

Abstract
Bruton's tyrosine kinase (BTK) is a key regulator of B cell receptor and Fc receptor signaling, and a rational therapeutic target for autoimmune diseases. This first‐in‐human phase I, double‐blind, placebo‐controlled trial investigated the safety, tolerability, pharmacokinetics (PK), target occupancy, and effects on QT interval of evobrutinib, a highly selective, oral inhibitor of BTK, in healthy subjects. This dose escalation trial consisted of two parts. Part 1 included 48 subjects in 6 ascending dose cohorts (25, 50, 100, 200, 350, and 500 mg) randomized to a single dose of evobrutinib or placebo. Part 2 included 36 subjects in 3 ascending dose cohorts (25, 75, and 200 mg/day) randomized to evobrutinib or placebo once daily for 14 days. Safety and tolerability, as well as PK and target occupancy (total and free BTK in peripheral blood mononuclear cells), were assessed following single and multiple dosing. PK parameters were determined by noncompartmental methods. QT interval was obtained from 12‐lead electrocardiogram recordings and corrected for heart rate by Fridericia's method (QTcF). Treatment‐emergent adverse events (TEAEs) were mostly mild, occurring in 25% of subjects after single dosing, and 48.1% after multiple dosing. There was no apparent dose relationship regarding frequency or type of TEAE among evobrutinib‐treated subjects. Absorption was rapid (time to reach maximum plasma concentration (Tmax) ~ 0.5 hour), half‐life short (~ 2 hours), and PK dose‐proportional, with no accumulation or time dependency on repeat dosing. BTK occupancy was dose‐dependent, reaching maximum occupancy of > 90% within ~ 4 hours after single doses ≥ 200 mg; the effect was long‐lasting (> 50% occupancy at 96 hours with ≥ 100 mg). After multiple dosing, full BTK occupancy was achieved with 25 mg, indicating slow turnover of BTK protein in vivo. Concentration‐QTcF analyses did not show any impact of evobrutinib concentration on corrected QT (QTc). In summary, evobrutinib was well‐tolerated, showed linear and time‐independent PK, induced long‐lasting BTK inhibition, and was associated with no prolongation of QT/QTc interval in healthy subjects. Evobrutinib is, therefore, suitable for investigation in autoimmune diseases.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Bruton's tyrosine kinase (BTK) inhibition represents a rational therapeutic approach to autoimmune disease through its potential to block B cell receptor‐dependent and Fc receptor‐dependent pathways. Evobrutinib is an oral BTK inhibitor that has demonstrated efficacy in preclinical autoimmune disease models.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This first‐in‐human phase I study examined the safety and tolerability of evobrutinib administered as either single or multiple ascending oral doses, compared with placebo, in healthy subjects. The pharmacokinetic (PK) profile, target occupancy, and effect of evobrutinib on QT interval were also investigated.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Single doses of evobrutinib up to 500 mg and multiple doses up to 200 mg for 14 days were safe and well‐tolerated in healthy subjects. PKs were linear and predictable with no accumulation or time dependency on repeat dosing. BTK occupancy was high and sustained after both single and multiple dosing. Single doses of evobrutinib up to 500 mg did not prolong QT/corrected QT interval.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Our findings in healthy subjects indicate that evobrutinib is a promising new BTK inhibitor suitable for further investigation in patients with autoimmune disease.
Bruton's tyrosine kinase (BTK) is a cytoplasmic tyrosine kinase expressed in cells of hematopoietic origin, including B cells and macrophages, but not T cells. As a member of the Tec kinase family, BTK is a key regulator of B cell receptor (BCR)‐mediated signaling, and plays pivotal roles in B cell development, differentiation, activation, class‐switching, proliferation, survival, and cytokine release.1, 2, 3, 4, 5 BTK also mediates Fc receptor (FcR) signaling6 and may have a role in toll‐like receptor signaling1 in myeloid cells. Small molecule inhibitors of BTK have emerged as a promising new class of therapeutics for B cell driven pathologies and have the potential to treat a broad spectrum of autoimmune, inflammatory, and oncologic disorders.7
Evobrutinib is a highly selective, oral inhibitor of BTK. In human cellular assays, evobrutinib potently inhibits B cell and basophil activation and a range of functions mediated by BCR and FcR induction.8 Moreover, evobrutinib has shown promising activity in preclinical models in a number of autoimmune diseases.8, 9, 10 In a mouse model of interferon‐accelerated systemic lupus erythematosus, evobrutinib was associated with robust reduction in disease activity which correlated with inhibition of B cell function.8 Similarly, in a collagen‐induced arthritis mouse model, rheumatoid arthritis‐like symptoms were inhibited by evobrutinib.8 Furthermore, in both B cell‐dependent9 and T cell‐dependent10 experimental autoimmune encephalomyelitis mouse models, evobrutinib limited central nervous system inflammation and disease severity suggesting a potentially broader therapeutic benefit in multiple sclerosis (MS) compared with therapeutic agents with modes of action involving B cell depletion alone.
This first‐in‐human phase I study was designed to examine the safety and tolerability, pharmacokinetics (PK), and target occupancy of evobrutinib when administered as either single or multiple ascending oral doses, compared with placebo, in healthy subjects. With atrial fibrillation and ventricular tachyarrhythmia reported in up to 6% and 1% of patients receiving the BTK inhibitor ibrutinib,11 and atrial fibrillation/flutter reported in 3% of those receiving acalabrutinib,12 our investigation took advantage of the wide range of doses administered in a placebo‐controlled, double‐blind fashion to perform an early corrected QT (QTc)‐exposure evaluation for evobrutinib, as recently recommended in the revised 2017 International Conference on Harmonization (ICH) E14 clinical guidance.13
Methods
Study design
This was a phase I, randomized, double‐blind, placebo‐controlled trial conducted in healthy volunteers between September 2014 and April 2015 at a single site in the United States (Quintiles Phase I Services, Overland Park, KS). The study was carried out in accordance with the principles of the ICH requirements for Good Clinical Practice and the Declaration of Helsinki, and was reviewed and approved by Midlands Independent Review Board, Overland Park, KS. Written informed consent was obtained from all participants. The primary end point was the safety and tolerability of evobrutinib.
The study comprised two distinct parts; part 1 examined single ascending doses (SADs) of evobrutinib and part 2 examined multiple ascending doses (MADs) of evobrutinib (Figure S1 ). Part 1 involved SAD cohorts of evobrutinib (25, 50, 100, 200, 350, and 500 mg) administered as an oral solution (2.5 mg/mL; see Supplementary Material S1 for justification of the starting dose.) In each dose cohort, eight subjects were randomized to receive a single administration of evobrutinib or placebo (6:2) using consecutive randomization codes. A sentinel dosing strategy was used in all SAD cohorts whereby the first 2 subjects were initially dosed with evobrutinib or placebo (1:1) on day 1 with the remainder (5:1) dosed after 24 hours if safety was deemed satisfactory. Subjects remained resident at the trial site until discharge on day 8 and returned for a follow‐up visit between days 11 and 13. Part 2 examined MAD cohorts of evobrutinib (25, 75, and 200 mg) administered once daily over 14 days. A sentinel dosing strategy was not used in part 2 as the results from at least two higher sentinel dosing groups were available from part 1 prior to starting treatment in the MAD cohorts. In each dose cohort, 12 subjects were randomized to receive evobrutinib or placebo (9:3). For both the SAD and MAD portions of the study, subjects were fasted and had no food intake until 4 hours after drug administration. Subjects remained resident at the trial site until discharge on day 18 and returned for a single follow‐up visit on day 28 ± 2 days.
Dose escalation criteria
Following completion of each dose cohort in both parts of the study, a safety monitoring committee, which consisted of the principal investigator and the sponsor's clinical pharmacologist and global drug safety product lead, decided whether protocol‐defined dose escalation to the next level was appropriate (to a maximum of 500 mg in part 1, or to the highest safe and tolerated dose or highest dose from part 1 in part 2), based on all safety data and other available safety, PK, and target occupancy data. If two or more subjects per cohort experienced a per‐protocol dose‐limiting event (DLE) related to evobrutinib (i.e., identification of a nontolerated dose), dose escalation was to be terminated. DLEs were defined as follows: lymphocyte count decreased to < 500/mm3 or increased to > 20,000/mm3; severe infection requiring antibiotic and/or antimycotic treatment; alanine aminotransferase or aspartate aminotransferase > 3 times the upper limit of normal; adverse events considered related to evobrutinib or placebo with grade 3/4 toxicity, as specified by the Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventative Vaccine Clinical Trials.14 Other safety data (e.g., hematology parameters) were also taken into consideration for dose escalation decisions as a matter of course.
The decision whether to initiate part 2 of the study, as well as the MAD starting dose, was determined by the safety monitoring committee based on review of safety data from part 1, with PK and pharmacodynamic (PD) data also used to inform the decision. Dose‐limiting criteria and termination/stopping rules were as per part 1 of the study. Multiple doses were not to be escalated if three or more subjects per cohort experienced a DLE related to evobrutinib and the maximum daily dose was not to exceed the highest dose given in part 1. Ultimately, dose levels of 25, 75, and 200 mg were selected.
Subjects
Study participants were healthy male and female subjects aged 18–55 years with a body mass index between 19.0 and 30.0 kg/m2 who had been stable nonsmokers for at least 6 months, had no significant clinical abnormalities, and were in good general health. Subjects were required to abstain from consumption of xanthine‐containing food or beverages and to stop consuming caffeine from 48 hours prior to first administration of study drug until collection of the last PK sample in each period. See Supplementary Material S2 for the inclusion and exclusion criteria.
Safety and tolerability assessments
In both SAD and MAD cohorts, safety assessments included physical examination, vital signs, 12‐lead electrocardiogram (ECG), 24‐hour ECG telemetry and Holter monitoring for QT assessment, clinical laboratory assessments (hematology, biochemistry, coagulation, and urinalysis), and assessment of the humoral immune response (immunoglobulin G subclasses). Treatment‐emergent adverse events (TEAEs) and serious adverse events (SAEs) were recorded from time of informed consent until study completion (the last visit of the last volunteer).
Safety and tolerability were assessed in all subjects who had received at least one dose of evobrutinib or placebo with TEAEs summarized by treatment group, evobrutinib dose level, and preferred term (Medical Dictionary for Regulatory Activities (MedDRA) version 17.0, MedDRA MSSO, McLean, VA). Assessment of severity utilized the Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventative Vaccine Clinical Trials. Data were summarized using descriptive statistics.
PK assessments and methods
PK analysis was performed in all subjects who received at least one dose of evobrutinib and had at least one primary PK parameter evaluable by noncompartmental methods using Phoenix WinNonlin version 6.3 (Certara LP, Princeton, NJ).
Serial blood samples were obtained on day 1 predose and 0.25, 0.5, 1.0, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 16, 24 (day 2), 36 (day 2), and 48 hours (day 3) postdose during part 1 and used to prepare plasma samples for evobrutinib PK analysis. A similar sampling schedule was undertaken in part 2 on days 1 and 14, with additional trough samples obtained on days 5, 8, and 11, as well as days 17 and 18.
PK samples were analyzed by Quintiles Bioanalytical and ADME Laboratory (Ithaca, NY) using a validated bioanalytical liquid chromatographic assay with tandem mass spectrometric detection to determine evobrutinib concentration; the lower limit of quantification (LLOQ) of the assay was 0.1 ng/mL. Plasma evobrutinib concentrations and PK parameters were summarized descriptively.
To test dose proportionality, PK parameters were analyzed graphically as area under the concentration curve (AUC)/dose and maximum observed concentration (Cmax)/dose vs. dose and via a power model (PK parameter = α*doseβ) approach with log‐transformed PK parameters as the dependent variable and log‐transformed dose as the independent variable. Accumulation of evobrutinib and time dependency of PK parameters were evaluated for each dose level during part 2 using a linear mixed‐effect analysis of variance for log‐transformed PK parameters with a fixed‐repeated effect for day and random effect for each subject.
Target occupancy
Target occupancy was assessed in all subjects who received at least one dose of evobrutinib or placebo and had at least one evaluable measurement at one time point. BTK occupancy in peripheral blood mononuclear cells (PBMCs) was used as a surrogate marker of inhibition of BTK activity. To determine the degree of BTK occupancy by evobrutinib, both total BTK (BTKT) and free BTK (BTKF) were measured in PBMC protein lysates.
During part 1, blood samples were obtained on day 1 predose and postdose at 1, 2, 4, and 6 hours. Further blood samples were then obtained every 24 hours after the first day 1 dose, up until day 5. During part 2, blood samples were obtained on day 1 predose and postdose at 2 and 4 hours, then predose on days 2 and 8. Further blood samples were obtained on day 14 predose and postdose at 2 and 4 hours, and every 24 hours thereafter until day 18. Samples were analyzed at Cambridge Biomedical (Boston, MA).
BTKT was measured using a sandwich enzyme‐linked immunosorbent assay with mouse monoclonal anti‐BTK (BD Biosciences, San Jose, CA; catalog number 611117) as capture antibody and rabbit anti‐BTK (Cell Signaling Technologies, Danvers, MA; catalog number 8547BF) with goat anti‐rabbit horseradish peroxidase (Millipore, Danvers, MA; catalog number AP132P) as detection antibody. BTKF was measured using a biotinylated probe (Merck Patent GmbH, Darmstadt, Germany; June 2016) that irreversibly binds to the active site of BTK. Labeled BTK was captured from the lysate using streptavidin‐coated enzyme‐linked immunosorbent assay plates and detected using a different anti‐BTK antibody (Pierce; catalog number PA5‐20085) than in the BTKT assay. Total protein was measured and lysates were adjusted to 0.5 ng/nL total protein before measuring BTKT and BTKF. PD parameters of target occupancy were determined by noncompartmental methods using elapsed time from dosing (when available) and were summarized using arithmetic mean (SD) of percentage BTK occupancy. A standard curve was constructed for both assays using recombinant human BTK protein (Invitrogen; catalog number PR5442A). The LLOQ of the assays for BTKT and BTKF were 1.9 and 0.6 ng/mL, respectively, with the upper limit of quantification of 450 and 200 ng/mL, respectively, in PBMC lysate. The fraction of occupied BTK was calculated using the following formula: relative % occupancy = (1−(BTKF/BTKT)/(BTKF predose/BTKT predose))*100. Maximum effect (Emax) and time to maximum effect (Etmax) were determined for BTK occupancy after each dose administration.
ECG assessments
Holter ECG recordings of 24 hours duration were performed on day 1 in parts 1 and 2 of the study using digital 12‐lead Holter devices (GI M12R, Manlius, NY) and analyzed in the Quintiles central ECG laboratory. Subjects rested in beds for at least 10 minutes in a quiet environment to control for factors impacting heart rate when ECG assessments were conducted. Ten‐second 12‐lead ECGs were extracted in triplicate using Antares ECG extraction software version 2.15.1.0 (AMPS‐LLC, New York, NY) for each PK time point (predose and 0.25, 0.5, 1.0, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 16, and 24 hours postdose) within a –5 to 0‐minute window prior to and a 5‐minute window after each sampling time point. The quality of extracted ECG snapshots was checked by a Holter associate/specialist using the Antares quality control review interface. The mean QT and RR values from the triplicate ECGs were used to calculate QT interval corrected for heart rate by Fridericia's method (QTcF) for each time point.
ECG methods
Statistical analyses of ECG data were performed using SAS software version 9.3 (SAS Institute, Cary, NC) in all subjects who received at least one dose of evobrutinib or placebo on day 1, and had Holter ECG recordings on day 1 and at least one scheduled postdose time point. The data set consisted of day 1 ECG and PK data from all subjects in parts 1 and 2 of the study. Baseline was at a single predose time point (not time‐matched). For the subjects who had received placebo, the evobrutinib concentration value was assumed to be equal to 0 at each PK time point.
The main end point for statistical analysis of ECG data was the relationship between mean change from baseline in the QT interval corrected by Fridericia's formula (ΔQTcF) and evobrutinib plasma concentration after a single dose, with adjustment for the effect of time‐matched placebo on ΔQTcF (i.e., ΔΔQTcF). In model‐based analyses, mean ΔΔQTcF was estimated on the basis of a contrast in model parameters. In non‐model‐based analyses, ΔΔQTcF for an evobrutinib‐treated individual at a particular time point was defined as the difference between the individual's ΔQTcF value and the mean ΔQTcF in the placebo group at that time point.
Hysteresis between the plasma concentration of evobrutinib and ΔQTcF was assessed visually by graphical means and tested statistically on the basis of the difference between ΔΔQTcF at Tmax (time to Cmax) and ΔΔQTcF at the time point corresponding to the largest mean ΔΔQTcF.15 Nonlinearity was assessed via visual inspection of the scatterplot of ΔQTcF vs. evobrutinib concentration for all time points, and a significance test based on a model for ΔQTcF quadratic in concentration. In the absence of hysteresis or nonlinearity, ΔQTcF was to be modeled via a linear mixed effects model, with concentration as a continuous covariate, treatment (active or placebo) and time point as categorical factors, and random intercept and slope per subject, based on previous publications.16, 17, 18 Additional covariates and interactions were considered, and the final model selected on the basis of Akaike information criterion. Model‐based predicted mean ΔΔQTcF was calculated for the geometric mean Cmax of each dose cohort. Two‐sided 90% confidence intervals (CIs) of the estimate were determined using bootstrapping (1,000 resamples).
Absolute and change from baseline values of QTcF and other parameters were summarized descriptively by time point and dose cohort. In a by‐time point analysis, mean and 90% CI for non‐model‐based ∆∆QTcF were provided for each evobrutinib dose cohort.
Results
Subject disposition
In total, 48 subjects (46 men and two women) were randomized into part 1: six subjects to each evobrutinib dose (25, 50, 100, 200, 350, and 500 mg), and 12 subjects to placebo. All 48 randomized subjects were treated and completed the trial. Thirty‐six subjects were randomized into part 2: nine subjects to each once daily evobrutinib dose cohort (25, 75, and 200 mg q.d.), and nine subjects to placebo. All 36 randomized subjects were treated according to protocol. However, two subjects discontinued prematurely from part 2. One subject on placebo withdrew consent postdose on day 14 and one subject on evobrutinib (75 mg) was withdrawn due to noncompliance postdose on day 13. Demographic data are summarized in Table S1 .
Safety and tolerability
All randomized subjects were included in the safety analysis. Analysis of TEAEs following single and multiple dosing are shown in Table 1. There were no deaths or SAEs related to treatment. A single SAE of multitrauma due to an automobile accident was experienced by a subject in the evobrutinib 350 mg SAD cohort 7 days after study administration but was unrelated to the study drug.
Table 1.
Treatment‐emergent adverse events
| SAD study | ||||||||
|---|---|---|---|---|---|---|---|---|
| Preferred term, n (%), (number of events) | Placebo (n = 12) | Evobrutinib | ||||||
| 25 mg (n = 6) | 50 mg (n = 6) | 100 mg (n = 6) | 200 mg (n = 6) | 350 mg (n = 6) | 500 mg (n = 6) | Pooled active (n = 36) | ||
| Overall total | 4 (33.3) (6) | 0 (0.0) | 3 (50.0) (6) | 1 (16.7) (1) | 2 (33.3) (4) | 1 (16.7) (1) | 2 (33.3) (3) | 9 (25.0) (15) |
| Headache | 1 (8.3) (1) | 0 (0.0) | 1 (16.7) (2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 2 (5.6) (3) |
| Contact dermatitis | 0 (0.0) | 0 (0.0) | 2 (33.3) (2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (5.6) (2) |
| Amylase increased | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 0 (0.0) | 1 (2.8) (1) |
| Back pain | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.8) (1) |
| Dizziness | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 0 (0.0) | 1 (2.8) (1) |
| Dry eye | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 1 (2.8) (1) |
| Dyspepsia | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.8) (1) |
| Excoriation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 0 (0.0) | 1 (2.8) (1) |
| Lipase increased | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 0 (0.0) | 1 (2.8) (1) |
| Multiple injuries | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 1 (2.8) (1) |
| Nasal congestion | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.8) (1) |
| Odynophagia | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) (1) | 1 (2.8) (1) |
| Abdominal pain | 1 (8.3) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Application site pruritus (due to ECG stickers) | 1 (8.3) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Dry mouth | 1 (8.3) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Noncardiac chest pain | 1 (8.3) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| URTI | 1 (8.3) (1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| MAD study | |||||
|---|---|---|---|---|---|
| Preferred term, n (%), (number of events) | Placebo (n = 9) | Evobrutinib | |||
| 25 mg (n = 9) | 75 mg (n = 9) | 200 mg (n = 9) | Pooled active (n = 27) | ||
| Overall total | 2 (22.2) (2) | 3 (33.3) (6) | 7 (77.8) (14) | 3 (33.3) (3) | 13 (48.1) (23) |
| Headache | 1 (11.1) (1) | 1 (11.1) (1) | 2 (22.2) (2) | 0 (0.0) | 3 (11.1) (3) |
| Application site irritation | 0 (0.0) | 0 (0.0) | 1 (11.1) (1) | 1 (11.1) (1) | 2 (7.4) (2) |
| Fatigue | 0 (0.0) | 0 (0.0) | 2 (22.2) (2) | 0 (0.0) | 2 (7.4) (2) |
| URTI | 0 (0.0) | 0 (0.0) | 1 (11.1) (1) | 1 (11.1) (1) | 2 (7.4) (2) |
| Abdominal pain | 0 (0.0) | 0 (0.0) | 1 (11.1) (2) | 0 (0.0) | 1 (3.7) (2) |
| Nausea | 0 (0.0) | 0 (0.0) | 1 (11.1) (2) | 0 (0.0) | 1 (3.7) (2) |
| Abdominal discomfort | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 0 (0.0) | 1 (3.7) (1) |
| Complex regional pain | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 0 (0.0) | 1 (3.7) (1) |
| Constipation | 0 (0.0) | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 1 (3.7) (1) |
| Dry throat | 0 (0.0) | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 1 (3.7) (1) |
| Excoriation | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 0 (0.0) | 1 (3.7) (1) |
| Muscle spasms | 0 (0.0) | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 1 (3.7) (1) |
| Muscle strain | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 0 (0.0) | 1 (3.7) (1) |
| Rhinorrhea | 1 (11.1) (1) | 1 (11.1) (1) | 0 (0.0) | 0 (0.0) | 1 (3.7) (1) |
| Sneezing | 0 (0.0) | 0 (0.0) | 1 (11.1) (1) | 0 (0.0) | 1 (3.7) (1) |
| Toothache | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (11.1) (1) | 1 (3.7) (1) |
ECG, electrocardiogram; MAD, multiple ascending dose; SAD, single ascending dose; URTI, upper respiratory tract infection.
Overall, the nature and incidence of TEAEs were similar in evobrutinib‐treated and placebo‐treated subjects after single dosing in part 1. In total, 15 TEAEs developed in nine subjects (25.0%) receiving evobrutinib and six TEAEs in four subjects (33.3%) on placebo. The most common TEAEs in subjects on evobrutinib were headache (three events in two subjects (5.6%)) and contact dermatitis at locations of ECG pads (two events in two subjects (5.6%)). Headache also occurred in one subject (8.3%) on placebo. All TEAEs were mild (grade 1) except in one subject in the 200 mg treatment group, who experienced a dose‐limiting TEAE of grade 4 increased lipase in combination with grade 3 increased amylase on day 11. However, there were no accompanying clinical signs and symptoms, ultrasound examination of the abdomen revealed no abnormality of the pancreas, and values returned rapidly to baseline by day 12. Lipase and amylase levels were assessed in all other subjects. Any post‐baseline shifts in toxicity grade were transient, and not associated with any clinical signs and symptoms.
In part 2, 23 TEAEs were reported in 13 subjects (48.1%) on evobrutinib and two TEAEs were reported in two subjects (22.2%) on placebo. The most frequently reported TEAEs on evobrutinib included headache (three events in three subjects (11.1%) vs. one event (11.1%) on placebo), and skin irritation due to ECG pads, fatigue, and upper respiratory tract infection (each two events in two subjects (7.4%)). Seven gastrointestinal TEAEs occurred in five subjects (18.5%) on evobrutinib treatment. No relation to dose was observed for gastrointestinal or other TEAEs. All TEAEs reported were mild and no dose‐limiting adverse events were reported.
There were no TEAEs leading to discontinuation and no clinically significant trends in vital signs, ECGs, laboratory values, or immunoglobulin G subclasses in either part of the study. Overall, evobrutinib seemed to be safe and well‐tolerated and no nontolerated dose could be defined after SAD or MAD administrations, supporting further clinical investigation of evobrutinib in forthcoming trials.
PK analyses
Following single dosing in part 1, evobrutinib was rapidly absorbed with a median Tmax of 0.5 to 1.0 hours across all dose cohorts (25–500 mg; Table 2). After reaching Cmax, the plasma concentration of evobrutinib declined rapidly and dose‐independently in a mono‐exponential fashion (Figure 1) to < 1% of Cmax within 8 hours. Apparent clearance was high (2,553–3,995 mL/minute) and independent of dose. At the two higher single doses (350 and 500 mg), evobrutinib concentrations above the LLOQ were still measurable up to 48 hours after dosing in some (n = 3) subjects, revealing an additional terminal phase in the elimination of evobrutinib. For this reason, the estimated geometric mean terminal half‐life (t1/2) was smaller with lower doses of evobrutinib (25–200 mg) compared with the two higher doses (1.8–2.6 vs. 6.6–6.8 hours, respectively). As a consequence of the longer terminal t1/2 at 350 mg and 500 mg, geometric mean apparent volume of distribution during the terminal phase was 1,796 and 1,485 L, respectively. Nevertheless, because mean evobrutinib terminal phase concentrations at higher doses were small (< 1% of Cmax 24 hours postdose), all concentration‐time profiles declined at the same rate within the first 8 hours, with a relevant t1/2 of ~ 2 hours for all doses.
Table 2.
Geometric mean (CV% GM) PK parameters of evobrutinib
| Following single‐dose administration | |||||||
|---|---|---|---|---|---|---|---|
| Dose (mg) | N | AUC0–∞ (ng*hour/mL) | Cmax (ng/mL) | Tmax a (hour) | t1/2 (hour) | CL/F (mL/minute) | Vz/F (L) |
| 25 | 6 | 104 (53.6) | 69.9 (58.1) | 0.8 (0.5–1) | 1.80 (12.8) | 4,000 (53.6) | 621 (60.2) |
| 50 | 6 | 326 (35.7) | 234 (42.1) | 0.5 (0.3–1) | 2.06 (36.8) | 2,550 (35.7) | 455 (24.5) |
| 100 | 6 | 434 (30.3) | 309 (47.5) | 0.5 (0.3–0.5) | 2.56 (39.0) | 3,840 (30.3) | 850 (48.5) |
| 200 | 6 | 856 (27.0) | 555 (37.2) | 0.5 (0.5–1) | 2.09 (5.30) | 3,900 (27.0) | 703 (24.7) |
| 350 | 6 | 1,910 (41.7) | 846 (44.7) | 1.0 (0.3–2) | 6.81 (95.0) | 3,050 (41.7) | 1,800 (136) |
| 500 | 6 | 3,220 (25.0) | 1,510 (34.7) | 0.5 (0.5–2) | 6.63 (69.9) | 2,590 (25.0) | 1,490 (64.6) |
| On day 1 and day 14 of the multiple‐dose study | |||||||
| Day 1 | |||||||
| 25 | 9 | 123 (38.7) | 99.7 (44.5) | 0.5 (0.3–0.5) | 1.70 (19.9) | 3,390 (38.7) | 498 (32.9) |
| 75 | 9 | 325 (37.5) | 254 (46.0) | 0.5 (0.3–1) | 1.86 (15.1) | 3,850 (37.5) | 620 (48.6) |
| 200 | 9 | 1,110 (24.6) | 797 (43.9) | 0.5 (0.5–1) | 2.49 (34.7) | 3,000 (24.6) | 647 (42.3) |
| Dose (mg) | N | AUC0–24 h (ng*hour/mL) | Cmax (ng/mL) | Tmax a (hour) | t1/2 (hour) | R acc(Cmax)b | R acc(AUC0–24 h)b |
|---|---|---|---|---|---|---|---|
| Day 14 | |||||||
| 25 | 9 | 126 (46.4) | 80.4 (64.9) | 0.5 (0.3–1) | 1.59 (18.2) | 0.807 (0.59–1.11) | 1.03 (0.89–1.17) |
| 75 | 8 | 345 (44.6) | 252 (60.3) | 0.5 (0.3–1) | 2.29 (18.9) | 1.000 (0.65–1.54) | 1.03 (0.81–1.31) |
| 200 | 9 | 1,210 (34.0) | 782 (60.1) | 0.5 (0.5–1) | 3.62 (70.5) | 0.982 (0.71–1.35) | 1.09 (0.94–1.25) |
Geometric mean (CV% GM) values are rounded to 3 significant digits.
AUC0–∞,area under the plasma concentration‐time curve from time zero extrapolated to infinity; AUC0–24 h, area under the plasma concentration‐time curve from time zero to 24 hours; CL/F, apparent clearance; Cmax, maximum observed plasma concentration; CV% GM, geometric coefficient of variation; PK, pharmacokinetic; Racc(AUC0–24), accumulation ratio for AUC; Racc(Cmax), accumulation ratio for Cmax; Tmax, time to reach maximum plasma concentration; t1/2, terminal half‐life; VZ/F, apparent volume of distribution during terminal phase.
Median and range; rounded to 1 significant digit.
Geometric mean and 95% confidence interval.
Figure 1.
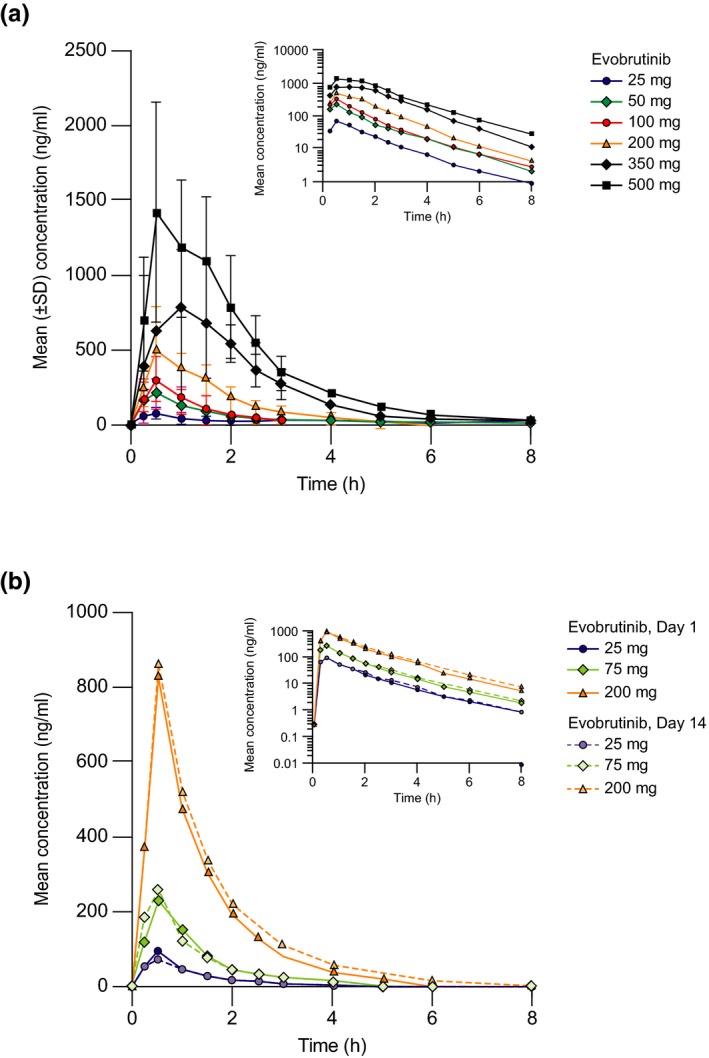
Pharmacokinetics of evobrutinib. (a) Arithmetic mean (SD) evobrutinib serum exposure vs. time after single dosing on day 1 of part 1 (single ascending dose study), and (b) mean evobrutinib serum exposure vs. time on day 1 and day 14 of part 2 (multiple ascending dose study).
In part 2, concentration‐time profiles were similar between day 1 and day 14 (Figure 1), indicating no accumulation with multiple dosing. This was confirmed by accumulation ratios for AUC (R acc(from time 0 to 24 hours (AUC0–24 h)) 1.03–1.09) and Cmax (R acc(Cmax) 0.81–1.00) and was in agreement with the observed short t1/2 (1.6–3.6 hours) after multiple dosing. In general, PK parameters determined on day 1 and day 14 of the multiple dose study agreed with those obtained after single dosing in part 1 (Table 2). Dose proportionality of AUC0–24h and Cmax was demonstrated following multiple dosing of 25–200 mg (Figure 2) and confirmed by the power model. Furthermore, no time dependency of the evobrutinib PK was noted based on the comparison of concentration‐time profiles. This was confirmed by comparisons between AUC0–24 h on day 14 and AUC from time zero extrapolated to infinity (AUC0–∞) on day 1, with 90% CIs including 100% across all comparisons.
Figure 2.
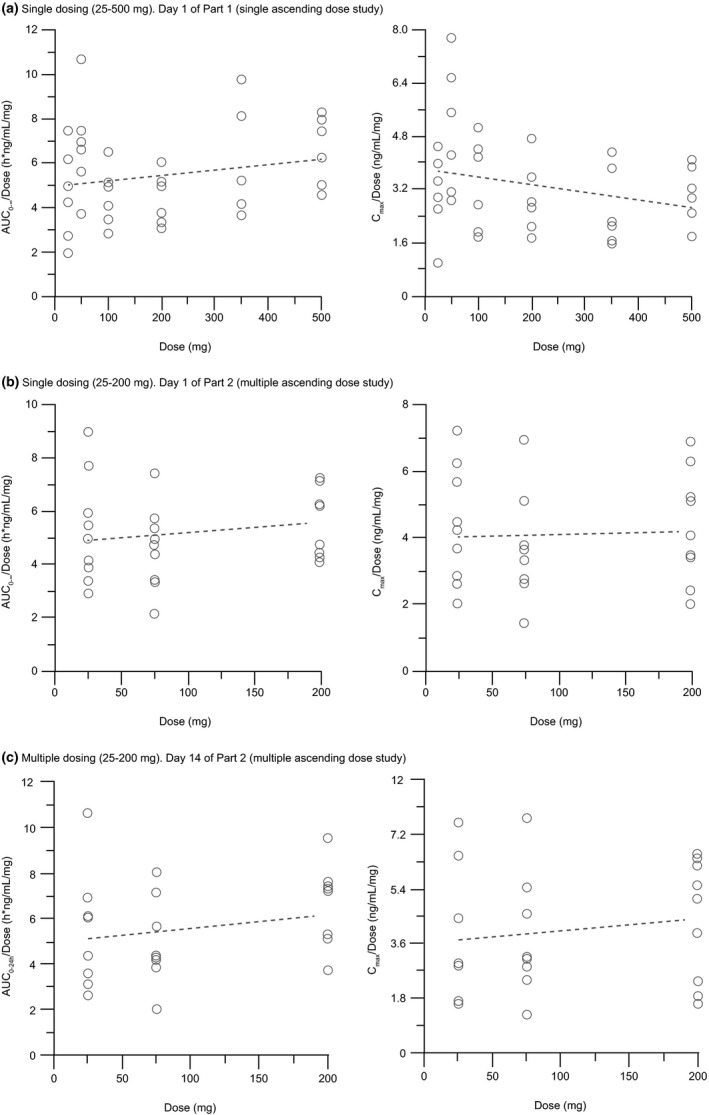
Individual dose‐normalized evobrutinib exposure (AUC and Cmax) and regression line after (a) single dosing on Day 1 of Part 1 (single ascending dose study); (b) single dosing on Day 1 of Part 2 (multiple ascending dose study), and (c) multiple dosing on Day 14 of Part 2 (multiple ascending dose study). Dotted line is linear regression through dose‐normalized parameters vs. dose. AUC0–24 h, area under the concentration curve from time 0 to 24 hours; AUC0–∞, area under the concentration curve from time zero extrapolated to infinity; Cmax, peak plasma concentration.
Target occupancy
BTK occupancy in PBMC was used as a surrogate marker of inhibition of BTK activity by evobrutinib.
In part 1, a dose‐dependent increase in BTK occupancy was observed after single dose administration of 25–500 mg of evobrutinib (Figure 3 a). Median Emax was 58.00% at 25 mg, 72.85% at 50 mg, 91.90% at 100 mg, and 98.90% at 200 mg, with complete saturation (100%) at 350 mg and 500 mg single dose (Table S2 ). Maximum effect was achieved rapidly (median Etmax, 1.0–4.0 hours across all dose groups), before declining slowly. At 24 hours postdose, median BTK occupancy ranged from 51.1–91.7% across all dose groups. BTK occupancy was still detectable 96 hours postdose, in contrast to serum evobrutinib concentrations (no relevant concentrations observed 8 hours postdose; Figure 1), in line with the irreversible binding mode of evobrutinib to BTK and slow turnover of BTK in vivo.
Figure 3.
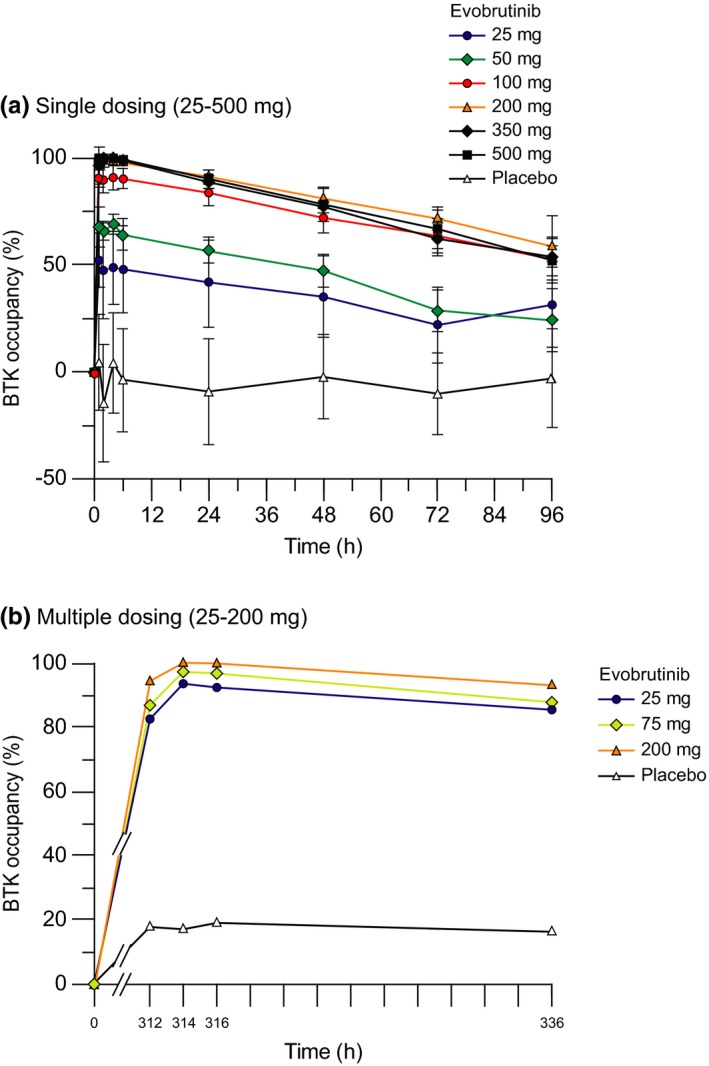
Arithmetic mean (SD) of % Bruton's tyrosine kinase (BTK) receptor occupancy* vs. time after administration of evobrutinib for (a) single dosing and (b) multiple dosing. *% BTK occupancy = (1‐(BTKF/BTKT)/(BTKF pre‐dose/BTKT pre‐dose))*100. BTKT, total Bruton's tyrosine kinase; BTKF, free Bruton's tyrosine kinase.
In part 2, following multiple dosing, median (range) Emax for the 25 mg dose increased from 70.4% (51.6–88.9%) on day 1 to 94.7% (91.8–98.3%) on day 14. In the 75 and 200 mg cohorts, Emax on day 14 reached 98.2% (94.0–98.8%) and 100% (100–100%), respectively (Table S2 ). Median BTK occupancy of ≥ 86% was maintained for 24 hours after the day 14 dose across all dose groups (Figure 3 b).
In general, there was clear discrimination between placebo and evobrutinib treatment groups in terms of BTK occupancy at all time points in both part 1 and part 2. However, low median levels of BTK occupancy were also observed in the placebo group. These probably represent artificial values due to assay variability in the measurement of BTKT and BTKF, which was observed to vary by up to 30% during assay validation, rather than actual BTK occupancy in nondosed subjects.
ECG analyses
ECG analyses were based on a data set of 83 evaluable subjects who had received a single dose of evobrutinib from 25–500 mg (analysis was performed for the first dose of subjects in part 2), which included two subjects with missing baseline data (1 of the 84 randomized subjects was excluded from the analysis due to the presence of flat T waves in all leads).
Concentration‐QT analysis
Categorical analysis of ECG parameters revealed no clinically significant change from baseline in heart rate, pulse rate, and QRS values postdose. Thus, QTcF was applied for the concentration‐QT analysis, which is also supported by the demonstration of independence of QTcF from heart rate (slope 0.0183; 95% CI –0.0519 to 0.0886; Figure S2 ).
Although the visual inspection of hysteresis indicated different profiles for evobrutinib concentration and mean ΔQTcF over time (Figure S3 a), examination of mean evobrutinib concentration vs. mean ΔΔQTcF over time plot did not point to meaningful changes in ΔΔQTcF with change in evobrutinib concentration (Figure S3 b). Accordingly, no statistically significant difference in time of maximum plasma concentration of evobrutinib (Tmax) compared with time of the maximum ΔΔQTcF (Umax) was observed. Thus, a hysteresis effect could not be concluded, and an effect compartment was not included in the model. Nonlinearity was not observed on visual inspection of the ΔQTcF vs. evobrutinib plasma concentration data or using a significance test. Consequently, modeling of the exposure‐effect analysis was conducted based on a linear mixed‐effects model for ΔQTcF. The final model included no additional covariates or interactions.
The slope of the relationship between placebo‐adjusted ΔQTcF and concentration was negative and very close to zero (–0.00027 ms/ng per mL; P = 0.86). Figure 4 shows model predictions for concentration‐QTc model, with mean ΔΔQTcF (90% CI) for each decile of PK concentration values. The predicted population mean ΔΔQTcF at geometric mean Cmax for the 25 mg group (86.5 ng/mL) was –0.78 ms with an upper limit of 2.71 ms for the 90% two‐sided bootstrapped CI (Table 3). For the 500 mg dose group, the predicted population mean ΔΔQTcF at geometric mean Cmax (1,512 ng/mL) was –1.16 ms with an upper limit of 3.26 ms for the 90% CI, which is well below the 10 ms threshold of regulatory concern (ICH‐E14 guidance).13
Figure 4.
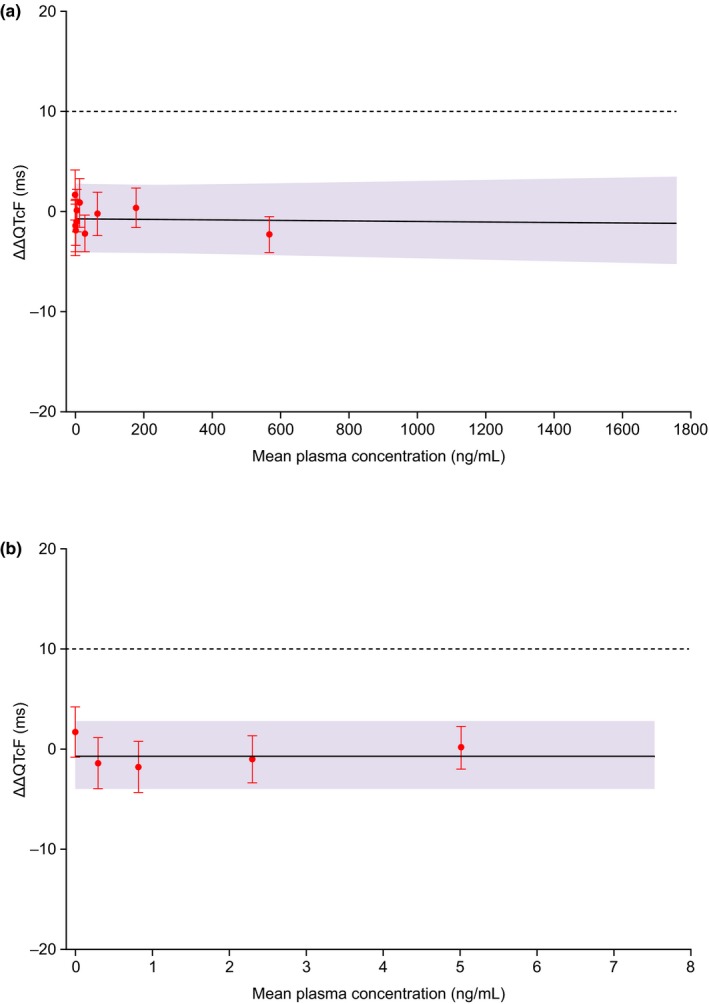
Model prediction of placebo‐adjusted change from baseline in QT interval corrected for heart rate by Fridericia's method (ΔΔQTcF; 90% confidence interval (CI)) from concentration‐QTc model, with mean ΔΔQTcF (90% CI) for each decile of pharmacokinetic (PK) concentration values. (a) entire concentration range and (b) the first five deciles of PK concentration values. Mean ΔΔQTcF values (ms) (denoted by circles) with two‐sided 90% CIs have been calculated for each decile of PK concentration values, and plotted at the corresponding median concentration value in each decile. The model‐derived predicted population mean ΔΔQTcF is shown as the continuous black line shaded with two‐sided 90% bootstrapped confidence limits of predicted mean ΔΔQTcF. The horizontal dotted line represents the regulatory threshold of concern of 10 ms.
Table 3.
Predicted values of mean ΔΔQTcF and associated two‐sided 90% CIs
| By evobrutinib plasma concentration | ||||
|---|---|---|---|---|
| Dose (mg) | Geometric mean Cmax (ng/mL) | Predicted mean ΔΔQTcFa (ms) | 90% CI of ΔΔQTcF (ms) (bootstrapped) | |
| Lower bound | Upper bound | |||
| 25 | 86.48 | −0.78 | −4.22 | 2.71 |
| 50 | 233.9 | −0.82 | −4.23 | 2.66 |
| 75 | 253.9 | −0.82 | −4.23 | 2.66 |
| 100 | 308.6 | −0.84 | −4.27 | 2.70 |
| 200 | 689.2 | −0.94 | −4.44 | 2.83 |
| 350 | 845.9 | −0.98 | −4.46 | 2.94 |
| 500 | 1,512.3 | −1.16 | −5.10 | 3.26 |
Based on the linear mixed model fitted to the data, the predicted values of population mean ΔΔQTcF and the associated two‐sided 90% bootstrapped CIs are reported at plasma evobrutinib concentration values corresponding to the observed geometric mean Cmax for different evobrutinib dose groups.
CI, confidence interval; Cmax, maximum observed plasma concentration; ΔΔQTcF, placebo‐adjusted change from baseline in QT interval corrected for heart rate by Fridericia's method.
Predicted population mean ΔΔQTcF was obtained from the original data set and not from bootstrapped data.
By‐time point analysis
By‐time point analysis showed that the maximum mean ΔΔQTcF for each of the evobrutinib dose groups ranged from –0.93 ms to 3.79 ms at all time points in the 25, 50, 75, 200, 350, and 500 mg dose groups. However, in the 100 mg dose group (n = 6), the mean ΔΔQTcF was 7.1 ms at the 6‐hour postdose time point (90% CI 3.8 to 10.4 ms), 5.6 ms at the 8‐hour postdose time point (90% CI –0.2 to 11.4 ms) and 7.2 ms at the 12‐hour postdose time point (90% CI 2.6 to 11.7 ms).
Categorical outlier analysis
The absolute QTcF value did not exceed 450 ms in any of the dose groups at any of the time points. There were no ECGs where the change from baseline in QTcF exceeded 60 ms. Change from baseline in QTcF between 30 ms and 60 ms was observed in 2 of 81 subjects (2.5%; both were in the 200 mg dose group), but the absolute QTcF value was ≤ 450 ms for both subjects. The number of ECGs with outlier values was extremely small and not clinically significant.
Treatment‐emergent T wave morphological abnormalities were reported in two evobrutinib‐treated subjects, one subject in the 50 mg dose group, and one subject in the 100 mg dose group, with abnormalities (flat T waves) observed at the day 1, 6‐hour and 8‐hour postdose time points. These abnormalities were not observed in either subject in ECGs acquired at previous and subsequent time points and did not seem to be dose‐dependent or concentration‐dependent.
Discussion
This first‐in‐human phase I study investigated the safety, tolerability, PK, and target occupancy of evobrutinib. Single doses up to 500 mg and multiple doses up to 200 mg for 14 days were safe and well‐tolerated. The PK profile of evobrutinib showed dose‐proportional and time‐independent exposure without accumulation after multiple dosing when administered once daily. A maximum BTK receptor occupancy of ≥ 90% (median) was achieved by single dose groups of ≥ 200 mg and by all multiple dose groups (25, 75, and 200 mg). No clinically relevant exposure‐effect relationship was detected between evobrutinib concentration and QTcF effect; single evobrutinib doses up to 500 mg with a peak concentration up to 1,512 ng/mL did not prolong QTcF.
TEAEs occurred in 25% of subjects after single dosing and in 48.1% after multiple dosing. The most common TEAEs after single dosing were headache and contact dermatitis, with headache, skin irritation (at locations of ECG stickers), fatigue, and upper respiratory tract infection most common after multiple dosing; the majority of events were of mild severity. With higher doses of evobrutinib, there was no apparent increase in frequency or type of adverse events. During part 1, one subject in the 200 mg treatment group experienced a DLE of grade 4 increased lipase in combination with grade 3 increased amylase. Although both events were considered related to treatment, they seemed to be isolated laboratory changes that resolved quickly with no accompanying clinical signs and symptoms or evidence of abnormality of the pancreas on ultrasound examination. Furthermore, the interval between the emergence of the first abnormal values on day 8 and the single dose itself casts doubt on any causal relationship, given the observed PK of evobrutinib. There were no other clinically relevant ECG changes or relevant cardiac or cardiovascular adverse events.
The PK profile of evobrutinib demonstrated rapid absorption with peak concentrations reached within ~ 0.5 hours, moderate‐to‐high plasma clearance, medium geometric mean apparent volume of distribution during the terminal phase, and short t1/2. PK were dose‐proportional over a 25–500 mg single‐dose range and a 25–200 mg once daily multiple‐dose range at steady‐state, with no accumulation and no time dependency observed after 14 days repeated daily dosing.
Target occupancy was dose‐dependent, approaching complete maximal BTK occupancy after single doses of evobrutinib ≥ 200 mg and multiple doses ≥ 75 mg, typically within 4 hours of administration. Median BTK occupancy was long‐lasting, remaining detectable 24 hours postdose on day 14 across the dose‐range of 25–200 mg, before declining slowly, consistent with irreversible binding, and a slow turnover of BTK protein (shown previously to be > 12 hours in human primary B cells19). These results are consistent with target occupancy data reported for other BTK inhibitors. Although no published target occupancy data are available for ibrutinib in healthy volunteers, median BTK occupancy of ≥ 95% was noted in patients with B cell lymphoma or chronic lymphocytic leukemia within 4 hours of administration of ibrutinib 2.5–12.5 mg/kg/day; BTK occupancy was maintained for at least 24 hours, consistent with the irreversible mechanism.20 Furthermore, following a single dose of CC‐292, five of six healthy subjects had > 98% BTK occupancy ≤ 4 hours after administration, with complete or near‐complete BTK occupancy sustained for 8–24 hours.21 These data supported the selection of recommended doses in the phase II program of evobrutinib and were successfully applied in a recent clinical study in MS.22
We found no evidence of a significant exposure‐effect relationship between evobrutinib concentration and QTcF. Over the range of doses evaluated, mean ΔΔQTcF was < 5 ms and the upper limit of the 90% two‐sided CI was well below the 10 ms threshold of regulatory concern specified in the ICH‐E14 guidance13 at all time points, with the exception of the 100 mg dose group, in which mean ΔΔQTcF ranged from 5.6 to 7.2 ms and the upper limit of the CI ranged from 10.4 ms to 11.7 ms at three time points. These findings are likely to be a chance occurrence given the results observed in higher‐dose groups. Indeed, our results are in line with those seen with other BTK inhibitors. Neither ibrutinib nor acalabrutinib have been associated with clinically relevant prolongation of the QTc interval at therapeutic or supratherapeutic doses during randomized, double‐blind, placebo‐controlled and positive‐controlled thorough QT studies.11, 12, 23
Evaluation of the relationship between concentration and QT/QTc using data collected in early phase clinical studies is a validated and US Food and Drug Administration‐accepted alternative strategy to a thorough QT study13, 15, 24 and has been widely used to reliably exclude relevant QTc effects during drug development,18, 25, 26 supporting waiver of the regulatory requirement for a thorough QT study in some cases.27 Although our concentration‐QT analysis is limited by the small number of subjects in each dose group and the small number of female subjects, pooling of data from parts 1 and 2 and the broad range of plasma concentrations obtained across dosage groups increase the reliability of our results. High‐resolution monitoring of ECGs over 24 hours permitted extraction of ECG snapshots at stable heart rates and recording of ECGs in triplicate and their central analysis further reduced any potential measurement error, helping to minimize within‐subject variability, and ultimately providing more robust statistical analyses.28
In summary, the central role of BTK in both FcR and BCR signaling makes BTK inhibition a promising approach for treatment of autoimmune diseases. The findings in healthy subjects reported herein indicate that evobrutinib is a promising new BTK inhibitor. Evobrutinib was well‐tolerated when administered as single (25–500 mg) or multiple (25–200 mg) ascending doses. PK were dose‐proportional after single and multiple dosing, with no time dependency or accumulation noted after 14 days' repeated once‐daily administration. High and sustained BTK occupancy was observed after both single and multiple dosing. Concentration‐response modeling of QT/QTc data revealed no prolongation of QT/QTc interval resulting from single doses of evobrutinib up to 500 mg. Further clinical studies designed to investigate the efficacy, safety, PD, and PK of evobrutinib in patients with systemic lupus erythematosus, rheumatoid arthritis, and relapsing MS are ongoing.
Funding
This study was sponsored by EMD Serono (a business of Merck KGaA, Darmstadt, Germany).
Conflict of Interest
A.D.B., J.L., and A.J. are employees of Merck KGaA. E.C.M., R.G., and A.T.B. are employees of EMD Serono, US (a business of Merck KGaA). H.M. was an employee of EMD Serono, US (a business of Merck KGaA) at the time of the study. D.Y.M. is an employee of Nuventra Pharma Sciences and a clinical pharmacology consultant working with Merck KGaA.
Author Contributions
All authors wrote the manuscript, designed the research, performed the research, and analyzed the data.
Supporting information
Figure S1. Study design.
Figure S2. Relationship between QT or QTcF and heart rate.
Figure S3. Assessment of hysteresis.
Table S1. Baseline demographics.
Table S2. Median (range) BTK receptor occupancy after single and multiple administration of evobrutinib.
Supinfo Supplementary Materials.
Acknowledgments
The authors would like to thank the volunteers and their families, investigators, co‐investigators, and the study teams at the participating center and acknowledge the significant contribution of Thomas Murtaugh, an employee at Quintiles at the time of the study. Statistical analysis was performed by Laurence Bernard (Cytel) and Indrasiri Fernando (IQVIA, India). Noncompartmental analyses of PK values were performed by Tobias Feige (Merck KGaA, Darmstadt, Germany). Concentration‐QT analyses were performed by Pramod Kadam and Khushboo Chaudhari (Cardiac Safety Services, IQVIA, India). Medical writing assistance was provided by Melanie Jones, Bioscript Science, Macclesfield, UK, and funded by Merck KGaA, Darmstadt, Germany.
Data were presented in part at the 2018 ECTRIMS Congress, October 10–12, Berlin, Germany, as a poster presentation.
EMD Serono Research & Development Institute, Inc., Billerica, MA, USA (a business of Merck KGaA, Darmstadt, Germany)
References
- 1. Crofford, L.J. , Nyhoff, L. , Sheehan, J. & Kendall, P. The role of Bruton's kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 12, 763–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de Gorter, D.J. et al Bruton's tyrosine kinase and phospholipase Cgamma2 mediate chemokine‐controlled B cell migration and homing. Immunity 26, 93–104 (2007). [DOI] [PubMed] [Google Scholar]
- 3. Petro, J.B. , Rahman, S.M. , Ballard, D.W. & Khan, W.N. Bruton's tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J. Exp. Med. 191, 1745–1754 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Craxton, A. , Jiang, A. , Kurosaki, T. & Clark, E.A. Syk and Bruton's tyrosine kinase are required for B cell antigen receptor‐mediated activation of the kinase Akt. J. Biol. Chem. 274, 30644–30650 (1999). [DOI] [PubMed] [Google Scholar]
- 5. Anderson, J.S. , Teutsch, M. , Dong, Z. & Wortis, H.H. An essential role for Bruton's [corrected] tyrosine kinase in the regulation of B‐cell apoptosis. Proc. Natl. Acad. Sci. USA 93, 10966–10971 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hendriks, R.W. Drug discovery: new Btk inhibitor holds promise. Nat. Chem. Biol. 7, 4–5 (2011). [DOI] [PubMed] [Google Scholar]
- 7. Corneth, O.B. , Klein Wolterinkm, R.G. & Hendriks, R.W. BTK signaling in B cell differentiation and autoimmunity. Curr. Top. Microbiol. Immunol. 393, 67–105 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Haselmayer, P. et al Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J. Immunol. 15, 2888–2906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boschert, U. et al T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by Evobrutinib (M2951) a novel Bruton's tyrosine kinase inhibitor. Presented at the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) Conference; October 25–28, 2017; Paris, France.
- 10. Torke, S. , Grenningloh, R. , Boschert, U. & Weber, M.S. B cell‐mediated experimental CNS autoimmunity is modulated by inhibition of Bruton's tyrosine kinase. The European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) Conference; October 25–28, 2017; Paris, France.
- 11. Imbruvica® (ibrutinib) capsules, for oral use. Highlights of prescribing information. Pharmacyclics LLC, July 2019 <https://www.imbruvica.com/docs/librariesprovider7/default-document-library/prescribing-information.pdf>. Accessed July 29, 2019.
- 12. CALQUENCE® (acalabrutinib) capsules, for oral use. Highlights of prescribing information. AstraZeneca, November 2017. <https://www.azpicentral.com/calquence/calquence.pdf>. Accessed July 29, 2019.
- 13. US Department of Health and Human Services, Food and Drugs Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) . E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs — questions and answers (R3). Guidance for Industry. June 2017, ICH. Revision 2. <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e14-clinical-evaluation-qtqtc-interval-prolongation-and-proarrhythmic-potential-non-antiarrhythmic-1>. Accessed July 29, 2019
- 14. US Food and Drug Administration . Guidance for industry — toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/toxicity-grading-scale-healthy-adult-and-adolescent-volunteers-enrolled-preventive-vaccine-clinical>. Accessed July 29, 2019.
- 15. Darpo, B. et al Results from the IQ‐CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin. Pharmacol. Ther. 97, 326–335 (2015). [DOI] [PubMed] [Google Scholar]
- 16. Garnett, C.E. et al Concentration‐QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J. Clin. Pharmacol. 48, 13–18 (2008). [DOI] [PubMed] [Google Scholar]
- 17. Zhang, J. , Chen, H. , Yi Tsong, Y. & Stockbridge, N. Lessons learned from hundreds of thorough QT studies. Ther. Innov. Regul. Sci. 49, 392–397 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Westerberg, G. et al. Safety, pharmacokinetics, pharmacogenomics and QT concentration – effect of modelling of the SirT1 inhibitor selisistat in healthy volunteers. Br. J. Clin. Pharmacol. 79, 477–491 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saffran, D.C. et al A point mutation in the SH2 domain of Bruton's tyrosine kinase in atypical X‐linked agammaglobulinemia. N. Engl. J. Med. 330, 1488–1491 (1994). [DOI] [PubMed] [Google Scholar]
- 20. Advani, R.H. et al Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J. Clin. Oncol. 31, 88–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evans, E.K. et al Inhibition of Btk with CC‐292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 346, 19–28 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Montalban, X. et al Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 380, 2406–2417 (2019). [DOI] [PubMed] [Google Scholar]
- 23. de Jong, J. et al Ibrutinib does not prolong the corrected QT interval in healthy subjects: results from a thorough QT study. Cancer Chemother. Pharmacol. 80, 1227–1237 (2017). [DOI] [PubMed] [Google Scholar]
- 24. Garnett, C. et al Scientific white paper on concentration‐QTc modelling. J. Pharmacokinet. Pharmacodyn. 45, 383–397 (2018). [DOI] [PubMed] [Google Scholar]
- 25. Li, Y. , Carayannopoulos, L.N. , Thomas, M. , Palmisano, M. & Zhou, S. Exposure‐response analysis to assess the concentration‐QTc relationship of CC‐122. Clin. Pharmacol. 8, 117–125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sparve, E. et al Prediction and modeling of effects on the QTc interval for clinical safety margin assessment, based on single‐ascending‐dose study data with AZD3839. J. Pharmacol. Exp. Ther. 350, 469–478 (2014). [DOI] [PubMed] [Google Scholar]
- 27. Mohamed, M.F. , Zeng, J. , Jiang, P. , Hosmane, B. & Othman, A.A. Use of early clinical trial data to support thorough QT study waiver for upadacitinib and utility of food effect to demonstrate ECG assay sensitivity. Clin. Pharmacol. Ther. 103, 836–842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patterson, S. et al Investigating drug‐induced QT and QTc prolongation in the clinic: a review of statistic design and analysis considerations: report from the Pharmaceutical Research and Manufacturers of America QT Statistics Expert Team. Drug Inf. J. 39, 243–265 (2005). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Study design.
Figure S2. Relationship between QT or QTcF and heart rate.
Figure S3. Assessment of hysteresis.
Table S1. Baseline demographics.
Table S2. Median (range) BTK receptor occupancy after single and multiple administration of evobrutinib.
Supinfo Supplementary Materials.