and 127. This trend did not hold, however, for all analogs. Perhaps most intriguingly, the para-trifluoromethoxy substituted compounds 130–132, which were completely nonselective and only modestly potent toward cryptococcal Hsp90, exhibited dramatic improvements in potency toward C. albicans, with EC50 values ranging from 16–134 nM and 15- to 18-fold Candida selectivity. These divergent structure-selectivity trends, wherein ortho/meta-methyl and meta-methoxy compounds exhibited high Cryptococcus selectivity and poor Candida selectivity, whereas para-trifluoromethoxy substitution rendered high Candida selectivity and poor Cryptococcus activity, are summarized in Figure 2.
Figure 2.

Summary of iterative progression to fungal selective inhibitors 106–132 with divergent patterns of species selectivity dependent on position of substitution on the aminopyrazole phenyl ring (red)
Relationship of fungal to human selectivity
To better understand the phylogenetic origins of the divergent selectivity between fungi, we performed protein::protein BLAST sequence alignments across the different species studied. This analysis indicated that C. neoformans and C. albicans share 69% sequence identity across the entire Hsp90 protein, and 71% identity across their NBD (residues 1–240). As a comparison, human Hsp90α and Hsp90β share 69% and 67% identity with C. albicans Hsp90 across their NBD, respectively. Thus, the two fungal species diverge in primary sequence as greatly from one another as they do from human Hsp90. In light of such sequence divergence, perhaps it is not surprising that while we set out to discriminate against human Hsp90, the potency and selectivity of our synthetic inhibitors also diverged between the two fungal species studied. A graphic summary of inhibitor potency/selectivity relationships found by screening compounds in C. neoformans lysate (Fig 3A) and C. albicans lysate (Fig. 3B) highlights the progress made toward our goal of achieving fungal selectivity, while the divergence between compound selectivity in regards to C. neoformans vs. C. albicans is best demonstrated by plotting the selectivity of compounds for one fungus vs. human against selectivity for the other (Fig. 3C). To more accurately define their potency and selectivity, the activity of 27 compounds with a screening EC50 < 1μM in lysate of either fungal species was confirmed by repeat testing in two additional experiments, with results provided in Supplemental Table 1.
Figure 3.

Scatter plots depict fungal potency (x-axis) vs. fungal selectivity (y-axis) relationships for all aminopyrazoles when screened using human cell lysate and lysate of either C. neoformans (Panel A) or C. albicans (Panel B). All potencies are reported as the inverse log10 of compound EC50 (pEC50 as measured by FP assay). The scatter plot in Panel C compares compound selectivity patterns between the two fungi. Key fungal-selective compounds for each species (112–114 and 130–132) are highlighted in color to underscore their divergence in potency and selectivity. Each point represents the mean of duplicate determinations in a single experiment.
Validation of whole cell lysate FP results
To confirm the FP results obtained in lysate for our most potent and selective compounds, we prepared recombinant Candida, Cryptococcus, and human Hsp90 NBDs by expression and purification in E. coli. Using recombinant proteins, we were able to define assay-independent nanomolar inhibitory constants (Ki) for these compounds (Figure S1). We also confirmed binding of the compounds to their relevant NBD by thermal shift denaturation assays (Figure S1). Thermal shift assays were performed under saturating ligand conditions, i.e. equimolar concentrations (10 μM) of protein and ligand. As a result, they can provide only qualitative evidence of target binding, but not a quantitative measurement of ligand affinity. This feature of the thermal shift method is well demonstrated in Supplemental Table 2, which presents Ki and thermal shift data for both high and low potency compounds. Here, compounds with Ki values of less than 50 nM for a particular NBD increase its ΔTm to a similar extent irrespective of absolute potency. In contrast, lower affinity compounds (Ki > 100 nM) fail to increase the Tm of the respective NBD.
As an orthogonal, highly quantitative approach to FP, we measured the binding affinities of our six lead compounds for C. albicans, C. neoformans and human Hsp90 NBDs by surface plasmon resonance (SPR, Supplemental Table 3). The affinity values determined for compounds varied by less than an order of magnitude between the two different experimental techniques. The same pattern of fungal selectivity for compounds demonstrated by FP assay in whole cell lysates was also seen by SPR. The magnitude of selectivity determined by SPR assays compared to FP assays in lysate, however, was reduced. Such a difference might be expected given the absence in SPR assays of native cochaperone-containing complexes and, in the case of human cell lysate, a biologically relevant mix of Hsp90 paralogs.
Whole cell antifungal activity
Having achieved promising potency and species-selectivity for several compounds at the level of fungal target engagement, we next examined the ability of these compounds to inhibit fungal growth. We found that minimal inhibitory concentrations (MICs) for most of the potent and selective analogs highlighted in Table 5 were much higher than their EC50 values in lysate, generally > 50 μM. The disparity between whole cell antifungal activity and the EC50 values we determined in FP assays is undoubtedly due to poor permeability/accumulation of the compounds in fungal cells. This common problem in the development of antifungals occurs because the fungal cell wall and membrane as well as the diverse drug efflux pumps expressed by fungi render it a challenge to achieve intracellular concentrations of experimental compounds sufficient to inhibit the function of their targets.
Of the fungal Hsp90-selective compounds tested, only the 14-fold C. neoformans-selective analog 106 inhibited growth of the organism below 10 μM (Fig. 4A). While triazole antifungals in current clinical use against Cryptococcus do have MICs in excess of this range, they also possess far greater selectivity than we have achieved so far and are much less toxic to human cells. As single agents, the MICs of all our Candida-selective compounds were >50 μM. To provide a more sensitive read-out, however, we took advantage of the well-established ability of Hsp90 inhibitors to potentiate the activity of conventional antifungals against drug-resistant isolates of C. albicans.5 Testing compounds 130 and 131 in combination with the widely used antifungal fluconazole, we found an MIC of 12.5 μM for the 15-fold Candida-selective analog 130 against a moderately fluconazole-resistant clinical isolate of C. albicans. This compound also converted the fungistatic activity of fluconazole to fungicidal against the same isolate, an effect consistent with Hsp90 inhibitory activity (Figure 4B).
Figure 4.

Biological activity of fungal-selective inhibitors.
Panel A: Growth inhibition by fungal-selective aminopyrazoles of C. neoformans reference strain H99 cultured in RPMI 1640 medium at 37 °C. Panel B: Growth inhibition by fungal-selective aminopyrazoles of a C. albicans clinical isolate (CaCi2) with or without a background concentration of 8 μg/mL fluconazole. The effect of 48-hour exposure to inhibitors over a twofold dilution series of concentrations is displayed in heat-map format. Color scale bar: no growth inhibition (green) to complete inhibition (black). Each colored box represents the mean of technical duplicates. The experiment was repeated as an independent biological replicate to confirm results. Following exposure to compounds, aliquots of the cultures in each well were spotted onto compound-free YPD agar and plates incubated at 30 °C for an additional 24 hours before imaging to assess fungicidal activity (Panel B, right).
Thus, although their potency and selectivity require further improvement, the whole cell activity of these resorcylate aminopyrazoles remains consistent with an Hsp90-targeted mode of action. Encouraged by this finding and to aid future efforts in developing the scaffold, we performed an initial evaluation of its stability to P450-mediated metabolism in liver microsomes, a major pharmacological liability of our previous fungal-selective macrocyclic oxime CMLD01307511. Although all the compounds tested suffered from relatively rapid metabolism (Fig. 5 and Supplemental Table 4), important insights were gained into the basis of their instability. Comparing the half-lives of cryptococcal-selective compounds 112–114 reveals an apparent stabilizing effect of the pyrazolopyrrolidine amide, which is consistent with the previously reported metabolic instability of isoindolines due to oxidation at the 5/6 position.24 The isoindoline was chosen for this study despite its known downstream pharmacological liabilities, as it represented a low molecular-weight starting point allowing for the methodical assessment of the relative potency and selectivity of different aminopyrazole substitutions. Assessment of additional analogs 105, 111, 129, and 131–132 indicate that additional metabolic liabilities are also likely present at the aminopyrazole, with the para-trifluoromethoxy substitution clearly inhibiting metabolism. Still, the relatively short half-life of compound 132 (31 minutes) underscores the need for further optimization of metabolic stability, in addition to fungal penetration, as we advance in future work to compounds with suitable properties for testing in vivo. Metabolic stability optimization for resorcylate Hsp90 inhibitors via modification of the amide is well-precedented;24, 50 similar strategies, paired with targeted alterations of the aminopyrazole aryl substituent, are currently under study in our laboratory and will be reported in due course.
Figure 5.

Microsomal stability (mouse liver microsomes) of a panel of fungal-selective inhibitors. Assays were performed by Charles River Laboratories (Worcester, MA).
The factors governing the ability of small molecules to cross cell wall and membrane barriers, avoid active efflux and accumulate within fungi are not well defined. To gain initial insights for our resorcylate aminopyrazoles, we expanded the scope of compounds tested in cellulo to include all biochemically active compounds (FP EC50 < 10 μM) irrespective of their selectivity in cell-free lysates. An additional 83 compounds with diverse physicochemical and structural properties were tested to identify several (21, 27, 29, 49, and 89) with single agent bioactivity against C. neoformans (Table 6).
Table 6.
Aminopyrazoles with whole cell anti-cryptococcal activity
| Entry | Compound | MIC (μM) | FP EC50 (nM) | FP Selectivity |
|---|---|---|---|---|
| 1 | 21 | 6.25 | 877 | 2.5 |
| 2 | 27 | 12.5 | 787 | 2.2 |
| 3 | 29 | 6.25 | 705 | 2.7 |
| 4 | 49 | 12.5 | 1026 | 1.4 |
| 5 | 89 | 25 | 560 | 0.2 |
Minimum inhibitory concentration (MIC) value for compounds against C. neoformans (Strain H99) was determined in dose-response format, in technical duplicate. Experiments were conducted in RPMI medium at 37 °C for 48 h. Relative viable cell number was measured by standard dye reduction (resazurin) assay.
The pattern of results suggests that enhancement of lipophilicity through the introduction of halogens or bulky aliphatic moieties can improve whole cell activity. To independently verify that the whole cell activity of these compounds was consistent with an ability to engage Hsp90, we complemented primary FP-based testing of 21, 27, 29, 49 and 89 and 106 with thermal shift assays using C. neoformans NBD (Supplemental Table 5). Whole cell testing of all biochemically active, but non-selective compounds also revealed three inhibitors (21, 41, and 89) with fungicidal activity in combination with fluconazole against the same clinical isolate of C. albicans used in Fig. 4 (Fig. S2). Analogous to our approach with Cryptococcus-active compounds, target engagement for Candida-active compounds was confirmed by thermal shift assay using C. albicans Hsp90 NBD (Supplemental Table 5).
CONCLUSION
Through the iterative design and optimization of a novel aminopyrazole-substituted resorcylate amide chemotype, we have identified advanced analogs with markedly improved potency and selectivity for binding to fungal Hsp90 isoforms as compared to their human counterparts. Interestingly, as fungal selectivity increased, a marked divergence in structure-activity relationship between C. albicans and C. neoformans became evident. Beyond potent and selective target engagement, the need to increase intracellular accumulation and activity against whole organisms remains to be addressed in future work if useful antifungals are to be developed. By investigating the bioactivity of the entire series of analogs, we have identified key physicochemical properties (e.g. structural modification and lipophilicity enhancement through the introduction of halogens or bulky aliphatic moieties) that appear to contribute to improved whole cell activity and metabolic stability. Targeted exploration of these identified modifications in the context of our fungal-selective aminopyrazole substitutions, as well as medicinal chemistry work to further optimize potency and selectivity of the top fungal-selective leads are ongoing in our efforts to cripple human fungal pathogens by selectively targeting Hsp90.
Experimental Section
Yeast strains and culture conditions.
Strains used in this study were C. albicans CaCi2 (clinical isolate 2),58 SC5314,59 and C. neoformans H99.60 Archives of all fungal strains were maintained at −80 °C in 25% glycerol. Active cultures were maintained on solid (2% agar) yeast extract peptone (YPD, 1% yeast extract, 2% bactopeptone, 2% glucose) at 4 °C for no more than one month. For growth experiments, strains were cultured in YPD medium or in RPMI medium 1640 (Gibco SKU#318000–089, 3.5% MOPS, 2% glucose, pH 7.0), as indicated in figure legends.
Antifungal sensitivity testing.
Minimum inhibitory concentrations (MICs) were determined in flat bottom, 96-well plate format using a modified broth microdilution protocol as previously described,6, 61 except relative viable cell number was monitored by standard dye reduction assay after a 3-hour incubation with resazurin at 37 °C. Radicicol and all synthetic analogs were formulated in dimethyl sulfoxide (DMSO, Sigma Aldrich Co.); fluconazole was dissolved in sterile ddH2O. Each compound was tested in duplicate in at least two independent experiments. Minimum inhibitory concentration (MIC) data were quantitatively displayed in heat-map format using the program Java TreeView 1.1.3 (http://jtreeview.sourceforge.net). To test for fungicidal activity, cultures from MIC plates were spotted on YPD agar plates using a spotter (Frogger, V&P Scientific, Inc). Plates were photographed after 24 h of incubation at 30 °C.
FP assays.
Whole cell lysates were prepared for FP assays as described previously.11 Total protein concentration of human and yeast lysates was determined by Bradford assay.6 Titrations of Cy3-labeled geldanamycin (Cy3-GdA) probe and lysate were evaluated to define conditions that resulted in 75% maximal probe polarization with no competitor present.11 Serial dilutions of test Hsp90 inhibitors were then assayed under these same conditions to monitor loss of fluorescence polarization as an indicator of probe displacement from Hsp90. All determinations were performed in duplicate wells using 384-well black flat-bottom microtiter plates (Greiner Bio-One; 655076). Titrations of test compound in 25 μL of binding buffer (supplemented with 0.1 mg/mL bovine gamma globulin), were mixed with an equal volume of freshly prepared whole-cell lysate spiked with Cy3-GdA (0.1 nM). Plates were incubated at room temperature for 4.5 h to achieve equilibrium binding for the geldanamycin-based probe. Signal in millipolarization (mP) units was measured at an excitation wavelength of 535 nm and emission wavelength of 595 nm in a SpectraMax i3 microplate reader (Molecular Devices) using Softmax Pro software (version 5.4.1). Non-linear 4-parameter curve fitting of raw displacement data was performed in GraphPad Prism 5.0 to determine EC50 values as a measure of relative Hsp90-binding affinity. Results were normalized to the value determined for GdA in lysate of each cell type. This experiment was repeated for a set of 27 key compounds for SAR in at least three independent experiments.
FP assays were also performed with purified C. albicans and C. neoformans Hsp90 NBD for the determination of inhibitory constants (Ki) for relevant fungal-selective compounds. Titrations of the Cy3-GdA probe and purified proteins were evaluated to define assay conditions and to determine the dissociation constant Kd of the probe for each NBD. Serial dilutions of test Hsp90 inhibitors were then assayed in triplicate wells under these conditions. Non-linear 4-parameter curve fitting of raw displacement data was performed in GraphPad Prism 5.0 to determine IC50 values. Finally, inhibitory constants (Ki) were calculated as described previously.11, 62
Protein thermal shift assays.
Thermal melting curves were determined using a Protein Thermal Shift Kit (ThermoFisher # 4462263), employing a CFX384 Real-Time PCR System (Bio-Rad, C1000 Touch Thermal Cycler). Reactions were performed in a final volume of 10 μL, and contained purified C. albicans or C. neoformans Hsp90 NBD diluted to 250 μg/mL in Buffer HBS-P (GE Healthcare Life Sciences, 0.01 M HEPES pH 7.4, 0.15 M NaCl, 0.005% v/v Surfactant P20) with 10 μM synthetic analog or DMSO control, and 1 × Sypro Orange dye solution. Samples were prepared in triplicate in 384-well white plates (Bio-Rad; HSP3805). The instrument was set to melt curve, step 1 (25 °C, 2 min) and step 2 (ramp to 98.6 °C, increasing 0.2 °C per 5 s cycle). The inflection point of each curve is defined as the protein melting temperature (Tm). Temperature shift, ΔTm, was recorded as the difference between the Tm of the fungal Hsp90 NBD with compound minus Tm of the protein without compound.
NBD expression and purification.
Recombinant Hsp90 NBDs were expressed and purified as previously described.11 Stock protein solutions in 50% glycerol were stored at −20 °C until dilution into relevant buffers and use for FP and thermal shift assays.
SPR assays.
For SPR experiments, Hsp90 NBD expression constructs were modified to encode a C-terminal AviTag™ for site-specific on-column biotinylation with a BirA biotin-ligase kit (Avidity LLC; BirA-500). SPR experiments were performed on a Biacore T200 instrument at 25 °C. Biotinylated Hsp90 NBD was diluted to 40 μg/mL and immobilized on a streptavidin chip (Sensor Chip SA, GE Healthcare) at a density of 2000 – 2500 response units (RU) on the biosensor surface. Binding experiments were done in HBS-P (0.01 M HEPES pH 7.4, 0.15 M NaCl, 0.005% v/v Surfactant P20, GE Healthcare) with 5% DMSO at a flow rate of 40 μL/min. Test compounds were injected in two dilutions series, with low concentrations ranging from 6 to 96 nM and high concentrations ranging from 60 to 960 nM, with a 60 s association time and 600 s dissociation time, with the exception of compound 130 for which the injection time was extended to 300 s after observing a very slow on-rate with this molecule. Resulting sensorgrams were analyzed with a fit to a 1:1 binding model, using BIA evaluation software.
Microsome stability testing.
The potential susceptibility of compounds to hepatic metabolism was assessed by Charles River Laboratories (Worcester, MA) using standard in-house protocols. Compounds were incubated at 1 μM concentration in mixed-gender CD-1 mouse liver microsomes (0.5 mg/mL) in the presence of 2 μM NADPH. Percent compound remaining was measured by LC/MS/MS at six timepoints (0, 15, 30, 60, 90 and 120 min) in duplicate. 7-ethoxycoumarin was utilized as a positive control. In addition, NADPH-free control samples were assessed at two timepoints (0 and 15 min) in duplicate to exclude non-CYP450-mediated decomposition. First-order half-lives are calculated from the equation T1/2 = −0.693/x, where x is the slope found in the linear fit for the plot of ln(% remaining) versus incubation time. Calculated mouse intrinsic hepatic clearance (CLint) in mL/min/kg is extrapolated63 based on 45 mg microsomes/g liver and 87.5 g liver/kg body weight.
Statistical methods.
For FP experiments in support of SAR studies, GraphPad Prism 5.0 was used to perform curve fitting and calculate the concentrations of compounds resulting in 50% reduction in maximal polarization signal (EC50). All curve fits demonstrated a correlation coefficient (R2) >0.95 The number of independent experiments performed and the number of technical replicates in each experiment are provided in the legends of figures and tables characterizing the biochemical and biological activities of compounds. In calculating the error of selectivity determinations, the fractional error of measurements in each species was summed to yield a composite error for the derived ratio.
Chemistry Methods.
General Methods.
All melting points are uncorrected. 1H NMR spectra were recorded at 400 or 500 MHz at ambient temperature. 13C NMR spectra were recorded at 101 or 126 MHz at ambient temperature. Chemical shifts are reported in parts per million. Data for 1H NMR are reported as follows: chemical shift, multiplicity (app = apparent, br = broad, s = singlet, d = doublet, t = triplet, q = quartet, sxt = sextet, hept = heptet, m = multiplet, ovrlp = overlap), coupling constants, and integration. All 13C NMR spectra were recorded with complete proton decoupling. Analytical thin layer chromatography was performed using 0.25 mm silica gel 60-F plates. Flash column chromatography was performed using 200–400 mesh silica gel (Sorbent Technologies, Inc.). Automated flash chromatography was performed using prepacked columns (SI-HC, puriFlash or Premium Universal, Yamazen) on either an Interchim puriFlash450 or Yamazen Smart Flash EPCLC W-Prep2XY system. All mass-guided preparative HPLC was performed using an acetonitrile:water gradient (mobile phase modified with 0.01% formic acid) on a Waters FractionLynx system equipped with a 600 HPLC pump, a micromass ZQ quadrapole, Waters 996 diode array, and Sedere Sedex 75 ELS detectors, using an XBridge Prep C18 5 μM OBD 19 mm diameter column of either 100 mm or 250 mm length. Isolated yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. All reactions were carried out in oven-dried glassware under an argon atmosphere unless otherwise noted. Analytical LC-MS experiments were performed using a Waters Acquity UPLC (ultraperformance liquid chromatography) with a binary solvent manager, SQ mass spectrometer, Waters 2996 PDA (photodiode array) detector, and evaporative light scattering detector (ELSD). All microwave experiments were performed on a CEM Discover microwave reactor, using a sealed 10 or 35 mL vessel with temperatures monitored by an external sensor. All compounds tested in biological assays were determined to be >95% pure by UPLC-MS-ELSD analysis.
General Procedure A: Synthesis of α-Formyl Nitriles.
All α-formyl nitriles used as synthetic precursors for aminopyrazoles 10 were synthesized via a procedure adapted from 64. To a suspension of potassium tert-butoxide in THF (2.2 equiv, 1.4 M solution in THF) at room temperature was added a mixture of the requisite nitrile (1 equiv) and ethyl formate (1.05 equiv) in THF (6.3 M relative to nitrile) dropwise. After stirring overnight at room temperature, the reaction mixture was diluted with CH2Cl2 and water. The resulting mixture was adjusted to pH = 4 using concentrated HCl (aq.). The layers were separated and aqueous layer was extracted twice with CH2Cl2. The combined organic layers were washed with brine and dried with anhydrous MgSO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography to give the intermediate α-formyl nitrile.
General Procedure B: Synthesis of α,β-unsaturated Nitriles.
All α,β-unsaturated nitriles used as synthetic precursors for aminopyrazoles 10 were generated from commercially-available aldehydes according to the following procedure: To a solution of potassium tert-butoxide (2 M in THF, 1.04 equiv) at 0 ºC was added diethyl cyanomethylphosphonate (1.1 equiv) dropwise. After stirring at 0 ºC for 1 h, the requisite aldehyde (1 equiv) was added dropwise and the reaction was allowed to warm to room temperature overnight. The reaction mixture was poured into saturated NH4Cl (aq.) and diluted with ethyl acetate. The layers were separated and the aqueous layer was extracted three times with ethyl acetate. The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and condensed in vacuo. The crude mixture was purified via automated flash chromatography to give the intermediate α,β-unsaturated nitrile.
General Procedures C: Syntheses of aminopyrazoles 10
C1: Procedure adapted from 65. A suspension of 3-aminocrotonitrile (1.08 equiv) and the requisite hydrazine hydrochloride (1 equiv) in 1 M HCl (aq.) (0.72 M concentration of hydrazine) was refluxed for 3 h. The resulting mixture was diluted with water and extracted twice with ethyl acetate. The aqueous layer was basicified with solid NaHCO3 until solid remained. The aqueous layer was extracted twice with ethyl acetate. The combined organic layers from each extraction sequence were separately washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and the mother liquors were combined and condensed in vacuo. The crude residues were purified via automated flash chromatography.
C2: A mixture of the requisite α-formyl nitrile and 4-(methoxybenzyl)hydrazine hydrochloride (1 equiv) was refluxed overnight in ethanol (0.36 M relative to α-formyl nitrile). The solution was cooled to room temperature and condensed in vacuo. The residue was diluted with CH2Cl2 and the organic layer was washed twice with saturated NaHCO3 (aq.) and brine. The organic layer was dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography.
C3: Procedure adapted from 65. To a solution of hydrazine monohydrate (1.03 equiv) in THF (5 M relative to hydrazine) at room temperature was added the requisite α,β-unsaturated nitrile (1.02 equiv) and heated to 40 ºC for 2 h. After cooling to room temperature, the requisite aldehyde (1 equiv) was added dropwise. The mixture was heated to 40 ºC for an additional 2 h. After cooling to room temperature, volatile materials were condensed in vacuo. The resulting residue was dissolved in iPrOH (4.5 M relative to benzaldehyde). Sodium tert-butoxide (1.03 equiv) was added to the reaction mixture and the resulting suspension was heated to 100 ºC for 2.5 h and then stirred overnight at room temperature. The reaction mixture was diluted with water and extracted twice with diethyl ether. The combined organic layers were washed twice with 1 M HCl. The combined 1 M HCl washes were basicified to pH = 14 with 50% NaOH (aq.) and extracted with diethyl ether. The second set of ether extractions were combined and washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography.
C4: A solution of requisite oxonitrile (1 equiv) and (4-methoxybenzyl)hydrazine hydrochloride (2 equiv) in EtOH (0.3 M relative to oxonitrile) was heated to reflux overnight. After cooling to room temperature, volatile materials were condensed in vacuo. The residue was dissolved in CH2Cl2 and saturated NaHCO3 (aq.). The layers were separated and the aqueous layer was extracted twice with CH2Cl2. The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography.
C5: A solution of requisite oxonitrile (1 equiv) and methylhydrazine (1 equiv) in methanol (2 M) were irradiated at 120 ºC for 40 min in a microwave reactor. After cooling to room temperature, volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography
General Procedures D. Pd-mediated coupling of aryl bromides to aminopyrazoles 10.
D1: Inside a nitrogen glovebox were combined aryl bromide (1 equiv), amine 10 (1.1 equiv), tris(dibenzylideneacetone)dipalladium (0.04 equiv), Xantphos (0.08 equiv), sodium phenoxide (1.5 equiv). Dioxane (0.13 M) was added to the mixture and the reaction vessel was capped and removed from the glovebox. After the reaction was heated in an oil bath at 120 ºC for 2 h, the reaction was cooled to room temperature and diluted with ethyl acetate. The resulting mixture was washed three times with saturated Na2CO3 (aq.), brine, then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography.
D2: Inside a nitrogen glovebox were combined aryl bromide (1 equiv), amine 10 (1.1 equiv), tris(dibenzylideneacetone)dipalladium (0.04 equiv), Xantphos (0.08 equiv), sodium phenoxide (1.5 equiv) in a 10 mL microwave reaction vessel. Dioxane (0.13 M) was added to the mixture and the reaction vessel was capped and removed from the glovebox. After the reaction was irradiated at 170 ºC for 2 h in a microwave reactor, the reaction was cooled to room temperature and diluted with ethyl acetate. The resulting mixture was washed three times with saturated Na2CO3 (aq.), brine, then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography.
General Procedure E. Hydrolysis conditions to generate crude acids 19.
To a solution of ester (1 equiv) in EtOH:water (1:1 ratio, 0.06 M) was added potassium hydroxide (9.2 equiv) and then heated to 95 ºC for 1h. After cooling to room temperature, volatile materials were condensed in vacuo. The residue was suspended in saturated NH4Cl (aq.) and CH2Cl2. The layers were separated and the aqueous layer was extracted three times with CH2Cl2. The combined organic layers were washed twice with water, brine and then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude acid 19 was used without further purification.
General Procedure F: Global MOM-deprotection.
To a solution of amide (1 equiv) in methanol (13.7 mM) was added HCl (2 M, 6.5 equiv). The resulting solution was stirred at 50 ºC overnight. After cooling to room temperature, volatile materials were condensed in vacuo. The residue was purified on mass-guided preparative HPLC.
General Procedure G: Amidation of acids 19.
To a suspension of crude carboxylic acid 19 (1 equiv) and amine (1.5 equiv) in CH2Cl2:THF (1:1 mixture, 0.08–0.09 M) was added triethylamine followed by HATU (1.2 equiv). The suspension was stirred overnight at room temperature and then diluted with CH2Cl2. The reaction mixture was washed with saturated NaHCO3 (aq.), brine and then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography.
General Procedures H:
Tandem PS-CDI-mediated amidation and MOM deprotection of crude acids 19.
H1: To a solution of crude carboxylic acid 19 (1 equiv) and isoindoline hydrochloride (1.5 equiv) in THF: CH2Cl2 (1:1 ratio, 77 mM) was added trimethylamine (4 equiv) followed by HOBt hydrate (1.2 equiv) and PS-Carbodiimide (1.18 mmol/g loading, 1.2 equiv). The suspension was shaken overnight at room temperature. The resin was removed via filtration and the resulting filtrate was washed twice with saturated NaHCO3 (aq.) and once with brine. The organic layer was dried with anhydrous sodium sulfate. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The resulting residue was dissolved in methanol (20 mM) and HCl (aq.) (2 M, 6.5 equiv) was added to the mixture. The resulting solution was stirred at 50 ºC overnight. After cooling to room temperature, volatile materials were condensed in vacuo. The residue was purified on mass-guided preparative HPLC.
H2: Identical to General Procedure H1, except using 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine instead of isoindoline hydrochloride.
H3: Identical to General Procedure H1, except using 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole instead of isoindoline hydrochloride.
1-(4-methoxybenzyl)-3-methyl-1H-pyrazol-5-amine (10a).
Synthesized using General Procedure C1 with (4-methoxybenzyl)hydrazine hydrochloride (250 mg, 1.33 mmol) and purified using automated flash chromatography (5% to 25% ethyl acetate in hexanes) to afford 189 mg of 10a as a white/orange solid (66% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.37 (s, 1H), 5.08 (s, 2H), 3.80 – 3.74 (m, 3H), 3.30 (s, 2H), 2.19 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.0, 147.4, 145.2, 129.0, 128.1, 114.2, 91.3, 55.2, 50.8, 13.9. LC/MS (m/z): 218.126 [M+H+]; UPLC tR 1.04 min.
1-(4-methoxybenzyl)-3-phenyl-1H-pyrazol-5-amine (10b).
A solution of benzoylacetonitrile (350 mg, 2.41 mmol) and (4-methoxybenzyl)hydrazine hydrochloride (910 mg, 4.82 mmol) in ethanol (8 mL) was heated to reflux overnight. After cooling to room temperature, the solution was condensed in vacuo. The residue was dissolved in CH2Cl2 and saturated NaHCO3 (aq.). The layers were separated and the aqueous layer was extracted twice with CH2Cl2. The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and the volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (1% to 5% ethyl acetate in CH2Cl2) to afford 498 mg of 10b (74% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.77 (dd, J = 8.2, 1.4 Hz, 2H), 7.38 (dd, J = 8.4, 6.9 Hz, 2H), 7.29 (d, J = 7.4 Hz, 1H), 7.18 (d, J = 8.4 Hz, 2H), 6.90 – 6.80 (m, 2H), 5.90 (s, 1H), 5.24 (s, 2H), 3.78 (s, 3H), 3.44 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 149.3, 144.5, 134.5, 128.5, 127.2, 125.5, 88.9, 56.2, 32.3, 25.8, 25.3. LC/MS (m/z): 281.203 [M+H+]; UPLC tR 1.64 min.
1-(4-methoxybenzyl)-4-methyl-1H-pyrazol-5-amine (10c).
2-methyl-3-oxopropanenitrile was synthesized using General Procedure A from propionitrile (0.82 mL, 11.4 mmol) in 6.7% yield after automated flash chromatography (20% to 60% ethyl acetate in hexanes). 2-methyl-3-oxopropanenitrile (64 mg, 0.73 mmol) was subjected to General Procedure C2 to afford 68 mg of 10c as an off-white solid (41% yield) after purification via automated flash chromatography (15% to 85% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.18 (s, 1H), 7.16 – 7.07 (m, 2H), 6.85 (d, J = 8.6 Hz, 2H), 5.14 (s, 2H), 3.78 (s, 3H), 3.11 (s, 1H), 1.90 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.1, 141.3, 138.7, 128.9, 128.3, 114.2, 100.5, 55.3, 51.4, 7.9. LC/MS (m/z): 218.17 [M+H+]; UPLC tR 1.11 min.
4-isopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10d).
2-formyl-3-methylbutanenitrile was synthesized using General Procedure A from isovaleronitrile (1.20 mL, 11.4 mmol) in 24% yield after automated flash chromatography (10% to 30% acetone in hexanes and 5% to 20% ethyl acetate in CH2Cl2).
2-formyl-3-methylbutanenitrile (291 mg, 2.62 mmol) was subjected to General Procedure C2 to afford 260 mg of 10d as a white/yellow solid (40% yield) after purification via automated flash chromatography (15% to 55% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.22 (s, 1H), 7.12 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 5.14 (s, 2H), 3.78 (s, 3H), 3.13 (s, 2H), 2.62 (p, J = 6.9 Hz, 1H), 1.19 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.1, 140.1, 135.8, 129.0, 128.3, 114.2, 112.4, 55.2, 51.2, 23.7, 23.3. Mp: 74–76 ºC. LC/MS (m/z): 245.916 [M+H+]; UPLC tR 1.30 min.
1-(4-Methoxybenzyl)-4-phenyl-1H-pyrazol-5-amine (10e).
Synthesized using General Procedure C2 from 3-oxo-2-phenylpropanenitrile (250 mg, 1.72 mmol) to afford 223 mg of 10e (46% yield) as an off-white solid after purification via automated flash chromatography (4% to 12% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H), 7.46 – 7.33 (m, 4H), 7.25 – 7.11 (m, 3H), 6.88 (d, J = 8.5 Hz, 2H), 5.21 (s, 2H), 3.79 (s, 3H), 3.61 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 159.3, 141.2, 137.3, 133.6, 129.0, 128.4, 128.3, 126.3, 125.6, 114.4, 106.9, 55.3, 51.6. Mp: 154–156 ºC. LC/MS (m/z): 281.159 [M+H+]; UPLC tR 1.68 min.
4-benzyl-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10f).
2-benzyl-3-oxopropanenitrile was synthesized using General Procedure A from 3-phenylpropionitrile (1.50 mL, 11.4 mmol) in 17% yield after automated flash chromatography (10% to 30% acetone in hexanes and 5% to 20% ethyl acetate in CH2Cl2). 2-benzyl-3-oxopropanenitrile (300 mg, 1.88 mmol) was subjected to General Procedure C2 to afford 152 mg of 10f (27% yield) as a white/brown solid after purification via automated flash chromatography (20% to 60% ethyl acetate in hexanes and 4% to 15% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.15 (m, 5H), 7.11 (d, J = 8.3 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 5.14 (s, 2H), 3.78 (s, 3H), 3.70 (s, 2H), 3.03 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 159.2, 141.7, 140.3, 138.7, 128.8, 128.6, 128.3, 126.2, 114.3, 103.9, 55.3, 51.4, 29.7. LC/MS (m/z): 295.186 [M+H+]; UPLC tR 1.54 min.
3-ethyl-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10g).
Synthesized using General Procedure C3 from pent-2-enenitrile (239 mg, 2.95 mmol) and p-anisaldehyde (0.353 mL, 2.90 mmol) to afford 131 mg of 10g (19% yield) after purification via automated flash chromatography (10% to 30% acetone in hexanes and 5% to 20% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.40 (s, 1H), 5.11 (s, 2H), 3.78 (s, 3H), 3.36 (s, 2H), 2.57 (q, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.1, 153.6, 144.9, 128.9, 128.1, 114.2, 89.9, 55.3, 50.9, 21.8, 14.0. LC/MS (m/z): 231.933 [M+H+]; UPLC tR 1.14 min.
3-isopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10h).
Synthesized using General Procedure C4 from 4-methyl-3-oxopentanenitrile (100 mg, 0.900 mmol) to afford 278 mg of 10h (>100% yield) as a yellow oil after purification via automated flash chromatography (7% to 20% ethyl acetate in CH2Cl2). Chromatographed product was impure and was carried forward to the next step without further purification.
3-(tert-butyl)-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10i).
Synthesized using General Procedure C4 from 4,4-dimethyl-3-oxopentanenitrile (200 mg, 1.60 mmol) and (4-methoxybenzyl)hydrazine hydrochloride (301 mg, 1.60 mmol) to afford 342 mg of 10i (83% yield) as an orange solid after purification via automated flash chromatography (10% to 35% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.13 – 7.00 (m, 2H), 6.85 (d, J = 8.5 Hz, 2H), 5.44 (s, 1H), 5.12 (s, 2H), 3.78 (s, 3H), 3.25 (s, 2H), 1.29 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 160.6, 159.0, 144.5, 129.2, 127.9, 114.2, 88.3, 55.2, 50.9, 32.1, 30.5. Mp: 72–74 ºC. LC/MS (m/z): 261.222 [M+H+]; UPLC tR 1.30 min.
3-cyclopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10j).
Synthesized using General Procedure C4 from 3-cyclopropyl-3-oxopropanenitrile (100 mg, 0.916 mmol) to afford 176 mg of 10j (79% yield) as an off-white solid after purification via automated flash chromatography (7% to 20% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.20 (s, 1H), 5.10 (s, 2H), 3.78 (s, 3H), 3.35 (s, 2H), 1.93 – 1.81 (m, 1H), 0.96 – 0.82 (m, 2H), 0.75 – 0.59 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 159.0, 154.0, 145.0, 128.9, 128.1, 114.2, 87.4, 55.2, 50.9, 9.5, 7.7. Mp: 113–114 ºC. LC/MS (m/z): 245.21 [M+H+]; UPLC tR 1.19 min.
3-cyclopentyl-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10k).
3-cyclopentylacrylonitrile was synthesized using General Procedure A from cyclopentanecarboxaldehyde (0.50 mL, 4.7 mmol) to afford 390 mg (68% yield) of a 1:1.4 mixture of E:Z isomers as a colorless oil after purification via automated flash chromatography (1% to 5% ethyl acetate in hexanes). 3-cyclopentylacrylonitrile (387 mg, 3.19 mmol) was subjected to General Procedure C3 with p-anisaldehyde (0.381 mL, 3.14 mmol) to afford 68.7 mg of 10k (8.2% yield) after purification via automated flash chromatography (3% to 15% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.10 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.39 (s, 1H), 5.09 (s, 2H), 3.77 (s, 3H), 3.31 (s, 2H), 3.06 – 2.89 (m, 1H), 2.09 – 1.94 (m, 2H), 1.83 – 1.53 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 156.4, 144.9, 129.1, 128.2, 114.3, 88.9, 55.3, 51.0, 39.6, 33.5, 25.5. LC/MS (m/z): 272.426 [M+H+]; UPLC tR 1.08 min.
3-(furan-3-yl)-1-(4-methoxybenzyl)-1H-pyrazol-5-amine (10l).
To a solution of potassium tert-butoxide (2 M in THF, 1.04 equiv) at 0 ºC was added diethyl cyanomethylphosphonate (1.1 equiv) dropwise. After stirring at 0 ºC for 1 h, 3-furancarboxaldehyde (0.50 mL, 5.8 mmol, 1 equiv) was added dropwise and the reaction was allowed to warm to room temperature overnight. The reaction mixture was poured into saturated NH4Cl (aq.) and diluted with ethyl acetate. The layers were separated and the aqueous layer was extracted three times with ethyl acetate. The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography(4% to 12% ethyl acetate in CH2Cl2) to afford 3-(furan-3-yl)acrylonitrile 600 mg (88% yield) as an oil in a 3.3:1 mixture of E:Z isomers. 3-(furan-3-yl)acrylonitrile was subjected to General Procedure C3 using p-anisaldehyde (0.605 mL, 4.98 mmol) to afford 292 mg of 10l (22% yield) as a beige solid after purification via automated flash chromatography (3% to 15% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 7.44 (t, J = 1.7 Hz, 1H), 7.18 (d, J = 8.1 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 6.76 (s, 1H), 5.69 (s, 1H), 5.23 (s, 1H), 3.79 (s, 3H), 3.47 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 161.1, 159.2, 145.3, 143.1, 139.0, 130.1, 128.5, 128.1, 120.3, 114.3, 108.8, 89.3, 55.3, 51.3. Mp: 140–142 ºC. LC/MS (m/z): 270.176 [M+H+]; UPLC tR 1.45 min.
3-methyl-1-(4-methylbenzyl)-1H-pyrazol-5-amine (10m).
Synthesized using General Procedure C3 from crotononitrile (0.70 mL, 8.6 mmol) and p-tolualdehyde (1.0 mL, 8.5 mmol) to afford 610 mg of 10m (36% yield) as a yellow solid after purification via automated flash chromatography (5% to 20% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.13 (d, J = 7.9 Hz, 2H), 7.06 (d, J = 7.8 Hz, 2H), 5.37 (s, 1H), 5.11 (s, 2H), 3.29 (s, 2H), 2.32 (s, 3H), 2.19 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 147.4, 145.6, 137.1, 134.0, 129.4, 126.7, 90.9, 50.8, 21.1, 13.9. Mp: 102–104 ºC. LC/MS (m/z): 202.158 [M+H+]; UPLC tR 1.13 min.
3-methyl-1-(2-methylbenzyl)-1H-pyrazol-5-amine (10n).
Synthesized using General Procedure C1 from (2-methylbenzyl)hydrazine hydrochloride (180 mg, 1.33 mmol) to afford 125 mg of 10n (47% yield) as a white solid after purification via automated flash chromatography (5% to 20% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.22 – 7.08 (m, 3H), 6.74 (d, J = 7.3 Hz, 1H), 5.41 (s, 1H), 5.13 (s, 2H), 3.36 – 3.18 (m, 2H), 2.33 (s, 3H), 2.20 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 147.6, 145.6, 135.4, 135.0, 130.4, 127.5, 126.5, 126.1, 91.1, 74.1, 49.4, 19.1, 14.0. Mp: 84–87 ºC. LC/MS (m/z): 202.202 [M+H+]; UPLC tR 1.10 min.
1-(2-chlorobenzyl)-3-methyl-1H-pyrazol-5-amine (10o).
Synthesized using General Procedure C1 from (2-chlorobenzyl)hydrazine dihydrochloride (300 mg, 1.31 mmol) to afford 245 mg of 10o (85% yield) as a white solid after purification via automated flash chromatography (3% to 15% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.33 (m, 1H), 7.24 – 7.15 (m, 2H), 6.85 – 6.72 (m, 1H), 5.43 (s, 1H), 5.23 (s, 2H), 3.42 (s, 2H), 2.20 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 148.2, 145.6, 134.7, 131.9, 129.3, 128.7, 127.9, 127.3, 91.1, 48.3, 14.0. Mp: 97–99 ºC. LC/MS (m/z): 222.14 [M+H+]; UPLC tR 1.12 min.
1-methyl-3-phenyl-1H-pyrazol-5-amine (10p).
Synthesized using General Procedure C5 from benzoylacetonitrile (250 mg, 1.72 mmol) to afford 221 mg of 10p (74% yield) as a white solid after purification via automated flash chromatography (25% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.78 – 7.67 (m, 2H), 7.36 (td, J = 7.2, 6.4, 1.3 Hz, 2H), 7.31 – 7.21 (m, 1H), 5.83 (s, 1H), 3.68 (s, 3H), 3.56 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 149.7, 145.6, 133.8, 128.5, 127.5, 125.3, 88.5, 34.4. Mp: 127–128 ºC. LC/MS (m/z): 174.103 [M+H+]; UPLC tR 1.14 min.
1-(tert-butyl)-3-phenyl-1H-pyrazol-5-amine (10q).
Synthesized using General Procedure C4 from benzoylacetonitrile (250 mg, 1.72 mmol) and tert-butylhydrazine hydrochloride (429 mg, 3.44 mmol) to afford 278 mg of 10q (85% yield) as a yellow solid after purification via automated flash chromatography (7% to 20% ethyl acetate in hexanes). 1H NMR (400 MHz, (CD3)2SO) δ 7.62 (d, J = 7.5 Hz, 2H), 7.38 – 7.27 (m, 2H), 7.26 – 7.13 (m, 1H), 5.76 (d, J = 1.6 Hz, 1H), 4.95 (s, 2H), 1.55 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 147.5, 145.6, 134.4, 128.5, 127.1, 125.3, 91.3, 58.8, 29.4. Mp: 103–104 ºC. LC/MS (m/z): 217.2 [M+H+]; UPLC tR 1.63 min.
1-cyclohexyl-3-phenyl-1H-pyrazol-5-amine (10r).
Synthesized using General Procedure C4 from benzoylacetonitrile (250 mg, 1.72 mmol) and cyclohexylhydrazine hydrochloride (519 mg, 3.44 mmol) to afford 326 mg of 10r (79% yield) as a yellow solid after purification via automated flash chromatography (7% to 20% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.6 Hz, 2H), 7.36 (t, J = 7.5 Hz, 2H), 7.30 – 7.25 (m, 1H), 5.88 (s, 1H), 3.99 (s, 1H), 3.62 (s, 1H), 2.17 – 1.84 (m, 7H), 1.72 (d, J = 11.5 Hz, 1H), 1.48 – 1.11 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 149.3, 144.5, 134.5, 128.5, 127.2, 125.5, 88.9, 56.2, 32.3, 25.8, 25.3. Mp: 126–128 ºC. LC/MS (m/z): 243.225 [M+H+]; UPLC tR 1.68 min.
1-isobutyl-3-phenyl-1H-pyrazol-5-amine (10s).
Synthesized using General Procedure C4 from benzoylacetonitrile (100 mg, 0.689 mmol) and isobutylhydrazine hydrochloride (172 mg, 1.38 mmol) to afford 102 mg of 10s (69% yield) after purification via automated flash chromatography (15% to 40% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.80 – 7.70 (m, 2H), 7.41 – 7.32 (m, 2H), 7.32 – 7.23 (m, 1H), 5.87 (s, 1H), 3.80 (d, J = 7.5 Hz, 2H), 3.50 (s, 2H), 2.29 (dt, J = 13.8, 6.9 Hz, 1H), 0.97 (d, J = 6.7 Hz, 6H).
3-isopropyl-1-methyl-1H-pyrazol-5-amine (10t).
Synthesized using General Procedure C5 from 4-methyl-3-oxopentanenitrile (200 mg, 1.80 mmol) to afford 205 mg of 10s (82% yield) as a purple solid after purification via automated flash chromatography (2% to 6% methanol in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.38 (s, 1H), 3.61 (s, 3H), 3.42 (s, 2H), 2.83 (p, J = 7.0 Hz, 1H), 1.21 (d, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 157.9, 144.7, 87.8, 33.9, 28.1, 22.9. Mp: 105–107 ºC. LC/MS (m/z): 140.358 [M+H+].; UPLC tR 0.37 min.
3-cyclohexyl-1-methyl-1H-pyrazol-5-amine (10u).
Synthesized using General Procedure C5 from 3-cyclohexyl-3-oxopropanenitrile (253 mg, 1.67 mmol) to afford 161 mg of 10u (54% yield) as a clear crystalline solid after recrystallization of the crude material from an ethyl acetate in CH2Cl2 mixture. 1H NMR (400 MHz, CD3OD) δ 5.27 (s, 1H), 3.51 (s, 3H), 2.47 – 2.32 (m, 1H), 1.93 – 1.66 (m, 5H), 1.43 – 1.12 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 157.1, 144.6, 88.2, 38.0, 33.9, 33.3, 26.4, 26.1. Mp: 170–171 ºC. LC/MS (m/z): 181.205 [M+H+]; UPLC tR 1.16 min.
1-methyl-3-(o-tolyl)-1H-pyrazol-5-amine (10v).
Synthesized using General Procedure C5 from 3-(2-methylphenyl)-3-oxopropanenitrile (256 mg, 1.61 mmol) to afford 186 mg of 10v (62% yield) as a brown solid after purification via automated flash chromatography (15% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.55 – 7.45 (m, 1H), 7.23 – 7.17 (m, 3H), 5.73 (s, 1H), 3.74 (s, 3H), 3.51 (s, 2H), 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 150.1, 144.7, 135.9, 133.8, 130.6, 129.0, 127.4, 125.7, 91.8, 34.2, 21.1. Mp: 69–72 ºC. LC/MS (m/z): 189.145 [M+H+]; UPLC tR 1.14 min.
1-methyl-3-(m-tolyl)-1H-pyrazol-5-amine (10w).
Synthesized using General Procedure C5 from 3-(3-methylphenyl)-3-oxopropanenitrile (278 mg, 1.75 mmol) to afford 258 mg of 10w (79% yield) as a white solid after purification via automated flash chromatography (15% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.31 – 7.20 (m, 1H), 7.14 – 7.03 (m, 1H), 5.86 (s, 1H), 3.73 (d, J = 0.8 Hz, 3H), 3.53 (s, 2H), 2.37 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 149.8, 145.7, 138.1, 133.7, 128.4, 128.2, 125.9, 122.5, 88.5, 34.3, 21.5. Mp: 103–104 ºC. LC/MS (m/z): 188.396 [M+H+]; UPLC tR 1.27 min.
3-(3-methoxyphenyl)-1-methyl-1H-pyrazol-5-amine (10x).
Synthesized using General Procedure C5 from 3-(3-methoxyphenyl)-3-oxopropanenitrile (306 mg, 1.75 mmol) to afford 279 mg of 10x (79% yield) as a yellow solid after purification via automated flash chromatography (15% to 45% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.24 (m, 3H), 6.86 – 6.80 (m, 1H), 5.86 (s, 1H), 3.85 (s, 3H), 3.73 (s, 3H), 3.53 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 159.8, 149.5, 145.8, 135.3, 129.5, 117.9, 113.4, 110.3, 88.6, 74.1, 55.3, 34.3. Mp: 91–92 ºC. LC/MS (m/z): 205.158 [M+H+]; UPLC tR 1.21 min.
1-methyl-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-amine (10y).
Synthesized using General Procedure C5 from 3-(trifluoromethyl)benzoylacetonitrile (373 mg, 1.75 mmol) to afford 334 mg of 10y (79% yield) as a white/beige solid after purification via automated flash chromatography (15% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 2.2 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.48 (dt, J = 15.3, 7.7 Hz, 2H), 5.90 (s, 1H), 3.74 (s, 3H), 3.57 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 148.2, 146.0, 134.7, 130.8 (q, 2JC-F = 31.9 Hz), 129.0, 128.4, 128.4, 124.3 (q, 1JC-F = 272.4 Hz), 123.9 (q, 3JC-F = 4.1 Hz), 121.9 (q, 3JC-F = 4.1 Hz), 88.4, 74.1, 34.3. 19F NMR (376 MHz, CDCl3) δ −62.7. Mp: 87–88 ºC. LC/MS (m/z): 243.137 [M+H+]; UPLC tR 1.60 min.
1-methyl-3-(p-tolyl)-1H-pyrazol-5-amine. (10z).
Synthesized using General Procedure C5 with 3-(4-methylphenyl)-3-oxopropanenitrile (278 mg, 1.75 mmol) to afford 238 mg of 10z (73% yield) as a white solid after purification via automated flash chromatography (15% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.2 Hz, 2H), 7.22 – 7.09 (m, 2H), 5.84 (s, 1H), 3.72 (s, 3H), 3.52 (s, 2H), 2.35 (s, 3H). Mp: 139–140 ºC. LC/MS (m/z): 188.396 [M+H+]; UPLC tR 1.25 min.
3-(4-methoxyphenyl)-1-methyl-1H-pyrazol-5-amine (10aa).
Synthesized using General Procedure C5 with 3-(4-methoxyphenyl)-3-oxopropanenitrile (306 mg, 1.75 mmol) to afford 238 mg of 10aa (67% yield) as an off-white/brown crystalline solid after purification via automated flash chromatography (15% to 45% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, (CD3)2SO) δ 7.60 – 7.47 (m, 2H), 6.91 – 6.80 (m, 2H), 5.57 (s, 1H), 5.18 (s, 2H), 3.73 (s, 3H), 3.51 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.1, 149.6, 145.5, 126.7, 126.5, 113.9, 88.1, 55.3, 34.3. Mp: 139–142 ºC. LC/MS (m/z): 204.364 [M+H+]; UPLC tR 1.15 min.
1-methyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-amine (10ab).
Synthesized using General Procedure C5 from 4-(trifluoromethyl)benzoylacetonitrile (373 mg, 1.75 mmol) to afford 326 mg of 10ab (77% yield) as a white solid after purification via automated flash chromatography (15% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CD3OD) δ 7.84 (d, J = 8.1 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 5.89 (s, 1H), 3.67 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 149.8, 149.7, 138.8, 130.1 (q, 2JC-F = 32.2 Hz), 126.6, 125.8 (q, 1JC-F = 271.1 Hz), 126.4 (q, 3JC-F = 3.9 Hz), 88.5, 34.4. 19F NMR (376 MHz, CD3OD) δ −64.0. Mp: 170–172 ºC. LC/MS (m/z): 243.137 [M+H+]; UPLC tR 1.61 min.
3-(4-(tert-butyl)phenyl)-1-methyl-1H-pyrazol-5-amine (10ac).
Synthesized using General Procedure C5 from 3-(4-tert-butylphenyl)-3-oxopropanenitrile (253 mg, 1.26 mmol) to afford 222 mg of 10ac (77% yield) as a white solid after purification via automated flash chromatography (15% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.5 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 5.85 (s, 1H), 3.72 (s, 3H), 1.33 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 150.4, 149.7, 145.5, 131.1, 125.4, 125.0, 88.4, 74.1, 34.6, 34.3, 31.4. Mp: 143–145 ºC. LC/MS (m/z): 231.183 [M+H+]; UPLC tR 1.59 min.
1-methyl-3-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-5-amine (10ad).
Synthesized using General Procedure C5 from 4-(trifluoromethoxy)benzoyl acetonitrile (400 mg, 1.75 mmol) to afford 362 mg of 10ad (81% yield) as a purple solid after purification via automated flash chromatography (15% to 40% ethyl acetate in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.7 Hz, 2H), 7.20 (d, J = 7.8 Hz, 2H), 5.85 (s, 1H), 3.73 (s, 3H), 3.55 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 148.4, 146.0, 132.7, 126.5, 120.5 (q, 1JC-F = 256.8 Hz) 121.0, 88.3, 34.2. 19F NMR (376 MHz, CDCl3) δ −57.8. Mp: 97–99 ºC. LC/MS (m/z): 259.105 [M+H+]; UPLC tR 1.62 min.
2-Bromo-4,6-bis(methoxymethoxy)benzoic acid (12).
To a suspension of 2-bromo-4,6-dimethoxybenzaldehyde (3.0 g, 12 mmol) in CH2Cl2 (40 mL) was added a freshly-prepared solution of boron tribromide (3.5 mL, 37 mmol) in CH2Cl2 (10 mL) via cannula over 15 minutes. The reaction was warmed to room temperature and stirred overnight. The reaction mixture was poured into 200 mL ice water and the resulting mixture was extracted four times with ethyl acetate. The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (5% to 20% acetone in hexanes) to afford 1.8 g of 2-bromo-4,6-dihydroxybenzaldehyde as a white solid (81% yield). 1H NMR (400 MHz, (CD3)2SO) δ 12.18 (s, 1H), 11.33 (s, 1H), 9.96 (s, 1H), 6.69 (d, J = 2.2 Hz, 1H), 6.29 (d, J = 2.2 Hz, 1H). 13C NMR (101 MHz, (CD3)2SO) δ 194.5, 165.6, 165.4, 128.2, 113.9, 111.5, 102.3.
To a solution of 2-bromo-4,6-dihydroxybenzaldehyde (1.5 g, 6.9 mmol) and N,N-diisopropylethylamine (4.8 mL, 28 mmol) in DMF (20 mL) at room temperature was chloromethyl methyl ether (2.1 mL, 28 mmol) dropwise. The reaction was stirred at room temperature overnight. The reaction mixture was poured into water and the resulting mixture was extracted four times with Et2O. The combined organic layers were washed twice with water and once with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (5% to 25% ethyl acetate in hexanes) to afford 2.1 g of 2-bromo-4,6-bis(methoxymethoxy)benzaldehyde as a white solid (93% yield). 1H NMR (400 MHz, CDCl3) δ 10.34 (s, 1H), 7.01 (d, J = 2.3 Hz, 1H), 6.83 (d, J = 2.2 Hz, 1H), 5.26 (s, 2H), 5.20 (s, 2H), 3.51 (s, 3H), 3.48 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 189.1, 161.9, 161.2, 126.3, 118.3, 115.3, 102.9, 95.0, 94.3, 56.7, 56.5. Mp: 60–64 ºC.
To a solution of 2-bromo-4,6-bis(methoxymethoxy)benzaldehyde (350 mg, 1.15 mmol) in tBuOH (3.6 mL) and THF (1.3 mL) at room temperature was added a solution of sodium chlorite (80%, 260 mg, 2.29 mmol) and sodium phosphate monobasic monohydrate (791 mg, 5.74 mmol) in water (1.9 mL) dropwise. To the yellow solution was added 2-methyl-2-butene (90%, 1.08 mL, 9.18 mmol). After 25 minutes, the orange solution became faint yellow/colorless and was diluted with ethyl acetate. The layers were separated, and the organic layer was washed three times with saturated NH4Cl (aq.). The organic layer was dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude carboxylic acid 12 (364 mg, 99% crude yield) was used in the next step without further purification.
(2-bromo-4,6-bis(methoxymethoxy)phenyl)(isoindolin-2-yl)methanone (13).
To a suspension of benzoic acid 12 (320 mg, 0.997 mmol) and isoindoline hydrochloride (156 mg, 1.49 mmol) in THF (2.9 mL) and CH2Cl2 (2.9 mL) at room temperature was added trimethylamine (0.420 mL, 2.99 mmol) followed by HATU (451 mg, 1.20 mmol). After stirring the suspension was stirred at room temperature, the reaction mixture was diluted with CH2Cl2. The resulting mixture was washed with saturated NaHCO3 (aq.), brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (20% to 50% ethyl acetate in hexanes) to afford 266 mg of 13 as a white solid (63% yield). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.27 (m, 3H), 7.19 – 7.14 (m, 1H), 6.99 (d, J = 2.1 Hz, 1H), 6.85 (d, J = 2.1 Hz, 1H), 5.21 – 5.10 (m, 4H), 5.07 – 4.94 (m, 2H), 4.68 – 4.48 (m, 2H), 3.49 (s, 3H), 3.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 165.6, 158.8, 154.8, 136.3, 136.2, 127.7, 127.5, 123.1, 123.0, 122.5, 119.8, 113.3, 103.1, 94.9, 94.4, 56.4, 56.2, 53.1, 51.7. Mp: 102–104 ˚C. LC/MS (m/z): 422.128 and 424.133 [M+H+]; UPLC tR 1.97 min.
Isoindolin-2-yl(2-((1-(4-methoxybenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (14a).
Synthesized using General Procedure D1 from 13 (45 mg, 110 μmol) and 10a (25 mg, 170 μmol). Following silica gel flash chromatography (12% to 40% acetone in hexanes), TMT (18 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 52 mg of 14a (87% yield). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.24 (m, 3H), 7.16 (d, J = 7.1 Hz, 1H), 7.06 (d, J = 8.6 Hz, 2H), 6.64 (d, J = 8.7 Hz, 2H), 6.44 – 6.35 (m, 2H), 6.22 (d, J = 2.1 Hz, 1H), 5.85 (s, 1H), 5.15 (q, J = 6.7 Hz, 2H), 5.07 (d, J = 1.3 Hz, 2H), 5.03 (s, 2H), 4.99 – 4.41 (m, 4H), 3.66 (s, 3H), 3.45 (s, 2H), 3.44 (s, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 159.7, 158.9, 155.3, 147.5, 143.6, 139.3, 136.5, 136.1, 128.6, 128.6, 128.6, 127.7, 127.6, 122.9, 122.5, 113.8, 107.7, 98.5, 96.4, 95.3, 95.0, 94.2, 56.5, 56.2, 55.1, 52.9, 52.0, 51.2, 14.1. LC/MS (m/z): 559.299 [M+H+]; UPLC tR 1.66 min.
Isoindolin-2-yl(2-((1-(4-methoxybenzyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (14b).
Synthesized using General Procedure D1 from 13 (40 mg, 95 μmol) and 10b (25 mg, 170 μmol). Following silica gel flash chromatography (5% to 30% acetone in hexanes), TMT (18 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 52.6 mg of 14b (89% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 – 7.76 (m, 2H), 7.43 – 7.23 (m, 5H), 7.18 (d, J = 7.2 Hz, 1H), 7.16 – 7.09 (m, 2H), 6.70 – 6.61 (m, 2H), 6.48 (s, 1H), 6.42 (d, J = 2.1 Hz, 1H), 6.39 (s, 1H), 6.29 (d, J = 2.1 Hz, 1H), 5.16 (d, J = 7.2 Hz, 4H), 5.06 (s, 2H), 5.01 – 4.49 (m, 4H), 3.67 (s, 3H), 3.45 (s, 3H), 3.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 159.8, 159.0, 155.3, 150.0, 143.5, 140.0, 136.5, 136.1, 133.7, 128.7, 128.5, 128.3, 127.7, 127.6, 127.5, 125.3, 122.9, 122.5, 113.8, 107.8, 96.5, 96.3, 95.3, 95.2, 94.2, 56.5, 56.2, 55.1, 52.9, 52.0, 51.8. LC/MS (m/z): 621.311 [M+H+]; UPLC tR 2.03 min.
Isoindolin-2-yl(2-((1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (14c).
Synthesized using General Procedure D2 from 13 (45 mg, 110 μmol) and 1-[(4-methoxyphenyl)methyl]-1H-pyrazol-5-amine (24 mg, 170 μmol). Following silica gel flash chromatography (10% to 45% ethyl acetate in hexanes), TMT (22 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 66 mg of 14c (108% yield). 1H NMR (400 MHz, CDCl3) δ 7.31 (ddd, J = 16.1, 7.2, 2.0 Hz, 3H), 7.17 (d, J = 7.3 Hz, 1H), 7.09 (d, J = 8.7 Hz, 2H), 6.66 (d, J = 8.7 Hz, 2H), 6.45 (s, 1H), 6.40 (d, J = 2.2 Hz, 1H), 6.19 (d, J = 2.1 Hz, 1H), 6.05 (d, J = 2.0 Hz, 1H), 5.16 (dd, J = 9.7, 5.0 Hz, 2H), 5.11 (s, 2H), 5.07 – 5.01 (m, 2H), 4.99 – 4.79 (m, 3H), 4.56 (d, J = 14.6 Hz, 1H), 3.68 (s, 3H), 3.48 – 3.39 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 166.9, 159.8, 159.0, 155.4, 143.8, 138.9, 138.8, 136.6, 136.2, 128.9, 128.4, 128.3, 127.7, 127.6, 123.0, 122.5, 114.3, 113.9, 107.7, 99.2, 96.2, 95.3, 95.3, 94.3, 56.5, 56.2, 55.1, 53.0, 52.1, 51.6. LC/MS (m/z): 545.185 [M+H+]; UPLC tR 1.67 min.
Isoindolin-2-yl(2-((1-(4-methoxybenzyl)-4-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (14d).
Synthesized using General Procedure D2 from 13 45 mg, 110 μmol) and 10c (25 mg, 120 μmol). Following silica gel flash chromatography (10% to 40% acetone in hexanes), TMT (17 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 49 mg of 14d (82% yield). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.28 (m, 4H), 7.20 (d, J = 7.2 Hz, 1H), 7.07 – 7.01 (m, 2H), 6.66 – 6.54 (m, 2H), 6.36 (d, J = 2.1 Hz, 1H), 6.09 (s, 1H), 5.71 (d, J = 2.1 Hz, 1H), 5.16 (s, 2H), 5.10 – 4.81 (m, 7H), 4.61 (d, J = 14.7 Hz, 1H), 3.68 (s, 3H), 3.46 (s, 3H), 3.42 (s, 3H), 1.86 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.0, 159.9, 158.9, 155.4, 144.6, 139.2, 136.6, 136.3, 135.5, 128.9, 128.9, 127.8, 127.7, 123.1, 122.6, 113.8, 111.5, 107.1, 95.5, 95.3, 94.5, 94.2, 56.6, 56.2, 55.1, 53.1, 52.1, 51.8, 8.3. LC/MS (m/z): 559.166 [M+H+]; UPLC tR 1.81 min.
Isoindolin-2-yl(2-((4-isopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (14e).
Synthesized using General Procedure D2 from 13 (45 mg, 110 μmol) and 10d (29 mg, 120 μmol). Following silica gel flash chromatography (10% to 30% acetone in hexanes), TMT (15 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 54 mg of 14e (86% yield). 1H NMR (400 MHz, CDCl3) δ 7.41 (s, 1H), 7.39 – 7.27 (m, 3H), 7.21 (d, J = 7.2 Hz, 1H), 7.02 (d, J = 8.1 Hz, 2H), 6.58 (d, J = 8.1 Hz, 2H), 6.35 (d, J = 2.2 Hz, 1H), 6.13 (s, 1H), 5.65 (d, J = 2.1 Hz, 1H), 5.17 (s, 2H), 5.11 – 4.83 (m, 7H), 4.62 (d, J = 14.7 Hz, 1H), 3.66 (s, 3H), 3.46 (s, 3H), 3.39 (s, 3H), 2.67 (p, J = 6.9 Hz, 1H), 1.13 (d, J = 6.9 Hz, 3H), 1.08 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.1, 159.9, 158.9, 155.5, 145.4, 136.6, 136.6, 136.3, 133.9, 129.0, 128.9, 127.7, 127.7, 123.7, 123.1, 122.6, 113.7, 106.9, 95.5, 95.3, 94.5, 94.2, 56.6, 56.1, 55.1, 53.1, 52.1, 51.6, 23.7, 23.6, 23.3. LC/MS (m/z): 587.262 [M+H+]; UPLC tR 1.96 min.
Isoindolin-2-yl(2-((1-(4-methoxybenzyl)-4-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (14f).
Synthesized using General Procedure D1 from 13 (40 mg, 95 μmol) and 10e (29 mg, 100 μmol) for 4 h. Following silica gel flash chromatography (5% to 35% acetone in hexanes), TMT (13 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 25 mg of 14f (43% yield). 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H), 7.41 (dt, J = 6.2, 1.3 Hz, 2H), 7.38 – 7.26 (m, 3H), 7.22 – 7.02 (m, 6H), 6.65 (d, J = 8.3 Hz, 2H), 6.47 (s, 1H), 6.36 (d, J = 2.1 Hz, 1H), 5.72 (d, J = 2.1 Hz, 1H), 5.24 – 4.94 (m, 6H), 4.93 – 4.81 (m, 3H), 4.58 (d, J = 14.7 Hz, 1H), 3.68 (s, 3H), 3.47 (s, 3H), 3.34 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.0, 159.9, 159.1, 155.5, 144.3, 137.7, 136.6, 136.2, 134.3, 132.1, 129.2, 128.5, 128.4, 127.8, 127.7, 126.3, 126.1, 123.1, 122.5, 116.7, 113.9, 107.3, 95.8, 95.4, 94.8, 94.2, 56.6, 56.2, 55.2, 53.0, 52.2, 51.9. LC/MS (m/z): 621.311 [M+H+]; UPLC tR 1.92 min
(2-((4-Benzyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)(isoindolin-2-yl)methanone (14g).
Synthesized using General Procedure D1 from 13 (40 mg, 95 μmol) and 10f (31 mg, 100 μmol) for 4 h. Following silica gel flash chromatography (10% to 35% acetone in hexanes), TMT (17 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 45 mg of 14g (75% yield). 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.27 (m, 4H), 7.23 – 6.96 (m, 7H), 6.68 – 6.56 (m, 2H), 6.34 (d, J = 2.1 Hz, 1H), 6.19 (s, 1H), 5.65 (d, J = 2.1 Hz, 1H), 5.16 (d, J = 2.4 Hz, 2H), 5.11 – 4.82 (m, 7H), 4.53 (d, J = 14.7 Hz, 1H), 3.69 (s, 3H), 3.61 (d, J = 3.5 Hz, 2H), 3.46 (s, 3H), 3.41 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.0, 159.8, 159.0, 155.4, 144.8, 140.2, 139.0, 136.6, 136.3, 135.4, 129.1, 128.7, 128.4, 128.2, 127.7, 127.6, 125.9, 123.0, 122.6, 115.6, 113.8, 106.9, 95.4, 95.3, 94.7, 94.1, 56.6, 56.2, 55.2, 53.0, 52.1, 51.8, 29.8. LC/MS (m/z): 635.292 [M+H+]; UPLC tR 1.96 min.
(2-((3-(tert-Butyl)-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)(isoindolin-2-yl)methanone (14h)
Synthesized using General Procedure D1 from 13 (40 mg, 95 μmol) and 10i (27 mg, 100 μmol) for 4 h. Following silica gel flash chromatography (5% to 25% acetone in hexanes), TMT (18 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 45 mg of 14h (79% yield). 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.27 (m, 3H), 7.16 (d, J = 7.2 Hz, 1H), 7.03 (d, J = 8.5 Hz, 2H), 6.64 (d, J = 8.6 Hz, 2H), 6.38 (d, J = 2.1 Hz, 1H), 6.33 – 6.23 (m, 2H), 5.92 (s, 1H), 5.14 (q, J = 6.5 Hz, 2H), 5.08 (s, 2H), 5.05 (s, 2H), 4.91 (d, J = 14.8 Hz, 1H), 4.84 (s, 2H), 4.51 (d, J = 14.6 Hz, 1H), 3.66 (s, 3H), 3.45 (s, 3H), 3.44 (s, 3H), 1.30 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 166.8, 160.8, 159.8, 158.8, 155.3, 143.7, 138.8, 136.6, 136.2, 128.9, 128.4, 127.7, 127.6, 123.0, 122.5, 113.8, 107.8, 96.5, 95.3, 95.2, 95.1, 94.4, 56.5, 56.2, 55.1, 53.0, 52.0, 51.4, 32.3, 30.5. LC/MS (m/z): 601.331 [M+H+]; UPLC tR 2.04 min.
(2,4-Bis(methoxymethoxy)-6-((1-methyl-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (14i).
Synthesized using General Procedure D2 from 13 (45 mg, 110 μmol) and 1-methyl-1H-pyrazol-5-amine (11 mg, 120 μmol). Following silica gel flash chromatography (30% to 70% ethyl acetate in CH2Cl2), TMT (19 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 38 mg of 14i (82% yield). 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 2.0 Hz, 1H), 7.38 – 7.27 (m, 3H), 7.19 (d, J = 6.9 Hz, 1H), 6.64 (s, 1H), 6.43 (d, J = 2.2 Hz, 1H), 6.08 (d, J = 2.1 Hz, 1H), 6.01 (d, J = 2.0 Hz, 1H), 5.21 – 5.10 (m, 3H), 5.08 (s, 2H), 5.05 – 4.84 (m, 2H), 4.63 (d, J = 14.7 Hz, 1H), 3.68 (s, 3H), 3.45 (s, 3H), 3.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.1, 159.9, 155.5, 144.4, 139.3, 138.6, 136.6, 136.1, 127.8, 127.6, 123.1, 122.5, 107.6, 99.1, 96.2, 95.4, 95.2, 94.2, 56.6, 56.3, 53.1, 52.2, 35.0. LC/MS (m/z): 439.33 [M+H+]; UPLC tR 1.48 min.
Isoindolin-2-yl(2-((1-(4-isopropylbenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (14j).
Synthesized using General Procedure D2 from 13 (40 mg, 95 μmol) and 1-([4-(propan-2-yl)phenyl]methyl)-1H-pyrazol-5-amine (22 mg, 100 μmol). Following silica gel flash chromatography (10% to 30% acetone in hexanes), TMT (15 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 42 mg of 14j (80% yield). 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 2.0 Hz, 1H), 7.35 – 7.28 (m, 3H), 7.18 (d, J = 7.3 Hz, 1H), 7.06 (d, J = 8.1 Hz, 2H), 6.98 (d, J = 8.2 Hz, 2H), 6.43 (s, 1H), 6.40 (d, J = 2.1 Hz, 1H), 6.20 (d, J = 2.1 Hz, 1H), 6.06 (d, J = 2.0 Hz, 1H), 5.23 – 5.07 (m, 5H), 5.07 – 5.00 (m, 2H), 5.00 – 4.76 (m, 3H), 4.57 (d, J = 14.7 Hz, 1H), 3.45 (s, 6H), 2.76 (p, J = 6.9 Hz, 1H), 1.14 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 166.9, 159.8, 155.4, 148.2, 143.9, 139.0, 138.9, 136.6, 136.2, 133.6, 127.7, 127.6, 127.5, 127.5, 126.7, 123.0, 122.5, 107.7, 99.4, 96.2, 95.3, 95.2, 94.3, 56.6, 56.3, 53.0, 52.1, 51.9, 33.7, 23.9, 23.8. LC/MS (m/z): 557.27 [M+H+]; UPLC tR 1.93 min
(2,4-Bis(methoxymethoxy)-6-((3-methyl-1-(4-methylbenzyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (14k).
Synthesized using General Procedure D1 from 13 (45 mg, 110 μmol) and 10m (24 mg, 120 μmol) and heated for 3 h. Following silica gel flash chromatography (10% to 35% acetone in hexanes), TMT (15 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 40 mg of 14k (70% yield). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.27 (m, 3H), 7.16 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 7.8 Hz, 2H), 6.88 (d, J = 7.8 Hz, 2H), 6.39 (d, J = 2.1 Hz, 1H), 6.32 (s, 1H), 6.24 (d, J = 2.1 Hz, 1H), 5.86 (s, 1H), 5.14 (q, J = 6.5 Hz, 2H), 5.07 (d, J = 1.3 Hz, 2H), 5.06 (s, 2H), 4.90 (d, J = 14.7 Hz, 1H), 4.83 (s, 2H), 4.48 (d, J = 14.6 Hz, 1H), 3.46 (s, 3H), 3.43 (s, 3H), 2.24 (s, 3H), 2.17 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 159.8, 155.3, 147.6, 143.7, 139.4, 137.0, 136.6, 136.2, 133.6, 129.2, 127.7, 127.6, 127.0, 122.9, 122.5, 107.8, 98.6, 96.4, 95.3, 95.1, 94.3, 56.5, 56.3, 52.9, 52.0, 51.7, 21.0, 14.2. LC/MS (m/z): 543.332 [M+H+]; UPLC tR 1.76 min.
(2-((1-(2-Chlorobenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)(isoindolin-2-yl)methanone (14l).
Inside a nitrogen glovebox were combined aryl bromide 13 (45 mg, 110 μmol), amine 10o (26 mg, 170 μmol), tris(dibenzylideneacetone)dipalladium (3.9 mg, 4.3 μmol), Xantphos (6.2 mg, 11 μmol), and sodium phenoxide (19 mg, 160 μmol). Dioxane (0.8 mL) was added to the mixture and the reaction vessel was capped and removed from the glovebox. The reaction vessel was heated at 60 ºC for 90 min, 90 ºC for 90 min, and then 120 ºC for 2.5 h. After cooling to room temperature, the reaction was diluted with ethyl acetate. The resulting mixture was washed three times with saturated Na2CO3 (aq.), brine, then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. Following silica gel flash chromatography (10% to 30% acetone in hexanes), TMT (20 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 47 mg of 14l (78% yield). 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.27 (m, 3H), 7.16 (s, 1H), 7.09 – 6.93 (m, 3H), 6.72 – 6.62 (m, 1H), 6.39 (d, J = 2.1 Hz, 1H), 6.37 (s, 1H), 6.20 (d, J = 2.1 Hz, 1H), 5.93 (s, 1H), 5.28 – 5.01 (m, 6H), 4.86 (d, J = 14.5 Hz, 1H), 4.77 (s, 2H), 4.38 (d, J = 14.6 Hz, 1H), 3.46 (s, 3H), 3.42 (s, 3H), 2.26 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 159.8, 155.4, 148.4, 143.8, 140.1, 136.6, 136.1, 134.6, 131.8, 129.1, 128.4, 127.7, 127.6, 127.5, 127.0, 122.9, 122.6, 107.9, 99.4, 96.5, 95.3, 95.2, 94.3, 56.5, 56.3, 52.9, 52.0, 49.1, 14.3. LC/MS (m/z): 563.224 [M+H+]; UPLC tR 1.73 min.
(2,4-Bis(methoxymethoxy)-6-((3-methyl-1-(2-methylbenzyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (14m).
Inside a nitrogen glovebox were combined aryl bromide 13 (45 mg, 110 μmol), amine 10n (28 mg, 140 μmol), tBuXPhos Palladacycle Gen. 1 (7.3 mg, 11 μmol), tBuXphos (4.5 mg, 11 μmol), sodium tert-butoxide (22 mg, 220 μmol). tert-Butanol (0.8 mL) was added to the mixture and the reaction vessel was capped and removed from the glovebox. After stirring at room temperature for 2.5 h, the reaction was quenched with saturated NH4Cl (aq.). The resulting mixture was extracted four times with ethyl acetate. The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. Following silica gel flash chromatography (40% to 80% ethyl acetate in hexanes), TMT (21 mg) was added to the isolated residue; the mixture was suspended in toluene (1.5 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 25 mg of 14m (52% yield). 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.28 (m, 3H), 7.21 – 7.09 (m, 1H), 7.03 – 6.88 (m, 3H), 6.65 (d, J = 7.6 Hz, 1H), 6.39 (d, J = 2.1 Hz, 1H), 6.33 – 6.26 (m, 2H), 5.91 (s, 1H), 5.16 – 5.03 (m, 7H), 4.85 (d, J = 14.7 Hz, 1H), 4.73 (d, J = 9.2 Hz, 2H), 4.38 (d, J = 14.7 Hz, 1H), 3.46 (s, 3H), 3.42 (s, 3H), 2.25 (s, 3H), 2.18 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.7, 159.7, 155.3, 147.8, 143.6, 140.1, 136.5, 136.2, 135.1, 134.8, 130.2, 127.7, 127.5, 127.2, 126.4, 126.2, 123.0, 122.5, 107.9, 98.3, 96.5, 95.3, 95.2, 94.3, 56.5, 56.3, 52.8, 51.9, 49.6, 19.0, 14.3. LC/MS (m/z): 543.288 [M+H+]; UPLC tR 1.75 min.
(2,4-Bis(methoxymethoxy)-6-((1-methyl-3-phenyl-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (14n).
Synthesized using General Procedure D2 from 13 (60 mg, 140 μmol) and 10p (27 mg, 160 μmol) and purified via automated flash chromatography (30% to 80% ethyl acetate in CH2Cl2). To QuadraPure™ MPA resin (1.5 mmol/g loading, 68 mg) soaked in CH2Cl2 (1 mL) for 90 min was transferred the purified product using CH2Cl2 (3 mL) and shaken overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 65 mg of 14n (89% yield). 1H NMR (400 MHz, CDCl3) δ 7.79 – 7.70 (m, 2H), 7.41 – 7.27 (m, 6H), 7.20 (d, J = 7.1 Hz, 1H), 6.72 (s, 1H), 6.45 (d, J = 2.1 Hz, 1H), 6.34 (s, 1H), 6.18 (d, J = 2.1 Hz, 1H), 5.24 – 5.11 (m, 3H), 5.09 (s, 2H), 5.06 – 4.86 (m, 2H), 4.65 (d, J = 14.7 Hz, 1H), 3.73 (s, 3H), 3.46 (d, J = 0.8 Hz, 3H), 3.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.1, 160.0, 155.6, 150.0, 144.3, 140.5, 136.6, 136.2, 133.6, 128.6, 128.4, 127.8, 127.6, 127.6, 127.4, 125.2, 123.1, 122.5, 107.7, 96.5, 96.4, 95.4, 95.2, 94.3, 56.6, 56.3, 53.1, 52.3, 35.2. LC/MS (m/z): 516.34 [M+H+]; UPLC tR 1.88 min.
(2-Bromo-4,6-bis(methoxymethoxy)phenyl)(5-((1-methylpiperidin-4-yl)amino)isoindolin-2-yl)methanone (15a).
Inside a glovebox under a nitrogen atmosphere were combined tert-butyl 5-bromoisoindoline-2-carboxylate (85 mg, 0.29 mmol), tris(dibenzylideneacetone)dipalladium (13 mg, 0.014 mmol), Johnphos (8.5 mg, 0.029 mmol) and sodium tert-butoxide (38 mg, 0.40 mmol) and suspended in toluene (4 mL). 4-Amino-1-methylpiperidine (39 mg mL, 0.34 mmol) was added to the mixture and the reaction vessel was sealed and removed from the glovebox. The reaction mixture was irradiated at 120 ºC for 30 minutes in a microwave reactor. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate, washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via silica gel flash chromatography (95:5:1 CH2Cl2:methanol:concentrated NH4OH (aq.)) to afford 47 mg of tert-butyl 5-((1-methylpiperidin-4-yl)amino)isoindoline-2-carboxylate (50% yield). 1H NMR (400 MHz, CDCl3) δ 7.01 (dd, J = 20.7, 8.1 Hz, 1H), 6.63 – 6.40 (m, 2H), 4.65 – 4.42 (m, 4H), 3.33 – 3.17 (m, 1H), 2.82 (d, J = 11.3 Hz, 2H), 2.31 (s, 3H), 2.22 – 2.02 (m, 4H), 1.50 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.5, 154.4, 146.6, 146.6, 138.4, 138.0, 125.6, 125.3, 123.3, 123.0, 113.1, 106.6, 106.5, 79.2, 54.4, 52.3, 52.0, 51.6, 51.3, 46.1, 32.3, 28.4. LC/MS (m/z): 332.193 [M+H+]; UPLC tR 1.19 min
To a solution of tert-butyl 5-((1-methylpiperidin-4-yl)amino)isoindoline-2-carboxylate (47 mg, 0.14 mmol) from above in CH2Cl2 (0.28 mL) at room temperature was added HCl (4 M in dioxane, 0.45 mL, 1.8 mmol) and stirred overnight. The reaction was then triturated with ether. The suspension was filter and the resulting solid washed with ether to afford 29 mg of N-(1-methylpiperidin-4-yl)isoindolin-5-amine dihydrogenchloride as a viscous gum (66% based on crude mass).
Benzoic acid 12 (68 mg, 0.21 mmol), N-(1-methylpiperidin-4-yl)isoindolin-5-amine dihydrogenchloride salt (64 mg, 0.21 mmol) from above, trimethylamine (0.12 mL, 0.84 mmol) and HATU (95 mg, 0.25 mmol) were reacted using the same procedure for the synthesis of amide 13 to afford 57 mg of 15a (51% yield) after purification via automated flash chromatography (1% to 10% methanol in CH2Cl2). 1H NMR (400 MHz, CD3OD) δ 7.15 – 6.87 (m, 3H), 6.71 – 6.38 (m, 2H), 5.32 – 5.13 (m, 4H), 4.89 (s, 3H), 4.86 – 4.74 (m, 2H), 4.45 (d, J = 14.7 Hz, 2H), 3.48 (d, J = 1.2 Hz, 2H), 3.42 – 3.38 (m, 3H), 3.35 (s, 1H), 3.02 (t, J = 13.6 Hz, 2H), 2.44 (d, J = 11.0 Hz, 4H), 2.17 – 1.97 (m, 2H), 1.69 – 1.42 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 166.7, 166.7, 159.3, 159.3, 155.0, 147.8, 147.6, 136.7, 136.5, 123.5, 123.4, 123.1, 122.9, 122.2, 122.1, 119.2, 113.9, 113.7, 112.9, 112.8, 106.4, 106.3, 102.8, 102.8, 94.8, 94.7, 94.3, 55.4, 55.2, 53.7, 53.3, 52.7, 51.6, 51.0, 44.1, 30.7. LC/MS (m/z): 534.114 and 536.099 [M+H+]; UPLC tR 1.31 min.
(2-Bromo-4,6-bis(methoxymethoxy)phenyl)(5-(2-(dimethylamino)ethoxy)isoindolin-2-yl)methanone (15b).
A suspension of tert-butyl 5-hydroxyisoindoline-2-carboxylate (250 mg, 1.06 mmol), 2-chloro-N,N-dimethylethylamine hydrochloride (367 mg, 2.55 mmol), and cesium carbonate (1.73 g, 5.31 mmol) in MeCN (4 mL) was heated overnight at 90 ºC. The mixture was cooled to room temperature and diluted with 15% methanol in CH2Cl2. The resulting mixture was washed twice with water and once with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (5% to 10% methanol in CH2Cl2) to afford 165 mg of tert-butyl 5-(2-(dimethylamino)ethoxy)isoindoline-2-carboxylate (51% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (dd, J = 20.9, 8.3 Hz, 1H), 6.88 – 6.72 (m, 2H), 4.60 (t, J = 15.2 Hz, 4H), 4.05 (td, J = 5.7, 2.0 Hz, 2H), 2.74 (t, J = 5.7 Hz, 2H), 2.35 (s, 6H), 1.51 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 158.4, 158.4, 154.3, 154.3, 138.5, 138.1, 129.2, 128.8, 123.2, 123.0, 114.3, 114.0, 108.3, 108.2, 79.4, 66.0, 58.1, 52.2, 52.0, 51.6, 51.2, 45.7, 28.4. LC/MS (m/z): 307.139 [M+H+]; UPLC tR 1.18 min.
To a solution of tert-butyl 5-(2-(dimethylamino)ethoxy)isoindoline-2-carboxylate from above (135 mg, 0.441 mmol) in CH2Cl2 (0.88 mL) at room temperature was added HCl (4 M in dioxane, 1.4 mL, 5.6 mmol) and stirred overnight. The reaction was then triturated with ether. The suspension was filter and the resulting solid washed with ether to afford 103 mg 2-(Isoindolin-5-yloxy)-N,N-dimethylethan-1-amine dihydrochloride as a solid (84% based on crude mass).
Benzoic acid 12 (38 mg, 0.12 mmol), 2-(isoindolin-5-yloxy)-N,N-dimethylethan-1-amine dihydrochloride salt from above (33 mg, 0.12 mmol), trimethylamine (0.066 mL, 0.47 mmol) and HATU (54 mg, 0.14 mmol) were reacted using the same procedure for the synthesis of amide 13 to afford 60 mg of 15b (75% yield) after purification via automated flash chromatography (1% to 8% methanol in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.14 (dd, J = 75.4, 8.4 Hz, 1H), 6.98 (t, J = 2.0 Hz, 1H), 6.94 – 6.66 (m, 3H), 5.25 – 5.05 (m, 4H), 4.92 (dd, J = 7.9, 3.4 Hz, 2H), 4.50 (q, J = 13.3 Hz, 2H), 4.09 (dt, J = 20.4, 5.5 Hz, 2H), 3.57 – 3.45 (m, 3H), 3.41 (s, 3H), 3.18 (q, J = 7.3 Hz, 4H), 2.87 (dt, J = 16.3, 5.6 Hz, 2H), 2.46 (s, 3H), 2.42 (s, 3H), 1.34 (t, J = 7.3 Hz, 4H). LC/MS (m/z): 509.062 and 511.047 [M+H+]; UPLC tR 1.29 min.
(2-Bromo-4,6-bis(methoxymethoxy)phenyl)(5-(4-methylpiperazin-1-yl)isoindolin-2-yl)methanone (15c).
Procedure adapted from 66. Inside a glovebox under a nitrogen atmosphere were combined tert-butyl 5-bromoisoindoline-2-carboxylate (300 mg, 1.01 mmol), tris(dibenzylideneacetone)dipalladium (46.1 mg, 0.0503 mmol), Xantphos (29.1 mg, 0.0503 mmol) and sodium tert-butoxide (145 mg, 1.51 mmol) and suspended in toluene (3 mL). 1-Methylpiperazine (0.134 mL, 1.21 mmol) was added to the mixture and the reaction vessel was sealed and removed from the glovebox. After heating at 100 ºC overnight, the reaction mixture was cooled to room temperature and diluted with ethyl acetate. The organic mixture was washed with brine and dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (1% to 7% methanol in CH2Cl2) to afford 259 mg of tert-butyl 5-(4-methylpiperazin-1-yl)isoindoline-2-carboxylate (81% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (dd, J = 20.8, 8.3 Hz, 1H), 6.92 – 6.75 (m, 2H), 4.70 – 4.46 (m, 4H), 3.19 (d, J = 5.1 Hz, 4H), 2.58 (t, J = 5.0 Hz, 4H), 2.35 (s, 3H), 1.51 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 154.3, 154.3, 151.0, 138.1, 137.7, 128.2, 127.9, 122.9, 122.7, 115.8, 115.6, 110.0, 109.8, 79.2, 54.8, 52.3, 52.0, 51.6, 51.3, 49.4, 49.3, 45.9, 28.3. LC/MS (m/z): 318.167 [M+H+]; UPLC tR 1.04 min.
To a solution of tert-butyl 5-(4-methylpiperazin-1-yl)isoindoline-2-carboxylate from above (259 mg, 0.816 mmol) in CH2Cl2 (1.6 mL) at room temperature was added HCl (4 M in dioxane, 2.59 mL, 10.4 mmol) and stirred overnight. The reaction was then triturated with ether. The suspension was filter and the resulting solid washed with ether to afford 262 mg of crude 5-(4-methylpiperazin-1-yl)isoindoline dihydrochloride salt (110% crude yield).
Benzoic acid 12 (133 mg, 0.374 mmol), 5-(4-methylpiperazin-1-yl)isoindoline dihydrochloride salt from above (106 mg, 0.365 mmol), trimethylamine (0.208 mL, 1.49 mmol) and HATU (169 mg, 0.448 mmol) were reacted using the same procedure for the synthesis of amide 13 to afford 132 mg of 15c (68% yield) after purification via automated flash chromatography (1% to 8% methanol in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 8.6 Hz, 1H), 7.04 (d, J = 8.3 Hz, 1H), 6.98 (dd, J = 2.1, 1.5 Hz, 1H), 6.96 – 6.82 (m, 3H), 5.30 (s, 1H), 5.23 – 5.07 (m, 4H), 4.93 (d, J = 13.0 Hz, 2H), 4.61 – 4.39 (m, 2H), 3.49 (d, J = 0.9 Hz, 3H), 3.41 (d, J = 2.0 Hz, 3H), 3.27 – 3.12 (m, 5H), 2.64 (dt, J = 9.5, 4.7 Hz, 4H), 2.40 (d, J = 7.0 Hz, 3H), 1.39 (t, J = 7.3 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 165.7, 165.6, 158.8, 154.8, 151.3, 151.2, 137.2, 137.2, 127.1, 127.1, 123.5, 123.0, 122.7, 119.7, 116.5, 116.0, 113.2, 113.2, 110.4, 109.8, 103.1, 103.0, 94.9, 94.8, 94.4, 77.2, 56.4, 56.2, 54.8, 53.4, 52.7, 51.9, 51.2, 49.1, 49.1, 47.0, 47.0, 45.7, 8.7. LC/MS (m/z): 520.089 and 522.073 [M+H+]; UPLC tR 1.25 min.
N-benzyl-2-bromo-4,6-bis(methoxymethoxy)-N-methylbenzamide (15d).
The product was synthesized using the same procedure for the synthesis of amide 13. The reaction with benzoic acid 12 (0.68 g, 2.1 mmol), N-benzylmethylamine (0.41 mL, 3.2 mmol), trimethylamine (0.59 mL, 4.2 mmol) and HATU (0.96 g, 2.5 mmol) to afford 0.55 g of 15d as a colorless oil (61% yield) after purification via automated flash chromatography (20% to 40% ethyl acetate in hexanes and 5% to 10% ethyl acetate in CH2Cl2). Isomer 1: 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.5 Hz, 1H), 7.32 (dt, J = 20.9, 8.2 Hz, 4H), 6.96 (d, J = 2.1 Hz, 1H), 6.79 (d, J = 2.1 Hz, 1H), 5.29 – 5.08 (m, 6H), 5.03 – 4.59 (m, 2H), 3.47 (s, 3H), 3.43 (s, 3H), 2.75 (s, 3H). Isomer 2: 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.5 Hz, 1H), 7.32 (dt, J = 20.9, 8.2 Hz, 4H), 6.96 (d, J = 2.1 Hz, 1H), 6.82 (d, J = 2.2 Hz, 1H), 5.29 – 5.08 (m, 5H), 4.52 – 4.25 (m, 2H), 3.45 (s, 3H), 3.43 (s, 3H), 3.01 (s, 3H). LC/MS (m/z): 424.069 and 426.010 [M+H+]; UPLC tR 1.69 min.
(2-((1-(4-Methoxybenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)(5-((1-methylpiperidin-4-yl)amino)isoindolin-2-yl)methanone (16a).
Synthesized using General Procedure D2 from 15a (56.9 mg, 106 μmol) and 10a (25.4 mg, 117 μmol) in dioxane. The crude mixture was purified via automated flash chromatography (2% to 10% methanol in CH2Cl2). To QuadraPure™ MPA resin (1.5 mmol/g loading, 45 mg) soaked in CH2Cl2 (2 mL) for 30 min was transferred the purified product using CH2Cl2 (3 mL) and shaken overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 41.8 mg of 16a (59% yield). 1H NMR (400 MHz, CDCl3) δ 7.14 – 6.91 (m, 3H), 6.66 (dd, J = 8.6, 1.5 Hz, 2H), 6.53 (q, J = 8.9, 8.2 Hz, 2H), 6.41 – 6.16 (m, 3H), 5.84 (s, 1H), 5.23 – 5.09 (m, 2H), 5.09 – 4.97 (m, 4H), 4.88 – 4.67 (m, 3H), 4.43 (t, J = 13.1 Hz, 1H), 3.68 (d, J = 1.5 Hz, 3H), 3.51 – 3.39 (m, 6H), 3.26 (d, J = 28.2 Hz, 1H), 2.83 (d, J = 13.5 Hz, 2H), 2.30 (d, J = 8.8 Hz, 4H), 2.24 (s, 3H), 2.18 – 1.95 (m, 4H), 1.49 (d, J = 11.8 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 166.8, 166.7, 159.7, 158.9, 155.3, 147.6, 147.6, 147.1, 146.9, 143.7, 139.3, 137.8, 137.5, 128.7, 128.7, 128.7, 124.8, 124.5, 123.6, 123.2, 113.9, 113.9, 113.5, 113.4, 108.0, 107.9, 106.8, 106.3, 98.7, 98.6, 96.4, 96.4, 95.3, 95.3, 95.1, 95.0, 94.3, 56.5, 56.5, 56.2, 55.1, 55.1, 54.5, 53.2, 52.6, 52.2, 51.6, 51.3, 49.6, 46.2, 46.2, 32.4, 14.2. LC/MS (m/z): 671.282 [M+H+]; UPLC tR 1.45 min.
(5-(2-(Dimethylamino)ethoxy)isoindolin-2-yl)(2-((1-(4-methoxybenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)methanone (16b).
Synthesized using General Procedure D2 from 15b (41.9 mg, 82.3 μmol) and 10a (19.7 mg, 90.5 μmol) in dioxane (0.8 mL). The crude mixture was purified via automated flash chromatography (1% to 8% methanol in CH2Cl2). To QuadraPure™ MPA resin (1.5 mmol/g loading, 34 mg) soaked in CH2Cl2 (2 mL) for 30 min was transferred the purified product using CH2Cl2 (3 mL) and shaken overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated in vacuo to afford 37.7 mg of 16b (71% yield). 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.4 Hz, 1H), 7.05 (t, J = 8.8 Hz, 2H), 6.90 – 6.79 (m, 2H), 6.72 – 6.59 (m, 2H), 6.37 (dd, J = 7.7, 3.6 Hz, 2H), 6.22 (d, J = 2.4 Hz, 1H), 5.84 (s, 1H), 5.14 (q, J = 7.1 Hz, 2H), 5.04 (d, J = 12.2 Hz, 4H), 4.92 – 4.67 (m, 3H), 4.46 (t, J = 12.8 Hz, 1H), 4.05 (dt, J = 18.9, 5.7 Hz, 2H), 3.67 (s, 3H), 3.44 (s, 3H), 3.43 (s, 3H), 2.74 (dt, J = 11.6, 5.6 Hz, 2H), 2.35 (s, 3H), 2.33 (s, 3H), 2.23 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 166.8, 159.7, 158.9, 158.9, 158.8, 155.3, 147.6, 143.7, 139.3, 137.9, 137.5, 128.7, 128.6, 128.5, 128.2, 127.7, 126.4, 125.8, 125.3, 123.7, 123.3, 121.5, 120.7, 114.8, 114.5, 113.9, 108.6, 108.5, 107.8, 98.6, 96.4, 95.3, 95.3, 95.1, 94.3, 66.3, 66.2, 58.2, 58.2, 56.5, 56.5, 56.2, 55.1, 53.1, 52.5, 52.2, 51.5, 51.3, 45.8, 45.8, 14.2. LC/MS (m/z): 646.275 [M+H+]; UPLC tR 1.47 min.
(2-((1-(4-Methoxybenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)phenyl)(5-(4-methylpiperazin-1-yl)isoindolin-2-yl)methanone (16c).
Synthesized using General Procedure D2 from 15c (41 mg, 79 μmol) and 10a (19 mg, 87 μmol) in dioxane (0.8 mL). The crude mixture was purified via automated flash chromatography (1% to 7% methanol in CH2Cl2). To QuadraPure™ MPA resin (1.5 mmol/g loading, 34 mg) soaked in CH2Cl2 (2 mL) for 30 min was transferred the purified product using CH2Cl2 (3 mL) and shaken overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated in vacuo to afford 39 mg of 16c (75% yield). 1H NMR (400 MHz, CDCl3) δ 7.22 – 6.99 (m, 3H), 6.94 – 6.82 (m, 2H), 6.71 – 6.58 (m, 2H), 6.40 – 6.31 (m, 2H), 6.21 (dd, J = 5.1, 2.1 Hz, 1H), 5.84 (s, 1H), 5.19 – 5.08 (m, 2H), 5.08 – 4.97 (m, 4H), 4.91 – 4.71 (m, 3H), 4.46 (t, J = 13.0 Hz, 1H), 3.67 (d, J = 3.6 Hz, 3H), 3.51 – 3.40 (m, 6H), 3.29 – 3.10 (m, 4H), 2.59 (dt, J = 10.0, 4.7 Hz, 4H), 2.36 (d, J = 7.7 Hz, 3H), 2.24 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 166.7, 159.7, 158.9, 155.3, 151.5, 151.4, 147.6, 143.7, 139.3, 137.7, 137.3, 128.7, 128.7, 128.7, 127.5, 127.3, 123.4, 123.0, 116.4, 116.1, 113.9, 110.3, 109.8, 107.9, 107.8, 98.7, 98.6, 96.4, 95.3, 95.3, 95.1, 95.0, 94.3, 56.5, 56.2, 55.1, 55.0, 55.0, 53.2, 52.6, 52.3, 51.6, 51.3, 49.5, 49.5, 46.1, 46.1, 14.2. LC/MS (m/z): 657.301 [M+H+]; UPLC tR 0.89 min.
N-Benzyl-2-((1-(4-methoxybenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)-N-methylbenzamide (16d).
Synthesized using General Procedure D2 from 15d (30 mg, 71 μmol) and 10a (17 mg, 78 μmol). Following silica gel flash chromatography (10% to 35% acetone in hexanes), TMT (15 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 39 mg 16d (98% yield). 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.07 (m, 8H), 7.06 – 6.97 (m, 1H), 6.87 – 6.74 (m, 2H), 6.43 – 6.30 (m, 1H), 6.23 (dd, J = 15.2, 2.1 Hz, 1H), 5.84 (d, J = 12.4 Hz, 1H), 5.19 – 4.96 (m, 7H), 4.65 – 4.17 (m, 1H), 3.72 (d, J = 6.6 Hz, 3H), 3.46 – 3.35 (m, 6H), 2.91 – 2.78 (m, 3H), 2.27 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.0, 168.0, 159.6, 159.6, 159.0, 155.2, 155.0, 147.7, 147.6, 144.2, 144.1, 139.9, 139.5, 136.8, 136.4, 128.9, 128.8, 128.7, 128.7, 128.6, 128.6, 128.1, 127.7, 127.6, 127.4, 114.2, 114.1, 114.0, 106.9, 106.5, 98.4, 97.5, 97.0, 96.4, 95.0, 94.9, 94.9, 94.3, 94.3, 94.2, 91.4, 56.5, 56.3, 56.2, 56.2, 55.3, 55.2, 54.8, 51.2, 51.0, 50.9, 50.4, 35.8, 32.4, 14.3. LC/MS (m/z): 561.284 [M+H+]; UPLC tR 1.80 min.
Methyl 2-bromo-4,6-bis(methoxymethoxy)benzoate (17).
To a suspension of benzoic acid 12 (1.26 g, 3.93 mmol) and K2CO3 (0.951 g, 6.88 mmol) in DMF (39 mL) at room temperature was added iodomethane (0.428 mL, 6.88 mmol) dropwise. The suspension was heated to 80 ºC and stirred for 1 h. After cooling to room temperature, the reaction was quenched with saturated NH4Cl (aq.). The resulting mixture was extracted 4 times with ether. The combined organic layers were washed twice with water, brine and then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (5% to 20% ethyl acetate in hexanes twice) to afford 1.02 g of 17 (77% yield) as a clear colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.94 (d, J = 2.1 Hz, 1H), 6.79 (d, J = 2.1 Hz, 1H), 5.15 (s, 2H), 5.14 (s, 2H), 3.92 (s, 3H), 3.46 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 166.5, 159.0, 155.6, 120.7, 119.9, 113.0, 102.8, 94.7, 94.4, 56.3, 56.2, 52.6. LC/MS (m/z): [M+H+]; UPLC tR min (dH-109-763).
Methyl 2-((1-(4-methoxybenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18a).
Synthesized using General Procedure D2 from 17 (130 mg, 390 μmol) and 10a (93 mg, 430 μmol). Following silica gel flash chromatography (20% to 60% ethyl acetate in hexanes and 15% to 50% ethyl acetate in hexanes), TMT (59 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 161 mg of 18a (88% yield). 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.17 – 7.10 (m, 2H), 6.82 – 6.73 (m, 2H), 6.24 (d, J = 2.3 Hz, 1H), 6.10 (d, J = 2.3 Hz, 1H), 5.88 (s, 1H), 5.17 (s, 2H), 5.06 (s, 2H), 5.03 (s, 2H), 3.84 (s, 3H), 3.76 (s, 3H), 3.52 (s, 3H), 3.42 (s, 3H), 2.27 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.3, 159.7, 158.9, 149.2, 147.7, 138.9, 128.9, 128.8, 113.9, 99.9, 99.5, 95.2, 95.0, 95.0, 93.9, 56.4, 56.2, 55.1, 51.8, 51.1, 14.2. LC/MS (m/z): 473.16 [M+H+]; UPLC tR 1.70 min.
Methyl 2-((1-(4-methoxybenzyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18b).
Synthesized using General Procedure D2 from 17 (203 mg, 606 μmol) and 10b (186 mg, 666 μmol). Following silica gel flash chromatography (7% to 25% ethyl acetate in hexanes), TMT (115 mg) was added to the isolated residue; the mixture was suspended in toluene (4 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 270 mg of 18b (84% yield). 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 7.85 – 7.76 (m, 2H), 7.44 – 7.36 (m, 2H), 7.31 (d, J = 7.4 Hz, 1H), 7.20 (d, J = 8.7 Hz, 2H), 6.80 (d, J = 8.7 Hz, 2H), 6.42 (d, J = 0.7 Hz, 1H), 6.26 (d, J = 2.3 Hz, 1H), 6.15 (d, J = 2.3 Hz, 1H), 5.19 (s, 2H), 5.18 (s, 2H), 5.01 (s, 2H), 3.85 (s, 3H), 3.76 (s, 3H), 3.53 (s, 3H), 3.41 (s, 3H), 1.56 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.4, 159.7, 159.0, 150.2, 149.0, 139.6, 133.7, 128.9, 128.5, 128.5, 127.5, 125.3, 113.9, 100.1, 97.3, 95.2, 95.2, 95.1, 93.9, 56.4, 56.3, 55.1, 51.8, 51.6. LC/MS (m/z): 535.438 [M+H+]; UPLC tR 2.01 min
Methyl 2-((3-ethyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18c).
Synthesized using General Procedure D2 from 17 (100 mg, 298 μmol) and 10g (75.9 mg, 328 μmol). Following silica gel flash chromatography (15% to 40% ethyl acetate in hexanes), TMT (40 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 123 mg of 18c (85% yield). 1H NMR (400 MHz, CDCl3) δ 7.16 (s, 2H), 6.80 (d, J = 8.3 Hz, 2H), 6.25 (s, 1H), 6.14 (s, 1H), 5.93 (s, 1H), 5.17 (s, 2H), 5.11 (s, 2H), 5.03 (s, 2H), 3.83 (s, 3H), 3.76 (s, 3H), 3.52 (s, 3H), 3.42 (s, 3H), 2.65 (q, J = 7.7 Hz, 2H), 1.25 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.4, 159.7, 159.0, 153.9, 149.2, 138.9, 128.9, 128.9, 113.9, 100.0, 98.0, 95.3, 95.1, 94.0, 56.4, 56.3, 55.2, 51.8, 51.2, 22.1, 13.9. LC/MS (m/z): 487.318 [M+H+]; UPLC tR 1.80 min.
Methyl 2-((3-isopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18d).
Synthesized using General Procedure D2 from 17 100 mg, 298 μmol) and 10h (80.5 mg, 328 μmol). Following silica gel flash chromatography (12% to 35% ethyl acetate in hexanes), TMT (42 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 112 mg of 18d (75% yield). 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 7.14 (d, J = 8.2 Hz, 2H), 6.86 – 6.71 (m, 2H), 6.24 (d, J = 2.2 Hz, 1H), 6.13 (s, 1H), 5.93 (s, 1H), 5.17 (s, 2H), 5.10 (s, 2H), 5.01 (s, 2H), 3.82 (s, 3H), 3.76 (s, 3H), 3.51 (s, 3H), 3.42 (s, 3H), 2.97 (p, J = 6.9 Hz, 1H), 1.27 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.6, 161.4, 159.7, 159.0, 158.3, 149.2, 138.8, 129.0, 128.8, 113.9, 100.1, 96.5, 95.3, 95.2, 95.2, 94.0, 56.4, 56.2, 55.2, 51.8, 51.2, 28.3, 22.9. LC/MS (m/z): 501.344 [M+H+]; UPLC tR 1.89 min.
Methyl 2-((3-cyclopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18e).
Synthesized using General Procedure D2 from 17 (100 mg, 298 μmol) and 10j (80.0 mg, 328 μmol). Following silica gel flash chromatography (8% to 50% ethyl acetate in hexanes), TMT (44 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 133 mg of 18e (89% yield). 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.15 (d, J = 8.2 Hz, 2H), 6.85 – 6.73 (m, 2H), 6.24 (d, J = 2.2 Hz, 1H), 6.11 (s, 1H), 5.72 (s, 1H), 5.16 (s, 2H), 5.07 (s, 2H), 5.02 (s, 2H), 3.82 (s, 3H), 3.76 (s, 3H), 3.51 (s, 3H), 3.42 (s, 3H), 1.93 (dq, J = 8.8, 5.1, 4.4 Hz, 1H), 0.90 (dd, J = 7.7, 5.5 Hz, 2H), 0.77 – 0.63 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.4, 159.7, 159.0, 154.3, 149.1, 138.9, 128.9, 128.9, 113.9, 100.0, 95.7, 95.3, 95.2, 94.0, 56.4, 56.3, 55.2, 51.8, 51.2, 9.7, 7.9. LC/MS (m/z): 499.315 [M+H+]; UPLC tR 1.81 min.
Methyl 2-((3-cyclopentyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18f).
Synthesized using General Procedure D2 from 17 (81 mg, 240 μmol) and 10k (67 mg, 240 μmol) in dioxane (1.2 mL). Following silica gel flash chromatography (8% to 25% ethyl acetate in hexanes), TMT (36 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 90 mg of 18f (71% yield). 1H NMR (400 MHz, CDCl3) δ 8.60 (s, 1H), 7.15 (d, J = 8.2 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 6.24 (s, 1H), 6.15 (s, 1H), 5.92 (s, 1H), 5.17 (s, 2H), 5.10 (s, 2H), 5.02 (s, 2H), 3.82 (s, 3H), 3.76 (s, 3H), 3.51 (s, 3H), 3.42 (s, 3H), 3.08 (t, J = 8.1 Hz, 1H), 2.06 (s, 2H), 1.83 – 1.45 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 168.6, 161.4, 159.7, 159.0, 156.6, 149.2, 138.9, 129.0, 128.8, 113.9, 100.1, 97.0, 95.3, 95.2, 95.2, 94.0, 56.4, 56.3, 55.2, 51.8, 51.2, 39.6, 33.4, 25.4. LC/MS (m/z): 527.366 [M+H+]; UPLC tR 2.02 min.
Methyl 2-((3-(furan-3-yl)-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18g).
Synthesized using General Procedure D2 from 17 (88.4 mg, 328 μmol) and 10l (88.4 mg, 328 μmol). Following silica gel flash chromatography (12% to 35% ethyl acetate in hexanes), TMT (44 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 111 mg of 18g (71% yield). 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 7.77 (dd, J = 1.6, 0.9 Hz, 1H), 7.45 (t, J = 1.7 Hz, 1H), 7.17 (d, J = 8.6 Hz, 2H), 6.85 – 6.78 (m, 2H), 6.76 (dd, J = 1.9, 0.9 Hz, 1H), 6.26 (d, J = 2.3 Hz, 1H), 6.22 – 6.18 (m, 1H), 6.14 (d, J = 2.2 Hz, 1H), 5.18 (s, 2H), 5.16 (s, 2H), 5.02 (s, 2H), 3.84 (s, 3H), 3.76 (s, 3H), 3.52 (s, 3H), 3.41 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.4, 159.8, 159.1, 149.0, 143.7, 143.2, 139.5, 139.1, 128.9, 128.6, 120.2, 113.9, 108.8, 100.2, 97.6, 95.3, 95.3, 95.2, 94.0, 56.5, 56.3, 55.2, 51.9, 51.6. LC/MS (m/z): 524.279 [M+H+]; UPLC tR 1.81 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-1H-pyrazol-5-yl)amino)benzoate (18h).
Synthesized using General Procedure D2 from 17 (100 mg, 207 μmol) and 1-methyl-1H-pyrazol-5-amine (31.9 mg, 328 μmol). Following silica gel flash chromatography (10% to 35% ethyl acetate in hexanes), TMT (30 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 80.6 mg of 18h (77% yield). 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 7.47 (d, J = 2.0 Hz, 1H), 6.27 (d, J = 2.3 Hz, 1H), 6.08 (dd, J = 8.0, 2.1 Hz, 2H), 5.18 (s, 2H), 5.07 (s, 2H), 3.91 (s, 3H), 3.73 (s, 3H), 3.52 (s, 3H), 3.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.1, 161.6, 160.0, 149.5, 139.0, 138.6, 99.4, 95.3, 95.2, 94.6, 93.9, 56.5, 56.3, 52.0, 35.0. LC/MS (m/z): 353.233 [M+H+]; UPLC tR 1.34 min.
Methyl 2-((1-isopropyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18i).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 1-(propan-2-yl)-1H-pyrazol-5-amine (61.6 mg, 492 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (47 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 124 mg of 18i (73% yield). 1H NMR (400 MHz, CDCl3) δ 8.84 (s, 1H), 7.53 (d, J = 1.9 Hz, 1H), 6.24 (d, J = 2.3 Hz, 1H), 6.05 (dd, J = 5.1, 2.0 Hz, 2H), 5.18 (s, 2H), 5.05 (s, 2H), 4.48 (p, J = 6.6 Hz, 1H), 3.91 (s, 3H), 3.52 (d, J = 1.2 Hz, 3H), 3.40 (d, J = 0.8 Hz, 3H), 1.45 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.6, 160.0, 150.4, 138.6, 137.4, 100.2, 99.4, 95.3, 95.1, 94.5, 93.9, 56.4, 56.2, 51.9, 48.6, 22.4. LC/MS (m/z): 381.329 [M+H+]; UPLC tR 1.61 min.
Methyl 2-((1-isobutyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18j).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 1-(2-methylpropyl)-1H-pyrazol-5-amine (68.3 mg, 492 μmol). Following silica gel flash chromatography (12% to 33% ethyl acetate in hexanes), TMT (50 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 136 mg of 18j (77% yield). 1H NMR (400 MHz, CDCl3) δ 8.93 (s, 1H), 7.50 (d, J = 2.0 Hz, 1H), 6.25 (d, J = 2.3 Hz, 1H), 6.18 (d, J = 2.3 Hz, 1H), 6.07 (d, J = 1.9 Hz, 1H), 5.18 (s, 2H), 5.06 (s, 2H), 3.90 (s, 3H), 3.80 (d, J = 7.4 Hz, 2H), 3.52 (s, 3H), 3.42 (s, 3H), 2.22 (hept, J = 7.0 Hz, 1H), 0.90 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 169.1, 161.6, 160.0, 149.7, 138.9, 138.6, 99.6, 99.2, 95.3, 95.2, 94.7, 93.9, 56.4, 56.2, 55.1, 51.9, 29.4, 19.9. LC/MS (m/z): 395.355 [M+H+]; UPLC tR 1.73 min.
Methyl 2-((1-(cyclohexylmethyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18k).
Synthesized using General Procedure D2 from 17 (139 mg, 415 μmol) and 1-(cyclohexylmethyl)-1H-pyrazol-5-amine (81.8 mg, 456 μmol). Following silica gel flash chromatography (8% to 25% ethyl acetate in hexanes), TMT (44 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 130 mg of 18k (72% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 7.49 (d, J = 2.0 Hz, 1H), 6.25 (d, J = 2.3 Hz, 1H), 6.16 (d, J = 2.3 Hz, 1H), 6.09 – 5.97 (m, 1H), 5.18 (s, 2H), 5.06 (s, 2H), 3.91 (s, 3H), 3.82 (d, J = 7.3 Hz, 2H), 3.53 (s, 3H), 3.41 (s, 3H), 1.91 (tt, J = 7.5, 3.7 Hz, 1H), 1.75 – 1.54 (m, 6H), 1.32 – 1.07 (m, 3H), 0.96 (q, J = 11.8 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 169.1, 161.6, 159.9, 149.7, 139.0, 138.6, 99.7, 99.1, 95.3, 95.2, 94.7, 93.9, 56.4, 56.3, 53.9, 51.9, 38.5, 30.6, 26.3, 25.7. LC/MS (m/z): 435.405 [M+H+]; UPLC tR 1.93 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-phenyl-1H-pyrazol-5-yl)amino)benzoate (18l).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 1-phenyl-1H-pyrazol-5-amine (78.4 mg, 492 μmol). Following silica gel flash chromatography (12% to 33% ethyl acetate in hexanes), TMT (47 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 132 mg of 18l (71% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.99 (s, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.60 – 7.53 (m, 2H), 7.49 – 7.40 (m, 2H), 7.38 – 7.32 (m, 1H), 6.46 (d, J = 2.3 Hz, 1H), 6.30 – 6.19 (m, 2H), 5.16 (s, 2H), 5.09 (s, 2H), 3.79 (s, 3H), 3.50 (s, 3H), 3.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.6, 161.5, 159.7, 148.2, 140.3, 139.3, 138.5, 129.2, 127.6, 124.0, 100.5, 99.1, 95.7, 95.2, 95.2, 94.0, 56.4, 56.3, 51.9. LC/MS (m/z): 415.292 [M+H+]; UPLC tR 1.67 min.
Methyl 2-((1-cyclohexyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18m).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 1-cyclohexyl-1H-pyrazol-5-amine (81.4 mg, 492 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (60 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 152 mg of 18m (81% yield) as a clear yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.84 (s, 2H), 7.51 (d, J = 1.9 Hz, 2H), 6.24 (d, J = 2.3 Hz, 2H), 6.13 – 5.96 (m, 1H), 5.19 (s, 1H), 5.05 (s, 2H), 4.09 – 3.97 (m, 1H), 3.91 (s, 3H), 3.53 (d, J = 1.0 Hz, 3H), 3.40 (s, 3H), 1.99 – 1.81 (m, 7H), 1.72 – 1.64 (m, 1H), 1.46 – 1.06 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.6, 160.0, 150.4, 138.5, 137.6, 99.9, 99.5, 95.3, 95.1, 94.5, 93.8, 56.4, 56.3, 56.2, 51.9, 32.7, 25.6, 25.2. LC/MS (m/z): 421.379 [M+H+]; UPLC tR 1.83 min.
Methyl 2-((1-benzyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18n).
Synthesized using General Procedure D2 from 17 (140 mg, 418 μmol) and 1-benzyl-1H-pyrazol-5-amine (79.6 mg, 460 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (44 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 116 mg of 18n (65% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.31 – 7.23 (m, 3H), 7.22 – 7.17 (m, 2H), 6.25 (d, J = 2.3 Hz, 1H), 6.11 (d, J = 2.0 Hz, 1H), 6.09 (d, J = 2.3 Hz, 1H), 5.20 (s, 2H), 5.17 (s, 2H), 5.01 (s, 2H), 3.82 (s, 3H), 3.51 (s, 3H), 3.41 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.4, 159.8, 149.3, 139.2, 138.9, 136.5, 128.6, 127.7, 127.6, 100.2, 100.0, 95.3, 95.3, 94.9, 93.9, 56.5, 56.3, 52.0, 51.9. LC/MS (m/z): 429.362 [M+H+]; UPLC tR 1.71 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-(pyridin-3-ylmethyl)-1H-pyrazol-5-yl)amino)benzoate (18o).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 1-(pyridin-3-ylmethyl)-1H-pyrazol-5-amine (85.8 mg, 492 μmol) and purified via silica gel flash chromatography (10% to 30% acetone in CH2Cl2). To QuadraPure™ MPA resin (1.5 mmol/g loading, 192 mg) soaked in CH2Cl2 (3 mL) for 30 min was transferred the purified product using CH2Cl2 (2 mL) and shaken overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 135 mg of 18o (71% yield) as a brown oil. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 7.31 (dd, J = 8.5, 7.3 Hz, 2H), 7.18 (dd, J = 8.5, 1.3 Hz, 2H), 7.07 – 6.99 (m, 1H), 6.59 (d, J = 2.2 Hz, 1H), 6.26 (d, J = 2.3 Hz, 1H), 5.18 (s, 2H), 5.08 (s, 2H), 3.88 (s, 3H), 3.52 (s, 3H), 3.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.8, 160.9, 159.4, 148.1, 141.1, 129.3, 123.0, 121.5, 101.8, 95.7, 95.2, 95.2, 94.0, 56.4, 56.2, 51.9. LC/MS (m/z): 429.582 [M+H+]; UPLC tR 1.16 min.
Methyl 2-((1-(furan-2-ylmethyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18p).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 1-(furan-2-ylmethyl)-1H-pyrazol-5-amine (80.3 mg, 492 μmol). Following silica gel flash chromatography (12% to 35% ethyl acetate in hexanes), TMT (60 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 145 mg of 18p (78% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 7.51 (d, J = 1.9 Hz, 1H), 7.41 – 7.33 (m, 1H), 6.35 – 6.28 (m, 2H), 6.28 (d, J = 2.2 Hz, 1H), 6.17 (d, J = 2.2 Hz, 1H), 6.09 (d, J = 2.0 Hz, 1H), 5.19 (s, 4H), 5.05 (s, 2H), 3.90 (s, 3H), 3.52 (d, J = 0.9 Hz, 3H), 3.42 (d, J = 0.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.8, 161.4, 159.8, 149.4, 149.2, 142.8, 139.3, 139.0, 110.4, 108.7, 100.2, 99.9, 95.4, 95.3, 95.0, 93.9, 56.4, 56.3, 51.9, 44.7. LC/MS (m/z): 419.35 [M+H+]; UPLC tR 1.57 min.
Methyl 2-((1-(4-isopropylbenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18q).
Synthesized using General Procedure D2 from 17 (80 mg, 240 μmol) and 1-([4-(propan-2-yl)phenyl]methyl)-1H-pyrazol-5-amine (57 mg, 270 μmol). Following silica gel flash chromatography (7% to 25% ethyl acetate in hexanes), TMT (21 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 55 mg of 18q (49% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 7.52 (d, J = 2.0 Hz, 1H), 7.14 (s, 4H), 6.24 (d, J = 2.3 Hz, 1H), 6.10 (t, J = 2.2 Hz, 2H), 5.17 (s, 2H), 5.16 (s, 2H), 5.00 (s, 2H), 3.83 (s, 3H), 3.52 (s, 3H), 3.41 (s, 3H), 2.85 (p, J = 7.0 Hz, 1H), 1.20 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.4, 159.8, 149.3, 148.3, 139.1, 138.8, 133.8, 127.7, 126.7, 100.1, 100.0, 95.3, 95.3, 94.9, 93.9, 56.4, 56.3, 51.9, 51.7, 33.8, 23.9. LC/MS (m/z): 470.381 [M+H+]; UPLC tR 1.96 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-(4-(trifluoromethyl)benzyl)-1H-pyrazol-5-yl)amino)benzoate (18r).
Inside a nitrogen glovebox were combined aryl bromide 17 (145 mg, 432 μmol), 1-([4-(trifluoromethyl)phenyl]methyl)-1H-pyrazol-5-amine hydrochloride (100 mg, 360 μmol), tris(dibenzylideneacetone)dipalladium (16.5 mg, 18.0 μmol). Xantphos (25.0 mg, 43.2, μmol) and sodium phenoxide (155 mg, 1.33 mmol). Dioxane (3.4 mL) was added to the mixture and the reaction vessel was capped and removed from the glovebox. After the reaction was irradiated at 170 ºC for 2 h in a microwave reactor, the reaction was cooled to room temperature and diluted with ethyl acetate. The resulting mixture was washed three times with saturated Na2CO3 (aq.), brine, then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. Following silica gel flash chromatography (10% to 40% ethyl acetate in hexanes), TMT (30 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 66 mg of 18r (37% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1H), 7.55 (d, J = 1.9 Hz, 1H), 7.53 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 6.25 (d, J = 2.3 Hz, 1H), 6.13 (d, J = 1.9 Hz, 1H), 6.04 (d, J = 2.3 Hz, 1H), 5.26 (s, 2H), 5.17 (s, 2H), 5.01 (s, 2H), 3.83 (s, 3H), 3.51 (s, 3H), 3.40 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 168.9, 161.5, 159.9, 149.2, 140.4, 140.4, 139.5, 139.1, 129.9 (q, 2JC-F = 32.6 Hz), 127.9, 125.6 (q, 3JC-F = 3.7 Hz), 124.0 (q, 1JC-F = 272.0 Hz), 100.4, 99.9, 95.4, 95.3, 94.8, 93.9, 56.4, 56.3, 51.9, 51.5. 19F NMR (376 MHz, CDCl3) δ −62.6. LC/MS (m/z): 497.33 [M+H+]; UPLC tR 1.88 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-3-phenyl-1H-pyrazol-5-yl)amino)benzoate (18s).
Synthesized using General Procedure D2 from 17 (50 mg, 150 μmol) and 10p (28 mg, 160 μmol). Following silica gel flash chromatography (10% to 35% MTBE in hexanes), TMT (21 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 55 mg of 18s (86% yield). 1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H), 7.81 – 7.72 (m, 2H), 7.48 – 7.34 (m, 2H), 7.34 – 7.27 (m, 1H), 6.51 – 6.36 (m, 1H), 6.29 (d, J = 2.3 Hz, 1H), 6.18 (d, J = 2.2 Hz, 1H), 5.20 (s, 2H), 5.08 (s, 2H), 3.92 (s, 3H), 3.78 (s, 3H), 3.53 (s, 3H), 3.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.7, 160.1, 150.1, 149.4, 140.1, 133.6, 128.6, 127.6, 125.3, 99.8, 96.6, 95.3, 95.3, 94.9, 94.0, 56.5, 56.3, 52.0, 35.2. LC/MS (m/z): 429.23 [M+H+]; UPLC tR 1.76 min.
Methyl 2-((1-(tert-butyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18t).
Synthesized using General Procedure D2 from 17 (69.4 mg, 207 μmol) and 10q (49.0 mg, 227 μmol) in dioxane (1.6 mL). Following silica gel flash chromatography (7% to 20% MTBE in hexanes), TMT (29 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 65.5 mg of 18t (68% yield). 1H NMR (400 MHz, CDCl3) δ 8.91 (s, 1H), 7.83 – 7.73 (m, 2H), 7.47 – 7.35 (m, 2H), 7.33 – 7.22 (m, 1H), 6.47 – 6.38 (m, 1H), 6.28 – 6.19 (m, 2H), 5.19 (s, 2H), 5.05 (s, 2H), 3.91 (s, 3H), 3.54 (s, 3H), 3.40 (s, 3H), 1.68 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 169.3, 161.6, 160.0, 150.4, 147.8, 139.2, 134.1, 128.5, 127.3, 125.2, 100.0, 99.3, 95.4, 95.0, 94.7, 94.0, 59.8, 56.5, 56.3, 51.9, 29.8. LC/MS (m/z): 471.263 [M+H+]; UPLC tR 2.15 min.
Methyl 2-((1-cyclohexyl-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18u).
Synthesized using General Procedure D2 from 17 (100 mg, 298 μmol) and 10r (79.2 mg, 323 μmol). Following silica gel flash chromatography (8% to 25% MTBE in hexanes), TMT (44 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 111 mg of 18u (75% yield). 1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 7.84 – 7.75 (m, 2H), 7.38 (t, J = 7.7 Hz, 2H), 7.30 – 7.27 (m, 1H), 6.40 – 6.33 (m, 1H), 6.26 (d, J = 2.3 Hz, 1H), 6.17 (d, J = 2.3 Hz, 1H), 5.20 (s, 2H), 5.05 (s, 2H), 4.07 (td, J = 11.2, 5.5 Hz, 1H), 3.92 (s, 3H), 3.54 (s, 3H), 3.40 (s, 3H), 2.12 – 1.87 (m, 6H), 1.74 – 1.66 (m, 1H), 1.47 – 1.16 (m, 3H).
Methyl 2-((1-isobutyl-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18v).
Synthesized using General Procedure D2 from 17 (100 mg, 298 μmol) and 10s (66.4 mg, 308 μmol). Following silica gel flash chromatography (7% to 20% ethyl acetate in hexanes), TMT (42 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 118 mg of 18v (84% yield). 1H NMR (400 MHz, CDCl3) δ 9.05 (s, 1H), 7.84 – 7.78 (m, 2H), 7.40 (t, J = 7.6 Hz, 2H), 7.30 (t, J = 7.3 Hz, 1H), 6.40 (s, 1H), 6.29 (d, J = 2.1 Hz, 2H), 5.20 (s, 2H), 5.08 (s, 2H), 3.92 (s, 3H), 3.88 (d, J = 7.4 Hz, 2H), 3.54 (s, 3H), 3.42 (s, 3H), 2.40 – 2.21 (m, 1H), 0.94 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.6, 160.0, 150.0, 149.6, 140.0, 133.8, 128.6, 127.5, 125.4, 99.7, 96.4, 95.3, 95.2, 95.0, 94.0, 56.5, 56.3, 55.2, 52.0, 29.5, 20.0. LC/MS (m/z): 471.307 [M+H+]; UPLC tR 2.05 min.
Methyl 2-((3-isopropyl-1-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18w).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) an amine 10t (68.5 mg, 492 μmol). Following silica gel flash chromatography (15% to 45% ethyl acetate in hexanes), TMT (55 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 129 mg of 18w (78% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 6.26 (d, J = 2.3 Hz, 1H), 6.14 (d, J = 2.2 Hz, 1H), 5.89 (s, 1H), 5.18 (s, 2H), 5.08 (s, 2H), 3.89 (d, J = 0.9 Hz, 3H), 3.66 (s, 3H), 3.52 (d, J = 0.6 Hz, 3H), 3.43 (d, J = 0.6 Hz, 3H), 2.92 (p, J = 6.9 Hz, 1H), 1.26 (d, J = 7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 169.1, 161.6, 159.9, 158.1, 149.5, 139.1, 99.7, 95.8, 95.3, 95.1, 94.9, 94.0, 56.4, 56.2, 51.9, 34.6, 28.3, 22.8. LC/MS (m/z): 395.355 [M+H+]; UPLC tR 1.68 min.
Methyl 2-((3-cyclohexyl-1-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18x).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) an amine 10u (88.3 mg, 492 μmol). Following silica gel flash chromatography (15% to 40% ethyl acetate in hexanes), TMT (42 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 108 mg of 18x (56% yield) as a clear yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 6.26 (d, J = 2.3 Hz, 1H), 6.12 (d, J = 2.3 Hz, 1H), 5.87 (s, 1H), 5.18 (s, 2H), 5.07 (s, 2H), 3.89 (s, 3H), 3.66 (s, 3H), 3.52 (s, 3H), 3.43 (s, 3H), 2.64 – 2.50 (m, 1H), 2.05 – 1.90 (m, 2H), 1.86 – 1.75 (m, 2H), 1.75 – 1.62 (m, 1H), 1.50 – 1.15 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 169.0, 161.6, 159.9, 157.3, 149.5, 139.0, 99.7, 96.1, 95.3, 95.1, 94.9, 94.0, 56.4, 56.2, 51.9, 38.1, 34.6, 33.2, 26.4, 26.1. LC/MS (m/z): 435.405 [M+H+]; UPLC tR 1.97 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-3-(o-tolyl)-1H-pyrazol-5-yl)amino)benzoate (18y).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10v (92.2 mg, 492 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (48 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 139 mg of 18y (70% yield) as a clear yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.59 (s, 1H), 7.27 – 7.21 (m, 2H), 6.30 – 6.16 (m, 3H), 5.20 (s, 2H), 5.08 (s, 2H), 3.92 (s, 3H), 3.79 (s, 3H), 3.53 (s, 3H), 3.42 (s, 3H), 2.48 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.0, 150.4, 149.5, 139.2, 135.8, 133.5, 130.7, 129.0, 127.6, 125.8, 99.7, 95.3, 95.3, 94.7, 94.0, 56.5, 56.3, 52.0, 35.1, 21.1. LC/MS (m/z): 443.388 [M+H+]; UPLC tR 1.93 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-3-(m-tolyl)-1H-pyrazol-5-yl)amino)benzoate (18z).
Synthesized using General Procedure D2 from 17 (170 mg, 570 μmol) and 10w (104 mg, 558 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (66 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 155 mg of 18z (69% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.99 (s, 1H), 7.63 (s, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.29 (d, J = 7.7 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 6.42 – 6.32 (m, 1H), 6.29 (d, J = 2.3 Hz, 1H), 6.17 (d, J = 2.3 Hz, 1H), 5.20 (s, 2H), 5.08 (s, 2H), 3.92 (s, 3H), 3.77 (s, 3H), 3.53 (s, 3H), 3.42 (s, 3H), 2.39 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.1, 150.2, 149.5, 140.1, 138.2, 133.5, 128.5, 128.4, 125.8, 122.5, 99.8, 96.7, 95.3, 95.2, 94.9, 94.0, 56.5, 56.3, 52.0, 35.1, 21.5.
Methyl 2,4-bis(methoxymethoxy)-6-((3-(3-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)benzoate (18aa)
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10x (100 mg, 492 μmol). Following silica gel flash chromatography (10% to 40% ethyl acetate in hexanes), TMT (64 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 161 mg of 18aa (79% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 9.02 (s, 1H), 7.39 – 7.29 (m, 3H), 6.86 (dd, J = 7.8, 2.2 Hz, 1H), 6.39 (s, 1H), 6.30 (dd, J = 2.3, 1.0 Hz, 1H), 6.18 (t, J = 2.4 Hz, 1H), 5.20 (s, 2H), 5.08 (s, 2H), 3.92 (s, 3H), 3.87 (s, 3H), 3.78 (d, J = 2.1 Hz, 3H), 3.53 (s, 3H), 3.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.1, 159.9, 149.9, 149.4, 140.1, 135.0, 129.6, 117.9, 113.7, 110.2, 99.8, 96.8, 95.3, 95.2, 94.9, 93.9, 56.5, 56.3, 55.3, 52.0, 35.2. LC/MS (m/z): 459.399 [M+H+]; UPLC tR 1.85 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)benzoate (18ab).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10y (119 mg, 492 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (65 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 165 mg of 18ab (74% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 8.04 (s, 1H), 7.95 (d, J = 7.5 Hz, 1H), 7.58 – 7.43 (m, 2H), 6.47 – 6.40 (m, 1H), 6.31 (d, J = 2.3 Hz, 1H), 6.20 (d, J = 2.3 Hz, 1H), 5.20 (s, 2H), 5.09 (s, 2H), 3.92 (s, 3H), 3.79 (s, 3H), 3.53 (s, 3H), 3.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.1, 149.2, 148.6, 140.6, 134.5, 130.9 (q, 2JC-F = 32.2 Hz), 129.0, 128.4, 124.2 (q, 1JC-F = 272.4 Hz), 124.1 (q, 3JC-F = 3.8 Hz), 122.0 (q, 3JC-F = 3.9 Hz), 99.8, 96.5, 95.4, 95.3, 94.9, 94.0, 56.4, 56.3, 52.0, 35.2. 19F NMR (376 MHz, CDCl3) δ −62.7. LC/MS (m/z): 497.286 [M+H+]; UPLC tR 2.09 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-3-(p-tolyl)-1H-pyrazol-5-yl)amino)benzoate (18ac).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10z (92.2 mg, 492 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (59 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 160 mg of 18ac (81% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.66 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 7.9 Hz, 2H), 6.36 (s, 1H), 6.29 (d, J = 2.2 Hz, 1H), 6.18 (d, J = 2.3 Hz, 1H), 5.19 (s, 2H), 5.08 (s, 2H), 3.91 (d, J = 0.9 Hz, 3H), 3.76 (s, 3H), 3.53 (s, 3H), 3.42 (s, 3H), 2.37 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.1, 150.2, 149.5, 140.0, 137.3, 130.8, 129.3, 125.2, 99.8, 96.4, 95.3, 95.2, 94.9, 94.0, 56.5, 56.3, 52.0, 35.1, 21.2. LC/MS (m/z): 443.388 [M+H+]; UPLC tR 1.94 min.
Methyl 2,4-bis(methoxymethoxy)-6-((3-(4-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)benzoate (18ad).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10aa (100 mg, 492 μmol). Following silica gel flash chromatography (15% to 45% ethyl acetate in hexanes), TMT (57 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 161 mg of 18ad (78% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.70 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 8.6 Hz, 2H), 6.32 (s, 1H), 6.29 (d, J = 2.3 Hz, 1H), 6.18 (d, J = 2.3 Hz, 1H), 5.19 (s, 2H), 5.08 (s, 2H), 3.92 (s, 3H), 3.84 (s, 3H), 3.75 (s, 3H), 3.53 (s, 3H), 3.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.1, 161.7, 160.1, 159.3, 150.0, 149.5, 140.0, 126.5, 126.5, 114.0, 99.7, 96.1, 95.3, 95.2, 94.9, 94.0, 56.5, 56.3, 55.3, 52.0, 35.0. LC/MS (m/z): 459.354 [M+H+]; UPLC tR 1.80 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)benzoate (18ae).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10ab (119 mg, 492 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (60 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 180 mg of 18ae (81% yield) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 7.88 (d, J = 8.1 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 6.45 (s, 1H), 6.31 (d, J = 2.3 Hz, 1H), 6.19 (d, J = 2.2 Hz, 1H), 5.20 (s, 2H), 5.09 (s, 2H), 3.92 (s, 3H), 3.79 (s, 3H), 3.53 (s, 3H), 3.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.1, 149.2, 148.6, 140.5, 137.0, 137.0, 129.3 (q, 2JC-F = 32.3 Hz), 128.8, 125.53 (q, 3JC-F = 3.8 Hz), 124.3 (q, 1JC-F = 271.8 Hz), 123.0, 120.3, 99.8, 96.8, 95.4, 95.3, 94.9, 94.0, 56.5, 56.3, 52.0, 35.3. 19F NMR (376 MHz, CDCl3) δ −62.4. LC/MS (m/z): 497.33 [M+H+]; UPLC tR 2.06 min.
Methyl 2-((3-(4-(tert-butyl)phenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoate (18af).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10ac (113 mg, 492 μmol). Following silica gel flash chromatography (10% to 30% ethyl acetate in hexanes), TMT (58 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 174 mg of 18af (80% yield) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.42 (d, J = 8.2 Hz, 2H), 6.37 (s, 1H), 6.28 (d, J = 2.2 Hz, 1H), 5.19 (s, 2H), 5.07 (s, 2H), 3.92 (s, 2H), 3.76 (s, 3H), 3.53 (s, 3H), 3.42 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.0, 150.6, 150.1, 149.5, 140.0, 130.8, 129.0, 128.2, 125.5, 125.3, 125.0, 99.7, 96.5, 95.3, 95.2, 94.9, 94.0, 56.5, 56.3, 52.0, 35.1, 34.6, 31.3. LC/MS (m/z): 485.377 [M+H+]; UPLC tR 2.20 min.
Methyl 2,4-bis(methoxymethoxy)-6-((1-methyl-3-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)benzoate (18ag).
Synthesized using General Procedure D2 from 17 (150 mg, 448 μmol) and 10ad (127 mg, 492 μmol). Following silica gel flash chromatography (12% to 33% ethyl acetate in hexanes), TMT (66 mg) was added to the isolated residue; the mixture was suspended in toluene (3 mL) and stirred overnight. The suspension was filtered through a plug of Celite® and the filtrate was concentrated using a rotary evaporator to afford 183 mg of 18ag (80% yield) as a white/yellow solid. 1H NMR (400 MHz, CDCl3) δ 9.04 (s, 1H), 7.83 – 7.71 (m, 2H), 7.25 – 7.19 (m, 2H), 6.41 – 6.34 (m, 1H), 6.30 (d, J = 2.3 Hz, 1H), 6.17 (d, J = 2.2 Hz, 1H), 5.20 (s, 2H), 5.08 (s, 2H), 3.92 (s, 3H), 3.77 (s, 3H), 3.53 (s, 3H), 3.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.7, 160.1, 149.3, 148.8, 140.4, 132.5, 126.6, 121.1, 120.5 (q, J = 256.9 Hz), 99.8, 96.5, 95.3, 95.3, 94.9, 94.0, 56.4, 56.3, 52.0, 35.2. 19F NMR (376 MHz, CDCl3) δ −57.8. LC/MS (m/z): 513.296 [M+H+]; UPLC tR 2.04 min.
2-((1-(4-Methoxybenzyl)-3-methyl-1H-pyrazol-5-yl)amino)-4-(methoxymethoxy)-6-((methoxymethyl)peroxy)benzoic acid (19a).
Ester 18a (105 mg, 0.222 mmol) was hydrolyzed using General Procedure E to afford 95.1 mg of crude acid 19a (93% crude yield).
2-((1-(4-Methoxybenzyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4-(methoxymethoxy)-6-((methoxymethyl)peroxy)benzoic acid (19b).
Ester 18b (267 mg, 500 μmol) was hydrolyzed using General Procedure E to afford 235 mg of crude acid 19b (90% crude yield).
2-((3-Ethyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19c).
Ester 18c (123 mg, 253 μmol) was hydrolyzed using General Procedure E to afford 112 mg of crude acid 19c (94% crude yield).
2-((3-Isopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19d)
Ester 18d (112 mg, 224 μmol) was hydrolyzed using General Procedure E to afford 111 mg of crude acid 19d (102% crude yield).
2-((3-Cyclopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19e)
Ester 18e (132 mg, 265 μmol) was hydrolyzed using General Procedure E to afford 118 mg of crude acid 19e (92% crude yield).
2-((3-Cyclopentyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19f).
Ester 18f (90.1 mg, 171 μmol) was hydrolyzed using General Procedure E to afford 90 mg of crude acid 19f (103% crude yield).
2-((3-(Furan-3-yl)-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19g).
Ester 18g (110 mg, 210 μmol) was hydrolyzed using General Procedure E to afford 104 mg of crude acid 19g (97% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-1H-pyrazol-5-yl)amino)benzoic acid (19h).
Ester 18h (79.8 mg, 227 μmol) was hydrolyzed using General Procedure E to afford 27.6 mg of crude acid 19h (36% crude yield).
2-((1-Isopropyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19i).
Ester 18i (124 mg, 327 μmol) was hydrolyzed using General Procedure E to afford 98.8 mg of crude acid 19i (83% crude yield).
2-((1-Isobutyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19j).
Ester 18j (135 mg, 343 μmol) was hydrolyzed using General Procedure E to afford 119 mg of crude acid 19j (91% crude yield).
2-((1-(Cyclohexylmethyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19k).
Ester 18k (125 mg, 288 μmol) was hydrolyzed using General Procedure E to afford 105 mg of crude acid 19k (87% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-phenyl-1H-pyrazol-5-yl)amino)benzoic acid (19l).
Ester 18l (131 mg, 317 μmol) was hydrolyzed using General Procedure E to afford 114 mg of crude acid 19l (90% crude yield).
2-((1-Cyclohexyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19m).
Ester 18m (152 mg, 362 μmol) was hydrolyzed using General Procedure E to afford 152 mg of crude acid 19m (103% crude yield).
2-((1-Benzyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19n).
Ester 18n (115 mg, 269 μmol) was hydrolyzed using General Procedure E to afford 106 mg of crude acid 19n (95% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-(pyridin-3-ylmethyl)-1H-pyrazol-5-yl)amino)benzoic acid (19o).
Ester 18o (135 mg, 315 μmol) was hydrolyzed using General Procedure E to afford 90.1 mg of crude acid 19o (69% crude yield).
2-((1-(Furan-2-ylmethyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19p).
Ester 18p (145 mg, 347 μmol) was hydrolyzed using General Procedure E to afford 128 mg of crude acid 19p (91% crude yield).
2-((1-(4-Isopropylbenzyl)-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19q).
Ester 18q (55.1 mg, 117 μmol) was hydrolyzed using General Procedure E to afford 47.9 mg of crude acid 19q (90% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-(4-(trifluoromethyl)benzyl)-1H-pyrazol-5-yl)amino)benzoic acid (19r)
Ester 18r (66 mg, 133 μmol) was hydrolyzed using General Procedure E to afford 57 mg of crude acid 19r (89% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-3-phenyl-1H-pyrazol-5-yl)amino)benzoic acid (19s).
Ester 18s (65.7 mg, 154 μmol) was hydrolyzed using General Procedure E to afford 64.1 mg of crude acid 19s (101% crude yield).
2-((1-(tert-Butyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19t).
Ester 18t (65.5 mg, 140 μmol) was hydrolyzed using General Procedure E to afford 62.2 mg of crude acid 19t (98% crude yield).
2-((1-Cyclohexyl-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19u).
Ester 18u (111 mg, 224 μmol) was hydrolyzed using General Procedure E to afford 104 mg of crude acid 19u (96% crude yield).
2-((1-Isobutyl-3-phenyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19v).
Ester 18v (118 mg, 251 μmol) was hydrolyzed using General Procedure E to afford 112 mg of crude acid 19v (98% crude yield).
2-((3-Isopropyl-1-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19w).
Ester 18w (124 mg, 316 μmol) was hydrolyzed using General Procedure E to afford 124 mg of crude acid 19w (104% crude yield).
2-((3-Cyclohexyl-1-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19x).
Ester 18x (105 mg, 242 μmol) was hydrolyzed using General Procedure E to afford 102 mg of crude acid 19x (101% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-3-(o-tolyl)-1H-pyrazol-5-yl)amino)benzoic acid (19y).
Ester 18y (138 mg, 313 μmol) was hydrolyzed using General Procedure E to afford 133 mg of crude acid 19y (100% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-3-(m-tolyl)-1H-pyrazol-5-yl)amino)benzoic acid (19z).
Ester 18z (154 mg, 349 μmol) was hydrolyzed using General Procedure E to afford 166 mg of crude acid 19z (112% crude yield).
2,4-Bis(methoxymethoxy)-6-((3-(3-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)benzoic acid (19aa)
Ester 18aa (161 mg, 352 μmol) was hydrolyzed using General Procedure E to afford 142 mg of crude acid 19aa (91% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)benzoic acid (19ab).
Ester 18ab (161 mg, 325 μmol) was hydrolyzed using General Procedure E to afford 152 mg of crude acid 19ab (97% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-3-(p-tolyl)-1H-pyrazol-5-yl)amino)benzoic acid (19ac).
Ester 18ac (146 mg, 331 μmol) was hydrolyzed using General Procedure E to afford 142 mg of crude acid 19ac (100% crude yield).
2,4-Bis(methoxymethoxy)-6-((3-(4-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)benzoic acid (19ad).
Ester 18ad (155 mg, 339 μmol) was hydrolyzed using General Procedure E to afford 144 mg of crude acid (96% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)benzoic acid (19ae).
Ester 18ae (174 mg, 351 μmol) was hydrolyzed using General Procedure E to afford 174 mg of crude acid 19ae (105% crude yield).
2-((3-(4-(tert-Butyl)phenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4,6-bis(methoxymethoxy)benzoic acid (19af).
Ester 18af (167 mg, 345 μmol) was hydrolyzed using General Procedure E to afford 138 mg of crude acid 19af (85% crude yield).
2,4-Bis(methoxymethoxy)-6-((1-methyl-3-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)benzoic acid (19ag).
Ester 18ag (175 mg, 342 μmol) was hydrolyzed using General Procedure E to afford 156 mg of crude acid 19ag (92% crude yield).
4-(2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (20).
Amide 14a (38 mg, 68 μmol) was deprotected using General Procedure F to afford 24 mg of 20 (74% yield). 1H NMR (400 MHz, (CD3)2SO) δ 9.66 (s, 1H), 9.31 (s, 1H), 7.45 – 7.13 (m, 3H), 7.13 – 6.92 (m, 3H), 6.68 (d, J = 8.5 Hz, 1H), 5.85 (d, J = 2.1 Hz, 1H), 5.78 (s, 1H), 5.71 (d, J = 2.0 Hz, 1H), 4.89 (s, 2H), 4.72 (s, 2H), 3.62 (s, 3H), 2.03 (s, 3H). 13C NMR (126 MHz, (CD3)2SO) δ 166.6, 159.2, 158.4, 155.4, 146.0, 143.8, 143.7, 140.1, 129.3, 128.8, 127.2, 122.8, 113.6, 103.7, 98.0, 94.3, 92.8, 55.0, 50.0, 40.4, 13.9. LC/MS (m/z): 471.207 [M+H+]; UPLC tR 1.31 min.
4-(2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (21).
Amide 14b was deprotected using General Procedure F (52.6 mg, 84.7 μmol) to afford 25.9 mg of 21 (57% yield). 1H NMR (400 MHz, CD3OD) δ 7.75 – 7.66 (m, 2H), 7.42 – 7.32 (m, 3H), 7.32 – 7.21 (m, 5H), 7.06 (d, J = 8.7 Hz, 2H), 6.65 (d, J = 8.7 Hz, 2H), 6.46 (s, 1H), 5.93 (dd, J = 14.2, 2.1 Hz, 2H), 5.17 (s, 2H), 4.96 – 4.59 (m, 4H), 3.63 (s, 3H). 13C NMR (126 MHz, (CD3)2SO) δ 166.6, 159.4, 158.5, 155.6, 148.7, 143.6, 141.2, 136.6 (br), 133.5, 129.0, 128.8, 128.5, 127.4, 127.3, 124.8, 122.8, 113.7, 104.0, 96.1, 94.6, 93.0, 55.0, 50.6, 40.4. LC/MS (m/z): 533.257 [M+H+]; UPLC tR 1.63 min.
5-((1-((4-Methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (22).
Crude acid 19a (31.7 mg, 69.3 μmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (20.1 mg, 104 μmol), and triethylamine (72 μL, 520 μmol) using General Procedure G to give 21.8 mg of MOM-protected intermediate (56% yield) after purification via automated flash chromatography (10% to 50% acetone in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.56 – 8.42 (m, 1H), 7.67 – 7.43 (m, 1H), 7.21 (ddd, J = 13.1, 7.7, 4.9 Hz, 1H), 7.06 (ddd, J = 9.9, 6.0, 2.6 Hz, 2H), 6.70 – 6.57 (m, 2H), 6.45 (d, J = 2.5 Hz, 1H), 6.39 (dd, J = 7.5, 2.1 Hz, 1H), 6.23 (dd, J = 13.2, 2.1 Hz, 1H), 5.84 (s, 1H), 5.30 – 4.72 (m, 9H), 4.61 – 4.41 (m, 1H), 3.68 (d, J = 2.8 Hz, 3H), 3.44 (dd, J = 4.3, 2.0 Hz, 6H), 2.23 (d, J = 3.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.2, 167.2, 159.9, 159.0, 158.9, 157.6, 157.2, 155.4, 155.4, 149.4, 149.3, 147.6, 147.6, 143.9, 143.8, 139.4, 139.2, 131.0, 130.6, 130.3, 129.9, 128.8, 128.6, 128.5, 122.5, 122.4, 113.9, 113.8, 107.1, 98.5, 98.5, 96.5, 95.4, 95.3, 95.1, 95.1, 94.3, 94.2, 56.6, 56.5, 56.3, 56.2, 55.2, 55.1, 53.8, 53.4, 52.6, 51.4, 51.3, 51.3, 50.5, 29.3, 14.2, 14.2. LC/MS (m/z): 560.225 [M+H+]; UPLC tR 1.41 min
The MOM-protected intermediate (21.8 mg, 39.0 μmol) was deprotected using General Procedure F to afford 3.0 mg of 22 (16% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (d, J = 5.0 Hz, 1H), 7.76 (s, 1H), 7.40 – 7.30 (m, 1H), 7.00 (d, J = 8.7 Hz, 2H), 6.66 (d, J = 8.7 Hz, 2H), 5.96 – 5.85 (m, 2H), 5.83 (d, J = 2.1 Hz, 1H), 5.04 (s, 2H), 4.96 – 4.43 (m, 4H), 3.65 (s, 3H), 2.13 (s, 3H). LC/MS (m/z): 472.234 [M+H+]; UPLC tR 1.12 min.
5-((1-((4-Methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (23).
Acid 19b (31.7 mg, 69.3 μmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (20.1 mg, 104 μmol), and triethylamine (72 μL, 520 μmol) using General Procedure G to give 43.2 mg of MOM-protected intermediate (77% yield) after purification via automated flash chromatography (4% to 40% acetone in hexanes and 0% to 3% methanol in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.57 – 8.40 (m, 1H), 7.78 (dq, J = 6.4, 1.4 Hz, 2H), 7.66 – 7.46 (m, 1H), 7.42 – 7.34 (m, 2H), 7.33 – 7.18 (m, 2H), 7.18 – 7.06 (m, 2H), 6.73 – 6.58 (m, 2H), 6.53 (d, J = 4.4 Hz, 1H), 6.46 – 6.34 (m, 2H), 6.30 (dd, J = 18.9, 2.1 Hz, 1H), 5.16 (d, J = 17.6 Hz, 4H), 5.06 (d, J = 3.8 Hz, 2H), 5.02 – 4.78 (m, 3H), 4.56 (d, J = 16.2 Hz, 1H), 3.69 (d, J = 3.1 Hz, 3H), 3.52 – 3.40 (m, 6H). LC/MS (m/z): 622.237 [M+H+]; UPLC tR 1.71 min.
The MOM-protected intermediate (43.2 mg, 69.5 μmol) was deprotected using General Procedure F to afford 11.5 mg of 23 (31% yield) after purification using mass-guided preparative HPLC. 1H NMR (500 MHz, CD3OD) δ 8.40 (d, J = 5.1 Hz, 1H), 7.75 (d, J = 7.5 Hz, 1H), 7.72 – 7.63 (m, 2H), 7.38 – 7.30 (m, 2H), 7.30 – 7.20 (m, 1H), 7.15 – 7.04 (m, 2H), 6.73 – 6.66 (m, 2H), 6.45 (s, 1H), 6.01 – 5.90 (m, 1H), 5.19 (s, 2H), 4.99 – 4.49 (m, 4H), 3.65 (s, 2H), 2.65 (s, 3H). LC/MS (m/z): 535.173 [M+H+]; UPLC tR 1.40 min.
5-((1-((4-Methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (24).
Acid 19a (39 mg, 85 μmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (14 mg, 130 μmol), and triethylamine (24 μL, 170 μmol) using General Procedure G to give 28.1 mg of MOM-protected intermediate (60% yield) after purification via automated flash chromatography (15% to 60% acetone in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.17 (m, 2H), 7.13 – 6.98 (m, 2H), 6.73 – 6.61 (m, 2H), 6.43 – 6.31 (m, 2H), 6.20 (d, J = 2.1 Hz, 1H), 5.84 (d, J = 2.3 Hz, 1H), 5.20 – 5.10 (m, 2H), 5.09 – 4.97 (m, 4H), 4.77 – 4.56 (m, 3H), 4.31 (dd, J = 13.6, 7.2 Hz, 1H), 3.68 (d, J = 1.0 Hz, 3H), 3.48 – 3.38 (m, 6H), 2.23 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.8, 159.7, 159.0, 158.9, 155.2, 147.6, 143.6, 139.4, 139.3, 128.7, 128.6, 128.6, 128.6, 113.9, 113.9, 107.7, 107.6, 98.7, 98.6, 96.5, 96.4, 95.2, 95.1, 95.1, 94.2, 56.5, 56.5, 56.2, 55.1, 55.1, 53.7, 51.2, 51.2, 46.6, 46.4, 45.6, 45.3, 29.2, 14.1. LC/MS (m/z): 549.199 [M+H+]; UPLC tR 1.28 min.
The MOM-protected intermediate (28.1 mg, 51.2 μmol) was deprotected using General Procedure F to afford 3.6 mg of 24 (15% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.44 (s, 1H), 7.06 – 6.95 (m, 2H), 6.75 – 6.65 (m, 2H), 5.95 – 5.87 (m, 2H), 5.82 (d, J = 2.1 Hz, 1H), 5.03 (s, 2H), 4.77 – 4.22 (m, 4H), 3.68 (s, 3H), 2.65 (s, 3H), 2.15 (s, 3H). LC/MS (m/z): 461.207 [M+H+]; UPLC tR 0.94 min.
5-((1-((4-Methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (25).
Acid 19b (90 mM in 1:1 CH2Cl2:THF, 1.0 mL, 90 μmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (15 mg, 140 μmol), and triethylamine (25 μL, 180 μmol) using General Procedure G to give 33 mg of MOM-protected intermediate (61% yield) after purification via automated flash chromatography (10% to 40% acetone in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.85 – 7.75 (m, 2H), 7.39 (t, J = 7.6 Hz, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.16 (d, J = 8.2 Hz, 2H), 6.76 – 6.65 (m, 2H), 6.50 – 6.35 (m, 3H), 6.29 (dd, J = 8.7, 1.9 Hz, 1H), 5.15 (s, 1H), 5.05 (s, 2H), 4.83 – 4.50 (m, 3H), 4.35 (t, J = 12.4 Hz, 1H), 3.71 (dd, J = 2.1, 0.9 Hz, 3H), 3.45 (s, 3H), 3.44 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.8, 159.8, 159.0, 158.9, 155.2, 150.1, 143.4, 143.4, 140.1, 140.0, 133.6, 128.7, 128.5, 128.3, 128.2, 127.6, 125.3, 113.9, 113.9, 107.7, 107.7, 96.6, 96.4, 95.3, 95.2, 94.2, 56.5, 56.5, 56.2, 55.1, 55.1. LC/MS (m/z): 611.255 [M+H+]; UPLC tR 1.61 min.
The MOM-protected intermediate (33.5 mg, 54.9 μmol) was deprotected using General Procedure F to afford 7.2 mg of 25 (25% yield) after purification using mass-guided preparative HPLC. 1H NMR (500 MHz, CD3OD) δ 7.71 (dd, J = 8.0, 1.4 Hz, 2H), 7.46 – 7.31 (m, 3H), 7.30 – 7.21 (m, 1H), 7.13 – 7.00 (m, 2H), 6.75 – 6.68 (m, 2H), 6.46 (s, 1H), 5.92 (dd, J = 17.4, 2.1 Hz, 2H), 5.18 (s, 2H), 4.77 – 4.17 (m, 4H), 3.68 (d, J = 0.6 Hz, 3H). LC/MS (m/z): 523.132 [M+H+]; UPLC tR 1.31 min.
4-(4-Fluoro-2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (26).
Acid 19a (30 mg, 66 μmol) was coupled with 4-fluoroisoindoline (13 mg, 98 μmol), and triethylamine (18 μL, 130 μmol) using General Procedure G to give 38 mg of MOM-protected intermediate (78% yield) after purification via automated flash chromatography (10% to 45% acetone in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.30 (dd, J = 7.9, 5.2 Hz, 1H), 7.17 – 7.03 (m, 2H), 7.03 – 6.88 (m, 1H), 6.72 – 6.61 (m, 2H), 6.46 – 6.33 (m, 2H), 6.25 (dd, J = 13.2, 2.1 Hz, 1H), 5.85 (d, J = 2.5 Hz, 1H), 5.16 (dd, J = 9.5, 3.3 Hz, 2H), 5.06 (d, J = 10.6 Hz, 4H), 5.00 – 4.76 (m, 3H), 4.53 (d, J = 14.9 Hz, 1H), 3.68 (d, J = 7.4 Hz, 3H), 3.49 – 3.38 (m, 6H), 2.24 (s, 3H). 19F NMR (376 MHz, CDCl3) δ −117.44 (dd, J = 9.1, 5.1 Hz), −117.90 (dd, J = 9.1, 5.0 Hz). 13C NMR (101 MHz, CDCl3) δ 167.0, 166.8, 159.9, 159.9, 159.0, 158.9, 156.8, 156.5, 155.3, 147.6, 147.6, 143.8, 143.7, 140.0, 139.9, 139.5, 139.5, 139.3, 139.3, 129.9, 129.9, 129.8, 128.7, 128.6, 128.5, 123.4, 118.7, 118.6, 118.2, 114.3, 114.1, 114.1, 114.0, 113.9, 113.9, 107.5, 107.2, 98.5, 98.3, 96.5, 95.4, 95.3, 95.1, 95.1, 94.3, 56.6, 56.3, 55.1, 55.1, 53.0, 52.3, 51.4, 51.3, 50.0, 49.2, 14.2. LC/MS (m/z): 578.22 [M+H+]; UPLC tR 1.65 min.
The MOM-protected intermediate (27.5 mg, 47.7 μmol) was deprotected using General Procedure F to afford 6.9 mg of 26 (30% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.33 (q, J = 7.7 Hz, 1H), 7.09 (s, 1H), 7.04 – 6.94 (m, 3H), 6.66 (d, J = 8.7 Hz, 2H), 5.96 – 5.87 (m, 2H), 5.84 (d, J = 2.1 Hz, 1H), 5.03 (s, 2H), 4.92 – 4.53 (m, 4H), 3.64 (s, 3H), 2.13 (s, 3H). LC/MS (m/z): 489.214 [M+H+]; UPLC tR 1.36 min.
4-(4-Fluoro-2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (27).
Acid 19b (90 mM in 1:1 CH2Cl2:THF, 1.0 mL, 90 μmol) was coupled with 4-fluoroisoindoline (17 mg, 140 μmol), and triethylamine (25 μL, 180 μmol) using General Procedure G to give 42 mg of MOM-protected intermediate (72% yield) after purification via automated flash chromatography (8% to 30% acetone in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.86 – 7.73 (m, 2H), 7.39 (t, J = 7.7 Hz, 2H), 7.35 – 7.23 (m, 2H), 7.19 – 7.08 (m, 3H), 6.98 (dt, J = 12.5, 8.4 Hz, 1H), 6.73 – 6.65 (m, 2H), 6.52 (d, J = 3.3 Hz, 1H), 6.46 – 6.37 (m, 2H), 6.32 (dd, J = 16.3, 2.1 Hz, 1H), 5.29 (s, 1H), 5.25 – 5.11 (m, 4H), 5.07 (s, 2H), 5.03 – 4.81 (m, 3H), 4.56 (dd, J = 14.7, 5.9 Hz, 1H), 3.69 (d, J = 8.1 Hz, 3H), 3.48 – 3.43 (m, 6H). 19F NMR (376 MHz, CDCl3) δ −117.44 (dd, J = 9.1, 4.9 Hz), −117.88 (dd, J = 9.4, 5.1 Hz). 13C NMR (101 MHz, CDCl3) δ 166.9, 166.8, 160.0, 159.9, 159.3, 159.1, 159.0, 156.8, 155.4, 150.1, 150.1, 143.7, 143.5, 140.0, 140.0, 139.9, 139.5, 133.7, 130.0, 129.9, 129.8, 128.7, 128.7, 128.6, 128.4, 128.2, 127.6, 125.4, 123.6, 123.4, 123.3, 123.1, 118.6, 118.2, 114.3, 114.2, 114.1, 114.0, 113.9, 113.9, 107.6, 107.3, 96.6, 96.3, 96.0, 95.4, 95.3, 95.3, 95.3, 94.3, 94.3, 56.6, 56.3, 55.1, 55.1, 53.1, 52.3, 51.9, 51.8, 50.0, 49.3. LC/MS (m/z): 639.306 [M+H+]; UPLC tR 2.00 min.
The MOM-protected intermediate (41.7 mg, 65.3 μmol) was deprotected using General Procedure F to afford 19.2 mg of 27 (53% yield) after purification using mass-guided preparative HPLC. 1H NMR (500 MHz, CD3OD) δ 7.76 – 7.52 (m, 3H), 7.41 – 7.20 (m, 4H), 7.06 (d, J = 8.5 Hz, 3H), 6.98 (t, J = 8.8 Hz, 1H), 6.74 – 6.62 (m, 2H), 6.46 (d, J = 0.9 Hz, 1H), 5.93 (ddd, J = 14.0, 2.1, 0.9 Hz, 2H), 5.18 (s, 2H), 4.93 – 4.53 (m, 4H), 3.64 (d, J = 0.9 Hz, 3H). 13C NMR (126 MHz, (CD3)2SO) δ 166.6, 159.5, 158.5, 157.2 (d, 1JC-F = 244.1 Hz), 155.7, 148.7, 143.7, 141.2, 133.5, 129.9 (d, 3JC-F = 4.8 Hz), 129.0, 128.7, 128.5, 127.3, 124.7, 123.1 (br), 119.1, 113.67 (app d, ovrlp), 113.63, 103.7, 96.2, 94.6, 93.2, 54.9, 50.5, 40.4. LC/MS (m/z): 551.250 [M+H+]; UPLC tR 1.65 min.
4-(5-Fluoro-2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (28).
Acid 19a (31.1 mg, 68.0 μmol) was coupled with 5-fluoroisoindoline hydrochloride (17.7 mg, 102 μmol), and triethylamine (28 μL, 204 μmol) using General Procedure G to give 33.3 mg of MOM-protected intermediate (85% yield) after purification via automated flash chromatography (20% to 50% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.17 – 6.80 (m, 4H), 6.66 (dd, J = 8.6, 1.8 Hz, 2H), 6.43 – 6.34 (m, 2H), 6.24 (dd, J = 2.1, 0.7 Hz, 1H), 5.85 (s, 1H), 5.15 (q, J = 6.6 Hz, 2H), 5.05 (dd, J = 11.3, 1.7 Hz, 4H), 4.96 – 4.74 (m, 3H), 4.55 – 4.40 (m, 1H), 3.68 (s, 3H), 3.51 – 3.37 (m, 6H), 2.24 (s, 3H). 19F NMR (376 MHz, CDCl3) δ −114.63 – −114.72 (m), −114.72 – −114.83 (m). 13C NMR (101 MHz, CDCl3) δ 166.9, 166.8, 163.9, 163.8, 161.4, 161.3, 159.9, 159.9, 158.9, 155.3, 155.3, 147.6, 143.8, 143.7, 139.3, 138.6, 138.6, 138.3, 138.2, 132.0, 131.7, 128.7, 128.7, 128.6, 124.3, 124.2, 123.8, 123.7, 115.1, 115.0, 114.9, 114.8, 113.9, 110.3, 110.1, 109.8, 109.6, 107.5, 107.5, 98.5, 98.4, 96.5, 95.4, 95.3, 95.1, 94.3, 56.6, 56.3, 55.1, 55.1, 52.9, 52.3, 52.0, 52.0, 51.5, 51.3, 14.2. LC/MS (m/z): 577.206 [M+H+]; UPLC tR 1.71 min.
The MOM-protected intermediate (29.9 mg, 51.9 μmol) was deprotected using General Procedure F to afford 9.1 mg of 28 (36% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.25 (s, 1H), 7.07 – 6.91 (m, 4H), 6.72 – 6.57 (m, 2H), 5.93 – 5.84 (m, 2H), 5.83 (d, J = 2.1 Hz, 1H), 5.03 (s, 2H), 4.91 – 4.48 (m, 4H), 3.65 (s, 3H), 2.14 (d, J = 6.9 Hz, 4H). LC/MS (m/z): 489.244 [M+H+]; UPLC tR 1.34 min
4-(5-Fluoro-2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (29).
Acid 19b (90 mM in 1:1 CH2Cl2:THF, 1.0 mL, 90 μmol) was coupled with 5-fluoroisoindoline hydrochloride (23 mg, 140 μmol), and triethylamine (38 μL, 270 μmol) using General Procedure G to give 39 mg of MOM-protected intermediate (68% yield) after purification via automated flash chromatography (8% to 30% acetone in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.83 – 7.74 (m, 2H), 7.38 (dd, J = 8.4, 6.9 Hz, 2H), 7.32 – 7.27 (m, 1H), 7.12 (ddd, J = 11.7, 7.9, 3.6 Hz, 2H), 7.04 – 6.80 (m, 2H), 6.72 – 6.59 (m, 2H), 6.48 (d, J = 5.9 Hz, 1H), 6.43 – 6.36 (m, 2H), 6.30 (t, J = 1.9 Hz, 1H), 5.16 (d, J = 9.5 Hz, 4H), 5.06 (s, 2H), 4.98 – 4.73 (m, 3H), 4.57 – 4.43 (m, 1H), 3.69 (s, 3H), 3.50 – 3.38 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 166.8, 166.8, 163.9, 163.8, 161.5, 161.4, 159.9, 159.9, 159.0, 155.4, 155.4, 150.1, 143.6, 143.6, 140.0, 140.0, 138.6, 138.5, 138.3, 138.2, 133.7, 132.0, 132.0, 131.7, 131.7, 128.7, 128.6, 128.6, 128.4, 128.3, 127.6, 125.4, 124.3, 124.2, 123.9, 123.8, 115.1, 115.0, 114.9, 114.8, 113.9, 110.3, 110.1, 109.8, 109.6, 107.6, 107.6, 96.7, 96.2, 96.2, 95.4, 95.4, 95.3, 94.3, 56.6, 56.3, 55.1, 55.1, 52.9, 52.4, 52.0, 51.8, 51.5. LC/MS (m/z): 639.306 [M+H+]; UPLC tR 1.98 min.
The MOM-protected intermediate (39.3 mg, 61.5 μmol) was deprotected using General Procedure F to afford 6.6 mg of 29 (19% yield) after purification using mass-guided preparative HPLC. 1H NMR (500 MHz, CD3OD) δ 7.72 – 7.60 (m, 2H), 7.34 (dd, J = 8.4, 6.9 Hz, 2H), 7.28 – 7.22 (m, 1H), 7.21 (s, 1H), 7.08 – 7.00 (m, 2H), 6.98 (dd, J = 9.0, 7.0 Hz, 2H), 6.72 – 6.64 (m, 2H), 6.45 (s, 1H), 5.93 (dd, J = 12.7, 2.1 Hz, 2H), 5.17 (s, 2H), 4.97 – 4.43 (m, 4H), 3.65 (s, 3H). 13C NMR (126 MHz, (CD3)2SO) δ 166.5, 161.8 (d, 1JC-F = 241.3 Hz), 159.4, 158.5, 155.6, 148.7, 143.6, 141.2, 133.4, 129.0, 128.8, 128.5, 127.3, 124.7, 124.5 (d, 3JC-F = 9.5 Hz), 114.3 (d, 2JC-F = 22.9 Hz), 113.6, 110.0 (d, 2JC-F = 22.9 Hz), 103.9, 96.2, 94.6, 93.1, 55.0, 50.5, 40.4. LC/MS (m/z): 551.250 [M+H+]; UPLC tR 1.64 min.
N-(Cyclopropylmethyl)-2,4-dihydroxy-6-((1-((4-methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)-N-methylbenzamide (30).
Acid 19a (49 mg, 110 μmol) was coupled with (cyclopropylmethyl)methylamine (27 mg, 320 μmol), and triethylamine (30 μL, 210 μmol) using General Procedure G to give 43 mg of MOM-protected intermediate (76% yield) after purification via automated flash chromatography (10% to 35% acetone in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.19 – 7.07 (m, 2H), 6.80 (t, J = 8.6 Hz, 2H), 6.43 – 6.26 (m, 1H), 6.25 – 6.15 (m, 1H), 5.84 (d, J = 8.5 Hz, 1H), 5.19 – 4.92 (m, 6H), 3.75 (d, J = 4.1 Hz, 3H), 3.50 – 3.30 (m, 7H), 3.10 – 2.87 (m, 3H), 2.31 – 2.17 (m, 3H), 1.07 – −0.15 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 167.5, 167.2, 159.5, 159.4, 159.0, 159.0, 155.0, 154.7, 147.7, 147.6, 143.9, 143.9, 139.7, 139.6, 128.9, 128.8, 128.8, 128.7, 114.0, 113.9, 107.4, 107.2, 98.3, 97.9, 96.6, 96.4, 95.0, 95.0, 94.8, 94.7, 94.3, 94.3, 56.4, 56.3, 56.2, 56.2, 55.4, 55.2, 55.2, 51.1, 51.0, 36.2, 32.4, 14.2, 9.9, 9.2, 3.9, 3.5, 3.4, 3.4. LC/MS (m/z): 525.249 [M+H+]; UPLC tR 1.66 min
The MOM-protected intermediate (42.7 mg, 81.4 μmol) was deprotected using General Procedure F to afford 19.2 mg of 30 (54% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.07 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 5.89 (s, 1H), 5.87 – 5.79 (m, 2H), 5.05 (s, 2H), 3.74 (s, 3H), 3.19 (dd, J = 6.9, 4.6 Hz, 2H), 3.02 (s, 3H), 2.20 (s, 4H), 0.99 – 0.82 (m, 1H), 0.53 – 0.32 (m, 2H), 0.12 (ddt, J = 37.2, 9.5, 4.8 Hz, 2H). LC/MS (m/z): 437.213 [M+H+]; UPLC tR 1.28 min.
N-(Cyclopropylmethyl)-2,4-dihydroxy-6-((1-((4-methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)-N-methylbenzamide (31).
Acid 19b (90 mM in 1:1 CH2Cl2:THF, 1.0 mL, 90 μmol) was coupled with (cyclopropylmethyl)methylamine (23 mg, 270 μmol), and triethylamine (25 μL, 180 μmol) using General Procedure G to give 40 mg of MOM-protected intermediate (75% yield) after purification via automated flash chromatography (8% to 30% acetone in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.80 (dt, J = 8.2, 1.6 Hz, 2H), 7.39 (t, J = 7.6 Hz, 2H), 7.33 – 7.27 (m, 1H), 7.22 (dd, J = 8.8, 2.6 Hz, 2H), 6.87 – 6.71 (m, 2H), 6.41 – 6.31 (m, 2H), 6.30 – 6.24 (m, 1H), 5.29 – 5.09 (m, 4H), 5.03 (s, 2H), 3.76 (d, J = 4.5 Hz, 3H), 3.50 – 3.30 (m, 7H), 3.17 – 2.92 (m, 4H), 1.04 – −0.10 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 167.4, 167.2, 159.5, 159.4, 159.1, 159.1, 155.1, 154.8, 150.1, 150.1, 143.8, 143.7, 140.5, 140.3, 133.8, 133.7, 128.9, 128.9, 128.5, 128.5, 128.4, 127.6, 125.4, 114.0, 114.0, 107.5, 107.3, 96.8, 96.6, 96.0, 95.5, 95.1, 95.0, 94.8, 94.3, 94.3, 56.4, 56.4, 56.3, 56.2, 55.2, 55.2, 51.6, 51.6, 51.1, 36.2, 32.5, 10.0, 9.3, 3.9, 3.5, 3.5, 3.4. LC/MS (m/z): 587.306 [M+H+]; UPLC tR 1.95 min
The MOM-protected intermediate (39.8 mg, 54.9 μmol) was deprotected using General Procedure F to afford 5.7 mg of 19b (17% yield) after purification using mass-guided preparative HPLC. 1H NMR (500 MHz, CD3OD) δ 7.78 – 7.73 (m, 2H), 7.38 (dd, J = 8.3, 7.0 Hz, 2H), 7.32 – 7.26 (m, 1H), 7.18 – 7.12 (m, 2H), 6.87 – 6.81 (m, 2H), 6.45 (s, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.20 (s, 2H), 3.75 (s, 3H), 3.26 – 3.15 (m, 2H), 3.03 (s, 3H), 0.99 – 0.84 (m, 1H), 0.50 – 0.33 (m, 2H), 0.12 (ddq, J = 42.7, 9.6, 4.8 Hz, 2H). LC/MS (m/z): 499.182 [M+H+]; UPLC tR 1.60 min
5-((1-((4-methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)-4-(5-((1-methylpiperidin-4-yl)amino)-2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (32).
Amide 16a (41.8 mg, 62.3 μmol) was deprotected using General Procedure F to afford 3.3 mg of 32 (9.1% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.52 (s, 1H), 7.08 – 6.94 (m, 3H), 6.63 (d, J = 8.7 Hz, 4H), 5.91 (d, J = 2.1 Hz, 1H), 5.88 (s, 1H), 5.82 (d, J = 2.0 Hz, 1H), 5.01 (s, 3H), 4.84 – 4.29 (m, 4H), 3.64 (s, 3H), 3.51 (s, 1H), 3.03 – 2.81 (m, 4H), 2.72 (s, 3H), 2.65 (s, 1H), 2.14 (s, 5H), 1.66 (s, 1H). LC/MS (m/z): 583.336 [M+H+]; UPLC tR 0.88 min.
5-((1-((4-Methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4-(5-((1-methylpiperidin-4-yl)amino)-2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (33).
Acid 19b 77 mM in 1:1 CH2Cl2:THF, 1.0 mL, 77 μmol) was coupled with N-(1-methylpiperidin-4-yl)isoindolin-5-amine dihydrogenchloride (25 mg, 81 μmol), and triethylamine (85 μL, 610 μmol) using General Procedure G to give 35 mg of MOM-protected intermediate (59% yield) after purification via silica gel flash chromatography (96:4:1 CH2Cl2:methanol:conc. NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 7.6 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.30 (d, J = 7.2 Hz, 1H), 7.21 – 7.05 (m, 2H), 6.71 – 6.61 (m, 2H), 6.60 – 6.46 (m, 1H), 6.40 (dd, J = 12.6, 10.4 Hz, 3H), 6.26 (dd, J = 8.6, 2.1 Hz, 1H), 5.15 (d, J = 6.9 Hz, 4H), 5.05 (d, J = 2.0 Hz, 2H), 4.81 (dt, J = 21.1, 14.4 Hz, 3H), 3.68 (d, J = 1.6 Hz, 3H), 3.47 – 3.35 (m, 6H), 2.81 (s, 1H), 2.30 (d, J = 8.6 Hz, 3H), 2.20 – 1.92 (m, 3H), 1.49 (d, J = 11.6 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 166.8, 166.7, 159.7, 159.0, 155.4, 150.1, 147.1, 147.0, 143.5, 140.0, 137.8, 137.5, 133.7, 128.8, 128.5, 128.4, 127.6, 125.4, 124.8, 124.5, 123.7, 123.2, 113.9, 113.6, 113.4, 106.8, 106.3, 96.5, 96.4, 95.3, 94.3, 56.5, 56.3, 55.1, 54.5, 52.6, 52.2, 51.8, 51.6, 46.2, 32.4. LC/MS (m/z): 733.603 [M+H+]; UPLC tR 1.45 min
The MOM-protected intermediate (34.7 mg, 47.4 μmol) was deprotected using General Procedure F to afford 7.7 mg of 33 (25% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.52 (s, 1H), 7.76 – 7.63 (m, 2H), 7.42 – 7.22 (m, 3H), 7.10 – 6.94 (m, 2H), 6.71 – 6.60 (m, 3H), 6.55 (s, 1H), 6.44 (s, 1H), 5.92 (dd, J = 16.0, 2.1 Hz, 1H), 5.15 (s, 2H), 4.83 – 4.43 (m, 4H), 3.64 (s, 3H), 3.52 (s, 1H), 3.36 (d, J = 14.5 Hz, 2H), 2.99 (s, 2H), 2.76 (s, 3H), 2.18 (d, J = 14.2 Hz, 2H), 1.68 (s, 2H). LC/MS (m/z): 645.481 [M+H+]; UPLC tR 1.21 min
4-(5-(2-(Dimethylamino)ethoxy)-2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (34).
Amide 16b (37.7 mg, 58.4 μmol) was deprotected using General Procedure F to afford 8.2 mg of 34 (25% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.52 (s, 1H), 7.19 (s, 1H), 7.02 – 6.96 (m, 2H), 6.93 (d, J = 8.7 Hz, 2H), 6.65 (d, J = 8.7 Hz, 2H), 5.92 (d, J = 2.1 Hz, 1H), 5.88 (s, 1H), 5.83 (d, J = 2.1 Hz, 1H), 5.02 (s, 3H), 4.85 – 4.45 (m, 3H), 4.22 (t, J = 5.3 Hz, 2H), 3.65 (s, 3H), 3.25 – 3.18 (m, 2H), 2.70 (s, 7H), 2.65 (s, 5H), 2.13 (s, 3H). LC/MS (m/z): 558.328 [M+H+]; UPLC tR 0.90 min.
4-(5-(2-(Dimethylamino)ethoxy)-2,3-dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (35).
To a suspension of crude carboxylic acid 19b (57 mg, 110 μmol) and 2-(isoindolin-5-yloxy)-N,N-dimethylethan-1-amine dihydrochloride (24 mg, 86 μmol) in CH2Cl2 (0.7 mL) and THF (0.7 mL) was added triethylamine (60 μL, 430 μmol) followed by HATU (26 mg, 69 μmol). After the suspension was stirred overnight at room temperature, additional 2-(isoindolin-5-yloxy)-N,N-dimethylethan-1-amine dihydrochloride (12 mg, 43 μmol), triethylamine (60 μL, 430 μmol) and HATU (13 mg, 34 μmol) were added to the reaction. After stirring overnight, the reaction was diluted with CH2Cl2. The reaction mixture was washed with saturated NaHCO3 (aq.), brine and then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude mixture was purified via automated flash chromatography (2% to 5% methanol in CH2Cl2) to afford 47 mg of MOM-protected intermediate (60% yield). 1H NMR (400 MHz, CDCl3) δ 7.82 – 7.73 (m, 2H), 7.38 (t, J = 7.6 Hz, 2H), 7.30 (d, J = 7.1 Hz, 1H), 7.23 – 7.01 (m, 3H), 6.91 – 6.80 (m, 1H), 6.66 (d, J = 8.6 Hz, 2H), 6.45 (d, J = 5.8 Hz, 1H), 6.42 – 6.33 (m, 2H), 6.28 (dd, J = 3.4, 2.1 Hz, 1H), 5.16 (d, J = 7.4 Hz, 4H), 5.05 (d, J = 1.9 Hz, 2H), 4.95 – 4.73 (m, 3H), 4.55 – 4.43 (m, 1H), 4.07 (dt, J = 17.1, 5.6 Hz, 2H), 3.68 (d, J = 1.0 Hz, 3H), 3.44 (t, J = 1.4 Hz, 6H), 2.80 (dt, J = 9.2, 5.5 Hz, 2H), 2.40 (s, 3H), 2.38 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.8, 166.8, 159.8, 159.1, 158.8, 158.7, 155.4, 150.1, 143.6, 140.1, 138.0, 137.6, 133.8, 128.7, 128.5, 128.4, 127.5, 125.4, 123.7, 123.3, 114.8, 114.6, 114.0, 108.8, 108.6, 108.0, 96.7, 96.4, 95.4, 94.4, 66.0, 58.1, 56.5, 56.2, 55.1, 53.1, 52.5, 52.2, 51.8, 51.5, 45.7. LC/MS (m/z): 708.551 [M+H+]; UPLC tR 1.45 min.
To a solution of the resulting MOM-protected intermediate (46.7 mg, 66.0 μmol) in methanol (6.4 mL) at room temperature was added HCl (aq.) (2 M, 0.21 mL, 420 μmol) and stirred at 50 ºC overnight. Additional HCl (aq.) (2 M, 0.21 mL, 420 μmol) was added to the reaction mixture and stirred at 50 ºC overnight. The reaction was cooled the room temperature and volatile materials were condensed in vacuo. The crude residue was purified using mass-guided preparative HPLC to afford 29.2 mg of 35 (71% yield). 1H NMR (400 MHz, CD3OD) δ 7.68 (dt, J = 6.4, 1.3 Hz, 2H), 7.37 – 7.28 (m, 2H), 7.28 – 7.20 (m, 1H), 7.16 (d, J = 8.6 Hz, 1H), 7.10 – 6.99 (m, 2H), 6.97 – 6.79 (m, 2H), 6.74 – 6.59 (m, 2H), 6.43 (s, 1H), 5.94 (dd, J = 14.2, 2.0 Hz, 2H), 5.16 (s, 2H), 4.85 – 4.45 (m, 4H), 4.25 (t, J = 5.1 Hz, 2H), 3.64 (s, 3H), 3.41 (t, J = 4.9 Hz, 2H), 3.34 (s, 1H), 2.84 (s, 6H). LC/MS (m/z): 620.473 [M+H+]; UPLC tR 1.20 min.
5-((1-((4-Methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)-4-[5-(4-methylpiperazin-1-yl)-2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (36).
Amide 16c (38.8 mg, 59.1 μmol) was deprotected using General Procedure F to afford 14.6 mg of 36 (43% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.04 – 6.92 (m, 4H), 6.68 – 6.58 (m, 2H), 5.92 (d, J = 2.1 Hz, 1H), 5.88 (s, 1H), 5.83 (d, J = 2.1 Hz, 1H), 5.01 (s, 3H), 4.68 (d, J = 59.6 Hz, 4H), 3.63 (s, 3H), 3.30 (dt, J = 3.7, 1.9 Hz, 4H), 3.01 (d, J = 5.1 Hz, 4H), 2.66 – 2.61 (m, 3H), 2.13 (s, 3H). LC/MS (m/z): 569.311 [M+H+]; UPLC tR 0.77 min.
N-Benzyl-2,4-dihydroxy-6-((1-((4-methoxyphenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)-N-methylbenzamide (37).
Amide 16d (39.1 mg, 69.7 μmol) was deprotected using General Procedure F to afford 12.6 mg of 37 (38% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.19 (q, J = 4.2, 3.4 Hz, 5H), 7.10 – 7.02 (m, 2H), 6.83 – 6.73 (m, 2H), 5.95 – 5.82 (m, 3H), 5.04 (s, 2H), 4.53 (d, J = 14.7 Hz, 2H), 3.70 (s, 3H), 2.84 (s, 3H), 2.21 (s, 3H). LC/MS (m/z): 473.16 [M+H+]; UPLC tR 1.49 min.
5-((1-((4-Methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4-(pyrrolidine-1-carbonyl)benzene-1,3-diol (38).
Acid 19b (31.5 mg, 60.6 μmol) was coupled with pyrrolidine (6.5 mg, 91 μmol), and triethylamine (17 μL, 120 μmol) using General Procedure G to give 22.6 mg of MOM-protected intermediate (65% yield) after purification via automated flash chromatography (30% to 60% ethyl acetate in hexanes, 10% to 20% ethyl acetate in CH2Cl2, and 10% to 30% acetone in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.83 – 7.76 (m, 2H), 7.39 (t, J = 7.6 Hz, 2H), 7.29 (t, J = 7.3 Hz, 1H), 7.25 – 7.16 (m, 2H), 6.86 – 6.74 (m, 2H), 6.69 (s, 1H), 6.40 – 6.32 (m, 2H), 6.28 (d, J = 2.1 Hz, 1H), 5.26 – 5.09 (m, 4H), 5.03 (s, 2H), 3.76 (s, 3H), 3.47 (s, 6H), 3.42 (s, 3H), 3.21 (s, 1H), 2.80 (s, 3H), 1.90 (d, J = 6.3 Hz, 3H), 1.76 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 166.1, 159.5, 159.1, 155.3, 150.1, 143.5, 140.4, 133.8, 129.0, 128.5, 127.6, 125.4, 114.0, 108.5, 96.6, 95.7, 95.3, 95.2, 94.3, 56.4, 56.2, 55.2, 51.6, 47.5, 45.6, 25.8, 24.5. LC/MS (m/z): 573.457 [M+H+]; UPLC tR 1.87 min
The MOM-protected intermediate (22.6 mg, 39.5 μmol) was deprotected using General Procedure F to afford 8.5 mg of 38 (44% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.82 – 7.66 (m, 2H), 7.38 (dd, J = 8.2, 6.8 Hz, 2H), 7.32 – 7.24 (m, 1H), 7.14 (s, 2H), 6.85 (d, J = 8.7 Hz, 2H), 6.45 (s, 1H), 5.89 (d, J = 4.1 Hz, 2H), 5.19 (s, 2H), 3.75 (s, 3H), 3.41 – 3.23 (m, 5H), 1.98 – 1.69 (m, 4H). LC/MS (m/z): 485.377 [M+H+]; UPLC tR 1.53 min.
N,N-Diethyl-2,4-dihydroxy-6-((1-((4-methoxyphenyl)methyl)-3-phenyl-1H-pyrazol-5-yl)amino)benzamide (39).
Acid 19b (35.6 mg, 68.5 μmol) was coupled with diethylamine (7.5 mg, 100 μmol), and triethylamine (19 μL, 140 μmol) using General Procedure G to give 25.2 mg of MOM-protected intermediate (64% yield) after purification via automated flash chromatography (20% to 50% ethyl acetate in hexanes and 10% to 30% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 7.85 – 7.76 (m, 2H), 7.43 – 7.36 (m, 2H), 7.33 – 7.28 (m, 1H), 7.24 – 7.14 (m, 2H), 6.81 (d, J = 8.7 Hz, 2H), 6.38 – 6.36 (m, 1H), 6.35 (d, J = 2.1 Hz, 1H), 6.27 (d, J = 2.1 Hz, 1H), 6.11 (s, 1H), 5.29 – 5.10 (m, 4H), 5.02 (s, 2H), 3.76 (s, 3H), 3.64 (dq, J = 13.7, 6.9 Hz, 1H), 3.46 (s, 3H), 3.42 (s, 3H), 3.33 (dp, J = 14.3, 7.1 Hz, 2H), 3.18 (dq, J = 14.3, 7.1 Hz, 1H), 1.14 (t, J = 7.1 Hz, 3H), 1.02 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.9, 159.3, 159.1, 154.7, 150.2, 143.4, 140.5, 133.7, 128.8, 128.5, 128.5, 127.6, 125.4, 114.1, 108.2, 96.8, 95.8, 95.1, 95.0, 94.3, 56.4, 56.2, 55.2, 51.5, 43.0, 39.0, 14.3, 12.9. LC/MS (m/z): 575.485 [M+H+]; UPLC tR 1.95 min.
The MOM-protected intermediate (25.4 mg, 44.2 μmol) was deprotected using General Procedure F to afford 13.5 mg of 39 (63% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.79 – 7.71 (m, 2H), 7.38 (td, J = 7.3, 6.4, 1.3 Hz, 2H), 7.32 – 7.26 (m, 1H), 7.21 – 7.10 (m, 2H), 6.94 – 6.78 (m, 2H), 6.44 (s, 1H), 5.91 (dd, J = 14.7, 2.1 Hz, 2H), 5.19 (s, 2H), 3.75 (s, 3H), 3.43 (dq, J = 14.1, 7.1 Hz, 2H), 3.36 – 3.24 (m, 2H), 1.07 (t, J = 7.1 Hz, 6H). LC/MS (m/z): 487.406 [M+H+]; UPLC tR 1.61 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (40).
Amide 14c (62.4 mg, 114 μmol) was deprotected using General Procedure F to afford 23.8 mg of 40 (46% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.43 (d, J = 2.1 Hz, 1H), 7.28 (s, 4H), 7.02 – 6.96 (m, 2H), 6.67 – 6.59 (m, 2H), 6.10 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.77 (d, J = 2.1 Hz, 1H), 5.10 (s, 2H), 4.98 – 4.56 (m, 4H), 3.63 (s, 3H). 13C NMR (126 MHz, (CD3)2SO) δ 166.6, 159.3, 158.5, 155.5, 143.7, 143.6, 139.73, 139.66, 138.1, 129.1, 128.8, 127.3, 122.8, 122.8, 113.6, 103.3, 98.8, 94.4, 92.6, 55.0, 50.4, 40.4. LC/MS (m/z): 457.227 [M+H+]; UPLC tR 1.30 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-4-methyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (41).
Amide 14d (49.1 mg, 87.9 μmol) was deprotected using General Procedure F to afford 12.2 mg of 41 (30% yield) after purification using mass-guided preparative HPLC 1H NMR (400 MHz, CD3OD) δ 7.31 (d, J = 14.5 Hz, 5H), 6.98 (d, J = 8.6 Hz, 2H), 6.65 – 6.54 (m, 3H), 5.87 (d, J = 2.1 Hz, 1H), 5.34 (d, J = 2.0 Hz, 1H), 5.03 (s, 2H), 4.94 – 4.73 (m, 4H), 3.63 (s, 3H), 1.90 (s, 3H). 13C NMR (126 MHz, (CD3)2SO) δ 166.6, 159.3, 158.4, 155.5, 144.4, 138.4, 136.7 (br), 136.6, 129.4, 128.9, 127.3, 122.9, 113.5, 109.9, 103.4, 93.8, 91.6, 55.0, 50.5, 40.4, 8.3. LC/MS (m/z): 471.251 [M+H+]; UPLC tR 1.34 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-4-(propan-2-yl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (42).
Amide 14e (53.7 mg, 91.5 μmol) was deprotected using General Procedure F to afford 22.4 mg of 42 (49% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.39 (s, 1H), 7.29 (s, 4H), 6.98 (d, J = 8.2 Hz, 2H), 6.62 – 6.50 (m, 3H), 5.88 (d, J = 2.1 Hz, 1H), 5.31 (d, J = 2.1 Hz, 1H), 5.00 (s, 2H), 4.97 – 4.70 (m, 4H), 3.61 (s, 3H), 2.73 (p, J = 6.9 Hz, 1H), 1.14 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 499.255 [M+H+]; UPLC tR 1.47 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-((4-methoxyphenyl)methyl)-4-phenyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (43).
Amide 14f (25.4 mg, 40.9 μmol) was deprotected using General Procedure F to afford 15.0 mg of 43 (69% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.81 (s, 1H), 7.50 – 7.44 (m, 2H), 7.33 – 7.16 (m, 9H), 7.10 (dd, J = 13.6, 7.6 Hz, 3H), 6.65 (d, J = 8.2 Hz, 2H), 5.87 (d, J = 2.1 Hz, 1H), 5.37 (d, J = 2.1 Hz, 1H), 5.12 (s, 2H), 3.65 (s, 3H). LC/MS (m/z): 533.257 [M+H+]; UPLC tR 1.53 min.
5-((4-Benzyl-1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (44).
Amide 14g (45.3 mg, 71.4 μmol) was deprotected using General Procedure F to afford 23.5 mg of 44 (60% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.29 (d, J = 4.5 Hz, 5H), 7.09 (d, J = 5.6 Hz, 4H), 7.04 – 6.95 (m, 3H), 6.67 – 6.56 (m, 3H), 5.88 (d, J = 2.1 Hz, 1H), 5.36 (d, J = 2.1 Hz, 1H), 5.05 (s, 2H), 4.96 – 4.57 (m, 4H), 3.68 – 3.59 (m, 5H). LC/MS (m/z): 547.17 [M+H+]; UPLC tR 1.70 min.
(5,7-dihydro-6H-pyrrolo[3,4-b]pyridin-6-yl)(2-((3-ethyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)methanone (45).
Acid 19c (51.7 mg, 110 μmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (31.7 mg, 164 μmol), and triethylamine (115 μL, 822 μmol) using General Procedure G to give 33.2 mg of MOM-protected intermediate (53% yield) after purification via silica gel flash chromatography (1% to 4% methanol in CH2Cl2) and manual flash chromatography (20:80:1 CH2Cl2:ethyl acetate:conc. NH4OH (aq.)) 1H NMR (400 MHz, CDCl3) δ 8.57 – 8.41 (m, 1H), 7.21 (ddd, J = 13.4, 7.6, 4.9 Hz, 1H), 7.08 (dd, J = 8.4, 5.9 Hz, 2H), 6.67 (dd, J = 8.7, 3.2 Hz, 2H), 6.47 (d, J = 2.8 Hz, 1H), 6.40 (dd, J = 8.3, 2.1 Hz, 1H), 6.26 (dd, J = 11.8, 2.1 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.22 – 5.00 (m, 6H), 4.99 – 4.78 (m, 3H), 4.52 (d, J = 15.5 Hz, 1H), 3.69 (d, J = 2.6 Hz, 3H), 3.45 (dd, J = 4.4, 1.7 Hz, 6H), 2.61 (qd, J = 7.7, 1.9 Hz, 2H), 1.29 – 1.14 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 167.2, 167.1, 159.9, 159.0, 158.9, 157.6, 157.2, 155.4, 155.3, 153.7, 153.7, 149.4, 149.3, 143.8, 143.7, 139.4, 139.2, 131.0, 130.6, 130.3, 129.9, 128.8, 128.6, 128.6, 128.5, 122.5, 122.4, 113.9, 113.8, 107.1, 96.9, 96.8, 96.6, 95.4, 95.2, 95.2, 95.1, 94.3, 94.3, 56.6, 56.5, 56.2, 56.2, 55.2, 55.1, 53.4, 52.5, 51.4, 51.4, 51.3, 50.5, 22.0, 13.9. LC/MS (m/z): 574.383 [M+H+]; UPLC tR 1.51 min.
The MOM-protected intermediate (33.2 mg, 57.9 μmol) was deprotected using General Procedure F to afford 22.9 mg of 45 (81% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.43 (dd, J = 5.0, 1.5 Hz, 1H), 7.74 (s, 1H), 7.33 (dd, J = 7.8, 5.0 Hz, 1H), 7.06 – 6.95 (m, 2H), 6.65 (d, J = 8.7 Hz, 2H), 5.93 (d, J = 3.1 Hz, 2H), 5.83 (d, J = 2.1 Hz, 1H), 5.05 (s, 2H), 4.97 – 4.48 (m, 4H), 3.64 (s, 3H), 2.51 (q, J = 7.6 Hz, 2H), 1.17 (t, J = 7.6 Hz, 3H). LC/MS (m/z): 486.259 [M+H+]; UPLC tR 1.16 min.
5-((3-Ethyl-1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (46).
Acid 19c (54.0 mg, 115 μmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (18.8 mg, 172 μmol), and triethylamine (32 μL, 230 μmol) using General Procedure G to give 26.9 mg of MOM-protected intermediate (42% yield) after purification via silica gel flash chromatography (12% to 35% acetone in CH2Cl2) and manual flash chromatography (96:4 CH2Cl2:methanol). 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.7 Hz, 2H), 6.38 (dd, J = 7.1, 2.1 Hz, 1H), 6.32 (d, J = 13.6 Hz, 1H), 6.24 (dd, J = 4.2, 2.1 Hz, 1H), 5.88 (d, J = 1.7 Hz, 1H), 5.15 (td, J = 7.3, 5.2 Hz, 2H), 5.06 (d, J = 2.5 Hz, 4H), 4.82 – 4.59 (m, 3H), 4.32 (t, J = 12.4 Hz, 1H), 3.71 (d, J = 1.8 Hz, 3H), 3.50 – 3.39 (m, 6H), 2.62 (q, J = 7.6 Hz, 2H), 1.23 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.8, 159.8, 159.0, 158.9, 155.2, 153.8, 143.6, 139.3, 139.2, 128.8, 128.6, 128.6, 118.3, 117.8, 113.9, 113.9, 107.7, 107.6, 97.1, 97.0, 96.6, 95.3, 95.2, 94.3, 56.5, 56.2, 55.2, 55.2, 51.3, 46.6, 46.4, 45.6, 45.3, 22.0, 13.9. LC/MS (m/z): 563.401 [M+H+]; UPLC tR 1.39 min.
The MOM-protected intermediate (26.9 mg, 47.8 μmol) was deprotected using General Procedure F to afford 15.3 mg of 46 (67% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.43 (s, 1H), 7.00 (d, J = 8.7 Hz, 2H), 6.69 (d, J = 8.7 Hz, 2H), 5.98 – 5.89 (m, 2H), 5.82 (d, J = 2.1 Hz, 1H), 5.04 (s, 2H), 4.83 – 4.28 (m, 4H), 3.67 (s, 3H), 2.53 (q, J = 7.6 Hz, 2H), 1.19 (t, J = 7.6 Hz, 3H). LC/MS (m/z): 475.321 [M+H+]; UPLC tR 1.04 min
5-((1-((4-Methoxyphenyl)methyl)-3-(propan-2-yl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (47).
Acid 19d (53.2 mg, 110 μmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (31.7 mg, 164 μmol), and triethylamine (115 μL, 822 μmol) using General Procedure G to give 44.1 mg of MOM-protected intermediate (68% yield) after purification via silica gel flash chromatography (1% to 4% methanol in CH2Cl2) and manual flash chromatography (20:80:1 CH2Cl2:ethyl acetate:conc. NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.59 – 8.42 (m, 1H), 7.56 (dd, J = 66.5, 7.7 Hz, 1H), 7.25 – 7.17 (m, 1H), 7.08 (dd, J = 8.3, 5.8 Hz, 2H), 6.68 (dd, J = 8.7, 2.9 Hz, 2H), 6.48 (d, J = 9.5 Hz, 1H), 6.40 (dd, J = 9.2, 2.1 Hz, 1H), 6.29 (dd, J = 10.5, 2.0 Hz, 1H), 5.89 (d, J = 3.2 Hz, 1H), 5.24 – 5.01 (m, 6H), 5.01 – 4.71 (m, 3H), 4.52 (d, J = 15.3 Hz, 1H), 3.69 (d, J = 2.7 Hz, 3H), 3.45 (d, J = 1.4 Hz, 3H), 3.44 (d, J = 1.9 Hz, 3H), 2.94 (p, J = 6.9 Hz, 1H), 1.25 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.2, 167.1, 160.0, 159.0, 158.8, 158.2, 158.2, 157.6, 157.2, 155.4, 155.3, 149.4, 149.3, 143.8, 143.7, 139.2, 139.0, 131.0, 130.6, 129.9, 128.8, 128.6, 128.5, 128.5, 122.5, 122.4, 113.9, 113.8, 107.2, 107.2, 96.7, 95.4, 95.3, 95.2, 95.2, 95.1, 94.4, 94.3, 56.6, 56.5, 56.2, 56.2, 55.2, 55.1, 53.4, 52.5, 51.4, 51.4, 51.3, 50.5, 28.3, 22.8. LC/MS (m/z): 588.408 [M+H+]; UPLC tR 1.61 min.
The MOM-protected intermediate (44.1 mg, 71.0 μmol) was deprotected using General Procedure F to afford 24.8 mg of 47 (66% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.43 (dd, J = 5.0, 1.5 Hz, 1H), 7.74 (s, 1H), 7.33 (dd, J = 7.8, 5.0 Hz, 1H), 6.98 (d, J = 8.7 Hz, 2H), 6.65 (d, J = 8.7 Hz, 2H), 5.99 – 5.87 (m, 2H), 5.83 (d, J = 2.1 Hz, 1H), 5.06 (s, 2H), 4.97 – 4.49 (m, 4H), 3.63 (s, 3H), 2.84 (hept, J = 6.9 Hz, 1H), 1.20 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 500.285 [M+H+]; UPLC tR 1.25 min.
5-((1-((4-Methoxyphenyl)methyl)-3-(propan-2-yl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (48).
Acid 19d (57.5 mg, 118 μmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (19.4 mg, 178 μmol), and triethylamine (33 μL, 240 μmol) using General Procedure G to give 23.2 mg of MOM-protected intermediate (34% yield) after purification via silica gel flash chromatography (12% to 35% acetone in CH2Cl2) and manual flash chromatography (96:4 CH2Cl2:methanol). 1H NMR (400 MHz, CDCl3) δ 7.40 – 7.19 (m, 1H), 7.09 (d, J = 8.1 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 6.44 – 6.27 (m, 2H), 6.26 (t, J = 2.1 Hz, 1H), 5.89 (d, J = 2.2 Hz, 1H), 5.21 – 5.00 (m, 6H), 4.78 – 4.57 (m, 3H), 4.38 – 4.22 (m, 1H), 3.71 (d, J = 1.4 Hz, 3H), 3.44 (d, J = 1.3 Hz, 6H), 2.94 (p, J = 6.9 Hz, 1H), 1.25 (dd, J = 7.0, 2.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.8, 159.8, 159.0, 158.9, 158.2, 155.2, 143.5, 143.4, 139.3, 139.2, 128.7, 128.6, 113.9, 113.9, 107.8, 107.7, 96.7, 96.6, 95.5, 95.4, 95.3, 95.2, 94.4, 56.5, 56.2, 55.2, 55.1, 51.3, 51.3, 46.6, 46.4, 45.6, 45.3, 28.3, 22.8. LC/MS (m/z): 577.382 [M+H+]; UPLC tR 1.47 min.
The MOM-protected intermediate (23.2 mg, 40.2 μmol) was deprotected using General Procedure F to afford 15.3 mg of 48 (78% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.43 (s, 1H), 7.05 – 6.92 (m, 2H), 6.74 – 6.63 (m, 2H), 5.99 – 5.88 (m, 2H), 5.82 (d, J = 2.1 Hz, 1H), 5.05 (s, 2H), 4.79 – 4.22 (m, 4H), 3.67 (s, 3H), 2.85 (h, J = 6.9 Hz, 1H), 1.21 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 489.303 [M+H+]; UPLC tR 1.13 min.
(2-((3-(tert-Butyl)-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(isoindolin-2-yl)methanone (49).
Amide 14h (45.0 mg, 74.9 μmol) was deprotected using General Procedure F to afford 12.5 mg of 49 (33% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.33 – 7.17 (m, 4H), 6.93 (d, J = 8.7 Hz, 2H), 6.61 (d, J = 8.7 Hz, 2H), 5.98 (s, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.82 (d, J = 2.1 Hz, 1H), 5.07 (s, 2H), 4.93 – 4.56 (m, 4H), 3.61 (s, 3H), 1.26 (s, 9H). 13C NMR (126 MHz, (CD3)2SO) δ 166.6, 159.32, 159.27, 158.4, 155.5, 143.7, 139.8, 136.7 (br), 129.4, 128.5, 127.2, 122.8, 113.6, 103.8, 94.6, 94.4, 92.9, 54.9, 50.2, 40.4, 31.9, 30.3. LC/MS (m/z): 513.208 [M+H+]; UPLC tR 1.73 min
(2-((3-cyclopropyl-1-(4-methoxybenzyl)-1H-pyrazol-5-yl)amino)-4,6-dihydroxyphenyl)(5,7-dihydro-6H-pyrrolo[3,4-b]pyridin-6-yl)methanone (50).
Acid 19e (52.5 mg, 109 μmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (31.5 mg, 163 μmol), and triethylamine (114 μL, 814 μmol) using General Procedure G to give 43.7 mg of MOM-protected intermediate (69% yield) after purification via automated flash system (1% to 4% methanol in CH2Cl2) and manual flash chromatography (20:80:1 CH2Cl2:ethyl acetate:conc. NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.56 – 8.44 (m, 1H), 7.68 – 7.40 (m, 1H), 7.25 – 7.16 (m, 1H), 7.08 (dd, J = 8.5, 5.0 Hz, 2H), 6.67 (dd, J = 8.7, 2.6 Hz, 2H), 6.48 (d, J = 7.3 Hz, 1H), 6.40 (dd, J = 8.9, 2.1 Hz, 1H), 6.29 – 6.20 (m, 1H), 5.69 (s, 1H), 5.24 – 4.97 (m, 7H), 5.00 – 4.79 (m, 3H), 4.51 (d, J = 15.8 Hz, 1H), 3.69 (d, J = 2.2 Hz, 3H), 3.45 (d, J = 1.5 Hz, 3H), 3.44 (d, J = 1.8 Hz, 3H), 1.89 (dtd, J = 8.9, 5.6, 5.2, 2.8 Hz, 1H), 0.89 (dd, J = 8.6, 2.0 Hz, 2H), 0.68 (dd, J = 5.2, 2.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 167.2, 167.1, 159.9, 159.0, 158.9, 157.5, 157.2, 155.4, 155.3, 154.2, 154.1, 149.4, 149.3, 143.6, 143.6, 139.5, 139.3, 131.0, 130.6, 130.3, 129.9, 128.7, 128.6, 128.5, 128.5, 122.5, 122.4, 113.9, 113.8, 107.2, 107.2, 96.7, 95.4, 95.3, 95.2, 95.2, 94.6, 94.5, 94.3, 94.3, 56.6, 56.5, 56.3, 56.2, 55.2, 55.1, 53.4, 52.5, 51.4, 51.4, 51.3, 50.5, 9.6, 7.9. LC/MS (m/z): 586.38 [M+H+]; UPLC tR 1.46 min.
The MOM-protected intermediate (43.7 mg, 74.6 μmol) was deprotected using General Procedure F to afford 25.3 mg of 50 (68% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.43 (dd, J = 5.1, 1.5 Hz, 1H), 7.74 (s, 1H), 7.33 (dd, J = 7.7, 5.0 Hz, 1H), 7.03 – 6.90 (m, 2H), 6.72 – 6.57 (m, 2H), 5.92 (d, J = 2.1 Hz, 1H), 5.81 (d, J = 2.1 Hz, 1H), 5.74 (s, 1H), 5.03 (s, 2H), 4.94 – 4.44 (m, 4H), 3.64 (s, 4H), 1.78 (tt, J = 8.4, 5.0 Hz, 1H), 0.83 (dd, J = 8.5, 2.1 Hz, 2H), 0.61 (dd, J = 5.1, 2.0 Hz, 2H). LC/MS (m/z): 498.3 [M+H+]; UPLC tR 1.19 min
5-((3-Cyclopropyl-1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (51).
Acid 19e (59.7 mg, 123 μmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (20.2 mg, 185 μmol), and triethylamine (34 μL, 250 μmol) using General Procedure G to give 31.4 mg of MOM-protected intermediate (44% yield) after purification via automated flash system (12% to 35% acetone in CH2Cl2) and manual flash chromatography (96:4 CH2Cl2:methanol). 1H NMR (400 MHz, CDCl3) δ 7.08 (d, J = 8.4 Hz, 2H), 6.75 – 6.63 (m, 2H), 6.38 (dd, J = 7.1, 2.1 Hz, 1H), 6.29 (d, J = 15.5 Hz, 1H), 6.22 (dd, J = 5.2, 2.1 Hz, 1H), 5.69 (s, 1H), 5.21 – 5.09 (m, 2H), 5.04 (d, J = 4.3 Hz, 5H), 4.78 – 4.65 (m, 2H), 4.64 (s, 1H), 4.30 (t, J = 12.6 Hz, 1H), 3.71 (d, J = 2.1 Hz, 3H), 3.48 – 3.37 (m, 6H), 1.89 (td, J = 8.6, 4.3 Hz, 1H), 0.89 (dd, J = 8.5, 2.2 Hz, 2H), 0.76 – 0.57 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.8, 159.8, 159.0, 158.9, 155.2, 154.2, 143.5, 139.4, 139.3, 128.7, 128.6, 128.6, 128.6, 118.2, 117.7, 113.9, 113.9, 107.7, 107.7, 96.6, 95.3, 95.2, 94.9, 94.8, 94.3, 56.5, 56.5, 56.2, 55.2, 55.1, 51.3, 46.6, 46.4, 45.6, 45.3, 9.7, 7.9. LC/MS (m/z): 575.397 [M+H+]; UPLC tR 1.41 min.
The MOM-protected intermediate (31.4 mg, 54.6 μmol) was deprotected using General Procedure F to afford 17.7 mg of 51 (67% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.43 (s, 1H), 6.99 (d, J = 8.7 Hz, 2H), 6.69 (d, J = 8.7 Hz, 2H), 5.90 (d, J = 2.1 Hz, 1H), 5.80 (d, J = 2.1 Hz, 1H), 5.74 (s, 1H), 5.02 (s, 2H), 4.77 – 4.18 (m, 4H), 3.67 (s, 4H), 1.82 (tt, J = 8.4, 5.0 Hz, 1H), 0.86 (dd, J = 8.5, 2.1 Hz, 2H), 0.64 (dd, J = 5.1, 2.1 Hz, 2H). LC/MS (m/z): 487.318 [M+H+]; UPLC tR 1.08 min.
5-((3-Cyclopentyl-1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (52).
Acid 19f (44.7 mg, 87.4 μmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (25.3 mg, 131 μmol), and triethylamine (91.4 μL, 655 μmol) using General Procedure G to give 41.2 mg of MOM-protected intermediate (77% yield) after purification via automated flash system (1% to 4% methanol in CH2Cl2) and manual flash chromatography (55:45:1 CH2Cl2:ethyl acetate:conc. NH4OH (aq.) to 40:60:1 CH2Cl2:ethyl acetate:conc. NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.58 – 8.38 (m, 1H), 7.68 – 7.38 (m, 1H), 7.24 – 7.15 (m, 1H), 7.08 (dd, J = 8.4, 5.7 Hz, 2H), 6.67 (dd, J = 8.7, 3.0 Hz, 2H), 6.46 (d, J = 6.2 Hz, 1H), 6.40 (dd, J = 8.6, 2.1 Hz, 1H), 6.29 (dd, J = 11.1, 2.1 Hz, 1H), 5.88 (d, J = 2.7 Hz, 1H), 5.27 – 4.98 (m, 6H), 4.98 – 4.78 (m, 3H), 4.51 (d, J = 15.6 Hz, 1H), 3.69 (d, J = 2.7 Hz, 3H), 3.45 (dd, J = 5.0, 1.7 Hz, 6H), 3.04 (t, J = 8.2 Hz, 1H), 2.03 (d, J = 9.1 Hz, 2H), 1.85 – 1.33 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 167.2, 159.9, 158.8, 157.6, 157.2, 156.5, 156.5, 155.4, 155.3, 149.4, 149.3, 143.8, 143.7, 139.3, 139.1, 131.0, 130.6, 130.3, 129.9, 128.8, 128.6, 128.6, 128.5, 122.5, 122.4, 113.9, 113.8, 107.1, 96.6, 95.8, 95.7, 95.4, 95.2, 95.2, 95.1, 94.4, 94.3, 56.6, 56.5, 56.2, 56.2, 55.2, 55.1, 53.4, 51.4, 51.3, 50.5, 39.5, 33.4, 25.4. LC/MS (m/z): 614.431 [M+H+]; UPLC tR 1.72 min
The MOM-protected intermediate (40.8 mg, 66.5 μmol) was deprotected using General Procedure F to afford 22.9 mg of 52 (66% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.43 (dd, J = 4.9, 1.5 Hz, 1H), 7.74 (s, 1H), 7.33 (dd, J = 7.8, 5.1 Hz, 1H), 6.98 (d, J = 8.7 Hz, 2H), 6.65 (d, J = 8.7 Hz, 2H), 5.92 (d, J = 2.6 Hz, 2H), 5.83 (d, J = 2.1 Hz, 1H), 5.05 (s, 2H), 4.88 (s, 9H), 3.64 (s, 3H), 2.92 (d, J = 8.1 Hz, 1H), 1.96 (s, 2H), 1.80 – 1.48 (m, 6H). 13C NMR (101 MHz, (CD3)2SO) δ 166.8, 159.5, 158.4, 157.3, 155.6, 154.8, 148.8, 143.9, 140.0, 131.3, 129.4, 128.7, 122.4, 113.6, 103.4, 95.8, 94.4, 93.0, 55.0, 50.1, 40.4, 32.7, 24.9 LC/MS (m/z): 526.308 [M+H+]; UPLC tR 1.36 min
5-((3-Cyclopentyl-1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (53).
Acid 19f (45.3 mg, 88.6 μmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (14.5 mg, 133 μmol), and triethylamine (35 μL, 180 μmol) using General Procedure G to give 27.1 mg of MOM-protected intermediate (51% yield) after purification via automated flash system (12% to 35% acetone in CH2Cl2) and manual flash chromatography (96:4 CH2Cl2:methanol). 1H NMR (400 MHz, CDCl3) δ 7.15 – 6.99 (m, 2H), 6.70 (d, J = 8.2 Hz, 2H), 6.44 – 6.29 (m, 2H), 6.27 (q, J = 1.9 Hz, 1H), 5.88 (d, J = 2.2 Hz, 1H), 5.24 – 4.95 (m, 6H), 4.78 – 4.58 (m, 3H), 4.31 (t, J = 12.4 Hz, 1H), 3.71 (t, J = 1.4 Hz, 3H), 3.44 (d, J = 1.5 Hz, 6H), 3.06 (q, J = 8.1 Hz, 1H), 2.04 (s, 2H), 1.82 – 1.48 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.8, 159.8, 159.0, 158.9, 156.5, 156.5, 155.2, 143.4, 139.4, 139.3, 128.7, 128.6, 113.9, 113.9, 107.8, 107.7, 96.7, 96.7, 95.9, 95.8, 95.3, 95.3, 94.4, 56.5, 56.2, 55.2, 55.1, 51.3, 51.3, 46.6, 46.4, 45.6, 45.3, 39.5, 33.4, 33.4, 25.4. LC/MS (m/z): 603.404 [M+H+]; UPLC tR 1.59 min.
The MOM-protected intermediate (27.1 mg, 45.0 μmol) was deprotected using General Procedure F to afford 16.1 mg of 53 (70% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.42 (s, 1H), 6.99 (d, J = 8.7 Hz, 2H), 6.69 (d, J = 8.7 Hz, 2H), 6.01 – 5.87 (m, 2H), 5.82 (d, J = 2.1 Hz, 1H), 5.05 (s, 2H), 4.82 – 4.24 (m, 4H), 3.67 (s, 3H), 2.92 (s, 1H), 1.98 (d, J = 10.7 Hz, 2H), 1.87 – 1.43 (m, 6H). LC/MS (m/z): 515.325 [M+H+]; UPLC tR 1.25 min.
5-((3-(Furan-3-yl)-1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (54).
Acid 19g (52.5 mg, 103 μmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (29.8 mg, 155 μmol), and triethylamine (108 μL, 773 μmol) using General Procedure G to give 48.6 mg of MOM-protected intermediate (77% yield) after purification via automated flash system (0% to 3% methanol in CH2Cl2) and manual flash chromatography (55:45:1 CH2Cl2:ethyl acetate:conc. NH4OH (aq.) to 40:60:1 CH2Cl2:ethyl acetate:conc. NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.51 (dd, J = 21.5, 4.9 Hz, 1H), 7.75 (q, J = 1.2 Hz, 1H), 7.70 – 7.61 (m, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.44 (q, J = 1.7 Hz, 1H), 7.24 – 7.16 (m, 1H), 7.12 (dd, J = 8.7, 6.7 Hz, 2H), 6.74 (dd, J = 1.9, 0.9 Hz, 1H), 6.73 – 6.59 (m, 2H), 6.53 (d, J = 5.5 Hz, 1H), 6.42 (dd, J = 7.9, 2.1 Hz, 1H), 6.29 (dd, J = 15.9, 2.1 Hz, 1H), 6.16 (d, J = 2.0 Hz, 1H), 5.16 (dd, J = 11.1, 7.1 Hz, 5H), 5.07 (d, J = 2.5 Hz, 3H), 5.01 – 4.78 (m, 4H), 4.53 (d, J = 15.1 Hz, 1H), 3.69 (d, J = 3.1 Hz, 4H), 3.47 – 3.42 (m, 6H), 2.80 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.2, 167.1, 160.0, 159.1, 159.0, 157.4, 157.1, 155.5, 155.4, 149.3, 149.2, 143.5, 143.5, 143.5, 143.2, 143.2, 140.0, 139.8, 139.1, 131.1, 130.7, 130.3, 129.9, 128.6, 128.6, 128.4, 128.2, 122.5, 122.5, 120.1, 113.9, 113.9, 108.7, 107.2, 96.7, 96.3, 95.4, 95.3, 95.3, 94.3, 94.2, 56.6, 56.6, 56.3, 56.3, 55.2, 55.2, 53.4, 52.5, 51.7, 51.6, 51.4, 50.6, 47.4, 38.6, 8.7. LC/MS (m/z): 612.358 [M+H+]; UPLC tR 1.55 min.
The MOM-protected intermediate (48.6 mg, 79.5 μmol) was deprotected using General Procedure F to afford 28.7 mg of 54 (69% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.39 (dd, J = 5.0, 1.5 Hz, 1H), 7.76 (dd, J = 1.6, 0.8 Hz, 1H), 7.70 (s, 1H), 7.46 (t, J = 1.7 Hz, 1H), 7.29 (dd, J = 7.8, 5.0 Hz, 1H), 7.04 (d, J = 8.7 Hz, 2H), 6.72 – 6.59 (m, 3H), 6.26 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.14 (s, 2H), 5.02 – 4.45 (m, 4H). LC/MS (m/z): 524.279 [M+H+]; UPLC tR 1.25 min.
5-((3-(Furan-3-yl)-1-((4-methoxyphenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (55).
Acid 19g (51.7 mg, 101 μmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (16.6 mg, 152 μmol), and triethylamine (28 μL, 200 μmol) using General Procedure G to give 33.3 mg of MOM-protected intermediate (55% yield) after purification via automated flash system (10% to 30% acetone in CH2Cl2) and manual flash chromatography (96:4 CH2Cl2:methanol). 1H NMR (400 MHz, CDCl3) δ 7.81 (s, 1H), 7.48 – 7.31 (m, 1H), 7.17 (d, J = 7.8 Hz, 2H), 6.80 – 6.66 (m, 3H), 6.43 (d, J = 8.6 Hz, 1H), 6.31 (s, 1H), 6.16 (d, J = 2.4 Hz, 1H), 5.16 (d, J = 11.2 Hz, 4H), 5.06 (s, 2H), 4.86 – 4.55 (m, 3H), 4.33 (s, 1H), 3.72 (d, J = 1.9 Hz, 3H), 3.51 – 3.35 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.8, 159.8, 159.1, 159.0, 155.3, 143.5, 143.3, 143.3, 140.0, 140.0, 139.1, 139.1, 128.7, 128.3, 128.2, 120.1, 114.0, 113.9, 108.8, 107.9, 107.8, 96.7, 96.6, 96.5, 95.4, 95.3, 94.3, 56.5, 56.3, 55.2, 55.2, 51.6, 46.6, 46.5, 45.6, 45.4. LC/MS (m/z): 601.331 [M+H+]; UPLC tR 1.35 min
The MOM-protected intermediate (32.9 mg, 55.8 μmol) was deprotected using General Procedure F to afford 18.9 mg of 55 (67% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.84 – 7.78 (m, 1H), 7.49 (t, J = 1.7 Hz, 1H), 7.41 (s, 1H), 7.05 (d, J = 8.7 Hz, 2H), 6.76 – 6.66 (m, 3H), 6.27 (s, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.14 (s, 2H), 4.79 – 4.27 (m, 4H), 3.67 (s, 3H). LC/MS (m/z): 513.296 [M+H+]; UPLC tR 1.15 min.
(2,4-dihydroxy-6-((1-methyl-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (56).
Amide 14i (37.0 mg, 84.4 μmol) was deprotected using General Procedure F to afford 17.1 mg of 56 (58% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.36 (d, J = 2.0 Hz, 1H), 7.28 (s, 4H), 6.03 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.61 (d, J = 2.1 Hz, 1H), 5.07 – 4.70 (m, 4H), 3.64 (s, 3H). LC/MS (m/z): 351.292 [M+H+]; UPLC tR 1.09 min.
5-((1-Methyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (57).
Acid 19h (27.6 mg, 81.8 μmmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (23.7 mg, 123 μmol), and triethylamine (86 μL, 610 μmol) using General Procedure G to give 13.3 mg of MOM-protected intermediate (37% yield) after purification via an automated flash system (1% to 5% methanol in CH2Cl2) and manual flash chromatography (60:40:1 CH2Cl2:acetone:conc. NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.52 (dd, J = 19.1, 5.0 Hz, 1H), 7.77 – 7.50 (m, 1H), 7.44 (d, J = 2.0 Hz, 1H), 7.26 (s, 1H), 6.79 (d, J = 15.3 Hz, 1H), 6.45 (dd, J = 10.5, 2.1 Hz, 1H), 6.13 (dd, J = 4.4, 2.1 Hz, 1H), 6.03 (d, J = 2.2 Hz, 1H), 5.18 (d, J = 9.2 Hz, 3H), 5.11 – 4.81 (m, 4H), 4.79 – 4.53 (m, 1H), 3.70 (s, 3H), 3.46 (s, 3H), 3.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.5, 160.1, 160.1, 157.6, 157.2, 155.7, 155.6, 149.5, 149.3, 144.6, 144.4, 138.6, 131.1, 130.6, 129.9, 122.6, 122.4, 106.9, 99.0, 96.3, 96.2, 95.5, 95.3, 95.3, 95.2, 94.3, 94.2, 56.6, 56.6, 56.3, 56.2, 53.5, 52.7, 51.5, 50.7, 35.1. LC/MS (m/z): 440.426 [M+H+]; UPLC tR 1.13 min
The MOM-protected intermediate (13.3 mg, 30.3 μmol) was deprotected using General Procedure F to afford 5.1 mg of 57 (48% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 5.1, 1.5 Hz, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.36 (d, J = 2.0 Hz, 2H), 6.03 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.62 (d, J = 2.1 Hz, 1H), 4.90 (s, 4H), 3.65 (s, 3H). LC/MS (m/z): 352.218 [M+H+]; UPLC tR 0.77 min.
5-((1-Methyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (58).
Acid 19h (16.8 mg, 49.8 μmmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (8.2 mg, 75 μmol), and triethylamine (14 μL, 100 μmol) using General Procedure G to afford 4.2 mg of MOM-protected intermediate (20% yield) after purification via automated flash system (1% to 5% methanol in CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 46.2 Hz, 1H), 6.59 (d, J = 7.2 Hz, 1H), 6.42 (dd, J = 8.2, 2.1 Hz, 1H), 6.07 (d, J = 2.1 Hz, 1H), 6.01 (t, J = 1.9 Hz, 1H), 5.16 (q, J = 6.3, 4.9 Hz, 2H), 5.07 (d, J = 2.5 Hz, 2H), 4.98 – 4.64 (m, 3H), 4.42 (dd, J = 13.5, 6.5 Hz, 1H), 3.67 (s, 3H), 3.45 (s, 3H), 3.43 (d, J = 1.6 Hz, 3H). LC/MS (m/z): 429.318 [M+H+]; UPLC tR 1.03 min.
The MOM-protected intermediate (4.2 mg, 9.8 μmol) was deprotected using General Procedure F to afford 2.3 mg of 58 (69% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.45 (s, 1H), 7.38 (d, J = 2.1 Hz, 1H), 6.04 (d, J = 2.1 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.60 (d, J = 2.1 Hz, 1H), 4.80 – 4.43 (m, 4H), 3.65 (s, 3H). LC/MS (m/z): 341.235 [M+H+]; UPLC tR 0.66 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-(propan-2-yl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (59).
To a mixture of carboxylic acid 19i (34.9 mg, 95.5 μmmol) and isoindoline hydrochloride (22.3 mg, 143 μmol) in THF (0.62 mL) and CH2Cl2 (0.62 mL) was added triethylamine (53 μL, 380 μmol) followed by PyBOP (59.7, 115 μmol). After the reaction was stirred at room temperature overnight, the reaction mixture was diluted with CH2Cl2. The reaction mixture was washed twice with saturated NaHCO3 (aq.), once with brine and then dried with anhydrous Na2SO4. The salts were removed via gravity filtration and volatile materials were condensed in vacuo. The crude residue was dissolved in methanol (4.8) and HCl (2 M, 310 μL, 620 μmol) was added to the resulting mixture. The reaction was stirred at 50 ºC overnight. After cooling to room temperature, volatile materials were condensed in vacuo. The crude residue was purified using mass-guided preparative HPLC to afford 8.7 mg of 59 (24% yield over 2 steps). 1H NMR (400 MHz, CD3OD) δ 7.43 (d, J = 2.0 Hz, 1H), 7.29 (d, J = 1.7 Hz, 4H), 6.03 (d, J = 2.0 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.60 – 5.55 (m, 1H), 4.90 (s, 4H), 4.55 (p, J = 6.7 Hz, 1H), 1.36 (d, J = 6.7 Hz, 6H). LC/MS (m/z): 379.344 [M+H+]; UPLC tR 1.25 min
(5,7-dihydro-6H-pyrrolo[3,4-b]pyridin-6-yl)(2,4-dihydroxy-6-((1-isopropyl-1H-pyrazol-5-yl)amino)phenyl)methanone (60).
Synthesized using the same procedure for the synthesis of 59 with 19i (37.6 mg, 103 μmol), 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (29.8 mg, 154 μmol) and triethylamine (110 μL, 770 μmol) followed by MOM deprotection with HCl (2 M, 330 μL, 670 μmol) in methanol at 50 ºC overnight to afford 6.6 mg of 60 (17% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.55 (s, 1H), 8.45 (d, J = 4.8 Hz, 1H), 7.82 (d, J = 7.4 Hz, 1H), 7.43 (d, J = 1.6 Hz, 1H), 7.36 (dd, J = 7.4, 4.8 Hz, 1H), 6.04 (d, J = 1.6 Hz, 1H), 5.90 (d, J = 2.0 Hz, 1H), 5.59 (d, J = 2.0 Hz, 1H), 4.65–4.53 (m, 2H), 1.67 (d, J = 6.6 Hz, 6H). LC/MS (m/z): 380.359 [M+H+]; UPLC tR 0.93 min.
5-((1-(Propan-2-yl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (61).
Synthesized using the same procedure for the synthesis of 59 with 19i (36.7 mg, 100 μmol), 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (16.4 mg, 151 μmol) and triethylamine (28 μL, 200 μmol) followed by MOM deprotection with HCl (2 M, 330 μL, 650 μmol) in methanol at 50 ºC overnight to afford 11.7 mg of 61 (32% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.45 (t, J = 2.8 Hz, 2H), 6.04 (d, J = 2.0 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.56 (d, J = 2.1 Hz, 1H), 4.80 – 4.42 (m, 5H), 1.37 (d, J = 6.6 Hz, 6H). LC/MS (m/z): 369.332 [M+H+]; UPLC tR 0.82 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-(2-methylpropyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (62).
Carboxylic acid 19j (45 mg, 120 μmol) was subjected to General Procedure H1 to afford 17 mg of 62 (37% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.40 (d, J = 2.0 Hz, 1H), 7.28 (s, 4H), 6.06 (d, J = 2.0 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.74 (d, J = 2.1 Hz, 1H), 5.14 – 4.69 (m, 4H), 3.75 (d, J = 7.6 Hz, 2H), 3.34 (s, 2H), 2.09 (dh, J = 12.5, 6.3, 5.9 Hz, 1H), 0.79 (d, J = 6.7 Hz, 6H). LC/MS (m/z): 393.238 [M+H+]; UPLC tR 1.38 min
5-((1-(2-Methylpropyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (63).
Carboxylic acid 19j (46 mg, 120 μmol) was subjected to General Procedure H2 to afford 4.7 mg of 63 (9.9% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 5.2, 1.5 Hz, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.35 (dd, J = 7.8, 5.0 Hz, 1H), 6.06 (d, J = 2.0 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.75 (d, J = 2.1 Hz, 1H), 5.04 – 4.57 (m, 4H), 3.77 (d, J = 7.5 Hz, 2H), 2.12 (hept, J = 7.0 Hz, 1H), 0.81 (d, J = 6.7 Hz, 6H). LC/MS (m/z): 394.341 [M+H+]; UPLC tR 1.08 min.
5-((1-(2-Methylpropyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (64).
Carboxylic acid 19j (46 mg, 120 μmol) was subjected to General Procedure H3 to afford 13.2 mg of 64 (28% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.46 (s, 1H), 7.42 (d, J = 2.0 Hz, 1H), 6.06 (d, J = 2.1 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.72 (d, J = 2.1 Hz, 1H), 4.88 (s, 4H), 3.76 (d, J = 7.5 Hz, 2H), 2.12 (p, J = 6.9 Hz, 1H), 0.81 (d, J = 6.7 Hz, 6H). LC/MS (m/z): 383.27 [M+H+]; UPLC tR 0.95 min.
5-((1-(Cyclohexylmethyl)-1H-pyrazol-5-yl)amino)-4-(2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (65).
Carboxylic acid 19k (30.9 mg, 73.4 μmol) was subjected to General Procedure H1 to afford 12.4 mg of 65 (39% yield) after purification using mass-guided preparative HPLC 1H NMR (400 MHz, CD3OD) δ 7.40 (d, J = 2.1 Hz, 1H), 7.29 (s, 5H), 6.06 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.0 Hz, 1H), 5.69 (d, J = 2.0 Hz, 1H), 5.10 – 4.65 (m, 4H), 3.75 (d, J = 7.4 Hz, 2H), 1.77 (ddd, J = 11.1, 7.5, 3.6 Hz, 1H), 1.63 – 1.40 (m, 5H), 1.16 – 0.72 (m, 5H). 13C NMR (126 MHz, 2:1 (CD3)2SO: CD3OD) δ 167.6, 159.8, 155.8, 144.4, 140.4, 138.1, 136.9, 127.7, 123.1, 104.0, 99.5, 94.6, 92.9, 53.7, 40.4, 38.3, 30.4, 26.1, 25.5. LC/MS (m/z): 433.596 [M+H+]; UPLC tR 1.46 min.
5-((1-(Cyclohexylmethyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (66).
Synthesized using General Procedure H2 from carboxylic acid 19k (36.4 mg, 86.8 μmol) was subjected to General Procedure H2 to afford 13.8 mg of 66 (37% yield) after purification using mass-guided preparative HPLC 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 5.0, 1.4 Hz, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.36 (dd, J = 7.8, 5.0 Hz, 1H), 6.06 (d, J = 2.0 Hz, 1H), 5.93 (d, J = 2.0 Hz, 1H), 5.71 (d, J = 2.1 Hz, 1H), 4.89 (s, 4H), 3.77 (d, J = 7.4 Hz, 2H), 1.86 – 1.72 (m, 1H), 1.68 – 1.39 (m, 5H), 1.18 – 0.74 (m, 5H). LC/MS (m/z): 434.39 [M+H+]; UPLC tR 1.22 min.
5-((1-(Cyclohexylmethyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (67).
The product 67 was synthesized following General Procedure H3 from carboxylic acid 19k (37.2 mg, 88.7 μmol) was subjected to General Procedure H3 to afford 14.8 mg of 67 (40% yield) after purification using mass-guided preparative HPLC 1H NMR (400 MHz, CD3OD) δ 7.46 (s, 1H), 7.41 (d, J = 2.0 Hz, 1H), 6.06 (d, J = 2.0 Hz, 1H), 5.92 (d, J = 2.0 Hz, 1H), 5.69 (d, J = 2.1 Hz, 1H), 5.04 – 4.38 (m, 4H), 3.76 (d, J = 7.4 Hz, 2H), 1.79 (ddq, J = 11.3, 7.4, 3.7 Hz, 1H), 1.67 – 1.43 (m, 5H), 1.18 – 0.76 (m, 5H). LC/MS (m/z): 423.363 [M+H+]; UPLC tR 1.08 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-phenyl-1H-pyrazol-5-yl)amino]benzene-1,3-diol (68).
Synthesized using General Procedure H1 from carboxylic acid 19l (42 mg, 110 μmol) was subjected to General Procedure H1 to afford 2.4 mg of 68 (5.5% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.61 (d, J = 2.0 Hz, 1H), 7.49 – 7.40 (m, 2H), 7.34 – 7.16 (m, 8H), 6.26 (d, J = 2.0 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.85 (d, J = 2.1 Hz, 1H), 4.98 – 4.42 (m, 4H). LC/MS (m/z): 413.307 [M+H+]; UPLC tR 1.39 min.
5-((1-Phenyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (69).
Synthesized using General Procedure H2 from carboxylic acid 19l (43 mg, 110 μmol) was subjected to General Procedure H2 to afford 5.1 mg of 69 (11% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.45 (d, J = 5.0 Hz, 1H), 7.77 (s, 1H), 7.61 (d, J = 2.0 Hz, 1H), 7.50 – 7.41 (m, 2H), 7.39 – 7.29 (m, 3H), 7.29 – 7.18 (m, 1H), 6.26 (d, J = 2.0 Hz, 1H), 5.91 – 5.84 (m, 2H), 5.12 – 4.44 (m, 4H). LC/MS (m/z): 414.277 [M+H+]; UPLC tR 1.08 min.
5-((1-Phenyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (70).
Synthesized using General Procedure H3 from carboxylic acid 19l (44 mg, 110 μmol) was subjected to General Procedure H3 to afford 6.6 mg of 70 (15% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.62 (d, J = 2.0 Hz, 1H), 7.52 – 7.41 (m, 3H), 7.39 – 7.24 (m, 3H), 6.26 (d, J = 2.0 Hz, 1H), 5.85 (dd, J = 17.7, 2.1 Hz, 2H), 4.73 – 4.07 (m, 4H). LC/MS (m/z): 403.295 [M+H+]; UPLC tR 0.97 min.
5-((1-Cyclohexyl-1H-pyrazol-5-yl)amino)-4-(2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (71).
Synthesized using General Procedure H1 from carboxylic acid 19m (48.8 mg, 120 μmol) was subjected to General Procedure H1 to afford 18.3 mg of 71 (36% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.41 (d, J = 2.0 Hz, 1H), 7.37 – 7.20 (m, 4H), 6.03 (d, J = 2.0 Hz, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.59 (d, J = 2.1 Hz, 1H), 5.03 – 4.76 (m, 4H), 4.09 (d, J = 11.4 Hz, 1H), 1.98 – 1.71 (m, 9H), 1.63 (d, J = 10.8 Hz, 1H), 1.23 (dd, J = 26.4, 15.1 Hz, 2H). LC/MS (m/z): 419.041 [M+H+]; UPLC tR 1.46 min
5-((1-Cyclohexyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (72).
Synthesized using General Procedure H2 from carboxylic acid 19m (54.4 mg, 134 μmol) was subjected to General Procedure H2 to afford 14.3 mg of 72 (25% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 5.1, 1.5 Hz, 1H), 7.86 – 7.74 (m, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.36 (dd, J = 7.8, 5.0 Hz, 1H), 6.03 (d, J = 2.0 Hz, 1H), 5.90 (d, J = 2.1 Hz, 1H), 5.61 (d, J = 2.1 Hz, 1H), 4.88 (s, 4H), 4.12 (dt, J = 11.3, 6.4 Hz, 1H), 1.96 – 1.71 (m, 7H), 1.71 – 1.59 (m, 1H), 1.44 – 1.08 (m, 3H). LC/MS (m/z): 420.32 [M+H+]; UPLC tR 1.18 min.
5-((1-Cyclohexyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (73).
Synthesized using General Procedure H3 from carboxylic acid 19m (58.8 mg, 145 μmol) was subjected to General Procedure H3 to afford 22.5 mg of 73 (38% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.46 (s, 1H), 7.43 (d, J = 2.0 Hz, 1H), 6.04 (d, J = 2.0 Hz, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.58 (d, J = 2.1 Hz, 1H), 4.80 – 4.43 (m, 4H), 4.11 (dt, J = 10.9, 6.4 Hz, 1H), 1.92 – 1.72 (m, 7H), 1.71 – 1.61 (m, 1H), 1.44 – 1.12 (m, 3H). LC/MS (m/z): 409.337 [M+H+]; UPLC tR 1.05 min.
5-((1-Benzyl-1H-pyrazol-5-yl)amino)-4-(2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (74).
Acid 19n (42.1 mg, 102 μmol) was subjected to General Procedure H1 to afford 3.3 mg of 74 (7.6% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.45 (d, J = 2.0 Hz, 1H), 7.34 – 7.18 (m, 4H), 7.10 (dd, J = 4.0, 2.5 Hz, 3H), 7.03 (dd, J = 6.8, 3.0 Hz, 2H), 6.12 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.79 (d, J = 2.1 Hz, 1H), 5.18 (s, 2H), 5.01 – 4.52 (m, 4H). LC/MS (m/z): 427.333 [M+H+]; UPLC tR 1.37 min.
5-((1-Benzyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (75).
Acid 19n (42.6 mg, 103 μmol) was subjected to General Procedure H2 to afford 2.9 mg of 75 (6.6% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (d, J = 5.1 Hz, 1H), 7.76 (s, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.41 – 7.31 (m, 1H), 7.17 – 6.99 (m, 5H), 6.13 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.79 (d, J = 2.1 Hz, 1H), 5.19 (s, 2H), 5.05 – 4.11 (m, 4H). LC/MS (m/z): 428.347 [M+H+]; UPLC tR 1.08 min.
5-((1-Benzyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (76).
Acid 19n (43 mg, 100 μmol) was subjected to General Procedure H3 to afford 5.0 mg of 76 (12% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.51 – 7.33 (m, 2H), 7.15 (dd, J = 5.2, 1.9 Hz, 4H), 7.06 (dd, J = 6.9, 2.7 Hz, 2H), 6.12 (d, J = 2.0 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 5.19 (s, 3H), 4.80 – 4.21 (m, 4H). LC/MS (m/z): 417.321 [M+H+]; UPLC tR 0.96 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-((pyridin-3-yl)methyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (77).
Acid 19o (41.5 mg, 100 μmol) was subjected to General Procedure H1 to afford 4.0 mg of 77 (9.3% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.32 (d, J = 6.5 Hz, 2H), 7.61 (d, J = 1.9 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.25 (d, J = 18.7 Hz, 6H), 6.13 (d, J = 2.0 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.64 (d, J = 2.1 Hz, 1H), 5.25 (s, 2H), 5.00 – 4.41 (m, 4H). LC/MS (m/z): 428.347 [M+H+]; UPLC tR 0.98 min.
5-((1-((Pyridin-3-yl)methyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (78).
Acid 19o (42.4 mg, 102 μmol) was subjected to General Procedure H2 to afford 2.5 mg of 78 (5.7% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (d, J = 5.1 Hz, 1H), 8.38 – 8.29 (m, 2H), 7.75 – 7.59 (m, 2H), 7.46 (d, J = 2.1 Hz, 1H), 7.41 – 7.32 (m, 1H), 7.32 – 7.19 (m, 2H), 6.13 (d, J = 2.0 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.64 (d, J = 2.0 Hz, 1H), 5.27 (s, 2H), 5.04 – 4.49 (m, 4H). LC/MS (m/z): 429.362 [M+H+]; UPLC tR 0.76 min.
5-((1-((Pyridin-3-yl)methyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (79).
Acid 19o (46.6 mg, 112 μmol) was subjected to General Procedure H3 to afford 4.2 mg of 79 (9.0% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.40 (dd, J = 5.0, 1.6 Hz, 1H), 8.39 – 8.31 (m, 2H), 7.67 – 7.56 (m, 1H), 7.49 (d, J = 2.1 Hz, 1H), 7.36 (ddd, J = 7.9, 4.9, 0.9 Hz, 1H), 6.09 (d, J = 2.0 Hz, 1H), 5.77 (s, 2H), 5.27 (s, 2H), 5.01 – 4.45 (m, 4H). LC/MS (m/z): 418.335 [M+H+]; UPLC tR 0.63 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-((furan-2-yl)methyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (80).
Acid 19p (47.1 mg, 117 μmol) was subjected to General Procedure H1 to afford 12.7 mg of 80 (26% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.40 (d, J = 2.0 Hz, 1H), 7.26 (d, J = 11.9 Hz, 5H), 6.23 (t, J = 1.6 Hz, 2H), 6.08 (d, J = 2.0 Hz, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.15 (s, 2H), 5.00 – 4.65 (m, 4H). LC/MS (m/z): 417.321 [M+H+]; UPLC tR 1.29 min.
5-((1-((Furan-2-yl)methyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (81).
Acid 19p (49.6 mg, 123 μmol) was subjected to General Procedure H2 to afford 7.6 mg of 81 (15% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 5.1, 1.4 Hz, 1H), 7.85 – 7.70 (m, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.35 (dd, J = 7.8, 5.0 Hz, 1H), 7.28 (dd, J = 1.8, 0.9 Hz, 1H), 6.25 (dd, J = 3.2, 1.3 Hz, 2H), 6.08 (d, J = 2.1 Hz, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.79 (d, J = 2.1 Hz, 1H), 5.16 (s, 2H), 5.03 – 4.72 (m, 4H). LC/MS (m/z): 418.291 [M+H+]; UPLC tR 0.98 min.
5-((1-((Furan-2-yl)methyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (82).
Acid 19p (55.5 mg, 138 μmol) was subjected to General Procedure H3 to afford 8.5 mg of 82 (15% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.45 (s, 1H), 7.41 (d, J = 2.1 Hz, 1H), 7.34 – 7.27 (m, 1H), 6.26 (t, J = 1.4 Hz, 2H), 6.08 (d, J = 2.0 Hz, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 5.16 (s, 2H), 4.80 – 4.33 (m, 4H). LC/MS (m/z): 407.308 [M+H+]; UPLC tR 0.89 min.
(2,4-Dihydroxy-6-((1-(4-isopropylbenzyl)-1H-pyrazol-5-yl)amino)phenyl)(isoindolin-2-yl)methanone (83).
Amide 14j (42.1 mg, 75.6 μmol) was deprotected using General Procedure F to afford 7.8 mg of 83 (22% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.45 (d, J = 2.1 Hz, 1H), 7.29 (s, 4H), 6.96 (d, J = 1.0 Hz, 4H), 6.11 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.75 (d, J = 2.1 Hz, 1H), 5.13 (s, 2H), 4.95 – 4.59 (m, 4H), 2.72 (p, J = 6.9 Hz, 1H), 1.11 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 469.234 [M+H+]; UPLC tR 1.59 min
5-((1-((4-(Propan-2-yl)phenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (84).
Acid 19q (24.6 mg, 54.0 μmmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (15.6 mg, 81.0 μmol), and triethylamine (57 μL, 410 μmol) using General Procedure G to give 20.7 mg of MOM-protected intermediate (69% yield) after purification via an automated flash system (25% to 70% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3) δ 8.51 (ddd, J = 19.6, 5.0, 1.5 Hz, 1H), 7.67 – 7.44 (m, 2H), 7.21 (ddd, J = 12.8, 7.7, 4.9 Hz, 1H), 7.07 (dd, J = 8.2, 3.7 Hz, 2H), 7.00 (dd, J = 8.2, 3.3 Hz, 2H), 6.51 (d, J = 6.0 Hz, 1H), 6.40 (dd, J = 6.7, 2.1 Hz, 1H), 6.20 (dd, J = 17.0, 2.1 Hz, 1H), 6.12 – 6.01 (m, 1H), 5.24 – 5.09 (m, 4H), 5.05 (d, J = 3.9 Hz, 2H), 4.93 (dt, J = 25.1, 13.4 Hz, 3H), 4.57 (dd, J = 15.1, 7.2 Hz, 1H), 3.44 (t, J = 1.9 Hz, 6H), 2.83 – 2.71 (m, 1H), 1.14 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.3, 167.2, 160.0, 157.6, 157.2, 155.5, 155.4, 149.4, 149.3, 148.5, 148.2, 144.0, 139.0, 138.9, 133.7, 133.6, 130.9, 130.6, 130.3, 129.9, 127.5, 126.7, 126.7, 126.6, 122.5, 122.4, 106.9, 99.3, 99.2, 96.3, 96.2, 95.4, 95.3, 95.3, 95.2, 94.3, 94.2, 56.6, 56.6, 56.3, 56.2, 53.4, 52.6, 51.9, 51.9, 51.5, 50.6, 33.7, 23.9. LC/MS (m/z): 558.417 [M+H+]; UPLC tR 1.60 min.
The MOM-protected intermediate (20 mg, 36 μmol) was deprotected using General Procedure F to afford 8.5 mg of 84 (50% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 5.1, 1.5 Hz, 1H), 7.77 (s, 1H), 7.44 (d, J = 2.1 Hz, 1H), 7.35 (dd, J = 7.8, 5.0 Hz, 1H), 6.99 (s, 4H), 6.11 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.73 (d, J = 2.1 Hz, 1H), 5.14 (s, 2H), 4.89 (s, 25H), 2.73 (p, J = 6.9 Hz, 1H), 1.11 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 470.381 [M+H+]; UPLC tR 1.36 min.
5-((1-((4-(Propan-2-yl)phenyl)methyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (85).
Acid 19q (27.7 mg, 60.8 μmmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (10 mg, 109 μmol), and triethylamine (17 μL, 120 μmol) using General Procedure G to give 17.2 mg of MOM-protected intermediate (52% yield) after purification via automated flash system (2% to 5% methanol in CH2Cl2) and manual flash chromatography (40:59:1 CH2Cl2:ethyl acetate: saturated NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 2.0 Hz, 1H), 7.32 (d, J = 43.7 Hz, 1H), 7.09 (d, J = 7.8 Hz, 2H), 7.04 (d, J = 8.1 Hz, 2H), 6.45 – 6.31 (m, 2H), 6.20 (dd, J = 3.7, 2.1 Hz, 1H), 6.07 (d, J = 1.9 Hz, 1H), 5.24 – 5.08 (m, 4H), 5.04 (s, 2H), 4.69 (d, J = 20.0 Hz, 2H), 4.36 (dd, J = 13.5, 8.5 Hz, 1H), 3.50 – 3.33 (m, 6H), 2.80 (q, J = 7.0, 6.5 Hz, 1H), 1.16 (dd, J = 7.0, 1.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.4, 159.9, 159.8, 155.3, 148.4, 148.2, 143.8, 139.1, 139.0, 138.9, 133.6, 133.6, 127.5, 126.7, 126.7, 107.6, 99.4, 99.3, 96.3, 95.3, 95.3, 94.2, 56.5, 56.2, 51.8, 46.6, 33.7, 23.9, 23.9. LC/MS (m/z): 547.434 [M+H+]; UPLC tR 1.53 min.
The MOM-protected intermediate (17.2 mg, 31.5 μmol) was deprotected using General Procedure F to afford 7.5 mg of 85 (52% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.45 (d, J = 2.0 Hz, 2H), 7.08 – 6.91 (m, 4H), 6.11 (d, J = 2.0 Hz, 1H), 5.91 (t, J = 1.4 Hz, 1H), 5.74 (d, J = 2.0 Hz, 1H), 5.14 (s, 2H), 4.80 – 4.36 (m, 4H), 2.77 (p, J = 6.9 Hz, 1H), 1.14 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 459.354 [M+H+]; UPLC tR 1.23 min.
4-(5H,6H,7H-Pyrrolo[3,4-b]pyridine-6-carbonyl)-5-((1-((4-(trifluoromethyl)phenyl)methyl)-1H-pyrazol-5-yl)amino]benzene-1,3-diol (86).
Acid 19r (35.1 mg, 65 μmol) was subjected to General Procedure H2 to afford 10.3 mg of 86 (32% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.43 (dd, J = 5.0, 1.4 Hz, 1H), 7.75 (s, 1H), 7.52 – 7.42 (m, 3H), 7.34 (dd, J = 7.8, 5.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 6.14 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.0 Hz, 1H), 5.68 (d, J = 2.1 Hz, 1H), 5.28 (s, 2H), 5.07 – 4.44 (m, 4H). LC/MS (m/z): 496.316 [M+H+]; UPLC tR 1.31 min
4-(1H,4H,5H,6H-Pyrrolo[3,4-c]pyrazole-5-carbonyl)-5-((1-((4-(trifluoromethyl)phenyl)methyl)-1H-pyrazol-5-yl)amino]benzene-1,3-diol (87).
Acid 19r (31.6 mg, 66 μmol) was subjected to General Procedure H3 to afford 10.6 mg of 87 (33% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.54 – 7.38 (m, 4H), 7.25 (d, J = 8.0 Hz, 2H), 6.14 (d, J = 2.0 Hz, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.67 (d, J = 2.1 Hz, 1H), 5.28 (s, 2H), 4.82 – 4.20 (m, 4H). 19F NMR (376 MHz, CD3OD) δ −64.1. LC/MS (m/z): 485.289 [M+H+]; UPLC tR 1.20 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((3-methyl-1-((4-methylphenyl)methyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (88).
Amide 14k (40.4 mg, 74.5 μmol) was deprotected using General Procedure F to afford 28.8 mg of 88 (85% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.29 (d, J = 8.8 Hz, 4H), 6.89 (s, 4H), 5.96 – 5.88 (m, 2H), 5.85 (d, J = 2.1 Hz, 1H), 5.05 (s, 2H), 4.95 – 4.46 (m, 4H), 2.14 (s, 3H), 2.13 (s, 3H). LC/MS (m/z): 455.208 [M+H+]; UPLC tR 1.55 min.
5-((1-((2-Chlorophenyl)methyl)-3-methyl-1H-pyrazol-5-yl)amino)-4-(2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (89).
Amide 14l (46.5 mg, 82.3 μmol) was deprotected using General Procedure F to afford 30.4 mg of 89 (78% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.31 – 7.19 (m, 4H), 7.17 – 7.11 (m, 1H), 7.10 – 6.99 (m, 2H), 6.64 – 6.53 (m, 1H), 5.97 (s, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 5.20 (s, 2H), 4.85 – 4.29 (m, 4H), 2.14 (s, 3H). 13C NMR (126 MHz, (CD3)2SO) δ 166.4, 159.2, 155.5, 146.7, 143.7, 141.3, 136.5, 135.0, 131.3, 129.0, 128.8, 128.6, 127.2, 127.1, 122.8, 104.1, 98.4, 94.5, 93.1, 48.0, 40.4, 13.9. LC/MS (m/z): 475.573 [M+H+]; UPLC tR 1.41 min
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((3-methyl-1-((2-methylphenyl)methyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (90).
Amide 14m (21.3 mg, 39.3 μmol) was deprotected using General Procedure F to afford 9.7 mg of 90 (54% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.28 (dt, J = 7.1, 3.6 Hz, 2H), 7.22 (d, J = 7.4 Hz, 2H), 7.01 – 6.94 (m, 2H), 6.91 – 6.83 (m, 1H), 6.49 (d, J = 7.6 Hz, 1H), 5.97 – 5.93 (m, 2H), 5.92 (d, J = 2.1 Hz, 1H), 5.11 (s, 2H), 4.82 – 4.32 (m, 4H), 2.16 (s, 3H), 2.14 (s, 3H). LC/MS (m/z): 455.164 [M+H+]; UPLC tR 1.51 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-phenyl-1H-pyrazol-5-yl)amino]benzene-1,3-diol (91).
Amide 14n (65.0 mg, 126 μmol) was deprotected using General Procedure F to afford 32.8 mg of 91 (61% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.70 – 7.53 (m, 2H), 7.34 – 7.27 (m, 2H), 7.24 (s, 5H), 6.35 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 5.03 – 4.71 (m, 4H), 3.69 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.2, 161.6, 157.5, 151.6, 145.8, 143.7, 137.7, 134.7, 129.7, 128.9, 128.8, 126.4, 123.9, 105.3, 98.3, 95.9, 95.2, 40.6, 35.3. LC/MS (m/z): 428.347 [M+H+]; UPLC tR 1.58 min
5-((1-Methyl-3-phenyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (92).
Acid 19s (44.1 mg, 107 μmmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (30.9 mg, 160 μmol), and triethylamine (110 μL, 800 μmol) using General Procedure G to give 48.3 mg of MOM-protected intermediate (88% yield) after purification via an automated flash system (0% to 4% methanol in CH2Cl2) and manual chromatography (70:30:1 CH2Cl2:acetone: saturated NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.61 – 8.46 (m, 1H), 7.75 (ddd, J = 8.3, 2.5, 1.3 Hz, 2H), 7.63 (dd, J = 51.9, 7.7 Hz, 1H), 7.38 (ddd, J = 7.9, 6.8, 2.1 Hz, 2H), 7.32 – 7.27 (m, 1H), 6.81 (s, 1H), 6.47 (dd, J = 7.4, 2.1 Hz, 1H), 6.34 (s, 1H), 6.26 – 6.16 (m, 1H), 5.19 (d, J = 10.6 Hz, 3H), 5.10 (d, J = 0.9 Hz, 3H), 4.99 – 4.83 (m, 1H), 4.75 – 4.51 (m, 1H), 3.74 (s, 3H), 3.47 (s, 3H), 3.46 – 3.42 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 169.2, 161.6, 160.0, 150.4, 149.7, 138.6, 134.1, 128.5, 127.3, 125.4, 99.6, 97.2, 95.4, 95.1, 94.8, 93.9, 56.7, 56.5, 56.3, 52.0, 32.7, 25.7, 25.2. LC/MS (m/z): 516.251 [M+H+]; UPLC tR 1.43 min.
The MOM-protected intermediate (48.3 mg, 94 μmol) was deprotected using General Procedure F to afford 25 mg of 92 (62% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.34 (dd, J = 5.0, 1.5 Hz, 1H), 7.68 (dd, J = 7.8, 1.4 Hz, 1H), 7.61 – 7.53 (m, 2H), 7.34 – 7.14 (m, 4H), 6.35 (s, 1H), 5.96 (d, J = 2.1 Hz, 1H), 5.82 (d, J = 2.1 Hz, 1H), 5.01 – 4.60 (m, 4H), 3.72 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.4, 161.8, 158.2, 157.6, 151.4, 149.6, 146.1, 144.0, 134.6, 133.4, 129.7, 128.8, 126.3, 124.3, 111.5, 104.9, 98.2, 96.0, 95.7, 40.6, 35.3. LC/MS (m/z): 428.259 [M+H+]; UPLC tR 1.11 min.
5-((1-Methyl-3-phenyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (93).
Acid 19s (64.1 mg, 155 μmmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (25 mg, 230 μmol), and triethylamine (43 μL, 310 μmol) using General Procedure G to give 17.2 mg of MOM-protected intermediate (52% yield) as a solid after purification via automated flash system (20% to 60% acetone in CH2Cl2). 1H NMR (400 MHz, Acetone-d6) δ 7.88 – 7.77 (m, 2H), 7.51 (d, J = 24.4 Hz, 1H), 7.43 – 7.32 (m, 2H), 7.30 – 6.88 (m, 1H), 6.49 (d, J = 5.2 Hz, 1H), 6.45 (dd, J = 5.0, 2.1 Hz, 1H), 6.18 (q, J = 2.1 Hz, 1H), 5.32 – 5.15 (m, 2H), 5.13 (d, J = 2.3 Hz, 2H), 4.82 – 4.48 (m, 3H), 4.43 (d, J = 13.0 Hz, 1H), 3.70 (d, J = 3.3 Hz, 3H), 3.43 (d, J = 2.5 Hz, 3H), 3.40 (s, 3H). LC/MS (m/z): 505.269 [M+H+]; UPLC tR 1.34 min.
The MOM-protected intermediate (41.6 mg, 82 μmol) was deprotected using General Procedure F to afford 24.5 mg of 93 (71% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.67 – 7.61 (m, 2H), 7.38 (s, 1H), 7.36 – 7.28 (m, 2H), 7.28 – 7.22 (m, 1H), 6.37 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.77 (d, J = 2.1 Hz, 1H), 4.78 – 4.44 (m, 4H), 3.71 (s, 3H). LC/MS (m/z): 417.233 [M+H+]; UPLC tR 1.01 min.
5-((1-tert-Butyl-3-phenyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (94).
Acid 19t (41.5 mg, 91.1 μmmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (26.4 mg, 140 μmol), and triethylamine (100 μL, 680 μmol) using General Procedure G to give 39.9 mg of MOM-protected intermediate (79% yield) after purification via an automated flash system (0% to 3% methanol in CH2Cl2) and manual chromatography (70:30:1 CH2Cl2:ethyl acetate: saturated NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.52 (dd, J = 18.4, 4.9 Hz, 1H), 7.84 – 7.74 (m, 2H), 7.60 (dd, J = 59.2, 7.8 Hz, 1H), 7.36 (t, J = 7.7 Hz, 2H), 7.27 – 7.17 (m, 2H), 6.75 (d, J = 7.3 Hz, 1H), 6.51 – 6.36 (m, 2H), 6.30 (dd, J = 9.4, 2.1 Hz, 1H), 5.32 – 5.01 (m, 6H), 4.94 (d, J = 16.5 Hz, 1H), 4.68 (d, J = 14.7 Hz, 1H), 3.47 (s, 3H), 3.44 (d, J = 1.9 Hz, 3H), 2.80 (s, 3H), 1.64 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 167.7, 167.6, 160.1, 160.1, 157.7, 157.2, 155.7, 155.6, 149.4, 149.3, 147.7, 147.7, 145.2, 145.2, 139.5, 139.4, 134.0, 131.1, 130.6, 129.9, 128.5, 127.2, 125.2, 122.5, 122.4, 106.5, 106.4, 99.3, 99.1, 96.5, 96.3, 95.5, 95.4, 94.6, 94.5, 94.3, 94.2, 59.7, 59.7, 56.7, 56.6, 56.3, 56.2, 53.5, 52.7, 51.5, 50.8, 38.6, 29.7. LC/MS (m/z): 558.328 [M+H+]; UPLC tR 1.84 min.
The MOM-protected intermediate (39.9 mg, 72 μmol) was deprotected using General Procedure F to afford 24.3 mg of 94 (72% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.42 (dd, J = 5.1, 1.5 Hz, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.74 – 7.65 (m, 2H), 7.41 – 7.26 (m, 3H), 7.26 – 7.18 (m, 1H), 6.45 (s, 1H), 5.89 (d, J = 2.1 Hz, 1H), 5.80 (d, J = 2.0 Hz, 1H), 5.08 – 4.70 (m, 4H), 1.63 (s, 9H). 13C NMR (101 MHz, CD3OD) δ 170.7, 161.8, 158.3, 157.4, 149.7, 149.5, 147.3, 142.2, 135.5, 133.4, 132.6, 129.6, 128.5, 126.3, 124.3, 103.8, 101.9, 95.1, 94.5, 61.2, 40.6, 30.4. LC/MS (m/z): 470.337 [M+H+]; UPLC tR 1.51 min.
5-((1-tert-Butyl-3-phenyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (95).
Acid 19t (62.2 mg, 137 μmmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (22.3 mg, 205 μmol), and triethylamine (38 μL, 270 μmol) using General Procedure G to give 44.4 mg of MOM-protected intermediate (59% yield) as a solid after purification via automated flash system (10% to 30% acetone in CH2Cl2). 1H NMR (400 MHz, (CD3)2SO) δ 12.70 (s, 1H), 7.80 – 7.71 (m, 3H), 7.71 – 7.62 (m, 1H), 7.53 (d, J = 31.5 Hz, 1H), 7.45 – 7.30 (m, 3H), 7.28 – 7.20 (m, 1H), 7.04 (d, J = 2.7 Hz, 1H), 6.60 (d, J = 9.9 Hz, 1H), 6.27 (t, J = 2.5 Hz, 1H), 5.85 (dd, J = 10.4, 2.1 Hz, 1H), 5.24 – 5.11 (m, 3H), 5.10 – 4.99 (m, 3H), 4.66 (dd, J = 14.4, 6.5 Hz, 1H), 4.56 – 4.39 (m, 3H), 4.34 (d, J = 12.9 Hz, 1H), 3.28 (s, 6H), 2.87 (s, 3H), 1.54 (d, J = 1.1 Hz, 9H). LC/MS (m/z): 547.346 [M+H+]; UPLC tR 1.60 min.
The MOM-protected intermediate (43.4 mg, 79 μmol) was deprotected using General Procedure F to afford 15.5 mg of 95 (43% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.78 – 7.70 (m, 2H), 7.45 (s, 1H), 7.39 – 7.29 (m, 2H), 7.27 – 7.21 (m, 1H), 6.46 (s, 1H), 5.87 (d, J = 2.1 Hz, 1H), 5.77 (t, J = 1.9 Hz, 1H), 4.84 – 4.44 (m, 4H), 1.63 (s, 9H). LC/MS (m/z): 459.266 [M+H+]; UPLC tR 1.40 min.
5-((1-Cyclohexyl-3-phenyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (96).
Acid 19u (51.9 mg, 108 μmmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (31.2 mg, 162 μmol), and triethylamine (110 μL, 810 μmol) using General Procedure G to give 39.5 mg of MOM-protected intermediate (63% yield) after purification via an automated flash system (0% to 3% methanol in CH2Cl2) and manual chromatography (65:35:1 CH2Cl2:ethyl acetate: saturated NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.58 – 8.44 (m, 1H), 7.83 – 7.74 (m, 2H), 7.61 (dd, J = 59.5, 7.7 Hz, 1H), 7.42 – 7.30 (m, 2H), 7.31 – 7.17 (m, 3H), 6.62 (d, J = 3.9 Hz, 1H), 6.43 (dd, J = 8.5, 2.1 Hz, 1H), 6.32 (d, J = 3.0 Hz, 1H), 6.22 (dd, J = 7.2, 2.1 Hz, 1H), 5.19 (d, J = 5.7 Hz, 3H), 5.08 (s, 3H), 4.96 (d, J = 14.1 Hz, 1H), 4.69 (d, J = 14.1 Hz, 1H), 4.03 (td, J = 11.2, 4.1 Hz, 1H), 3.47 (d, J = 1.0 Hz, 3H), 3.43 (d, J = 1.3 Hz, 3H), 2.80 (s, 3H), 2.09 – 1.75 (m, 6H), 1.75 – 1.57 (m, 1H), 1.25 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 167.5, 160.0, 157.6, 155.6, 155.5, 149.6, 149.5, 149.3, 145.1, 145.0, 138.9, 138.8, 134.0, 131.1, 130.6, 128.5, 127.3, 127.3, 125.3, 122.5, 106.9, 106.8, 96.8, 96.7, 96.5, 96.4, 95.5, 95.4, 95.1, 95.0, 94.2, 94.2, 56.8, 56.8, 56.6, 56.6, 56.2, 56.2, 53.6, 52.7, 51.6, 50.7, 38.6, 32.8, 32.4, 25.6, 25.2. LC/MS (m/z): 584.351 [M+H+]; UPLC tR 1.88 min.
The MOM-protected intermediate (43.4 mg, 79 μmol) was deprotected using General Procedure F to afford 15.5 mg of 96 (43% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.36 (dd, J = 5.1, 1.5 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.67 – 7.55 (m, 2H), 7.39 – 7.05 (m, 4H), 6.33 (s, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.79 (d, J = 2.1 Hz, 1H), 5.21 – 4.68 (m, 4H), 4.19 (p, J = 7.9 Hz, 1H), 2.08 – 1.76 (m, 6H), 1.72 – 1.59 (m, 1H), 1.49 – 1.20 (m, 3H). LC/MS (m/z): 496.271 [M+H+]; UPLC tR 1.54 min.
5-((1-Cyclohexyl-3-phenyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (97).
Acid 19u (54.8 mg, 114 μmmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (18.6 mg, 172 μmol), and triethylamine (32 μL, 230 μmol) using General Procedure G1. The resulting suspension was diluted with CH2Cl2 and saturated NaHCO3 (aq.). The layers were separated and the organic layer was washed with brine. The organic layer was dried with anhydrous sodium sulfate. The desired amide was collected along with sodium sulfate following vacuum filtration through Celite®. The desired product and residual Celite® was separated from sodium sulfate and used without purification.
To a mixture of the intermediate amide and residual Celite® in methanol (8.3 mL) was add HCl (2 M, 0.37 μL, 740 μmol). The resulting mixture was stirred at 50 ºC three nights. Additional HCl (2 M, 0.37 μL, 740 μmol) was added to the mixture and stirred at 50 ºC overnight. The mixture was cooled to room temperature and volatile material were condensed in vacuo. The residue was dissolved in DMSO and the residual Celite® was removed via filtration. The crude mixture was purified using mass-guided preparative HPLC to afford 13.7 mg of 97 (25% yield over 2 steps). 1H NMR (400 MHz, CD3OD) δ 7.73 – 7.61 (m, 2H), 7.41 (s, 1H), 7.33 (dd, J = 8.3, 6.7 Hz, 2H), 7.28 – 7.12 (m, 1H), 6.36 (s, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.73 (d, J = 2.1 Hz, 1H), 4.81 – 4.45 (m, 4H), 4.16 (p, J = 9.3, 8.7 Hz, 1H), 2.04 – 1.78 (m, 6H), 1.67 (d, J = 10.9 Hz, 1H), 1.48 – 1.10 (m, 3H). LC/MS (m/z): 485.245 [M+H+]; UPLC tR 1.41 min.
5-((1-(2-Methylpropyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (98).
Acid 19v (52.5 mg, 115 μmmol) was coupled with 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine dihydrochloride (33.4 mg, 173 μmol), and triethylamine (120 μL, 860 μmol) using General Procedure G to give 46.6 mg of MOM-protected intermediate (73% yield) after purification via an automated flash system (1% to 4% methanol in CH2Cl2) and manual chromatography (20:80:1 CH2Cl2:ethyl acetate: saturated NH4OH (aq.)). 1H NMR (400 MHz, CDCl3) δ 8.51 (dd, J = 21.5, 4.8 Hz, 1H), 7.80 – 7.71 (m, 2H), 7.71 – 7.45 (m, 1H), 7.37 (td, J = 7.4, 1.3 Hz, 2H), 7.33 – 7.15 (m, 2H), 6.79 (d, J = 7.2 Hz, 1H), 6.45 (dd, J = 8.4, 2.1 Hz, 1H), 6.38 – 6.30 (m, 2H), 5.14 (d, J = 32.1 Hz, 6H), 4.94 (d, J = 15.8 Hz, 1H), 4.67 (d, J = 14.2 Hz, 1H), 3.82 (dd, J = 7.5, 2.4 Hz, 2H), 3.47 (d, J = 0.9 Hz, 3H), 3.45 (d, J = 1.0 Hz, 3H), 2.80 (s, 3H), 2.32 – 2.17 (m, 1H), 0.88 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.6, 167.4, 160.1, 157.6, 157.2, 155.7, 155.6, 149.9, 149.5, 149.3, 144.4, 144.3, 140.3, 140.2, 133.8, 131.1, 130.6, 130.3, 129.8, 128.5, 127.5, 127.5, 125.3, 122.5, 122.5, 107.0, 106.9, 96.7, 96.6, 95.8, 95.7, 95.5, 95.4, 95.2, 95.1, 94.3, 94.3, 56.6, 56.6, 56.3, 56.2, 55.3, 55.2, 53.5, 52.7, 51.6, 50.8, 38.6, 29.4, 20.0.. LC/MS (m/z): 558.372 [M+H+]; UPLC tR 1.72 min.
The MOM-protected intermediate (46.6 mg, 79 μmol) was deprotected using General Procedure F to afford 26.0 mg of 98 (66% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.37 (dd, J = 5.0, 1.5 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.66 – 7.56 (m, 2H), 7.38 – 7.19 (m, 4H), 6.39 (s, 1H), 5.94 (dd, J = 12.6, 2.1 Hz, 2H), 5.16 – 4.61 (m, 4H), 3.83 (d, J = 7.5 Hz, 2H), 2.19 (hept, J = 6.9 Hz, 1H), 0.86 (d, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CD3OD) δ 170.5, 161.8, 158.2, 157.6, 151.5, 149.7, 146.1, 143.6, 134.8, 133.4, 132.5, 129.7, 128.8, 126.5, 124.3, 104.8, 98.1, 96.0, 95.4, 56.3, 40.6, 30.7, 20.4. LC/MS (m/z): 470.293 [M+H+]; UPLC tR 1.38 min.
5-((1-(2-Methylpropyl)-3-phenyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (99).
Acid 19v (59.0 mg, 130 μmmol) was coupled with 1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole (21.2 mg, 195 μmol), and triethylamine (36 μL, 260 μmol) using General Procedure G1. The reaction mixture was diluted with CH2Cl2 and saturated NaHCO3 (aq.). The layers were separated and brine was added to the organic layer. The combined mixture was filtered through a Celite® plug and the plug was washed with water, methanol and CH2Cl2. The organic layer from the combined filtrate evaporated to leave a fine powder. The remaining water layer was decanted from the solid. The solid was dried to afford 33.9 mg of impure MOM-protected amide which was used without further purification.
The impure MOM-protected amide from above was deprotected using General Procedure F to afford 15.3 mg of 99 (59% overall yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.73 – 7.64 (m, 2H), 7.41 (s, 1H), 7.38 – 7.31 (m, 2H), 7.30 – 7.17 (m, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.88 (d, J = 2.1 Hz, 1H), 4.67 (s, 4H), 3.82 (d, J = 7.5 Hz, 2H), 2.19 (hept, J = 6.9 Hz, 1H), 0.87 (d, J = 6.7 Hz, 6H). LC/MS (m/z): 459.31 [M+H+]; UPLC tR 1.27 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-(propan-2-yl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (100).
Acid 19w (45 mg, 120 μmol) was subjected to General Procedure H1 to afford 15.3 mg of 100 (33% yield) after purification using mass-guided preparative HPLC 1H NMR (400 MHz, CD3OD) δ 7.28 (s, 4H), 5.93 (dd, J = 12.6, 2.0 Hz, 1H), 5.84 (s, 1H), 5.67 (d, J = 2.1 Hz, 1H), 5.06 – 4.67 (m, 4H), 3.58 (s, 4H), 2.76 (p, J = 7.0 Hz, 1H), 1.15 (d, J = 7.0 Hz, 6H). LC/MS (m/z): 393.106 [M+H+]; UPLC tR 1.30 min.
5-((1-Methyl-3-(propan-2-yl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (101).
Acid 19w (45 mg, 120 μmol) was subjected to General Procedure H2 to afford 9.9 mg of 101 (21% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.44 (dd, J = 5.0, 1.5 Hz, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.35 (dd, J = 7.8, 5.0 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.85 (s, 1H), 5.68 (d, J = 2.1 Hz, 1H), 5.09 – 4.72 (m, 4H), 3.59 (s, 3H), 2.76 (hept, J = 6.6 Hz, 1H), 1.14 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 394.253 [M+H+]; UPLC tR 1.29 min.
5-((1-Methyl-3-(propan-2-yl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (102).
Acid 19w (45 mg, 120 μmol) was subjected to General Procedure H3 to afford 12.5 mg of 102 (28% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.45 (s, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.85 (s, 1H), 5.66 (d, J = 2.1 Hz, 1H), 4.85 – 4.44 (m, 4H), 3.59 (s, 3H), 2.78 (dq, J = 13.9, 6.9 Hz, 1H), 1.17 (d, J = 6.9 Hz, 6H). LC/MS (m/z): 383.314 [M+H+]; UPLC tR 0.92 min.
5-((3-Cyclohexyl-1-methyl-1H-pyrazol-5-yl)amino)-4-(2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (103).
Acid 19x (38.9 mg, 92.7 μmol) was subjected to General Procedure H1 to afford 16.1 mg of 103 (40% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.28 (s, 4H), 5.92 (d, J = 2.1 Hz, 1H), 5.80 (s, 1H), 5.69 (d, J = 2.1 Hz, 1H), 5.09 – 4.64 (m, 4H), 3.58 (s, 3H), 2.44 – 2.21 (m, 1H), 1.99 – 1.62 (m, 5H), 1.48 – 1.09 (m, 5H). LC/MS (m/z): 433.376 [M+H+]; UPLC tR 1.51 min.
5-((3-Cyclohexyl-1-methyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (104).
Acid 19x (39.9 mg, 95.1 μmol) was subjected to General Procedure H2 to afford 3.3 mg of 104 (8.0% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.54 (s, 1H), 8.44 (dd, J = 5.1, 1.5 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.35 (dd, J = 7.8, 5.0 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 5.81 (s, 1H), 5.71 (d, J = 2.1 Hz, 1H), 5.04 – 4.38 (m, 4H), 3.59 (s, 4H), 2.39 (d, J = 10.8 Hz, 1H), 1.73 (dq, J = 23.1, 11.6, 8.8 Hz, 6H), 1.27 (hept, J = 11.6 Hz, 4H). LC/MS (m/z): 434.346 [M+H+]; UPLC tR 1.32 min.
5-((3-Cyclohexyl-1-methyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (105).
Acid 19x (44 mg, 100 μmol) was subjected to General Procedure H3 to afford 12.5 mg of 105 (28% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.44 (s, 1H), 5.91 (d, J = 2.1 Hz, 1H), 5.81 (s, 1H), 5.68 (d, J = 2.1 Hz, 1H), 4.78 – 4.36 (m, 4H), 3.59 (s, 3H), 2.51 – 2.27 (m, 1H), 1.89 – 1.61 (m, 5H), 1.43 – 1.09 (m, 5H). LC/MS (m/z): 423.363 [M+H+]; UPLC tR 1.15 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-(2-methylphenyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (106).
Acid 19y (50.3 mg, 118 μmol) was subjected to General Procedure H2 to afford 18.8 mg of 106 (36% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.34 (dd, J = 7.1, 1.3 Hz, 1H), 7.27 (s, 4H), 7.22 – 7.17 (m, 2H), 7.17 – 7.09 (m, 1H), 6.17 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.75 (d, J = 2.1 Hz, 1H), 5.01 – 4.72 (m, 4H), 3.70 (s, 3H), 2.37 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.2, 161.6, 157.5, 151.9, 145.9, 142.8, 137.7, 137.2, 134.7, 131.7, 130.3, 129.0, 128.8, 126.9, 123.9, 105.3, 101.6, 95.9, 94.9, 40.6, 35.3, 21.3. LC/MS (m/z): 441.315 [M+H+]; UPLC tR 1.65 min.
5-((1-Methyl-3-(2-methylphenyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (107).
Acid 19y (50.4 mg, 118 μmol) was subjected to General Procedure H1 to afford 14.6 mg of 107 (28% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.40 (dd, J = 5.0, 1.5 Hz, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.44 – 7.27 (m, 2H), 7.27 – 7.16 (m, 2H), 7.16 – 7.08 (m, 1H), 6.17 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.77 (d, J = 2.1 Hz, 1H), 5.02 – 4.65 (m, 4H), 3.72 (s, 3H), 2.37 (s, 3H). LC/MS (m/z): 442.329 [M+H+]; UPLC tR 1.40 min.
5-((1-Methyl-3-(2-methylphenyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (108).
Acid 19y (50.4 mg, 118 μmol) was subjected to General Procedure H3 to afford 14.6 mg of 108 (28% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.42 (s, 1H), 7.36 (dd, J = 7.0, 1.6 Hz, 1H), 7.25 – 7.11 (m, 3H), 6.18 (s, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.74 (d, J = 2.1 Hz, 1H), 4.80 – 4.47 (m, 4H), 3.71 (s, 3H), 2.39 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.7, 161.6, 157.4, 152.0, 145.9, 142.9, 137.2, 134.7, 131.7, 130.3, 129.0, 126.9, 105.2, 101.7, 95.9, 95.0, 40.6, 35.3, 21.3. LC/MS (m/z): 431.347 [M+H+]; UPLC tR 1.31 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-(3-methylphenyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (109).
Acid 19z (54.1 mg, 127 μmol) was subjected to General Procedure H1 to afford 20.2 mg of 109 (36% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.49 – 7.36 (m, 2H), 7.24 (s, 4H), 7.18 (t, J = 7.6 Hz, 1H), 7.06 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 4.96 – 4.70 (m, 4H), 3.69 (s, 3H), 2.31 (s, 3H). LC/MS (m/z): 441.094 [M+H+]; UPLC tR 1.57 min.
5-((1-Methyl-3-(3-methylphenyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (110).
Acid 19z (56.7 mg, 133 μmol) was subjected to General Procedure H2 to afford 16.2 mg of 110 (28% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.34 (dd, J = 4.9, 1.4 Hz, 1H), 7.73 – 7.62 (m, 1H), 7.46 – 7.29 (m, 2H), 7.24 (dd, J = 7.7, 5.0 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.33 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.83 (d, J = 2.1 Hz, 1H), 5.02 – 4.65 (m, 4H), 3.72 (s, 3H), 2.31 (s, 3H). LC/MS (m/z): 442.329 [M+H+]; UPLC tR 1.29 min.
5-((1-Methyl-3-(3-methylphenyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (111).
Acid 19z (57.6 mg, 135 μmol) was subjected to General Procedure H3 to afford 20.3 mg of 111 (35% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.51 – 7.40 (m, 2H), 7.38 (s, 1H), 7.20 (t, J = 7.6 Hz, 1H), 7.13 – 7.05 (m, 1H), 6.34 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 4.79 – 4.45 (m, 4H), 3.70 (s, 3H), 2.33 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.6, 161.6, 157.4, 151.7, 145.8, 143.7, 139.4, 134.6, 129.6, 127.0, 123.6, 105.2, 98.4, 95.9, 95.3, 40.6, 35.3, 21.7. LC/MS (m/z): 431.347 [M+H+]; UPLC tR 1.22 min
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((3-(3-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (112).
Acid 19aa (48.6 mg, 110 μmol) was subjected to General Procedure H1 to afford 18.2 mg of 112 (36% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.54 (s, 1H), 7.34 – 7.09 (m, 7H), 6.82 (s, 1H), 6.36 (s, 1H), 5.94 (s, 1H), 5.77 (s, 1H), 5.17 – 4.70 (m, 4H), 3.80 (s, 3H), 3.70 (s, 3H). 13C NMR (101 MHz, (CD3)2SO) δ 166.4, 159.5, 159.3, 155.5, 148.1, 143.9, 143.8, 141.5, 141.4, 135.0, 129.6, 127.2, 122.9, 117.1, 113.1, 109.6, 104.0, 96.9, 94.3, 92.7, 55.0, 40.4, 35.0. LC/MS (m/z): 457.061 [M+H+]; UPLC tR 1.58 min.
5-((3-(3-Methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (113).
Acid 19aa (52.6 mg, 119 μmol) was subjected to General Procedure H2 to afford 6.7 mg of 113 (12% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.34 (dd, J = 5.0, 1.5 Hz, 1H), 7.67 (dd, J = 7.8, 1.4 Hz, 1H), 7.23 (dd, J = 7.8, 5.0 Hz, 1H), 7.21 – 7.07 (m, 3H), 6.78 (ddd, J = 7.9, 2.6, 1.3 Hz, 1H), 6.34 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.83 (d, J = 2.1 Hz, 1H), 4.98 – 4.63 (m, 4H), 3.79 (s, 3H), 3.72 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.4, 161.8, 161.5, 158.1, 157.6, 151.2, 149.6, 146.2, 144.0, 135.9, 133.3, 132.5, 130.7, 124.2, 118.8, 114.6, 111.4, 104.9, 98.3, 96.0, 95.9, 55.8, 40.6, 35.4. LC/MS (m/z): 458.296 [M+H+]; UPLC tR 1.30 min.
5-((3-(3-Methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (114)
Acid 19aa (55.5 mg, 125 μmol) was subjected to General Procedure H3 to afford 21.6 mg of 114 (37% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.38 (s, 1H), 7.30 – 7.15 (m, 3H), 6.82 (ddd, J = 7.6, 2.6, 1.7 Hz, 1H), 6.37 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.77 (d, J = 2.1 Hz, 1H), 4.80 – 4.47 (m, 4H), 3.80 (s, 3H), 3.71 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.7, 161.6, 161.5, 157.4, 151.4, 145.8, 143.7, 136.0, 130.8, 119.0, 114.7, 111.5, 98.6, 95.9, 95.3, 55.8, 40.6, 35.3. LC/MS (m/z): 447.313 [M+H+]; UPLC tR 1.25 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (115).
Acid 19ab (50.5 mg, 105 μmol) was subjected to General Procedure H1 to afford 22 mg of 115 (42% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.94 (s, 1H), 7.81 (d, J = 7.6 Hz, 1H), 7.49 (dt, J = 15.4, 7.8 Hz, 2H), 7.20 (s, 4H), 6.44 (s, 1H), 5.96 (d, J = 2.1 Hz, 1H), 5.81 (d, J = 2.1 Hz, 1H), 4.98 – 4.65 (m, 4H), 3.72 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 170.1, 161.6, 157.6, 149.7, 145.73, 145.66, 144.2, 137.6, 135.8, 132.0 (q, 2JC-F = 32.5 Hz), 130.6, 129.9, 128.8, 125.8 (q, 1JC-F = 270.0 Hz), 125.1 (q, 3JC-F = 3.8 Hz), 123.8, 122.6 (q, 3JC-F = 3.7 Hz), 105.5, 98.5, 96.1, 95.6, 40.6, 35.5. 19F NMR (376 MHz, CD3OD) δ −64.2. LC/MS (m/z): 495.301 [M+H+]; UPLC tR 1.78 min.
5-((1-Methyl-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (116).
Acid 19ab (56.4 mg, 117 μmol) was subjected to General Procedure H2 to afford 18.7 mg of 116 (32% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.30 (dd, J = 5.0, 1.4 Hz, 1H), 7.89 (d, J = 2.2 Hz, 1H), 7.79 (d, J = 7.5 Hz, 1H), 7.66 (dd, J = 7.8, 1.5 Hz, 1H), 7.54 – 7.40 (m, 2H), 7.20 (dd, J = 7.8, 5.0 Hz, 1H), 6.45 (s, 1H), 5.97 (d, J = 2.1 Hz, 1H), 5.85 (d, J = 2.1 Hz, 1H), 4.98 – 4.63 (m, 4H), 3.75 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 170.4, 161.8, 158.2, 157.7, 149.62, 149.59, 146.1, 144.5, 135.7, 133.3, 132.0 (q, 2JC-F = 31.5 Hz), 130.6, 129.8, 125.8 (q, 1JC-F = 267.0 Hz), 125.1 (q, 3JC-F = 3.8 Hz), 124.2, 122.5 (q, 3JC-F = 4.8 Hz), 105.1, 98.3, 96.2, 96.1, 40.6, 35.6. 19F NMR (376 MHz, CD3OD) δ −64.2. LC/MS (m/z): 496.316 [M+H+]; UPLC tR 1.52 min.
5-((1-Methyl-3-(3-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (117).
Acid 19ab (57.2 mg, 119 μmol) was subjected to General Procedure H3 to afford 18.9 mg of 117 (33% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.97 (s, 1H), 7.86 (d, J = 7.3 Hz, 1H), 7.60 – 7.47 (m, 2H), 7.36 (s, 1H), 6.47 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.80 (d, J = 2.1 Hz, 1H), 4.82 – 4.45 (m, 4H), 3.73 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 170.6, 161.6, 157.4, 149.8, 145.7, 144.2, 135.9, 132.1 (q, 2JC-F = 31.5 Hz), 130.6, 129.9, 125.8 (q, 1JC-F = 270.8 Hz), 125.2 (q, 3JC-F = 4.8 Hz), 122.7 (q, 3JC-F = 3.8 Hz), 105.4, 98.5, 96.1, 95.6, 40.6, 35.6. 19F NMR (376 MHz, CD3OD) δ −64.2. LC/MS (m/z): 485.289 [M+H+]; UPLC tR 1.45 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-(4-methylphenyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (118).
Acid 19ac (44.5 mg, 104 μmol) was subjected to General Procedure H1 to afford 18.2 mg of 118 (40% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.58 – 7.43 (m, 2H), 7.24 (s, 4H), 7.12 (d, J = 8.0 Hz, 2H), 6.31 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.76 (d, J = 2.1 Hz, 1H), 4.96 – 4.69 (m, 4H), 3.68 (s, 3H), 2.31 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.2, 161.6, 157.5, 151.7, 145.8, 143.6, 138.8, 137.7, 131.9, 130.3, 128.8, 126.4, 123.9, 105.2, 98.1, 95.9, 95.1, 40.6, 35.3, 21.4. LC/MS (m/z): 441.094 [M+H+]; UPLC tR 1.66 min.
5-((1-Methyl-3-(4-methylphenyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (119).
Acid 19ac (46.6 mg, 109 μmol) was subjected to General Procedure H2 to afford 14.2 mg of 119 (30% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.35 (dd, J = 5.0, 1.5 Hz, 1H), 7.69 (dd, J = 7.9, 1.5 Hz, 1H), 7.52 – 7.42 (m, 2H), 7.25 (dd, J = 7.8, 5.0 Hz, 1H), 7.09 (d, J = 7.9 Hz, 2H), 6.30 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.81 (d, J = 2.1 Hz, 1H), 4.99 – 4.64 (m, 4H), 3.70 (s, 3H), 2.31 (s, 3H). LC/MS (m/z): 442.329 [M+H+]; UPLC tR 1.40 min.
5-((1-Methyl-3-(4-methylphenyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (120).
Acid 19ac (50.8 mg, 119 μmol) was subjected to General Procedure H3 to afford 16.5 mg of 120 (32% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.58 – 7.46 (m, 2H), 7.38 (s, 1H), 7.14 (d, J = 7.9 Hz, 2H), 6.32 (s, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.76 (d, J = 2.1 Hz, 1H), 4.79 – 4.45 (m, 4H), 3.69 (s, 3H), 2.32 (s, 3H). LC/MS (m/z): 431.303 [M+H+]; UPLC tR 1.32 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((3-(4-methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)benzene-1,3-diol (121).
Acid 19ad (54.2 mg, 122 μmol) was subjected to General Procedure H1 to afford 21.4 mg of 121 (38% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.58 – 7.47 (m, 2H), 7.24 (s, 4H), 6.93 – 6.81 (m, 2H), 6.27 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.77 (d, J = 2.1 Hz, 1H), 4.99 – 4.72 (m, 4H), 3.78 (s, 3H), 3.67 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.2, 161.6, 161.0, 157.5, 151.5, 145.9, 143.6, 137.7, 128.8, 128.1, 127.7, 127.4, 123.9, 115.1, 105.3, 97.8, 95.9, 95.2, 55.8, 40.6, 35.2. LC/MS (m/z): 457.105 [M+H+]; UPLC tR 1.47 min.
5-((3-(4-Methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (122).
Acid 19ad (55.4 mg, 125 μmol) was subjected to General Procedure H2 to afford 14.4 mg of 122 (25% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.36 (dd, J = 5.1, 1.5 Hz, 1H), 7.69 (dd, J = 7.8, 1.5 Hz, 1H), 7.58 – 7.42 (m, 2H), 7.26 (dd, J = 7.8, 5.0 Hz, 1H), 6.88 – 6.74 (m, 2H), 6.26 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.82 (d, J = 2.1 Hz, 1H), 5.09 – 4.66 (m, 4H), 3.79 (s, 3H), 3.70 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 170.4, 161.8, 161.0, 158.2, 157.6, 151.4, 149.6, 146.2, 143.9, 133.4, 127.6, 127.3, 124.3, 115.1, 104.9, 97.7, 96.0, 95.7, 55.9, 40.6, 35.2. LC/MS (m/z): 458.241 [M+H+]; UPLC tR 1.04 min.
5-((3-(4-Methoxyphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (123).
Acid 19ad (55.6 mg, 125 μmol) was subjected to General Procedure H3 to afford 12.9 mg of 123 (23% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.61 – 7.51 (m, 2H), 7.39 (s, 1H), 6.93 – 6.82 (m, 2H), 6.28 (s, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.76 (d, J = 2.1 Hz, 1H), 4.85 – 4.41 (m, 4H), 3.79 (s, 3H), 3.68 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 170.7, 161.6, 161.1, 157.4, 151.6, 145.9, 143.7, 127.7, 127.4, 115.1, 105.3, 97.9, 95.9, 95.3, 55.9, 40.6, 35.2. LC/MS (m/z): 447.216 [M+H+]; UPLC tR 0.98 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (124).
Acid 19ae (61.4 mg, 128 μmol) was subjected to General Procedure H1 to afford 20.5 mg of 124 (32% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.81 – 7.71 (m, 2H), 7.56 (d, J = 8.2 Hz, 2H), 7.21 (s, 4H), 6.45 (s, 1H), 5.96 (d, J = 2.1 Hz, 1H), 5.81 (d, J = 2.1 Hz, 1H), 4.98 – 4.68 (m, 4H), 3.73 (s, 3H). 19F NMR (376 MHz, CD3OD) δ −64.1. LC/MS (m/z): 495.301 [M+H+]; UPLC tR 1.78 min.
5-((1-Methyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (125).
Acid 19ae (61.5 mg, 128 μmol) was subjected to General Procedure H2 to afford 15.2 mg of 125 (24% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.30 (d, J = 5.0 Hz, 1H), 7.76 – 7.69 (m, 2H), 7.65 (dd, J = 7.9, 1.4 Hz, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.20 (dd, J = 7.8, 5.0 Hz, 1H), 6.45 (s, 1H), 5.97 (d, J = 2.1 Hz, 1H), 5.85 (d, J = 2.1 Hz, 1H), 4.96 – 4.59 (m, 4H), 3.75 (s, 3H). 19F NMR (376 MHz, CD3OD) δ −64.0. LC/MS (m/z): 496.271 [M+H+]; UPLC tR 1.53 min.
5-((1-Methyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (126).
Acid 19ae (62.2 mg, 129 μmol) was subjected to General Procedure H3 to afford 15 mg of 126 (24% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.81 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 8.1 Hz, 2H), 7.37 (s, 1H), 6.47 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.79 (d, J = 2.1 Hz, 1H), 4.79 – 4.41 (m, 4H), 3.74 (s, 3H). 19F NMR (376 MHz, CD3OD) δ −64.0. LC/MS (m/z): 485.289 [M+H+]; UPLC tR 1.47 min.
5-((3-(4-tert-Butylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4-(2,3-dihydro-1H-isoindole-2-carbonyl)benzene-1,3-diol (127).
Acid 19af (59 mg, 130 μmol) was subjected to General Procedure H1 to afford 18.4 mg of 127 (30% yield) after purification using mass-guided preparative HPLC 1H NMR (400 MHz, CD3OD) δ 7.62 – 7.47 (m, 2H), 7.38 – 7.30 (m, 2H), 7.23 (s, 4H), 6.32 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 5.05 – 4.68 (m, 4H), 3.69 (s, 3H), 1.31 (s, 9H). LC/MS (m/z): 483.392 [M+H+]; UPLC tR 1.90 min.
5-((3-(4-tert-Butylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (128).
Acid 19af (59.8 mg, 127 μmol) was subjected to General Procedure H2 to afford 18.1 mg of 128 (29% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.33 (dd, J = 5.0, 1.5 Hz, 1H), 7.68 (dd, J = 7.9, 1.5 Hz, 1H), 7.54 – 7.42 (m, 2H), 7.36 – 7.28 (m, 2H), 7.23 (dd, J = 7.8, 5.0 Hz, 1H), 6.32 (s, 1H), 5.95 (d, J = 2.1 Hz, 1H), 5.83 (d, J = 2.1 Hz, 1H), 4.93 – 4.66 (m, 4H), 3.72 (s, 3H), 1.31 (s, 9H). LC/MS (m/z): 484.363 [M+H+]; UPLC tR 1.64 min.
5-((3-(4-tert-Butylphenyl)-1-methyl-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (129).
Acid 19af (61.7 mg, 131 μmol) was subjected to General Procedure H3 to afford 18.1 mg of 129 (29% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.57 (d, J = 8.5 Hz, 2H), 7.42 – 7.31 (m, 3H), 6.34 (s, 1H), 5.93 (d, J = 2.1 Hz, 1H), 5.76 (d, J = 2.1 Hz, 1H), 4.81 – 4.47 (m, 4H), 3.70 (s, 3H), 1.32 (s, 9H). LC/MS (m/z): 473.336 [M+H+]; UPLC tR 1.57 min.
4-(2,3-Dihydro-1H-isoindole-2-carbonyl)-5-((1-methyl-3-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)benzene-1,3-diol (130).
Acid 19ag (51.3 mg, 103 μmol) was subjected to General Procedure H1 to afford 21.3 mg of 130 (40% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.72 – 7.55 (m, 2H), 7.21 (s, 4H), 7.20 – 7.12 (m, 2H), 6.36 (s, 1H), 5.96 (d, J = 2.1 Hz, 1H), 5.80 (d, J = 2.1 Hz, 1H), 4.99 – 4.66 (m, 4H), 3.71 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 170.2, 161.6, 157.5, 150.0, 149.9, 145.8, 144.1, 137.7, 134.0, 128.8, 127.9, 123.9, 122.3, 122.1 (q, 1JC-F = 253.8 Hz), 105.5, 98.3, 96.1, 95.7, 40.6, 35.4. 19F NMR (376 MHz, CD3OD) δ −59.5. LC/MS (m/z): 511.162 [M+H+]; UPLC tR 1.61 min.
5-((1-Methyl-3-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)-4-(5H,6H,7H-pyrrolo[3,4-b]pyridine-6-carbonyl)benzene-1,3-diol (131).
Acid 19ag (53.1 mg, 107 μmol) was subjected to General Procedure H2 to afford 18.6 mg of 131 (34% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 8.31 (dd, J = 5.0, 1.5 Hz, 1H), 7.69 – 7.59 (m, 3H), 7.21 (dd, J = 7.8, 5.0 Hz, 1H), 7.19 – 7.11 (m, 2H), 6.37 (s, 1H), 5.96 (d, J = 2.1 Hz, 1H), 5.85 (d, J = 2.1 Hz, 1H), 4.97 – 4.64 (m, 4H), 3.73 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 170.4, 161.7, 158.1, 157.6, 149.9, 149.7, 149.4, 133.8, 133.5, 132.6, 127.7, 124.7, 122.1 (q, 1JC-F = 253.8 Hz), 105.1, 98.2, 96.24, 96.17, 40.6, 35.4. 19F NMR (376 MHz, CD3OD) δ −59.4. LC/MS (m/z): 512.326 [M+H+]; UPLC tR 1.48 min.
5-((1-Methyl-3-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-5-yl)amino)-4-(1H,4H,5H,6H-pyrrolo[3,4-c]pyrazole-5-carbonyl)benzene-1,3-diol (132).
Acid 19ag (54.4 mg, 109 μmol) was subjected to General Procedure H3 to afford 17 mg of 132 (31% yield) after purification using mass-guided preparative HPLC. 1H NMR (400 MHz, CD3OD) δ 7.77 – 7.67 (m, 2H), 7.37 (s, 1H), 7.28 – 7.17 (m, 2H), 6.39 (s, 1H), 5.94 (d, J = 2.1 Hz, 1H), 5.78 (d, J = 2.1 Hz, 1H), 4.79 – 4.45 (m, 4H), 3.72 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 170.6, 161.5, 157.4, 150.0, 149.9, 145.6, 144.1, 134.0, 127.9, 124.1, 122.3, 122.1 (q, 1JC-F = 255.0 Hz), 105.4, 98.2, 96.1, 95.6, 40.6, 35.4. 19F NMR (376 MHz, CD3OD) δ −59.5. LC/MS (m/z): 501.151 [M+H+]; UPLC tR 1.28 min.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the National Institutes of Health (Grant R01AI120958 to L.E.C., L.E.B., and D.K). L.E.C. holds a Canada Research Chair in Microbial Genomics & Infectious Disease and is Co-Director of the CIFAR Program, Fungal Kingdom: Threats & Opportunities. The authors thank Dr. Han Yueh for assistance in acquiring LC/MS purity data, Dr. Paul Ralifo for assistance with NMR analysis, Gaurang Patel and Adrian Sheldon (Charles River Laboratories) for assistance with microsomal stability assays, and the Ontario Institute for Cancer Research (M. Mohammed and R. Marcellus) for assistance with SPR assays.
ABBREVIATIONS
- Hsp90
heat shock protein 90
- Trap1
TNF receptor associated protein 1
- Grp94
94 kDa glucose-regulated protein
- NBD
nucleotide binding domain
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- DIPEA
N,N-diisopropylethylamine
- UPLC
ultraperformance liquid chromatography
- PS-CDI
polymer-supported carbonyldiimidazole
- TMT
trithiocyanuric acid
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The following files are available free of charge.
Supporting Information: Figures S1 & S2 and Supplementary Tables 1–5 (PDF)
Molecular formula strings and associated biological data (CSV)
L.E.C. is a co-founder, Chief Scientific Officer, and shareholder in Bright Angel Therapeutics, a platform company for the development of novel antifungal therapeutics. L.E.C. is a consultant for Boragen, a small molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health. L.W. is a co-founder and shareholder in Bright Angel Therapeutics. L.E.B. D.S.H., L.E.C. and L.W. are named as inventors on a provisional patent application pertaining to findings reported here.
REFERENCES
- 1.Brown GD; Denning DW; Gow NA; Levitz SM; Netea MG; White TC, Hidden killers: human fungal infections. Sci. Transl. Med 2012, 4 (165), 165rv113. [DOI] [PubMed] [Google Scholar]
- 2.Robbins N; Wright GD; Cowen LE, Antifungal drugs: the current armamentarium and development of new agents In The Fungal Kingdom, Heitman J; Howlett B; Crous P; Stukenbrock E; James T; Gow N, Eds. ASM Press: Washington DC., 2017; Vol. 4, pp 903–922. [DOI] [PubMed] [Google Scholar]
- 3.McKenzie CG; Koser U; Lewis LE; Bain JM; Mora-Montes HM; Barker RN; Gow NA; Erwig LP, Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect. Immun 2010, 78 (4), 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowen LE; Singh SD; Kohler JR; Collins C; Zaas AK; Schell WA; Aziz H; Mylonakis E; Perfect JR; Whitesell L; Lindquist S, Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Natl. Acad. Sci. USA 2009, 106 (8), 2818–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cowen LE; Lindquist S, Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science 2005, 309 (5744), 2185–2189. [DOI] [PubMed] [Google Scholar]
- 6.Singh SD; Robbins N; Zaas AK; Schell WA; Perfect JR; Cowen LE, Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog 2009, 5 (7), e1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee S; Tatu U, Heat shock protein 90 localizes to the surface and augments virulence factors of Cryptococcus neoformans. PLoS Negl. Trop. Dis 2017, 11 (8), e0005836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cordeiro Rde A; Evangelista AJ; Serpa R; Marques FJ; de Melo CV; de Oliveira JS; Franco Jda S; de Alencar LP; Bandeira Tde J; Brilhante RS; Sidrim JJ; Rocha MF, Inhibition of heat-shock protein 90 enhances the susceptibility to antifungals and reduces the virulence of Cryptococcus neoformans/Cryptococcus gattii species complex. Microbiology (Reading, England) 2016, 162 (2), 309–317. [DOI] [PubMed] [Google Scholar]
- 9.Ferraro M; D’Annessa I; Moroni E; Morra G; Paladino A; Rinaldi S; Compostella F; Colombo G, Allosteric modulators of HSP90 and HSP70: dynamics meets function through structure-based drug design. J. Med. Chem 2019, 62 (1), 60–87. [DOI] [PubMed] [Google Scholar]
- 10.Taipale M; Jarosz DF; Lindquist S, HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol 2010, 11, 515–528. [DOI] [PubMed] [Google Scholar]
- 11.Whitesell L; Robbins N; Huang DS; McLellan CA; Shekhar-Guturja T; LeBlanc EV; Nation CS; Hui R; Hutchinson A; Collins C; Chatterjee S; Trilles R; Xie JL; Krysan DJ; Lindquist S; Porco JA; Tatu U; Brown LE; Pizarro J; Cowen LE, Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat. Commun 2019, 10 (1), 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehn A; Moroni E; Zierer BK; Tippel F; Morra G; John C; Richter K; Colombo G; Buchner J, Allosteric regulation points control the conformational dynamics of the molecular chaperone Hsp90. J. Mol. Biol 2016, 428 (22), 4559–4571. [DOI] [PubMed] [Google Scholar]
- 13.Gewirth DT, Paralog specific Hsp90 inhibitors - a brief history and a bright future. Curr. Top. Med. Chem 2016, 16 (25), 2779–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee C; Park HK; Jeong H; Lim J; Lee AJ; Cheon KY; Kim CS; Thomas AP; Bae B; Kim ND; Kim SH; Suh PG; Ryu JH; Kang BH, Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J. Am. Chem. Soc 2015, 137 (13), 4358–4367. [DOI] [PubMed] [Google Scholar]
- 15.Crowley VM; Khandelwal A; Mishra S; Stothert AR; Huard DJE; Zhao J; Muth A; Duerfeldt AS; Kizziah JL; Lieberman RL; Dickey CA; Blagg BSJ, Development of glucose regulated protein 94-selective inhibitors based on the BnIm and radamide scaffold. J. Med. Chem 2016, 59 (7), 3471–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel PD; Yan P; Seidler PM; Patel HJ; Sun W; Yang C; Que NS; Taldone T; Finotti P; Stephani RA; Gewirth DT; Chiosis G, Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol 2013, 9, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duerfeldt AS; Peterson LB; Maynard JC; Ng CL; Eletto D; Ostrovsky O; Shinogle HE; Moore DS; Argon Y; Nicchitta CV; Blagg BS, Development of a Grp94 inhibitor. J. Am. Chem. Soc 2012, 134, 9796–9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stothert AR; Suntharalingam A; Tang X; Crowley VM; Mishra SJ; Webster JM; Nordhues BA; Huard DJE; Passaglia CL; Lieberman RL; Blagg BSJ; Blair LJ; Koren J; Dickey CA, Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma. Sci. Rep 2017, 7 (1), 17951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muth A; Crowley V; Khandelwal A; Mishra S; Zhao J; Hall J; Blagg BSJ, Development of radamide analogs as Grp94 inhibitors. Bioorg. Med. Chem 2014, 22 (15), 4083–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khandelwal A; Kent CN; Balch M; Peng S; Mishra SJ; Deng J; Day VW; Liu W; Subramanian C; Cohen M; Holzbeierlein JM; Matts R; Blagg BSJ, Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor. Nat. Commun 2018, 9 (1), 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohkubo S; Kodama Y; Muraoka H; Hitotsumachi H; Yoshimura C; Kitade M; Hashimoto A; Ito K; Gomori A; Takahashi K; Shibata Y; Kanoh A; Yonekura K, TAS-116, a highly selective inhibitor of heat shock protein 90α and β, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol. Cancer Ther 2015, 14 (1), 14–22. [DOI] [PubMed] [Google Scholar]
- 22.Ernst JT; Neubert T; Liu M; Sperry S; Zuccola H; Turnbull A; Fleck B; Kargo W; Woody L; Chiang P; Tran D; Chen W; Snyder P; Alcacio T; Nezami A; Reynolds J; Alvi K; Goulet L; Stamos D, Identification of novel HSP90α/β isoform selective inhibitors using structure-based drug design. Demonstration of potential utility in treating CNS disorders such as Huntington’s Disease. J. Med. Chem 2014, 57 (8), 3382–3400. [DOI] [PubMed] [Google Scholar]
- 23.Ernst JT; Liu M; Zuccola H; Neubert T; Beaumont K; Turnbull A; Kallel A; Vought B; Stamos D, Correlation between chemotype-dependent binding conformations of HSP90α/β and isoform selectivity—Implications for the structure-based design of HSP90α/β selective inhibitors for treating neurodegenerative diseases. Bioorg. Med. Chem. Lett 2014, 24 (1), 204–208. [DOI] [PubMed] [Google Scholar]
- 24.Woodhead AJ; Angove H; Carr MG; Chessari G; Congreve M; Coyle JE; Cosme J; Graham B; Day PJ; Downham R; Fazal L; Feltell R; Figueroa E; Frederickson M; Lewis J; McMenamin R; Murray CW; O’Brien MA; Parra L; Patel S; Phillips T; Rees DC; Rich S; Smith DM; Trewartha G; Vinkovic M; Williams B; Woolford AJ, Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydrois oindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J. Med. Chem 2010, 53, 5956–5969. [DOI] [PubMed] [Google Scholar]
- 25.Do K; Speranza G; Chang L-C; Polley EC; Bishop R; Zhu W; Trepel JB; Lee S; Lee M-J; Kinders RJ; Phillips L; Collins J; Lyons J; Jeong W; Antony R; Chen AP; Neckers L; Doroshow JH; Kummar S, Phase I study of the heat shock protein 90 (Hsp90) inhibitor onalespib (AT13387) administered on a daily for 2 consecutive days per week dosing schedule in patients with advanced solid tumors. Invest. New Drug 2015, 33 (4), 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner AJ; Agulnik M; Heinrich MC; Mahadevan D; Riedel RF; von Mehren M; Trent J; Demetri GD; Corless CL; Yule M; Lyons JF; Oganesian A; Keer H, Dose-escalation study of a second-generation non-ansamycin HSP90 inhibitor, onalespib (AT13387), in combination with imatinib in patients with metastatic gastrointestinal stromal tumour. Eur. J. Cancer 2016, 61, 94–101. [DOI] [PubMed] [Google Scholar]
- 27.Canella A; Welker AM; Yoo JY; Xu J; Abas FS; Kesanakurti D; Nagarajan P; Beattie CE; Sulman EP; Liu J; Gumin J; Lang FF; Gurcan MN; Kaur B; Sampath D; Puduvalli VK, Efficacy of onalespib, a long-acting second-generation HSP90 inhibitor, as a single agent and in combination with temozolomide against malignant gliomas. Clin. Cancer Res 2017, 23 (20), 6215–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stühmer T; Zöllinger A; Siegmund D; Chatterjee M; Grella E; Knop S; Kortüm M; Unzicker C; Jensen MR; Quadt C; Chène P; Schoepfer J; García-Echeverría C; Einsele H; Wajant H; Bargou RC, Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 2008, 22, 1604–1612. [DOI] [PubMed] [Google Scholar]
- 29.Jensen MR; Schoepfer J; Radimerski T; Massey A; Guy CT; Brueggen J; Quadt C; Buckler A; Cozens R; Drysdale MJ; Garcia-Echeverria C; Chène P, NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res 2008, 10 (2), R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doi T; Onozawa Y; Fuse N; Yoshino T; Yamazaki K; Watanabe J; Akimov M; Robson M; Boku N; Ohtsu A, Phase I dose-escalation study of the HSP90 inhibitor AUY922 in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol 2014, 74 (3), 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seggewiss-Bernhardt R; Bargou RC; Goh YT; Stewart AK; Spencer A; Alegre A; Bladé J; Ottmann OG; Fernandez-Ibarra C; Lu H; Pain S; Akimov M; Iyer SP, Phase 1/1B trial of the heat shock protein 90 inhibitor NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma. Cancer 2015, 121 (13), 2185–2192. [DOI] [PubMed] [Google Scholar]
- 32.Renouf DJ; Hedley D; Krzyzanowska MK; Schmuck M; Wang L; Moore MJ, A phase II study of the HSP90 inhibitor AUY922 in chemotherapy refractory advanced pancreatic cancer. Cancer Chemother. Pharmacol 2016, 78 (3), 541–545. [DOI] [PubMed] [Google Scholar]
- 33.Bendell JC; Bauer TM; Lamar R; Joseph M; Penley W; Thompson DS; Spigel DR; Owera R; Lane CM; Earwood C; Burris HA, A phase 2 study of the Hsp90 inhibitor AUY922 as treatment for patients with refractory gastrointestinal stromal tumors. Cancer Invest 2016, 34 (6), 265–270. [DOI] [PubMed] [Google Scholar]
- 34.Lin T-Y; Bear M; Du Z; Foley KP; Ying W; Barsoum J; London C, The novel HSP90 inhibitor STA-9090 exhibits activity against Kit-dependent and -independent malignant mast cell tumors. Exp. Hematol 2008, 36 (10), 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ying WW; Du ZJ; Sun LJ; Foley KP; Proia DA; Blackman RK; Zhou D; Inoue T; Tatsuta N; Sang J; Ye SX; Acquaviva J; Ogawa LS; Wada Y; Barsoum J; Koya K, Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol. Cancer Ther 2012, 11 (2), 475–484. [DOI] [PubMed] [Google Scholar]
- 36.Lock RB; Carol H; Maris JM; Kang MH; Reynolds CP; Kolb EA; Gorlick R; Keir ST; Billups CA; Kurmasheva RT; Houghton PJ; Smith MA, Initial testing (stage 1) of ganetespib, an Hsp90 inhibitor, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2013, 60 (7), E42–E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jhaveri K; Wang R; Teplinsky E; Chandarlapaty S; Solit D; Cadoo K; Speyer J; D’Andrea G; Adams S; Patil S; Haque S; O’Neill T; Friedman K; Esteva FJ; Hudis C; Modi S, A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer. Breast Cancer Res 2017, 19 (1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thakur MK; Heilbrun LK; Sheng S; Stein M; Liu G; Antonarakis ES; Vaishampayan U; Dzinic SH; Li X; Freeman S; Smith D; Heath EI, A phase II trial of ganetespib, a heat shock protein 90 Hsp90) inhibitor, in patients with docetaxel-pretreated metastatic castrate-resistant prostate cancer (CRPC)-a prostate cancer clinical trials consortium (PCCTC) study. Invest. New Drug 2016, 34 (1), 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goyal L; Wadlow RC; Blaszkowsky LS; Wolpin BM; Abrams TA; McCleary NJ; Sheehan S; Sundaram E; Karol MD; Chen J; Zhu AX, A phase I and pharmacokinetic study of ganetespib (STA-9090) in advanced hepatocellular carcinoma. Invest. New Drug 2015, 33 (1), 128–137. [DOI] [PubMed] [Google Scholar]
- 40.Socinski MA; Goldman J; El-Hariry I; Koczywas M; Vukovic V; Horn L; Paschold E; Salgia R; West H; Sequist LV; Bonomi P; Brahmer J; Chen L-C; Sandler A; Belani CP; Webb T; Harper H; Huberman M; Ramalingam S; Wong K-K; Teofilovici F; Guo W; Shapiro GI, A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non–small cell lung cancer. Clin. Cancer Res 2013, 19 (11), 3068–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jhaveri K; Chandarlapaty S; Lake D; Gilewski T; Robson M; Goldfarb S; Drullinsky P; Sugarman S; Wasserheit-Leiblich C; Fasano J; Moynahan ME; D’Andrea G; Lim K; Reddington L; Haque S; Patil S; Bauman L; Vukovic V; El-Hariry I; Hudis C; Modi S, A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin. Breast Cancer 2014, 14 (3), 154–160. [DOI] [PubMed] [Google Scholar]
- 42.Goldman JW; Raju RN; Gordon GA; El-Hariry I; Teofilivici F; Vukovic VM; Bradley R; Karol MD; Chen Y; Guo W; Inoue T; Rosen LS, A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer 2013, 13 (1), 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakashima T; Ishii T; Tagaya H; Seike T; Nakagawa H; Kanda Y; Akinaga S; Soga S; Shiotsu Y, New molecular and biological mechanism of antitumor activities of KW-2478, a novel nonansamycin heat shock protein 90 inhibitor, in multiple myeloma cells. Clin. Cancer Res 2010, 16 (10), 2792–2802. [DOI] [PubMed] [Google Scholar]
- 44.Ishii T; Seike T; Nakashima T; Juliger S; Maharaj L; Soga S; Akinaga S; Cavenagh J; Joel S; Shiotsu Y, Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J 2012, 2, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yong K; Cavet J; Johnson P; Morgan G; Williams C; Nakashima D; Akinaga S; Oakervee H; Cavenagh J, Phase I study of KW-2478, a novel Hsp90 inhibitor, in patients with B-cell malignancies. Br. J. Cancer 2015, 114, 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cavenagh J; Oakervee H; Baetiong-Caguioa P; Davies F; Gharibo M; Rabin N; Kurman M; Novak B; Shiraishi N; Nakashima D; Akinaga S; Yong K, A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br. J. Cancer 2017, 117, 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ritchie TJ; Macdonald SJF; Peace S; Pickett SD; Luscombe CN, The developability of heteroaromatic and heteroaliphatic rings – do some have a better pedigree as potential drug molecules than others? MedChemComm 2012, 3 (9), 1062–1069. [Google Scholar]
- 48.Khandelwal A; Crowley VM; Blagg BSJ, Resorcinol-based Grp94-selective inhibitors. ACS Med. Chem. Lett 2017, 8 (10), 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kung P-P; Funk L; Meng J; Collins M; Zhou JZ; Catherine Johnson, M.; Ekker A; Wang J; Mehta P; Yin M-J; Rodgers C; Davies JF; Bayman E; Smeal T; Maegley KA; Gehring MR, Dihydroxylphenyl amides as inhibitors of the Hsp90 molecular chaperone. Bioorg. Med. Chem. Lett 2008, 18 (23), 6273–6278. [DOI] [PubMed] [Google Scholar]
- 50.Kung P-P; Huang B; Zhang G; Zhou JZ; Wang J; Digits JA; Skaptason J; Yamazaki S; Neul D; Zientek M; Elleraas J; Mehta P; Yin M-J; Hickey MJ; Gajiwala KS; Rodgers C; Davies JF; Gehring MR, Dihydroxyphenylisoindoline amides as orally bioavailable inhibitors of the heat shock protein 90 (Hsp90) molecular chaperone. J. Med. Chem 2010, 53 (1), 499–503. [DOI] [PubMed] [Google Scholar]
- 51.Murray CW; Carr MG; Callaghan O; Chessari G; Congreve M; Cowan S; Coyle JE; Downham R; Figueroa E; Frederickson M; Graham B; McMenamin R; O’Brien MA; Patel S; Phillips TR; Williams G; Woodhead AJ; Woolford AJ, Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. J. Med. Chem 2010, 53, 5942–5955. [DOI] [PubMed] [Google Scholar]
- 52.Patel BH; Barrett AGM, Total synthesis of resorcinol amide Hsp90 inhibitor AT13387. J. Org. Chem 2012, 77 (24), 11296–11301. [DOI] [PubMed] [Google Scholar]
- 53.Ren J; Li J; Wang Y; Chen W; Shen A; Liu H; Chen D; Cao D; Li Y; Zhang N; Xu Y; Geng M; He J; Xiong B; Shen J, Identification of a new series of potent diphenol HSP90 inhibitors by fragment merging and structure-based optimization. Bioorg. Med. Chem. Lett 2014, 24 (11), 2525–2529. [DOI] [PubMed] [Google Scholar]
- 54.Moss TA; Addie MS; Nowak T; Waring MJ, Room-temperature palladium-catalyzed coupling of heteroaryl amines with aryl or heteroaryl bromides. Synlett 2012, 2012 (02), 285–289. [Google Scholar]
- 55.Schulte JP II; Tweedie SR, Palladium-catalyzed couplings of heteroaryl amines with aryl halides using sodium phenolate as the stoichiometric base. Synlett 2007, 2007 (15), 2331–2336. [Google Scholar]
- 56.Park SY; Oh YJ; Lho Y; Jeong JH; Liu K-H; Song J; Kim S-H; Ha E; Seo YH, Design, synthesis, and biological evaluation of a series of resorcinol-based N-benzyl benzamide derivatives as potent Hsp90 inhibitors. Eur. J. Med. Chem 2018, 143, 390–401. [DOI] [PubMed] [Google Scholar]
- 57.Ritchie TJ; Macdonald SJ, The impact of aromatic ring count on compound developability--are too many aromatic rings a liability in drug design? Drug Discov. Today 2009, 14 (21–22), 1011–1020. [DOI] [PubMed] [Google Scholar]
- 58.White TC, Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother 1997, 41 (7), 1482–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Odds FC; Brown AJ; Gow NA, Candida albicans genome sequence: a platform for genomics in the absence of genetics. Genome Biol 2004, 5 (7), 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Granger DL; Perfect JR; Durack DT, Virulence of Cryptococcus neoformans. Regulation of capsule synthesis by carbon dioxide. J. Clin. Invest 1985, 76 (2), 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.LaFayette SL; Collins C; Zaas AK; Schell WA; Betancourt-Quiroz M; Gunatilaka AA; Perfect JR; Cowen LE, PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog 2010, 6 (8), e1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rossi AM; Taylor CW, Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc 2011, 6 (3), 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Houston JB, Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol 1994, 47 (9), 1469–1479. [DOI] [PubMed] [Google Scholar]
- 64.Jung FH; Morgentin RR; Ple P Quinoline derivatives WO2007099323, 2007.
- 65.Nikolovska-Coleska Z; Abulwerdi F; Showalter H; Miao L; Stuckey J; Mady A Small molecule inhibitors of MCL-1 and uses thereof. WO2015153959, 2015.
- 66.Jefson MR; Lowe JA; Dey F; Bergmann A; Schoop A; Fuller NO Hetero-halo inhibitors of histone deacetylase. WO2017007756, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.