

Abstract
Schistosomiasis is a widespread human parasitic disease currently affecting over 200 million people. Chemotherapy for schistosomiasis relies exclusively on praziquantel. Although significant advances have been made in recent years to reduce the incidence and intensity of schistosome infections, these gains will be at risk should drug resistance parasites evolve. Thioredoxin glutathione reductase (TGR) is a selenoprotein of the parasite essential for the survival of schistosomes in the mammalian host. Several high-throughput screening campaigns have identified inhibitors of Schistosoma mansoni TGR. Follow up analyses of select active compounds form the basis of the present study. We identified eight compounds effective against ex vivo worms. Compounds 1 – 5 are active against all major species and development stages. Ability of these compounds to target immature worms is especially critical because praziquantel is poorly active against this stage. Compounds 1 – 5, 7, and 8 displayed schistosomicidal activity even after only one-hour incubation with the worms. Compounds 1 – 4 meet or exceed standards set by the World Health Organization for leads for schistosomiasis therapy activity. The mechanism of TGR inhibition was studied further with wild type and mutant TGR proteins. Compounds 4 – 6 were found to induce an nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity in TGR, leading to the production of superoxide and hydrogen peroxide. Collectively, this effort has identified several active compound series that may serve as the basis for the development of new schistosomicidal compounds.
Keywords: drug development, helminth, mechanism of inhibition, neglected disease, redox biology, high throughput screen
Graphical Abstract
Schistosomiasis, caused by blood-dwelling flukes of the genus Schistosoma, currently affects over 200 million people, mostly in developing countries.1–2 The global burden of schistosomiasis is estimated at 2.6 million disability-adjusted life years.2 Schistosomiasis ranks second only to malaria based on the World Health Organization’s assessment of parasitic diseases.3 Great efforts have been devoted to the development of antischistosomal drugs,2, 4 but since the mid-1980s the treatment of schistosomiasis relies on a single drug, praziquantel.5 Reports of praziquantel-resistant cases, as well as the generation of praziquantel-resistant parasites in the laboratory, highlight the need for new drugs to treat the disease.6
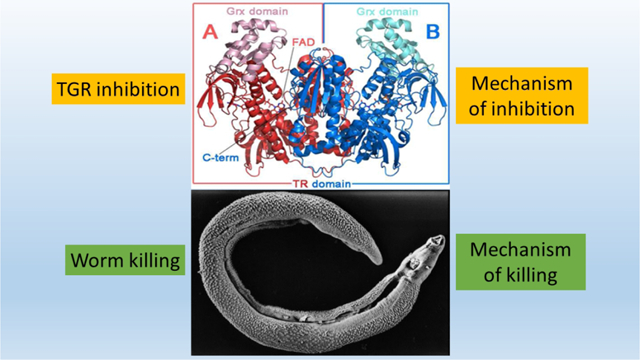
Living in the aerobic environment of its mammalian host, schistosomes have effective mechanisms to maintain cellular redox balance and to evade reactive oxygen species generated by the host’s immune response.7 Unlike their mammalian hosts, which have dedicated enzymes in the glutathione (GSH) and thioredoxin (Trx) pathways, the redox system of schistosomes depends on a unique, multifunctional enzyme, thioredoxin glutathione reductase (TGR), to maintain reduced forms of both GSH and Trx.8 RNA interference proved that TGR is essential for parasite survival and auranofin, a clinically used anti-inflammatory drug, found to inhibit TGR, was able to kill parasites rapidly in culture at physiological concentration (5 μM) and to partially cure infected mice, showing that TGR is drugable.9 These results show that TGR is a bottleneck in the maintenance of redox balance in schistosomes and have, in turn, made TGR an attractive antiparasitic target.10–12 The amino acid sequence and domain structure of schistosome TGR has similarities to mammalian forms of thioredoxin reductase (TrxR) and glutathione reductase (GR), with an additional amino-terminal extension of a glutaredoxin (Grx) domain.8, 13 TGR is a flavoenzyme, obtaining reducing equivalents from NADPH. Similar to mammalian TrxR, S. mansoni TGR is a selenoprotein and contains a selenocysteine (Sec/U) as the penultimate residue in the carboxyl terminal GCUG active site motif. Sec is highly reactive and sensitive to electrophilic attack.14 It is essential for TGR activity to maintain the redox state in worms, but also provides a good nucleophilic binding site for inhibitors.15
TGR has a complex domain structure and catalytic cycle (Scheme 1).13, 16–17 The flavin adenine dinucleotide (FAD) cofactor accepts electrons from NADPH. The electrons are then transferred to a proximal Cys couple. This reduced Cys couple then transfers electrons to the mobile C-terminal Cys-Sec pair of the symmetrical subunit in the homo dimer. The reduced C-terminal active site then moves to the protein surface where it can reduce either the Cys pair in the Grx domain belonging to the original subunit (from which the electrons initially came), where glutathione disulfide (GSSG) reduction occurs, or directly reduce oxidized Trx or other non-physiological compounds including 5,5’-dithio-bis-(2-nitrobenzoic acid) (DTNB).
Scheme 1. The catalytic cycle of TGR.

The reaction scheme was drawn following the proposed mechanism of SmTGR.16–17 When NADPH binds to dimeric oxidized TGR (A), FAD is reduced and then donates electrons to the proximal C154-C159 couple (B). The electrons are then transferred to the C596’-U597’ couple on the C-terminus of the other subunit (C). The reduced C-terminus transfers electrons to C28-C31 couple of Grx domain of the original subunit and additional NADPH reduces FAD and electrons are transferred to the proximal C154-C159 couple (D). The electrons are then transferred to the C596’-U597’ couple (E). This enzyme form can reduce both oxidized Trx (or DTNB) and GSSG.
Based on successful expression and purification of recombinant S. mansoni TGR, quantitative high throughput screens (qHTS) were carried out resulting in the identification of numerous TGR inhibitory compounds.18–20 To validate the qHTS actives and optimize druglike and worm permeability properties, a computational structure-activity relationship (SAR) study was conducted by searching for analogs and similar compounds among those identified in the screens and, when possible, purchased and retested in our biological pipeline. Confirmed TGR active compounds were advanced to worm studies. The compounds were screened against larval, juvenile, and adult S. mansoni worms and then screened further against S. japonicum and S. haematobium adult worms. Because previous studies found that inhibition of TrxR by electrophilic compounds can result in promotion of an NADPH oxidase activity of the otherwise inhibited enzyme,21–25 and that this gain of function is strictly related to their therapeutic effect,26 we assessed the NADPH oxidase activity of inhibited TGR. Collectively, this effort has identified several active compound series, which may serve as the basis for the development of new schistosomicidal compounds.
Results and Discussion
TGR high-throughput screening and confirmatory studies
Ninety-nine compounds identified in the screens, representing 24 loosely defined chemotypes, with IC50s in low or sub micro molar range, and 20 analogs were purchased and tested against TGR (Tables SI1 and SI2). Analogs are indicated by “n.d.” in the qHTS column of Supplemental Tables 1 and 2. Confirmatory assays identified 97 compounds with low micromolar TGR inhibitory activity representing 23 different chemotypes (Tables SI1 and SI2). These compounds were advanced to worm studies and screened against larval and/or adult S. mansoni. Compounds found to elicit > 50% killing at 12.5 μM in 5 days were tested against juvenile S. mansoni, and adult S. japonicum and S. haematobium worms. Compounds tested and found to have inadequate activity against worms or chemical liabilities were not investigated further (Table SI2). The eight compounds shown in Table 1 have schistosomicidal activity against all developmental stages and species.
Table 1:
Most active compounds identified in this study
| Compound | CID | Structure | TGR IC50 (μM) | Adult worms | Sm║ juvenile | Sm║ Somula | |||
|---|---|---|---|---|---|---|---|---|---|
| qHTS† | Confirm | Sm║ | Sj║ | Sh║ | |||||
| 1 | 3561200 | 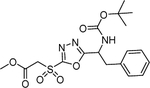 |
n. d.* | 0.33 | 10 μM 120 h 62% d‡ |
10 μM 120 h 65% d‡ |
10 μM 144 h 67% d‡ |
10 μM 120 h 39% d‡ |
10 μM 24 h 99% d‡ |
| 2 | 16012863 | 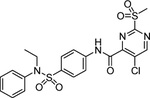 |
0.3981 | 0.21 | 10 μM 120 h 78% d‡ |
10 μM 120 h 50% d‡ |
10 μM 120 h 94% d‡ |
10 μM 24 h 100% d‡ |
10 μM 24 h 100% d‡ |
| 3 | 4412151 | 6.3^ | 2.84 | 10 μM 72 h 100% d‡ |
10 μM 120 h 70% d‡ |
10 μM 96 h 100% d‡ |
10 μM 48 h 100% d‡ |
10 μM 24 h 100% d‡ |
|
| 4€ | 2779853 | 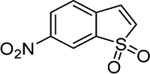 |
5.438^ | 2.01 | 10 μM 144 h 76% d‡ |
10 μM 120 h 69% d‡ |
10 μM 120 h 85% d‡ |
10 μM 72 h 100% d‡ |
10 μM 24 h 100% d‡ |
| 5& | 2801235 | 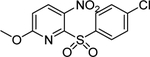 |
0.1 | 0.06 | 10 μM 120 h 67% d‡ |
10 μM 120 h 30% d‡ |
10 μM 120 h 48% d‡ |
10 μM 120 h 57% d‡ |
10 μM 24 h 100% d‡ |
| 6 | 659226 | 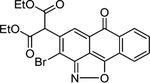 |
0.631 | 0.14 | 12.5 μM 120 h 28% d‡ |
12.5 μM 120 h 100% d‡ |
n. d.* | 12.5 μM 120 h 100% d‡ |
12.5 μM 120 h 96% d‡ |
| 7 | 1090309 | 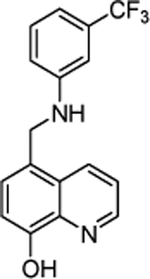 |
0.3162 | 0.09 | 12.5 μM 120 h 43% d‡ |
12.5 μM 120 h no killing |
n. d.* | 12.5 μM 120 h 8% d‡ |
12.5 μM 120 h 61% d‡ |
| 8 | 2943813 | 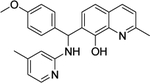 |
0.3981 | 0.13 | 12.5 μM 120 h 53% d‡ |
12.5 μM 120 h 56% d‡ |
n. d.* | 10 μM 120 h 24% d‡ |
10 μM 120 h 20% d‡ |
Commercially available analogs of the 8 compounds were tested and the results are shown in Table SI1. Their SAR against TGR is briefly summarized below. Compound 1 represents a chemotype containing a 1, 3, 4-oxadiazole-2-sulfone. Compounds with this group were reported to have antibacterial activity.28–29 Close analogs of 1 showed variable activity against TGR and were generally less potent than 1. Two compounds that do not contain sulfone but are otherwise similar to 1 were found to be at least 2.6-fold less potent against TGR. Compound 2 and its close analogs contain a 3- or 4-substituted phenylsulfoneamide moiety. Compounds with this chemotype displayed potent sub-micromolar inhibition of TGR, whereas compounds that did not contain these substituents or did not contain sulfone were at least 18.5-fold less potent. A pyridazine-3-one containing compound 318 is a moderately potent inhibitor of TGR with IC50 of 2.84 μM. Its close analogs did not inhibit TGR, whereas compounds similar but lacking the pyridazine-3-one portion showed activity that varied between 0.82 and 7.19 μM. Compound 4, which is also known as “Stattic”,30 inhibited TGR with IC50 of 2.01 μM. Consistent with the previously proposed mechanism of 4 (electrophile), its close analogs without electron withdrawing groups were poor inhibitors of TGR. Compound 5, a potent inhibitor of mammalian TrxR named TRi-1 having anticancer activity,26 displayed strong inhibition of TGR with IC50 of 0.06 μM. Its close analog had similar potency against TGR. Tetracyclic compound 6 showed potent 0.14 μM inhibition of TGR. The activity of somewhat similar tetracyclic compounds in this series varied but was generally 75.5-fold weaker. Considering rather large number of analogs, compounds containing 8-hydroxyquinoline scaffold were split into two series 7 and 8. Both 7 and 8 and their close analogs in Table SI1 displayed excellent inhibitory activity with IC50 between 0.03 and 0.15 μM. 8-Hydroxyquinolines were reported to have diverse biological activities,31 with at least some also exhibiting schistosomicidal activity.3 All of the 8-hydroxyquinoline analogs found to be active against TGR contained an aminomethylene substituent in either the para or ortho positions, suggesting that elimination of the amino substituent and formation of a reactive quinone methide may play a role in the mechanism of action of these compounds. Quinone methide forming compounds were recently found to have activity against cultured adult S. mansoni worms.32
TrxR and GR inhibitory activity comparison
TGR is responsible for reduction of both Trx and GSSG in schistosomes. To further investigate the TGR inhibitory activity of the eight best compounds, both DTNB and GSSG were used as substrates to assess inhibition of TGR (Table 2). For all of the compounds, inhibition of both activities was found to be almost identical. Both activities of TGR require the transfer of reducing equivalents from the FAD/C154-C159 redox center to the flexible C-terminal arm containing the Cys-Sec active site. The reduced C-terminal arm either directly reduces Trx (or DTNB) or reduces the Grx domain which, in turn, can reduce GSSG (Scheme 1).13, 17 During this electron pathway, either a modification of the reduced FAD proximal thiols or of the reduced C-terminal Cys-Sec pair can be responsible for the observed enzyme inhibition.
Table 2.
Inhibition of DNTB and GSSG reducing activities of TGR
| Compound | IC50 (DTNB) (μM) | IC50 (GSSG) (μM) |
|---|---|---|
| 1 | 0.33 ± 0.07 | 0.39 ± 0.07 |
| 2 | 0.21 ± 0.02 | 0.23 ± 0.03 |
| 3 | 2.84 ± 0.08 | 2.49 ± 0.11 |
| 4 | 2.01 ± 0.14 | 2.20 ± 0.20 |
| 5 | 0.06 ± 0.01 | 0.05 ± 0.01 |
| 6 | 0.14 ± 0.01 | 0.15 ± 0.02 |
| 7 | 0.09 ± 0.02 | 0.13 ± 0.03 |
| 8 | 0.13 ± 0.04 | 0.33 ± 0.05 |
Schistosomicidal activity studies in ex vivo worms
Compounds confirmed to have TGR inhibitory activity were screened against ex vivo S. mansoni adult and schistosomula (larval worms). Freshly perfused S. mansoni adult worms and newly transformed schistosomula were treated with different concentrations of compound for 5 days and survival scored. Compounds exhibiting ≥ 50% killing of worms in 5 days at 10 or 12.5 μM were further screened against S. mansoni juvenile worms (4 weeks post infection) and S. japonicum and S. haematobium adult worms (Figure 1). Compounds 1 – 5 exhibited potent schistosomicidal activity against all species of adult worms and all development stages of S. mansoni. It is notable that they have better killing activity against immature than adult S. mansoni worms. Schistosomula were highly susceptible; almost all were dead after the first day of incubation. The compounds were also potent against juvenile worms, which is important because praziquantel is much less effective against this development stage.33 Therefore, compounds with improved activity against juvenile worms could be used either alone or in combination with praziquantel to increase efficacy and prevent resistance. Treatment with 1 – 4 resulted in greater than 50% death of all three species of adult worms after 5 days incubation. Treatment of worms with 5 resulted in 67 % and 48 % killing of S. mansoni and S. haematobium adult worms, respectively, but only 30 % killing of S. japonicum adult worms. Compound 6 had killed 100 % of S. japonicum adult worms and S. mansoni larval and juvenile worms, but had weak killing activity against S. mansoni adult worms (28% killed). Unlike 1 – 5, 7 and 8 were less active against immature S. mansoni worms. The LD50 for compounds with potent activity against S. mansoni adult worms were determined (Figure 2). The results indicate that 1 – 5 have LD50 of 8.41, 9.07, 4.9, 7.08, and 10.1 μM respectively, approaching those determined here (not shown) of praziquantel (0.5 μM) and oltipraz (2 μM) a previously clinically used compound. Compounds 7 and 8 were not as potent as 1–5, displaying LD50 of 18.2 and 12.4 μM, respectively.
Figure 1. Compound activity against ex vivo worms.

 S. mansoni schistosomula,
S. mansoni schistosomula,  S. mansoni adult worms,
S. mansoni adult worms,  S. mansoni juvenile worms,
S. mansoni juvenile worms,  S. japonicum adult worms,
S. japonicum adult worms,  S. haematobium adult worms. Freshly perfused adult and juvenile worms and newly transformed larva were cultured overnight without compound addition. Compounds were added the following day and cultured for 5 days. The culture media was replaced every day with compounds added. Results show worm survival.
S. haematobium adult worms. Freshly perfused adult and juvenile worms and newly transformed larva were cultured overnight without compound addition. Compounds were added the following day and cultured for 5 days. The culture media was replaced every day with compounds added. Results show worm survival.
Figure 2. Determination of LD50 for the best hits.

Freshly perfused adult worms were cultured overnight and compounds were added and the culture continued for 5 days. The culture media and compounds were replaced daily. The result show worms’ survival after 5 days incubation in triplicate.
In the results shown in Figures 1 and 2, Table 1, and SI Tables 1 and 2, the worms were exposed to the compounds continuously; fresh compounds were added when media was replaced daily. To determine the required exposure time and to mimic in vivo conditions of drug metabolism and excretion, adult S. mansoni worms were exposed to different concentrations of the compounds for 1 hour. Then the media was removed, the worms were rinsed with fresh media, and incubated in media without compound. Worm survival was scored for 5 days (Figure 3). Treatment with compounds 1 and 2 resulted in 60 % and 40 % S. mansoni adult worm killing activity following 1 hr exposure at 20 μM. Treatment with compounds 3, 4, and 5 resulted in 80 %, 100 %, and 60 % S. mansoni adult worm killing activity after 1 hr exposure at 50 μM respectively, and about 10 % killing at 20 μM Compound 6 had no killing at all of concentrations tested. Compounds 7 and 8 were less potent than compounds 1 – 5 with 80 % and 30 % killing at 100 μM but still had 10 % killing activity at 20 μM By comparison with 1 – 5, 1 hr exposure to 100 μM praziquantel resulted in only 20% killing. After replacement with drug-free media, most of the worms recovered from the contractile state caused by incubation with praziquantel within 30 min of compound removal. It was previously determined that the after an overnight exposure to praziquantel the LD50 was 40 μM.34 Therefore, it is not surprising that a 1 hr exposure to 100 μM praziquantel resulted in little killing. We next determined the compounds cytotoxicity against mammalian cells. Most compounds have similar cytotoxicity to mammalian cells and schistosome worms (Table SI3). A notable exception is 7, an 8-hydroxyquinoline. It is worth noting that toxicity against cultured cells does not correspond well with whole animal toxicity; compounds 4 and 5 are well tolerated in animal models of different diseases.26, 35–38
Figure 3. Brief exposure to TGR inhibitors and worm survival.

 100 μM,
100 μM,  50 μM,
50 μM,  20 μM,
20 μM,  10 μM. Ex vivo S. mansoni adult worms were exposed to compounds for 1 hour. The worms were then washed and cultured in media without compounds for 5 days. Survival was scored daily. Compound 6 was also tested but no worm killing was seen at all concentrations tested.
10 μM. Ex vivo S. mansoni adult worms were exposed to compounds for 1 hour. The worms were then washed and cultured in media without compounds for 5 days. Survival was scored daily. Compound 6 was also tested but no worm killing was seen at all concentrations tested.
To determine if worm death was associated with TGR inhibition, S. mansoni adult worms were cultured in 50 μM of compound, harvested at indicated times before any worm death occurred, and TGR activities (NADPH-dependent DTNB and GSSG reduction) were determined in worm homogenates. Inhibition of 50 % or greater of both DTNB and GSSG reduction activities were observed in worms after treatment with all the compounds, including compound 6 that was not schistosomicidal (Figure 4). Compound 6 resulted in the lowest inhibition of TGR activity (~60%) of the compounds tested.
Figure 4. TGR activity in treated worms.

Worms were treated with compounds (50 μM) and collected at indicated times, homogenized, and homogenates were analyzed for TrxR activity (DTNB reduction,  ) and GR activity (GSSG reduction,
) and GR activity (GSSG reduction,  ). Assays were done in triplicate and compared to activities in untreated worms collected at the same times.
). Assays were done in triplicate and compared to activities in untreated worms collected at the same times.
Investigation of the mechanism of TGR inhibition by compounds 1 – 8
First, the effect of the redox state of TGR on the inhibitory activity of compounds was investigated. Incubation of TGR with NADPH results in the production of reduced thiols or selenols in the active sites of TGR (Scheme 1). In the reduced form, TGR is able to react with its natural substrates in order to reduce them, but also becomes susceptible towards inhibition by electrophiles. TGR was incubated with inhibitors with or without addition of NADPH for 30 min, followed by desalting to remove compounds. Then the activity of the enzyme was determined. TGR treated in the same fashion with the known inhibitor auranofin9 was tested for comparison. For compounds 1 – 8, TGR activity was recovered after incubation and desalting in the absence of NADPH (Figure 5). In the absence of NADPH, only auranofin was found to inhibit TGR. Therefore, the inhibitory activity of 1 – 8 is NADPH dependent, requiring TGR to be in a reduced state for their inhibitory activity to be expressed.
Figure 5. NADPH-dependence and reversibility of inhibition.

TGR (50 nM) and compounds at 20 times the IC50 incubated with or without NADPH for 15 minutes. Then the DTNB assay was performed. Samples were desalted to remove compounds by passing through a 7.0 kDa-cutoff spin desalting column and TGR activity was determined 30 minutes and 3 hours after desalting.
Next, the reversibility of inhibition was determined. TGR was incubated with inhibitors plus NADPH for 15 minutes. The TGR activity of an aliquot of the sample was determined. Unreacted compound was removed from the remainder of the sample by passing through a desalting column and the TGR activity was determined. Compounds 1 – 7 were found to be irreversible inhibitors of TGR, as was auranofin, while TGR inhibited by 8 was found to slowly recover activity after desalting, indicating reversible inhibition (Figure 5). The reversible inhibition of TGR by 8 may explain why it exerts minimal schistosomicidal activity after one-hour exposure of worms; TGR activity would recover in worms following removal of 8 from the media. However, TGR inhibition by 1 – 7 would continue after removal from the media due to their irreversible inhibitory activity.
To determine if the activity of the compounds against worms depends on their electrophilic potential, we calculated their lowest unoccupied molecular orbital (LUMO) energies ab initio using DFT(b3lyp)/SOLV QM method and 6–31G**++ QM basis set. The LUMO energies were calculated for the compounds in Table 1 and Table SI 1. If ex vivo schistosomicidal data were available, additional compounds with the chemotypes 1 – 8 found in Appendix 2 (Table SI4) were added to the calculations. Correlation between the LUMO energies and the schistosomicidal activity against S. mansoni adult is shown in Figure 6. The same data are also provided in the table format in the supplemental material (SI Table 3). Even with several outliers, the LUMO values of these compounds are inversely correlated (R2 = 0.48) with the percent of dead S. mansoni adult worms, suggesting that their mechanism of action is likely to be electrophilic, suggesting that they are likely to react with Sec or Cys residues of TGR. Considering that many other factors can affect their activity against TGR in ex vivo worms, this correlation is exceptionally strong (one-way ANOVA was performed in MS Excel using the Data Analysis pack. N=28, df=26, R=0.63, R2=0.48, p<0.0001).
Figure 6. Correlation between LUMO and schistosomicidal activity for compounds in this study.

Most of the compounds tested contain moieties generally recognized as electrophilic or able to generate electrophilic species in situ, suggesting that they may be scavenged by endogenously present nucleophiles and/or they may inhibit targets other than TGR in ex vivo worms. This was investigated by adding GSH to reactions and determining if this prevented inactivation of TGR. When the compounds were incubated together with reduced TGR and different concentrations of GSH, TGR activity was inhibited by these compounds even at the highest concentrations of GSH (SI Figure 1).
A related flavoenzyme in humans is GR (hGR). Inhibition of hGR would indicate that these compounds lack selectivity and, hence, signal potential toxicity. The compounds were tested for activity against hGR (Table SI5). Compounds 1, 2, 7, and 8 did not inhibit hGR at concentrations as high as 2 mM. Compounds 3 and 6 inhibited hGR at 19% and 25%, respectively, at 2 mM (IC50s for TGR are 2.84 μM and 0.14 μM, respectively). Compounds 4 and 5 had IC50s of 30.7 μM and 104 μM respectively, 15- and 1700-fold higher than for TGR. These results indicate selectivity of inhibition for TGR versus hGR.
Transformation of TGR from anti-oxidant to pro-oxidant enzyme
In order to better understand the mechanism of inhibition of TGR by the inhibitors, the ability of compounds 1–8 to induce an NADPH oxidase activity in TGR was investigated (Fig. 7 and 8). Control TGR showed low consumption of NADPH and low production of superoxide and H2O2 as measured by pyrogallol red and Amplex Red oxidation, respectively (Figure 7). It was found that after inhibition by 4 – 6, but not 1 – 3, 7, 8, and auranofin, NADPH consumption increased and superoxide was produced (Figure 7). Pyrogallol red is stoichiometrically oxidized by superoxide without reaction with other oxidants, including H2O2.39–40 Addition of superoxide dismutase (SOD) to the assay abolished the oxidation of pyrogallol red, verifying the formation of superoxide, but NADPH consumption was unchanged (not shown). Furthermore, compared with uninhibited TGR, NADPH consumption after treatment by 1 – 3, 7, or auranofin decreased by about 50%.
Figure 7. Conversion of TGR from antioxidant to pro-oxidant enzyme.

TGR (500 nM) was incubated with NADPH (100 μM) and compounds for 30 minutes. The inhibitor concentrations were 20 μM for 1, 2, 5 – 8 and 50 μM for 3 and 4. Samples were then desalted to remove unreacted compounds. NADPH consumption, superoxide production using pyrogallol red, and H2O2 production using Amplex Red and horseradish peroxidase were determined. Addition of 10 units of SOD abolished oxidation of pyrogallol red and addition of 20 units of catalase inhibited oxidation of Amplex Red (not shown).
Figure 8. NADPH oxidase activity of TGR mutants.

U597C and ΔCUG (each at 500 nM) were incubated with NADPH (100 μM) and compounds for 30 minutes. Samples were then desalted. NADPH consumption, superoxide production using pyrogallol red, and H2O2 production using Amplex Red and horseradish peroxidase, were determined. Addition of 10 units of SOD abolished oxidation of pyrogallol red and addition of 20 units of catalase inhibited oxidation of Amplex Red (not shown).
The mechanism of superoxide production by inhibited TGR could be interpreted by the model suggested for the NADPH oxidase activity of mammalian TrxR modified by dinitrochlorobenzene or juglone. These inhibitors were shown by mass spectrometric analysis to covalently modify Sec or Cys residues at the C-terminus of TrxR.21, 24 The modified TrxR at its C-terminus is capable of transferring a single electron from reduced FAD to O2 to produce superoxide. In this way, modified TrxR is converted to so-called “SecTRAP” forms of the enzyme25 and this gain of function is highly linked to the therapeutic activity of compounds targeting TrxR.26 This model mimics the catalytic process of TrxR with their substrates.16, 41 We hypothesize that a similar process occurs in TGR (Scheme 2). Based on this model, the susceptibility of the C-terminal motif to inhibitors is the pivotal factor for the induction of NADPH oxidase activity. The covalently linked inhibitor may also participate in electron shuttling. Modification of Sec by 4 – 6 would covalently link the C-terminus to their aromatic ring system, effectively sensitizing the resulting adduct for accepting an electron from the reduced flavin or proximal cysteine pair and transferring to oxygen. Compounds found to inhibit TGR but not induce TGR oxidase activity may not have this property and/or not specifically derivatize the Sec residue of the enzyme (see below). In addition, during the catalytic cycle the C-terminal tail moves to the FAD redox center of the other monomer to accept electrons; the bulk of the compound modifying the C-terminus motif may thus potentially impede movement of the C-terminus towards the internal flavin active site preventing the electron transfer. An alternative mechanism is that inhibition affects dimer assembly. Formation of the monomer could increase accessibility of the reduced flavin to directly react with oxygen. To test this, TGR was reacted with 4 and the reaction products were analyzed by native PAGE. No evidence for monomer formation was seen (Supplemental Figure 2).
Scheme 2. The proposed model of oxidase activity of C-terminus modified TGR.

The reaction scheme was drawn following the experimental results reported in this work and previous studies.21, 24 The nucleophilic Sec residue of reduced TGR could be covalently modified by electrophilic aromatic inhibitors (A). The modified C-terminus moves to FAD redox center of the other subunit (B). The electrophilic aromatic ring of bonded inhibitor accepts a single electron from the thiol of proximal C154 and then transfers it to oxygen molecule to produce superoxide (C – E). EWG = electron withdrawing group.
The production of H2O2 after inhibition was also determined using Amplex Red and horseradish peroxidase. TGR inhibited by 4 – 6 was found to produce more than 4 times the amount of H2O2 as untreated TGR (Figure 7). Addition of bovine catalase reduced the oxidation of Amplex Red, verifying the formation of H2O2 (not shown). TGR treated with 1 – 3, 8, or auranofin produced the same low amounts of H2O2 as untreated TGR. TGR treated with 7 produced less H2O2 than untreated TGR.
TGR inhibited by 7, an 8-hydroxyquinoline, showed lowest NADPH oxidase activity; it appears that all electron transport in the protein is blocked. This type of compound has been reported to generate a quinone methide.42 The resulting quinone methide may nonspecifically react with nucleophilic residues of TGR and inhibit all electron transfer. Compound 8, also an 8-hydroxyquinoline albeit with a 4-methoxyphenyl substituent, was found to be a reversible inhibitor. If the quinone methide is responsible for covalent, irreversible modification of TGR, the substituent in 8 may reduce the rate of transformation to the quinone methide and convert it to a reversible inhibitor.
To further investigate the contributions of the mobile C-terminus and the Sec residue to inhibition and oxidase activity, compounds 4 – 6 were incubated with the Sec to Cys mutant TGR (U597C) and TGR with terminal three amino acids deleted (ΔCUG). The consumption of NADPH and the oxidase activity of the resulting proteins were determined (Fig. 8). It was found that 4 – 6 could increase NADPH consumption and induce NADPH oxidase activity in U597C but not in the ΔCUG (Figure 8). These results indicate that the NADPH oxidase activity of TGR requires the presence of the redox active C-terminus but does not depend on selenocysteine residue. The same lack of dependence on a Sec residue was found for mammalian TrxR.43 Loss of function of the C-terminal reactive tail does not induce NADPH oxidase activity by itself, which is shown by the lack of oxidase activity of ΔCUG. Again, similar results have been shown for mammalian TrxR.24 Reduction of the flavin and adjacent redox Cys pair can still occur in ΔCUG, but no NADPH oxidase activity is induced in this enzyme either with or without inhibitor treatment. This indicates that compounds 4 – 6 inhibit TGR and induce NADPH oxidase activity through binding to the C-terminal active site motif, and/or (less likely) that the C-terminal motif is required for NADPH oxidase activity in compound-derivatized enzyme species.
Conclusions
Through several high-throughput screening campaigns and a “SAR by purchase” strategy, eight single-digit micromolar inhibitors of TGR 1 – 8 were identified. Compounds 1 – 5 showed uniform adult worm killing activity against S. monsoni, S. japonicum, and S. haematobium, and potent activity against immature worms. These five compounds showed LD50 ≤ 10 μM for S. monsoni adult worms, the target for lead compound activity specified by the World Health Organization.44 Brief, 1 hr exposures revealed their rapid and potent activity, which will be important in the presence of host detoxification systems and treatments limited to one or two administrations of compound, the reality of schistosomiasis control strategies. These compounds selectively inhibit TGR with minimal activity against human GR, suggesting potential selectivity for use in humans. TGR inhibited by compounds 4 – 6 was converted to an NADPH oxidase and produced superoxide and H2O2. The activation of this activity was not essential for worm killing; compounds 1 – 3 without NADPH oxidase induction were as potent in worm killing as 4 – 6. Compounds 1 – 4 were found to have schistosome killing activity that meets or exceeds standards set by the World Health Organization for leads for schistosomiasis therapy activity.44 Based on their potent parasite killing activity against all three major schistosome species and all developing stages, we envision that identification of these compounds will facilitate further development of anti-schistosomiasis drugs with novel mechanism of action.
Materials and Methods:
Reagents.
All compounds were purchased from commercial suppliers: 1, 2, and 6 (Vitas-M Laboratory), 3 (Scientific Exchange), 4 (Cayman Chemicals), 5 (Maybridge), 7 and 8 (Enamine), NADPH (Cayman Chemicals). DTNB, GSSG, pyrogallol red, SOD from bovine erythrocytes and horseradish peroxidase were purchased from Sigma. Amplex Red was from Thermo Fisher Scientific. RPMI medium 1640, M-199, Pen Strep (10000 units/ml penicillin, 10000 μg/ml streptomycin) and Antibiotic-Antimycotic (100×) were from Gibco. Fetal bovine serum was from Gemini. Percoll® was from GE Healthcare. HEPES was from Cellgro.
Compound selection from the quantitative high-throughput screening hits.
All of the compounds tested were selected from qHTS sorted by potency.18–20 To find compounds similar to the most active 24 chemotypes, the original qHTS data were downloaded from PubChem BioAssay AID485364,20 the structures were “washed” in MOE45 to remove salts, and the MOE-Similarity module in MOE were used to select compounds with Tanimoto overlap of 60% or more. Commercial availability of the primary hits and their not previously tested analogs were determined using a combination of PubChem and ZINC databases.46–47
Recombinant protein production.
Wild-type TGR and the U597C mutant were prepared as described.17, 48 The ΔCUG mutant was generated using the Q5® Site-Directed Mutagenesis Kit (New England Biolabs) with forward (AAAGGGCGAGCTCAACGATC) and reverse (TAACCGCTCACTATGGGCGA) primers to delete the three terminal amino acids in pET24a and add a termination codon. Protein expression was done in Escherichia coli strain BL21 (DE3) as described.48 The wild type protein was expressed with a His-SUMO tag, which was removed leaving the addition of Gly-Ser-His at the N-terminus of the protein with no additional amino acid changes. Sec incorporation with this system is close to 100%.49 The U597C and ΔCUG mutants were expressed and used as N-terminal His-tagged proteins. The codon-optimized open reading frame for hGR was synthesized by GenScript and cloned into pET-15b with an N-terminal 6 His tag and expressed in E. coli BL21 (DE3). An overnight culture in LB plus ampicillin was diluted 1:100 in the same medium and cultured to an OD600 = 0.6. Isopropyl-β-D-thiogalactoside (1 mM) and riboflavin (20 mg/ml) were added and cultured for an additional 3 hr, and was purified as described.48
TGR inhibition study.
Assays were performed in triplicate as described.9 The NADPH dependency and reversibility of inhibition was determined by incubation of TGR (50 nM) with inhibitors at 20 times the IC50 with or without NADPH for 30 minutes. To determine reversibility, inhibited samples were desalted using 7.0 kDa-cutoff spin Zeba desalting column (Thermo Scientific) and the DTNB assay was performed 30 minutes and 3 hours after desalting to determine the enzyme activity.
Ex Vivo Experiments with Adult Worms and Schistosomula.
Adult worms were isolated from infected mice as described50 and cultured in RPMI medium + 10% fetal calf serum, 10 mM glutamine, and 1× penicillin/streptomycin for 24 h before compound addition. Mice (Swiss-Webster) were infected with S. mansoni by percutaneous exposure to cercariae (NMRI strain) obtained from infected Biomphalaria glabrata snails for 1 hour.50 Mice infected with S. mansoni or S. japonicum and Syrian hamsters infected with S. haematobium were perfused at 7 weeks, 6 weeks, or 4 months, respectively, to obtain adult worms and mice infected with S. mansoni were perfused at 23 days for juvenile worms.48, 50 This study was approved by the Institutional Animal Care and Use Committee of Rush University Medical Center (17–053; Department of Health and Human Services animal welfare assurance number A-3120–01). About 10 worm pairs or 20 juvenile worms, were cultured in 1 mL of media in 24 well plates. Compounds were dissolved in DMSO and added at the indicated concentrations. The culture media was replaced every day and the fresh compounds added. Control worms were treated with DMSO alone. Worms were cultured for 5 days and observed and scored for motility and mortality daily. Worm viability was scored using published methods.51 The motility of worms was scored. Living parasites move smoothly. Decreased motility and a loss of plasticity led to a lower score. Pairing and egg production were scored. The morphology and the integrity of the tegument was observed. Worms were scored as dead if they displayed no movement over several minutes.
Schistosomula were prepared by mechanical transformation and Percoll gradient isolation from cercariae isolated from infected B. glabrata as described.50 Fifty schistosomula were cultured in M-199 + 10% fetal calf serum for 24 h before the addition of compounds. Compounds were dissolved in DMSO and added at the indicated concentrations. The culture media was replaced every day and the fresh compounds added. Control schistosomula were treated with DMSO alone. Compounds affecting schistosomula resulted in reduced transparency and granularity and changes in movement. Schistosomula were observed daily and scored as dead when no movement was seen.
For brief exposure, compounds (10, 20, 50, and 100 μM) were added to the media for 1 hour, after which the adult worms were washed 3 times with culture media and incubated in culture media without compounds for further 5 days. Worm motility and mortality were scored daily. Control worms were incubated with DMSO.
To determine TGR activity in adult worms, worms were collected after the indicated incubation periods, washed and homogenized by pestle motor mixer. Control worms collected at the same times were treated with DMSO alone. The homogenates were collected, centrifuged, protein concentration determined by Pierce® protein assay reagent and TGR activity determined as described.
LD50s were determined using the above described culture methods. Freshly perfused adult worms were cultured overnight in media alone and compounds were added the following day and the culture continued for 5 days. Compounds were used at 2.5, 5, 7.5, 10, 12.5, 15, and 20 μM for 1; 2.5, 5, 7.5, 10, 15, and 20 μM for 2 and 4; 1, 2, 5, 10, and 20 μM for 3; 2.5, 7.5, 10, 15, and 20 μM for 5, and 2, 5, 12.5, 25, and 50 μM for 6 – 8. The culture media and compounds were replaced daily. Worm survival after 5 days incubation was determined. Experiments were done in triplicate. Worms were scored as dead when no movement was observed.
Cytotoxicity in mammalian cells.
Mammalian cell cytotoxicity was determine using Vero cells as described,52 with the following modifications. Cells were exposed the compounds in DMSO for 1 hr. The media was removed and the cells were washed in fresh media without compounds. The cells were cultured in the absence of compounds for 5 days before viability was determined.
Ab initio calculations.
The geometry optimization of all the compounds in Tables 1, SI1, and SI2 and calculation of LUMO energies were performed with the quantum chemistry program Jaguar,53–54 the B3LYP/6–31G++ level of theory, and the Poisson d Boltzmann finite (PBF) implicit solvation model.55–57 A one-way ANOVA was performed in MS Excel using the Data Analysis pack. N=28, df=26, R=0.63, R2=0.48, p<0.0001.
Characterization of anti-oxidant to pro-oxidant enzyme transformation.
Wild type TGR or mutant TGRs (500 nM) were incubated with NADPH (100 μM) and inhibitors for 30 minutes. The inhibitor concentrations were 20 μM for 1, 2, 5, 6, 7 and 8 and 50 μM for 3 and 4. Then all the treated TGR solutions except for 8 were desalted. Protein concentrations of the desalted samples were determined.
For the detection of superoxide and NADPH consumption, 100 μl of a pyrogallol red (50 μM) and NADPH (300 μM) solution was mixed with 100 μl of each sample above and then monitored at 340 nm and 540 nm simultaneously for 2 hours.39–40 The same reactions were performed with addition of 10 units of SOD. NADPH consumption was determined using ε340nm = 6.22 mM−1cm−1 and superoxide production was determined by consumption of pyrogallol red by using ε540nm = 23.9 mM−1cm−1 with a stoichiometric relation of 1 molecule of pyrogallol red reacts with 2 molecules of superoxide.39–40
For the detection of H2O2, samples were diluted 10 times and incubated with 300 μM NADPH. Every 15 min over 2 hours, 2 μl was withdrawn, diluted 50 times and mixed with the same volume of solution containing 20 μM Amplex Red and 100 mU/ml horseradish peroxidase. After 30 minutes incubation at 37 °C the fluorescence intensity with λex = 550 nm and λem = 600 nm was measured.58 The same reactions were performed with addition of 20 units of catalase to each well. The H2O2 levels of reactions were quantified using an H2O2 standard curve.
Supplementary Material
ACKNOWLEDGEMENTS
DLW, PAP, FA, and GRJT received funding from NIH/NIAID grant R33AI127635. AS received funding from the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health. ESJA acknowledges funding from Karolinska Institutet, The Swedish Research Council, The Swedish Cancer Society and The Knut and Alice Wallenberg Foundations. The following reagents were provided by the NIAID Schistosomiasis Resource Center of the Biomedical Research Institute (Rockville, MD) through NIH-NIAID Contract HHSN272201700014I for distribution through BEI Resources: Schistosoma japonicum, Strain Philippine, Exposed Swiss Webster Mice, NR-34794, Biomphalaria glabrata snails exposed to Schistosoma mansoni (NMRI), and Schistosoma haematobium, Exposed LVG hamsters, NR-21966. We thank Rui Ma and Dr. Scott G. Franzblau (UIC) for performing the Vero cell cytotoxicity assays.
ABBREVIATIONS USED
- TGR
thioredoxin glutathione reductase
- NADPH
nicotinamide adenine dinucleotide phosphate
- GSH
glutathione
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- GR
glutathione reductase
- Grx
glutaredoxin
- Sec/U
selenocysteine
- FAD
flavin adenine dinucleotide
- GSSG
glutathione disulfide
- DTNB
5,5’-dithio-bis-(2-nitrobenzoic acid)
- qHTS
quantitative high throughput screens
- SAR
structure-activity relationship
- LUMO
lowest unoccupied molecular orbital
- SOD
superoxide dismutase
- PBF
Poisson d Boltzmann finite
Footnotes
Supporting Information
This manuscript is accompanied by supporting information. Table S1: TGR inhibition and worm killing activity of analogs of compounds 1–8. Table S2: compounds investigated in this study but failing to meet milestone criteria. Table S3: cytotoxicity of 1–8 against mammalian cells. Table S4: the correlation between LUMO and schistosomicidal activity for compounds in this study. Table S5: inhibition of human glutathione reductase by 1–8. Figure S1: inhibition of TGR by 1–8 in the presence of different concentrations of glutathione. Figure S2: analysis TGR dimer formation by native polyacrylamide gel electrophoresis after reaction with 4.
Conflict of Interest
The authors declare no competing financial interest.
This study was approved by the Institutional Animal Care and Use Committee of Rush University Medical Center (17–053; Department of Health and Human Services animal welfare assurance number A-3120-01).
References
- 1.Hotez PJ; Molyneux DH; Fenwick A; Ottesen E; Ehrlich Sachs S; Sachs JD, Incorporating a Rapid-Impact Package for Neglected Tropical Diseases with Programs for HIV/AIDS, Tuberculosis, and Malaria. PLOS Medicine 2006, 3 (5), e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lago EM; Xavier RP; Teixeira TR; Silva LM; da Silva Filho AA; de Moraes J, Antischistosomal agents: state of art and perspectives. Future Medicinal Chemistry 2017, 10 (1), 89–120. [DOI] [PubMed] [Google Scholar]
- 3.Allam G; Eweas AF; Abuelsaad ASA, In vivo schistosomicidal activity of three novels 8-hydroxyquinoline derivatives against adult and immature worms of Schistosoma mansoni. Parasitology Research 2013, 112 (9), 3137–3149. [DOI] [PubMed] [Google Scholar]
- 4.Xiao S-H; Keiser J; Chen M-G; Tanner M; Utzinger J, Chapter 9 - Research and Development of Antischistosomal Drugs in the People’s Republic of China: A 60-Year Review In Advances in Parasitology, Zhou X-N; Bergquist R; Olveda R; Utzinger J, Eds. Academic Press: 2010; Vol. 73, pp 231–295. [DOI] [PubMed] [Google Scholar]
- 5.Doenhoff M; Cioli D; Kimani G, Praziquantel and the Control of Schistosomiasis. Parasitology Today 2000, 16 (9), 364–366. [DOI] [PubMed] [Google Scholar]
- 6.Wang W; Wang L; Liang Y-S, Susceptibility or resistance of praziquantel in human schistosomiasis: a review. Parasitology Research 2012, 111 (5), 1871–1877. [DOI] [PubMed] [Google Scholar]
- 7.Alger HM; Sayed AA; Stadecker MJ; Williams DL, Molecular and enzymatic characterisation of Schistosoma mansoni thioredoxin. International Journal for Parasitology 2002, 32 (10), 1285–1292. [DOI] [PubMed] [Google Scholar]
- 8.Alger HM; Williams DL, The disulfide redox system of Schistosomamansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Molecular and Biochemical Parasitology 2002, 121 (1), 129–139. [DOI] [PubMed] [Google Scholar]
- 9.Kuntz AN; Davioud-Charvet E; Sayed AA; Califf LL; Dessolin J; Arnér ESJ; Williams DL, Thioredoxin Glutathione Reductase from Schistosoma mansoni: An Essential Parasite Enzyme and a Key Drug Target. PLOS Medicine 2007, 4 (6), e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sayed AA; Simeonov A; Thomas CJ; Inglese J; Austin CP; Williams DL, Identification of oxadiazoles as new drug leads for the control of schistosomiasis. Nat Med 2008, 14 (4), 407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prast-Nielsen S; Huang HH; Williams DL, Thioredoxin glutathione reductase: its role in redox biology and potential as a target for drugs against neglected diseases. Biochim Biophys Acta 2011, 1810 (12), 1262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams DL; Bonilla M; Gladyshev VN; Salinas G, Thioredoxin glutathione reductase-dependent redox networks in platyhelminth parasites. Antioxid Redox Signal 2013, 19 (7), 735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Angelucci F; Miele AE; Boumis G; Dimastrogiovanni D; Brunori M; Bellelli A, Glutathione reductase and thioredoxin reductase at the crossroad: The structure of Schistosoma mansoni thioredoxin glutathione reductase. Proteins: Structure, Function, and Bioinformatics 2008, 72 (3), 936–945. [DOI] [PubMed] [Google Scholar]
- 14.Johansson L; Gafvelin G; Arnér ESJ, Selenocysteine in proteins—properties and biotechnological use. Biochimica et Biophysica Acta (BBA) - General Subjects 2005, 1726 (1), 1–13. [DOI] [PubMed] [Google Scholar]
- 15.Angelucci F; Sayed AA; Williams DL; Boumis G; Brunori M; Dimastrogiovanni D; Miele AE; Pauly F; Bellelli A, Inhibition of Schistosoma mansoni thioredoxin glutathione reductase by auranofin: Structural and kinetic aspects. Journal of Biological Chemistry 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angelucci F; Dimastrogiovanni D; Boumis G; Brunori M; Miele AE; Saccoccia F; Bellelli A, Mapping the Catalytic Cycle of Schistosoma mansoni Thioredoxin Glutathione Reductase by X-ray Crystallography. Journal of Biological Chemistry 2010, 285 (42), 32557–32567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H-H; Day L; Cass CL; Ballou DP; Williams CH; Williams DL, Investigations of the Catalytic Mechanism of Thioredoxin Glutathione Reductase from Schistosoma mansoni. Biochemistry 2011, 50 (26), 5870–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li T; Ziniel PD; He P.-q.; Kommer VP; Crowther GJ; He M; Liu Q; Van Voorhis WC; Williams DL; Wang M-W, High-throughput screening against thioredoxin glutathione reductase identifies novel inhibitors with potential therapeutic value for schistosomiasis. Infectious Diseases of Poverty 2015, 4 (1), 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simeonov A; Jadhav A; Sayed AA; Wang Y; Nelson ME; Thomas CJ; Inglese J; Williams DL; Austin CP, Quantitative High-Throughput Screen Identifies Inhibitors of the Schistosoma mansoni Redox Cascade. PLOS Neglected Tropical Diseases 2008, 2 (1), e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.National Center for Biotechnology Information. PubChem Database. Source=NCGC, AID=485364, https://pubchem.ncbi.nlm.nih.gov/bioassay/485364 (accessed on July 23, 2019).
- 21.Nordberg J; Zhong L; Holmgren A; Arnér ESJ, Mammalian Thioredoxin Reductase Is Irreversibly Inhibited by Dinitrohalobenzenes by Alkylation of Both the Redox Active Selenocysteine and Its Neighboring Cysteine Residue. Journal of Biological Chemistry 1998, 273 (18), 10835–10842. [DOI] [PubMed] [Google Scholar]
- 22.Arnér ESJ; Björnstedt M; Holmgren A, 1-Chloro-2,4-dinitrobenzene Is an Irreversible Inhibitor of Human Thioredoxin Reductase: LOSS OF THIOREDOXIN DISULFIDE REDUCTASE ACTIVITY IS ACCOMPANIED BY A LARGE INCREASE IN NADPH OXIDASE ACTIVITY. Journal of Biological Chemistry 1995, 270 (8), 3479–3482. [DOI] [PubMed] [Google Scholar]
- 23.Fang J; Lu J; Holmgren A, Thioredoxin Reductase Is Irreversibly Modified by Curcumin: A NOVEL MOLECULAR MECHANISM FOR ITS ANTICANCER ACTIVITY. Journal of Biological Chemistry 2005, 280 (26), 25284–25290. [DOI] [PubMed] [Google Scholar]
- 24.Xu J; Cheng Q; Arnér ESJ, Details in the catalytic mechanism of mammalian thioredoxin reductase 1 revealed using point mutations and juglone-coupled enzyme activities. Free Radical Biology and Medicine 2016, 94, 110–120. [DOI] [PubMed] [Google Scholar]
- 25.Anestål K; Prast-Nielsen S; Cenas N; Arnér ESJ, Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PloS one 2008, 3 (4), e1846–e1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stafford WC; Peng X; Olofsson MH; Zhang X; Luci DK; Lu L; Cheng Q; Trésaugues L; Dexheimer TS; Coussens NP; Augsten M; Ahlzén H-SM; Orwar O; Östman A; Stone-Elander S; Maloney DJ; Jadhav A; Simeonov A; Linder S; Arnér ESJ, Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Science Translational Medicine 2018, 10 (428), eaaf7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schust J; Sperl B; Hollis A; Mayer TU; Berg T, Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 2006, 13 (11), 1235–42. [DOI] [PubMed] [Google Scholar]
- 28.Li P; Shi L; Yang X; Yang L; Chen X-W; Wu F; Shi Q-C; Xu W-M; He M; Hu D-Y; Song B-A, Design, synthesis, and antibacterial activity against rice bacterial leaf blight and leaf streak of 2,5-substituted-1,3,4-oxadiazole/thiadiazole sulfone derivative. Bioorganic & Medicinal Chemistry Letters 2014, 24 (7), 1677–1680. [DOI] [PubMed] [Google Scholar]
- 29.Xu W-M; Han F-F; He M; Hu D-Y; He J; Yang S; Song B-A, Inhibition of Tobacco Bacterial Wilt with Sulfone Derivatives Containing an 1,3,4-Oxadiazole Moiety. Journal of Agricultural and Food Chemistry 2012, 60 (4), 1036–1041. [DOI] [PubMed] [Google Scholar]
- 30.Schust J; Sperl B; Hollis A; Mayer TU; Berg T, Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chemistry & Biology 2006, 13 (11), 1235–1242. [DOI] [PubMed] [Google Scholar]
- 31.Song Y.n.; Xu H; Chen W; Zhan P; Liu X, 8-Hydroxyquinoline: a privileged structure with a broad-ranging pharmacological potential. MedChemComm 2015, 6 (1), 61–74. [Google Scholar]
- 32.Krieg R; Jortzik E; Goetz AA; Blandin S; Wittlin S; Elhabiri M; Rahbari M; Nuryyeva S; Voigt K; Dahse HM; Brakhage A; Beckmann S; Quack T; Grevelding CG; Pinkerton AB; Schonecker B; Burrows J; Davioud-Charvet E; Rahlfs S; Becker K, Arylmethylamino steroids as antiparasitic agents. Nat Commun 2017, 8, 14478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Utzinger J; Keiser J; Shuhua X; Tanner M; Singer BH, Combination chemotherapy of schistosomiasis in laboratory studies and clinical trials. Antimicrobial agents and chemotherapy 2003, 47 (5), 1487–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pica-Mattoccia L; Cioli D, Sex- and stage-related sensitivity of Schistosoma mansoni to in vivo and in vitro praziquantel treatment. Int J Parasitol 2004, 34 (4), 527–33. [DOI] [PubMed] [Google Scholar]
- 35.Adachi M; Cui C; Dodge CT; Bhayani MK; Lai SY, Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma. Oral Oncol 2012, 48 (12), 1220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q; Zhang D; Chen X; He L; Li T; Xu X; Li M, Nuclear PKM2 contributes to gefitinib resistance via upregulation of STAT3 activation in colorectal cancer. Sci Rep 2015, 5, 16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang R; Wang L; Sheng J; Huang Q; Pan D; Xu Y; Yan J; Wang X; Dong Z; Yang M, Combinatory effects of vaccinia virus VG9 and the STAT3 inhibitor Stattic on cancer therapy. Arch Virol 2019, 164 (7), 1805–1814. [DOI] [PubMed] [Google Scholar]
- 38.Wang CM; Hsu CT; Niu HS; Chang CH; Cheng JT; Shieh JM, Lung damage induced by hyperglycemia in diabetic rats: The role of signal transducer and activator of transcription 3 (STAT3). J Diabetes Complications 2016, 30 (8), 1426–1433. [DOI] [PubMed] [Google Scholar]
- 39.Cortés-Ríos J; Torres MJ; Campos-Bustamante MP; Romero-Parra J; Letelier ME; Pessoa-Mahana D; Chung H; Faúndez M, NADPH oxidase activity: Spectrophotometric determination of superoxide using pyrogallol red. Analytical Biochemistry 2017, 536, 96–100. [DOI] [PubMed] [Google Scholar]
- 40.Faúndez M; Rojas M; Bohle P; Reyes C; Letelier ME; Aliaga ME; Speisky H; Lissi E; López-Alarcón C, Pyrogallol red oxidation induced by superoxide radicals: Application to evaluate redox cycling of nitro compounds. Analytical Biochemistry 2011, 419 (2), 284–291. [DOI] [PubMed] [Google Scholar]
- 41.Fritz-Wolf K; Urig S; Becker K, The Structure of Human Thioredoxin Reductase 1 Provides Insights into C-terminal Rearrangements During Catalysis. Journal of Molecular Biology 2007, 370 (1), 116–127. [DOI] [PubMed] [Google Scholar]
- 42.Dufrasne F; Gelbcke M; Neve J; Kiss R; Kraus J-L, Quinone Methides and their Prodrugs: A Subtle Equilibrium Between Cancer Promotion, Prevention, and Cure. Current Medicinal Chemistry 2011, 18 (26), 3995–4011. [DOI] [PubMed] [Google Scholar]
- 43.Cheng Q; Antholine WE; Myers JM; Kalyanaraman B; Arnér ESJ; Myers CR, The Selenium-independent Inherent Pro-oxidant NADPH Oxidase Activity of Mammalian Thioredoxin Reductase and Its Selenium-dependent Direct Peroxidase Activities. Journal of Biological Chemistry 2010, 285 (28), 21708–21723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nwaka S; Ramirez B; Brun R; Maes L; Douglas F; Ridley R, Advancing drug innovation for neglected diseases-criteria for lead progression. PLoS Negl Trop Dis 2009, 3 (8), e440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molecular Operating Environment (MOE), 2019.0101, 2019.0101; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2019. [Google Scholar]
- 46.Sterling T; Irwin JJ, ZINC 15--Ligand Discovery for Everyone. J Chem Inf Model 2015, 55 (11), 2324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim S; Chen J; Cheng T; Gindulyte A; He J; He S; Li Q; Shoemaker BA; Thiessen PA; Yu B; Zaslavsky L; Zhang J; Bolton EE, PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 2019, 47 (D1), D1102–D1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silvestri I; Lyu H; Fata F; Boumis G; Miele AE; Ardini M; Ippoliti R; Bellelli A; Jadhav A; Lea WA; Simeonov A; Cheng Q; Arnér ESJ; Thatcher GRJ; Petukhov PA; Williams DL; Angelucci F, Fragment-Based Discovery of a Regulatory Site in Thioredoxin Glutathione Reductase Acting as “Doorstop” for NADPH Entry. ACS Chemical Biology 2018, 13 (8), 2190–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng Q; Arnér ESJ, Selenocysteine Insertion at a Predefined UAG Codon in a Release Factor 1 (RF1)-depleted Escherichia coli Host Strain Bypasses Species Barriers in Recombinant Selenoprotein Translation. Journal of Biological Chemistry 2017, 292 (13), 5476–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tucker MS; Karunaratne LB; Lewis FA; Freitas TC; Liang Y. s., Schistosomiasis. Current Protocols in Immunology 2013, 103 (1), 19.1.1–19.1.58. [DOI] [PubMed] [Google Scholar]
- 51.Lombardo FC; Pasche V; Panic G; Endriss Y; Keiser J, Life cycle maintenance and drug-sensitivity assays for early drug discovery in Schistosoma mansoni. Nat Protoc 2019, 14 (2), 461–481. [DOI] [PubMed] [Google Scholar]
- 52.Choules MP; Wolf NM; Lee H; Anderson JR; Grzelak EM; Wang Y; Ma R; Gao W; McAlpine JB; Jin YY; Cheng J; Lee H; Suh JW; Duc NM; Paik S; Choe JH; Jo EK; Chang CL; Lee JS; Jaki BU; Pauli GF; Franzblau SG; Cho S, Rufomycin Targets ClpC1 Proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrob Agents Chemother 2019, 63 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrödinger Release 2019–4: Jaguar, Schrödinger, LLC, New York, NY: 2019. [Google Scholar]
- 54.Bochevarov AD; Harder E; Hughes TF; Greenwood JR; Braden DA; Philipp DM; Rinaldo D; Halls MD; Zhang J; Friesner RA, Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem 2013, 113 (18), 2110–2142. [Google Scholar]
- 55.Cortis CM; Friesner RA, An automatic three-dimensional finite element mesh generation system for the Poisson-Boltzmann equation. J. Comput. Chem 1997, 18 (13), 1570–1590. [Google Scholar]
- 56.Cortis CM; Friesner RA, Numerical solution of the Poisson-Boltzmann equation using tetrahedral finite-element meshes. J. Comput. Chem 1997, 18 (13), 1591–1608. [Google Scholar]
- 57.Marten B; Kim K; Cortis C; Friesner RA; Murphy RB; Ringnalda MN; Sitkoff D; Honig B, New Model for Calculation of Solvation Free Eneries: Correction of Self-Consistent Reaction Field Continuum Dielectric Theory for Short-Range Hydrogen-Bonding Effects. J. Phys. Chem 1996, 100 (28), 11775–11788. [Google Scholar]
- 58.Gencheva R; Cheng Q; Arnér ESJ, Efficient selenocysteine-dependent reduction of toxoflavin by mammalian thioredoxin reductase. Biochimica et Biophysica Acta (BBA) - General Subjects 2018, 1862 (11), 2511–2517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.