

Abstract
Spinal cord microglia contribute to nerve injury‐induced neuropathic pain. We have previously demonstrated that toll‐like receptor 2 (TLR2) signaling is critical for nerve injury‐induced activation of spinal cord microglia, but the responsible endogenous TLR2 agonist has not been identified. Here, we show that nerve injury‐induced upregulation of sialyltransferase St3gal2 in sensory neurons leads to an increase in expression of the sialylated glycosphingolipid, GT1b. GT1b ganglioside is axonally transported to the spinal cord dorsal horn and contributes to characteristics of neuropathic pain such as mechanical and thermal hypersensitivity. Spinal cord GT1b functions as an TLR2 agonist and induces proinflammatory microglia activation and central sensitization. Pharmacological inhibition of GT1b synthesis attenuates nerve injury‐induced spinal cord microglia activation and pain hypersensitivity. Thus, the St3gal2‐GT1b‐TLR2 axis may offer a novel therapeutic target for the treatment of neuropathic pain.
Keywords: ganglioside, microglia, neuropathic pain, St3gal2, toll‐like receptor 2
Subject Categories: Immunology, Neuroscience
Upregulation of the sialyated glycosphingolipid GT1b in sensory neurons after peripheral nerve injury leads to TLR‐2 mediated microglia activation and nerve injury‐induced neuropathic pain.
Introduction
Neuropathic pain is a chronic pathological pain due to an injury or dysfunction in the peripheral or central nervous system. The clinical symptoms of neuropathic pain include spontaneous burning pain, enhanced pain in response to noxious stimuli (hyperalgesia), and pain in response to normally innocuous stimuli (allodynia) (Jensen & Finnerup, 2014). Studies over the last two decades have implicated spinal cord microglia activation as etiological factors that contribute to pain hypersensitivity at the spinal cord level (central sensitization) and consequent neuropathic pain (Inoue & Tsuda, 2009). In this regard, elucidating the molecular mechanisms by which peripheral nerve injury induces spinal cord microglia activation is critical to understanding the pathogenic mechanisms of neuropathic pain. Previously, we and others reported that toll‐like receptor 2 (TLR2) signaling is a significant contributor to spinal cord microglia activation due to peripheral nerve injury (Kim et al, 2007; Stokes et al, 2013), which suggested a presence of certain TLR2 endogenous agonist in the spinal cord. However, there is still no information as to the TLR2 endogenous agonist that drives spinal cord microglia activation after nerve injury.
The neuronal membrane consists of highly heterogeneous lipid components that are distinct from those in non‐neuronal cells. Specifically, neurons are enriched in glycosphingolipids or gangliosides, accounting for 10‐12% of the total membrane lipid content. Gangliosides localized in the outer leaflet of the membrane are constituents of lipid rafts and are implicated in neuronal communication with the extracellular microenvironment or contribute to neuron–glia interactions (Schnaar, 2016). Four major gangliosides (GM1, GD1a, GD1b, and GT1b) constitute up to 97% of gangliosides in the human brain (Tettamanti et al, 1973; Ando, 1983). Of note, the ganglioside profile is highly plastic, changing in brain development and pathophysiology (Olsen & Faergeman, 2017). For example, GD1a and GM1 levels increase at early brain developmental stages and contribute to neural differentiation and axonal outgrowth (Olsen & Faergeman, 2017). An increase of GT1b at a later stage contributes to axon–myelin stability of differentiated neurons (Olsen & Faergeman, 2017). Ganglioside composition also changes in several neuropathological conditions. For example, Alzheimer's disease is associated with increased brain levels of GM1 and GM2, whereas there is a decrease of GM1 content in Parkinson's disease (Molander‐Melin et al, 2005; Wu et al, 2012). Intriguingly, in patients with peripheral neuropathy or polyneuropathy, a significant increase of “b‐series” gangliosides (GD1b, GT1b, and GQ1b) was detected in the cerebrospinal fluid (CSF) (Trbojevic‐Cepe et al, 1991). In animal models, GM1 administration into spinal cord or GM3 synthase depletion exerted strong analgesic effects on the nerve injury‐induced neuropathic pain and diabetic neuropathy, respectively (Mao et al, 1992; Menichella et al, 2016), whereas intraplantar injection of GT1b produced nociceptive behavior (Watanabe et al, 2011).
Taken together with the reports of ganglioside profile change in different neuropathological conditions and the relationship between CSF/spinal cord ganglioside levels and peripheral neuropathy, these findings suggest that ganglioside profile changes may also be implicated in the central sensitization process that occurs after nerve damage and that underlies neuropathic pain development. To test this hypothesis, here we investigated the ganglioside profile in the spinal cord in a nerve injury‐induced mouse model of neuropathic pain. We found that GT1b is aberrantly upregulated in injured sensory neurons, transported to the spinal cord, and contributes to neuropathic pain. As to underlying mechanism, we discovered that GT1b in the spinal cord functions as an endogenous agonist of TLR2, which activates microglia and triggers the induction of proinflammatory genes that contribute to central sensitization and the neuropathic pain phenotype.
Results
Peripheral nerve injury upregulates GT1b in the spinal cord
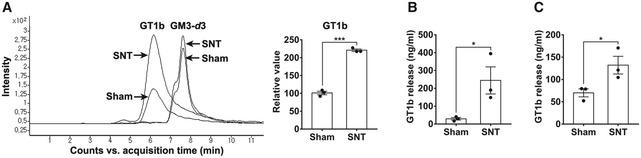
To investigate the ganglioside profile in an animal model of peripheral nerve injury‐induced neuropathic pain, we isolated spinal cord tissues of L5 spinal nerve‐transected or sham‐operated mice and searched for differentially regulated gangliosides using LC‐MRM/MS analysis. Among the four major gangliosides (GM1, GD1a, GD1b, and GT1b), we found that GT1b levels significantly increased in the spinal cord of nerve‐injured mice compared to sham‐operated mice (Fig 1A). Because GT1b is increased in the CSF of patients with peripheral neuropathy (Trbojevic‐Cepe et al, 1991), we next examined the level of GT1b in the interstitial fluid (ISF) of the spinal cord. In these studies, we implanted a microdialysis probe in the spinal cord dorsal horn of the nerve‐injured rat and collected ISF. Five days after the nerve injury, the concentration of GT1b in the ISF increased 8.4‐fold compared to the sham control, as assessed using ELISA (Fig 1B). Likewise, the GT1b level in the CSF increased to 132 ± 20 ng/ml in the nerve‐injured mice compared to 70 ± 9 ng/ml in the sham‐operated mice (Fig 1C). These data demonstrate that peripheral nerve injury increases GT1b levels in spinal cord tissue, where it is released into the ISF and CSF.
Figure 1. GT1b is upregulated and released in the spinal cord after nerve injury.
-
AA LC‐MRM/MS analysis of ganglioside GT1b species in pooled spinal cord tissues of Sham‐operated or nerve‐injured mice at 3 dpi. Representative traces of GT1b species (d18:1–18:0) are shown, and the intensity of the GT1b species is presented as the fold induction compared to the corresponding control. GM3‐d3 served as internal standard for normalization. d18:1–18:0 means the ceramide structure (carbon number: double bond number), and d3 means deuterated form.
-
B, CGT1b level in ISF (B) and CSF (C) was analyzed by ELISA. Data are represented as mean ± SEM (Student's t‐test, *P < 0.05, ***P < 0.001).
GT1b induces pain hypersensitivity and spinal cord microglia activation
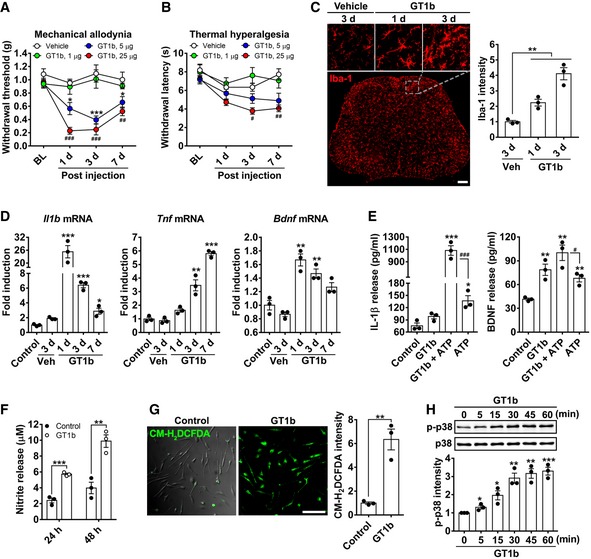
To examine the effects of GT1b increase in the spinal cord on the nociceptive processing, we assayed several nociceptive behaviors in mice upon intrathecal GT1b injection. Figure 2A and B shows that we observed significant decreases in thresholds to mechanical and thermal stimuli in GT1b‐injected mice, but not in vehicle‐injected mice. GT1b decreased the pain thresholds in a dose‐dependent manner. We next examined intrathecal GT1b effects on spinal cord microglia activation. Within 1 day of the intrathecal GT1b injection, we recorded a robust increase in Iba‐1‐immunoreactivity (IR) in the spinal cord, with even greater Iba‐1‐IR at day 3 (Fig 2C). Intrathecal GT1b administration also upregulated gene expression of Il1b, Tnf, and Bdnf, by 25‐, 6‐, and 1.6‐fold, respectively (Fig 2D). Note that each of these genes is expressed in activated spinal cord microglia after nerve injury and has been implicated in the central sensitization process that contributes to nerve injury‐induced neuropathic pain (Kawasaki et al, 2008b; Lu et al, 2009). These data demonstrate that GT1b induces spinal cord microglia activation and subsequent pain hypersensitivity in vivo. To test whether GT1b directly activates microglia, we also examined GT1b effect on microglia in vitro. In primary cultured microglia, GT1b induced production of IL‐1β and BDNF, nitric oxide, and intracellular reactive oxygen species, which were confirmed by ELISA, Griess assay, and CM‐H2DCFDA dye, respectively (Fig 2E–G). GT1b stimulation also induced p38 MAP kinase activation (Fig 2H), a key signaling event that has been implicated in proinflammatory gene expression in microglia (Ji & Suter, 2007). Taken together, these findings suggest that the nerve injury‐induced GT1b upregulation in spinal cord directly activates spinal cord microglia and thereby induces pain hypersensitivity in mice.
Figure 2. Intrathecal GT1b injection induced pain hypersensitivity and spinal cord microglia activation.
-
A, BMechanical allodynia (A) and thermal hyperalgesia (B) were assessed after intrathecal injection of GT1b (1 μg/5 μl, n = 6; 5 μg/5 μl, n = 7; 25 μg/5 μl, n = 6) or vehicle (n = 5; one‐way ANOVA, *P < 0.05; ***P < 0.001, # P < 0.05; ## P < 0.01; ### P < 0.001 vs. vehicle). Data are expressed as mean ± SEM.
-
CGT1b (25 μg/5 μl, n = 3/time point) or the vehicle (saline, n = 3) was intrathecally administrated, and lumbar spinal cord sections were immunostained with Iba‐1 antibody. Vehicle‐injected mice served as controls, and representative images are shown (scale bar, 100 μm). The fluorescence intensity of Iba‐1‐IR in spinal cord dorsal horn and CM‐H2DCFDA was measured and presented as the fold induction compared to the corresponding control. Data are represented as mean ± SEM (Student's t‐test, **P < 0.01).
-
DL4‐5 spinal cord tissues were removed after intrathecal injection of GT1b (25 μg/5 μl) or vehicle injection, and total RNA was isolated from the tissues. Transcript levels were measured by real‐time RT–PCR and presented as the fold induction compared to the corresponding level measured in control. Data are represented as mean ± SEM (Student's t‐test, *P < 0.05; **P < 0.01; ***P < 0.001, vs. control).
-
E, FIL‐1β, BDNF (E), and nitrite production (F) were measured in the conditioned media from primary microglia stimulated with GT1b. Data are expressed as mean ± SEM (Student's t‐test, *P < 0.05; **P < 0.01; ***P < 0.001, vs. control, # P < 0.05; ### P < 0.001).
-
GIntracellular reactive oxygen species production in primary microglia was measured using CM‐H2DCFDA after GT1b treatment. Data are expressed as mean ± SEM (Student's t‐test, **P < 0.01; scale bar, 100 μm).
-
HGlial cells were stimulated with 10 μg/ml of GT1b at the indicated time points, and p38 MAPK activation was determined using Western blot analysis. Data are expressed as mean ± SEM (Student's t‐test, *P < 0.05; **P < 0.01; ***P < 0.001, vs. control).
GT1b is transported from the injured sensory neurons and released to the spinal cord dorsal horn after nerve injury
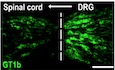
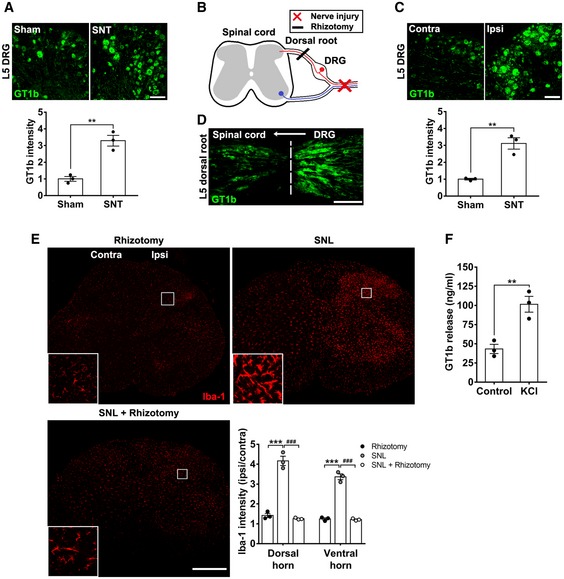
To determine whether the increased spinal cord levels of GT1b is derived from damaged sensory neurons, we examined the expression levels of GT1b in the dorsal root ganglion (DRG). We detected basal levels GT1b‐IR in sensory neurons of sham‐operated control mice, which was further upregulated in ipsilateral L5 DRG neurons after nerve injury (Fig 3A). The specificity of GT1b antibody was tested in previous studies using purified gangliosides and knockout mice (Gong et al, 2002; Schnaar et al, 2002). To examine whether sensory neuron‐derived GT1b is transported to the spinal cord, we ligated and transected the L5 dorsal root after L5 spinal nerve ligation in rats (Fig 3B). Similar to mice, GT1b was upregulated in the injured sensory neurons of the rat (Fig 3C). Figure 3D shows that dorsal rhizotomy followed by L5 spinal nerve ligation resulted in the accumulation of GT1b at the dorsal root transection site, indicating that GT1b is intra‐axonally transported from the DRG to the spinal cord (Fig 3D). Notably, the rhizotomy abrogated the nerve injury‐induced spinal cord microglia activation (Fig 3E). Taken together, these data indicate that nerve injury induces GT1b upregulation in injured sensory neurons and that GT1b axonally transported to the dorsal horn contributes to microglial activation.
Figure 3. GT1b is upregulated in DRG neurons and transported to spinal cord after nerve injury.
-
AL5 DRG tissue sections of sham‐operated or nerve‐injured mice (3 dpi) were immnostained with GT1b antibody. Representative images are shown (n = 3/group; scale bar, 100 μm).
-
BSchematic drawing of neuroanatomy and surgery.
-
C, DRat L5 DRG (C) and L5 dorsal root sections (D) with concurrent rhizotomy and L5 SNL (3 dpi) were immunostained with GT1b antibody. The dotted line denotes the transection site. Representative images are shown (n = 3; scale bar, 100 μm).
-
EL4‐5 spinal cord sections from rats with rhizotomy and SNL‐injured rats with or without rhizotomy (3 dpi) were immunostained with Iba‐1 antibody. Representative images are shown (n = 3/group; scale bar, 500 μm).
-
FThe GT1b level was measured using ELISA in conditioned media of F11 differentiated cells after 1‐h exposure to high concentration of potassium.
Although we attempted to localize GT1b in the spinal cord by immunostaining, our results were inconsistent, making it difficult to document the accumulation of GT1b in the spinal cord, possibly due to its release after axonal transport. For this reason, we turned to a more functional assessment of sensory neuron‐derived GT1b. Specifically, using the F11 sensory neuronal cell line in vitro, we asked whether GT1b is released in a neuronal activity‐dependent manner. F11 cells were differentiated with dibutyryl cAMP and then depolarized for 1 h with 50 mM potassium. Figure 3F demonstrates the depolarization‐induced accumulation of GT1b in the conditioned medium, indicating that GT1b can be released in a neuronal activity‐dependent manner. Taken together, we conclude that the nerve injury‐induced increase in spinal cord GT1b results from its increase in injured DRG neurons, followed by intra‐axonal transport of GT1b to and its release in the ipsilateral dorsal horn.
Nerve injury‐induced St3gal2 upregulation in sensory neurons is upstream of GT1b
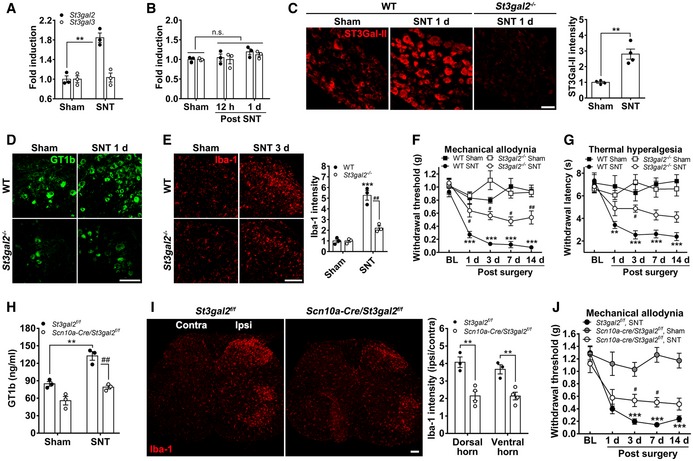
We next investigated the molecular mechanisms by which nerve injury increases GT1b in sensory neurons. GT1b is synthesized from a precursor molecule, GD1b, by enzymatic addition of single sialic acid functional group. As two enzymes, ST3Gal‐II and ST3Gal‐III, have been implicated (Sturgill et al, 2012), we examined St3gal2 and St3gal3 expression in DRG neurons after nerve injury. Figure 4A shows that peripheral nerve injury increased St3gal2 mRNA expression by 80% in the L5 DRG. St3gal3 mRNA did not change in the DRG, and neither St3gal2 nor St3gal3 mRNA increased in the spinal cord (Fig 4A and B). We also recorded an increase of ST3Gal‐II protein in the L5 DRG of mice, 1 day post‐injury (dpi) (Fig 4C). Specificity of the ST3Gal‐II antibody was confirmed by the absence of immunostaining in the L5 DRG of St3gal2 −/− mice (Fig 4C). To examine the relationship between ST3Gal‐II upregulation and the GT1b increase in L5 DRG after nerve injury, we next measured GT1b levels in St3gal2 −/− mice. Although significantly reduced, we nevertheless record basal levels of GT1b in the sensory neurons of the St3gal2 −/− mice. We assume that this reflects synthesis by ST3Gal‐III, an alternative GT1b‐synthesizing enzyme (Sturgill et al, 2012). Most importantly, in the St3gal2 −/− mice, GT1b was not further upregulated by the nerve injury, over the basal level (Fig 4D). As expected, the nerve injury‐induced spinal cord microglia activation, documented by Iba‐1‐IR, was reduced in St3gal2 −/− compared to wild‐type (WT) mice after the injury (Fig 4E). Furthermore, the nerve injury‐induced mechanical allodynia and thermal hyperalgesia were also reduced in the St3gal2 −/− compared to WT mice (Fig 4F and G). We conclude that St3gal2 is a critical contributor to the GT1b increase in sensory neurons, spinal cord microglia activation, and pain hypersensitivity after nerve injury. To conclusively demonstrate the contribution of sensory neuron‐specific ST3Gal‐II to GT1b increase in the spinal cord, we generated sensory neuron‐specific St3gal2 conditional knockout mice (Scn10a‐Cre/St3gal2 f/f). In these mice, GT1b increase in the spinal cord due to nerve injury was inhibited (Fig 4H). Accordingly, nerve injury‐induced spinal cord microglia activation and pain hypersensitivity were significantly reduced (Fig 4I and J). Taken together, these data suggest that nerve injury induces an increase in GT1b in axotomized sensory neurons secondary to upregulation of ST3Gal‐II. Thus, ST3Gal‐II lies upstream of GT1b in the activation of spinal cord microglia and the associated neuropathic pain phenotype.
Figure 4. St3gal2 upregulation in sensory neurons is responsible for nerve injury‐induced GT1b upregulation and subsequent spinal cord microglia activation and neuropathic pain.
-
A, BSt3gal2 and St3gal3 mRNA expression in the pooled L5 DRG (A) and spinal cord tissues (B) of sham‐operated and L5 SNT‐injured mice were measured using real time RT–PCR. Data are represented as mean ± SEM (n = 3/group, Student's t‐test, **P < 0.01).
-
CL5 DRG sections were prepared from WT mice (n = 4/group) with or without L5 SNT (1 dpi) and immunostained with ST3Gal‐II antibody. Representative images are shown (scale bar, 50 μm).
-
D, EL5 DRG (D) and L4‐5 spinal cord (E) sections from WT (n = 3/group) and St3gal2 −/− mice (n = 3/group) with or without L5 SNT were immunostained with GT1b or Iba‐1 antibody, respectively. Representative images are shown (scale bar, 100 μm).
-
F, GMechanical allodynia (F) and thermal hyperalgesia (G) were evaluated in sham‐operated or L5 SNT‐injured WT (sham, n = 5; SNT, n = 5) and St3gal2 −/− (sham, n = 5; SNT, n = 5) mice. Data are represented as mean ± SEM (one‐way ANOVA, **P < 0.01; ***P < 0.001, vs. sham control in WT, # P < 0.05; ## P < 0.01, vs. nerve‐injured WT in each time point).
-
HGT1b level in spinal cord tissues was analyzed by ELISA. Three mg of pooled ipsilateral L4‐5 spinal cord tissues of sham‐operated or L5 spinal nerve‐injured mice were used. Data are presented as mean ± SEM (Student's t‐test, **P < 0.01, ## P < 0.01).
-
IL4‐5 spinal cord sections of floxed‐St3gal2 (St3gal2 f/f, n = 3/group) and sensory neuron‐specific St3gal2 −/− mice (Scn10a‐Cre/St3gal2 f/f, n = 3/group) with L5 SNT at 3 dpi were immunostained with Iba‐1 antibody, and representative images are shown (scale bar, 100 μm).
-
JMechanical allodynia was performed in sham‐operated or L5 SNT‐injured St3gal2 f/f (SNT, n = 6) and Scn10a‐Cre/St3gal2 −/− (sham, n = 5; SNT, n = 7) mice (one‐way ANOVA, ***P < 0.001, vs. sham control, # P < 0.05; vs. nerve‐injured St3gal2 f/f at each time point).
GT1b is an endogenous agonist of microglial TLR2
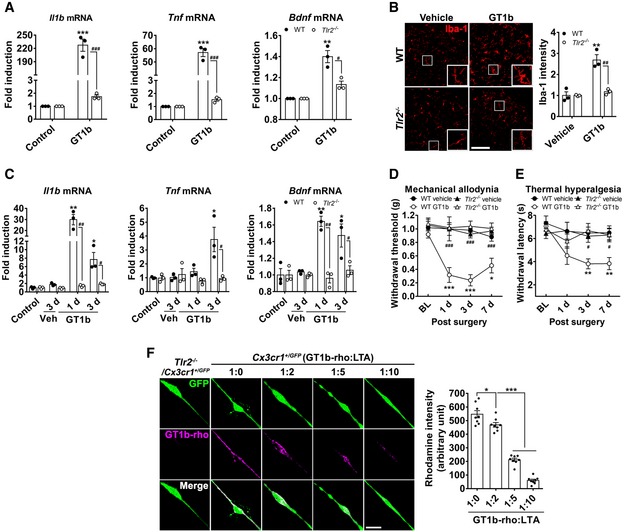
We previously reported that TLR2 is required for nerve injury‐induced spinal cord microglia activation (Kim et al, 2007). Here, we asked whether TLR2 is involved in the GT1b‐mediated spinal cord microglia activation. We first examined GT1b effects in vitro. We prepared glial cells from WT and Tlr2 −/− mice and found that GT1b‐induced expression of several genes implicated in persistent pain was almost completely abrogated in glial cells from Tlr2 −/− mice (Fig 5A). Next, we examined the contribution of Tlr2 in vivo. Figure 5B shows that spinal cord microglia activation after intrathecal GT1b, as assessed by Iba‐1 immunostaining, was almost entirely abolished in the Tlr2 −/− mice compared to WT mice. The GT1b‐induced expression of Il1b, Tnf, and Bdnf in the spinal cord was also prevented in the Tlr2 −/− mice (Fig 5C). We then tested the susceptibility of the Tlr2 −/− mice to the intrathecal GT1b‐induced neuropathic pain phenotype. Figure 5D and E shows that intrathecal GT1b provoked hypersensitivity to both mechanical and thermal stimuli in control mice, but this was almost completely prevented in the Tlr2 −/− mice. We conclude that the pronociceptive effects of GT1b are mediated by TLR2, which is expressed mainly on spinal cord microglia in vivo (Lim et al, 2013).
Figure 5. GT1b functions as an endogenous agonist of TLR2 to activate microglia.
-
AGlial cells cultured from WT and Tlr2 −/− mice were stimulated with 10 μg/ml of GT1b for 3 h, and transcript levels were measured using real‐time RT–PCR. Data are represented as mean ± SEM (Student's t‐test, **P < 0.01; ***P < 0.001, vs. control in WT, # P < 0.05; ### P < 0.001, vs. Tlr2 −/− mice).
-
BLumbar spinal cord sections prepared from WT (n = 3/group) and Tlr2 −/− mice (n = 3/group) 1 day after intrathecal injection of GT1b (25 μg/5 μl in saline) were immunostained with Iba‐1 antibody, and representative images are shown (scale bar, 100 μm). Vehicle‐injected mice served as controls. The fluorescence intensity of Iba‐1‐IR was measured and presented as the fold induction compared to the corresponding control. Data are represented as mean ± SEM (Student's t‐test, **P < 0.01, vs. control in WT, ## P < 0.01, vs. Tlr2 −/− mice).
-
CTranscript levels were measured using real‐time RT–PCR in L4‐5 spinal cord tissues of WT and Tlr2 −/− mice after intrathecal injection of GT1b (25 μg/5 μl, n = 3/group) or vehicle. Data are represented as mean ± SEM (n = 3/group, Student's t‐test, *P < 0.05; **P < 0.01, vs. control in WT, # P < 0.05; ## P < 0.01, vs. in Tlr2 −/− mice).
-
D, EMechanical allodynia (D) and thermal hyperalgesia (E) were assessed in WT (vehicle, n = 5; GT1b, n = 5) and Tlr2 −/− mice (vehicle, n = 6; GT1b, n = 7) after intrathecal injection of GT1b (25 μg/5 μl) or vehicle. Data are represented as mean ± SEM (one‐way ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001, vs. vehicle in WT, # P < 0.05; ## P < 0.01; ### P < 0.001, vs. WT GT1b group in each time point).
-
FPrimary microglia cultured from Cx3cr1 +/GFP and Tlr2 −/−/Cx3cr1 +/GFP mice were incubated with 5 μg/ml of GT1b‐rhodamine in the presence or absence of LTA (n = 8/group), and representative images are shown (scale bar, 20 μm). The fluorescence intensity of GT1b‐rhodamine was measured and presented as mean ± SEM (Student's t‐test, *P < 0.05; ***P < 0.001).
The TLR2 receptor mediates signaling by a variety tissue damage‐associated molecules (Yu et al, 2006; Kim et al, 2013). Taken together with our demonstration that TLR2 contributes to nerve injury‐induced microglia activation, we hypothesized that nerve injury must trigger exposure of a damage‐associated TLR2 endogenous agonist. Based on the requirement of TLR2 in GT1b‐induced microglia activation, we explored the possibility that GT1b functions as a TLR2 endogenous agonist, one that is particularly relevant in the context of neuropathic pain. We first asked whether there is a direct interaction between GT1b and TLR2. In these experiments, we incubated primary microglia cultured from Cx3cr1 +/GFP and Tlr2 −/−/Cx3cr1 +/GFP mice with rhodamine‐conjugated GT1b. Figure 5F demonstrates significant binding of the GT1b‐rhodamine to the GFP‐expressing microglia. In contrast, the rhodamine fluorescence signal could not be detected in microglia from Tlr2 −/−/Cx3cr1 +/GFP mice. Moreover, in a competitive binding assay between GT1b and lipoteichoic acid (LTA), a TLR2 agonist, we found that LTA pre‐treatment dose‐dependently inhibited the binding of GT1b‐rhodamine to microglia (Fig 5F). These data indicate that GT1b can indeed serve as an endogenous agonist of TLR2, activating spinal cord microglia and contributing to nerve injury‐induced neuropathic pain.
Pharmacological blockade of GT1b synthesis inhibits nerve injury‐induced spinal cord microglia activation and pain hypersensitivity
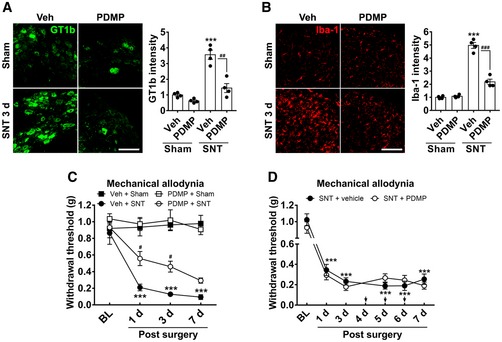
Finally, we asked whether pharmacological prevention of the GT1b increase would attenuate nerve injury‐induced spinal cord microglia activation and the associated neuropathic pain phenotype. To counteract the GT1b increase in the DRG, we intrathecally administrated D‐threo‐1‐phenyl‐2‐decanoylamino‐3‐morpholino‐1‐propanol·HCl (PDMP), a pharmacological inhibitor of ganglioside synthesis (Vunnam & Radin, 1980). We chose to administer PDMP intrathecally to ensure efficient access to the DRG (Wang et al, 2005; Kawasaki et al, 2008a). Figure 6A shows that intrathecal PDMP significantly reduced nerve injury‐induced GT1b expression in DRG neurons. PDMP injection also attenuated nerve injury‐induced spinal cord microglia activation (Fig 6B). Most importantly, the nerve injury‐induced mechanical hypersensitivity was attenuated by pre‐treatment with PDMP (Fig 6C), but not by post‐treatment with PDMP at 4–6 days post‐injury (Fig 6D). These results suggest that blocking GT1b synthesis at an early time point when microglia activation occurs inhibits the development of neuropathic pain caused by peripheral nerve injury but does not reverse the ongoing pain hypersensitivity.
Figure 6. Antagonizing St3gal2‐GT1b‐TLR2 signaling attenuates neuropathic pain.
-
A, BPDMP (10 μg/5 μl) was intrathecally injected once daily for 7 days prior to L5 SNT. L5 DRG (A) and L5 spinal cord tissue (B) sections of sham‐operated or SNT‐injured (3 dpi) WT mice were immunostained with GT1b or Iba‐1 antibody. Representative images are shown (n = 4/group; scale bar, 100 μm). The fluorescence intensity of GT1b‐IR and Iba‐1‐IR was measured and presented as the fold induction compared to that of the vehicle + sham control. Data are represented as mean ± SEM (Student's t‐test, ***P < 0.001, vs. vehicle + sham group, ## P < 0.01; ### P < 0.001, vs. PDMP + SNT).
-
CMechanical allodynia was assessed in sham‐operated WT mice with or without PDMP injection (Veh + Sham, n = 5; PDMP + Sham, n = 5) and L5 SNT‐injured WT mice with or without PDMP injection (Veh + SNT, n = 5; PDMP + SNT, n = 8; one‐way ANOVA, ***P < 0.001, vs. Veh + sham group, # P < 0.05, vs. Veh + SNT group at each time point). Data are presented as mean ± SEM.
-
DMechanical allodynia was evaluated in nerve‐injured mice with vehicle (n = 6) or PDMP (n = 7) injection. PDMP was intrathecally administrated once a day from 4 to 6 days after nerve injury. Data are presented as mean ± SEM (one‐way ANOVA, ***P < 0.001, vs. vehicle group at each time point).
Discussion
In this study, we revealed a novel mechanism by which GT1b induces spinal cord microglia activation and contributes to the development of nerve injury‐induced neuropathic pain: Injured sensory neurons upregulate GT1b ganglioside that is axonally transported to and released in the spinal cord. GT1b, in turn, functions as an endogenous agonist of TLR2 to activate microglia, which contributes to central sensitization and neuropathic pain (Fig 7).
Figure 7. Molecular mechanisms of nerve injury‐induced spinal cord microglia activation.
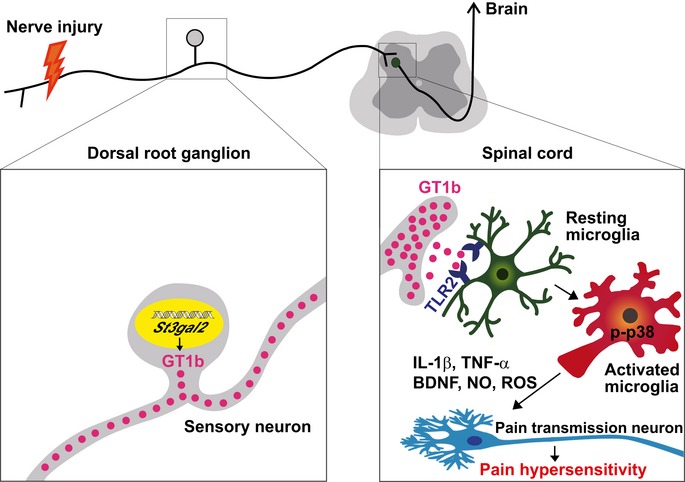
Peripheral nerve injury upregulates GT1b in the injured sensory neurons, which are intra‐axonally transported to and released in the spinal cord. In the spinal cord, GT1b functions as an endogenous agonist of microglial TLR2, activates p38 MAP kinase, and induces pain‐mediating proinflammatory cytokines (Il1b and Tnf), Bdnf, NO, and ROS production. These factors contribute to pain central sensitization.
Due to its clinical importance, great efforts have been made to understand the mechanisms of spinal cord microglia activation in neuropathic pain. Thus far, it has been proposed that ATP released from the central axons of the injured DRG neuron activates microglia in the spinal dorsal horn via the P2X4 receptor (Sawynok et al, 1993; Tsuda et al, 2003). P2X4 activation of microglia, in turn, induces release of BDNF that may induce pain central sensitization by disinhibiting inhibitory synaptic transmission (Coull et al, 2005). Meanwhile, a more recent study showed that ATP is released mainly from the spinal cord neurons, not from the injured sensory neurons (Masuda et al, 2016). This suggests that neuronal ATP might not be a primary signaling molecule mediating the injured sensory neuron‐to‐microglia signaling. Recently, colony‐stimulating factor 1 (CSF1) has been reported as a molecule that mediates injured neuron‐to‐microglia signaling (Guan et al, 2016; Okubo et al, 2016). CSF1 expressed in the injured sensory and motor neurons induced spinal cord microglia proliferation and DAP12‐dependent microglia activation. Intrathecal CSF1 administration upregulates mRNA expression of Itgam, Bdnf, and Ctss in the spinal cord, but with very limited induction of IL‐1β and TNF‐α (Guan et al, 2016). These proinflammatory cytokines are implicated in the central sensitization (Kawasaki et al, 2008b). Note, however, that these proinflammatory genes are significantly upregulated in the spinal cord microglia upon peripheral nerve injury (Jeong et al, 2016). Furthermore, nerve injury‐induced microglia activation was not completely abrogated in the Advillin‐Cre/Csf1 f/f mice after nerve injury (Guan et al, 2016). Taken together, those findings suggested that other nerve injury‐induced factors are required to fully activate spinal cord microglia. This appears to be particularly true for microglial proinflammatory gene expression.
Previously, we and others provided evidence that the TLR2 signaling pathway contributes to nerve injury‐induced spinal cord microglia activation and subsequent neuropathic pain (Kim et al, 2007, 2010; Lim et al, 2013; Stokes et al, 2013). However, how microglial TLR2 is engaged by peripheral nerve injury was not known. In the present studies, we provided evidences that GT1b serves as an endogenous agonist that exerts in action on microglia by binding to TLR2. First, intrathecal GT1b‐induced microglia activation and mechanical hypersensitivity are completely abrogated in Tlr2 −/− mice. Second, GT1b‐induced pain‐relevant gene expression such as Il1b, Tnf, and Bdnf is blocked in Tlr2 −/− mice in vivo and also in Tlr2‐deficient glial cells in vitro. Finally, in vitro data using rhodamine‐conjugated GT1b clearly demonstrate that GT1b directly binds to microglial TLR2. Taken together, our data support the hypothesis that GT1b is the long‐sought endogenous agonist of TLR2 that has been implicated in nerve injury‐induced neuropathic pain.
TLR2 is a pattern‐recognition receptor that detects danger‐associated and pathogen‐associated molecular patterns and activates innate immune cells upon tissue damage and infection, respectively (Kirschning & Schumann, 2002). In addition to neuropathic pain, TLR2 is implicated in glial cell activation and neuroinflammation in other neurological conditions including spinal cord injury (Kigerl et al, 2007), stroke (Tang et al, 2007), and neurodegenerative diseases (Jana et al, 2008). Thus far, studies have shown that HMGB1 and misfolded α‐synuclein may function as endogenous TLR2 agonists (Park et al, 2004; Kim et al, 2013). Our study found that GT1b is another TLR2 endogenous agonist that induces neuroinflammation. In addition to neuropathic pain, GT1b upregulation is detected in other neurological diseases including amyotrophic lateral sclerosis and epilepsy (Trbojevic‐Cepe et al, 1991). Notably, in these diseases, microglia activation and neuroinflammation are also observed (Beach et al, 1995; Henkel et al, 2009). Therefore, it may be worthwhile to investigate whether GT1b is also involved in microglia activation and neuroinflammation in these neurological disorders.
GT1b is one of the most abundant ganglioside in the brain (Tettamanti et al, 1973). GT1b localizes to the axonal membrane, where it interacts with myelin‐associated glycoprotein on Schwann cells or oligodendrocytes, and contributes to long‐term axon–myelin stability (Yang et al, 1996; Sheikh et al, 1999). Conceivably, the upregulation of GT1b by injured sensory neurons restores damaged axon–myelin integrity. Our data, however, point to a very different consequence of GT1b induction, microglia activation. As basal levels of GT1b, in the absence of nerve injury, do not activate microglia, it is not likely that membrane‐embedded GT1b directly activates TLR2. Rather, GT1b released from the membrane of damaged axons activates TLR2 on microglia. This is supported by the way TLR2 interacts with its agonist. LTA is comprised of two acyl chains attached to the hydrophilic glycan head group. Based on structural study, it was proposed that two acyl chains first bind to their binding pockets on TLR2 and the hydrophilic glycan head group induces dimerization of TLR2 with either TLR1 or TLR6 for intracellular signaling (Jin & Lee, 2008). Interestingly, GT1b, like LTA, consists of an oligosaccharide head group and two long saturated fatty acyl chains (Venkateshwari & Veluraja, 2012). Given the structural similarity between GT1b and LTA, it is conceivable that, once released, the exposed acyl chains of GT1b bind to TLR2 and the glycan head group of GT1b might induce dimerization with TLR1 or TLR6. Indeed, in our study we found that soluble GT1b levels are increased in the ISF and CSF upon peripheral nerve injury. Thus far, it is not clear how GT1b is aberrantly released from the neuronal plasma membrane upon peripheral nerve injury, which warrants future investigation.
In summary, we demonstrated a novel mechanism for spinal cord microglia activation after peripheral nerve injury: Nerve injury upregulates St3gal2 and subsequent GT1b synthesis in the injured DRG neurons. GT1b, in turn, is intra‐axonally transported and released in the spinal cord where it functions as a TLR2 endogenous agonist that activates microglia and contributes to nerve injury‐induced neuropathic pain. Thus, targeting St3gal2 upregulation in the DRG or GT1b binding to TLR2 in the spinal cord introduces potential novel therapeutic strategies to treat neuropathic pain.
Materials and Methods
Animals
All surgical and experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at Seoul National University. The animal treatments were performed in accordance with the guidelines of the International Association for the Study of Pain. Mice and rats were housed in a specific pathogen‐free vivarium with a 12‐h light–dark cycle and acclimated to the environment for at least 1 week before the experiments. All animals were allowed to access food and water ad libitum. WT mice with a C57BL/6 background and Sprague Dawley rats were purchased from Daehan Biolink (Eumsung, Korea). Sensory neuron‐specific St3gal2‐deficient mice were generated by breeding floxed‐St3gal2 mice and Scn10a‐Cre knockin mice expressing Cre recombinase under the control of an endogenous Nav1.8 promoter as previously described (Agarwal et al, 2004). Global St3gal2 −/− mice were generated as previously described (Ellies et al, 2002). Tlr2 −/−/Cx3cr1 +/GFP mice were generated by crossing Tlr2 −/− mice with knockin mice that express EGFP under the control of the endogenous Cx3cr1 locus. Transgenic Cx3cr1 +/GFP and floxed‐St3gal2 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Tlr2 −/− mice were kindly provided by Dr. S. Akira (Osaka University, Japan) (Takeuchi et al, 1999). All male genotypes were aged 8–10 weeks and weighed 21–25 g at the initiation of experimentation. Animals were assigned to the experimental groups using within‐cage randomization.
Neuropathic pain model and pain tests
To generate a persistent pain model, mice and rats were anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and L5 spinal nerve transection (SNT) was performed as previously described (Lim et al, 2017). For concurrent L5 dorsal root transection and L5 spinal nerve ligation (SNL), rats were anesthetized with enflurane (0.5–2%) and the L5 and L6 vertebra were surgically exposed. The L6 transverse process was removed, and the L5 spinal nerve was tightly ligated with 6‐0 silk thread. Then, an L5 laminectomy was carried out; the dura was opened, and the L5 dorsal root ipsilateral to the nerve injury was tightly ligated with 6‐0 silk thread and transected. Neuropathic pain tests were performed as previously reported by an observer blinded to group assignment and took place between 12:00 and 17:00 h (Lim et al, 2017).
LC–multiple‐reaction monitoring (MRM)–mass spectrometry (MS) analysis for gangliosides
All organic solvents of high‐performance liquid chromatography (HPLC) grade were purchased from J.T. Baker (Philipsburg, NJ, USA). Gangliosides were extracted from 10 mg of pooled ipsilateral L4‐5 spinal cord tissues of L5 spinal nerve‐injured mice. Ganglioside isolation was performed in accordance with established methods (Takamizawa et al, 1986) with minor modifications. Briefly, the tissue samples were pulverized in 400 μl methanol. Then, 10 μg of d18:1–18:0 GM3‐d3 (Matreya LLC, State College, PA, USA) was added to the samples as an internal standard and then were immediately mixed with 200 μl chloroform–methanol/chloroform (2:1 v/v). After vigorous mixing, the samples were placed on ice and incubated for 10 min. After incubation, the suspensions were mixed with equal volume of chloroform and 1.3× volumes of water. After mixing for 30 s, the suspensions were incubated at room temperature for 30 min. The samples were centrifuged at 2,500 × g for 10 min, and the upper phase was transferred into a new microtube and dried. For enrichment of gangliosides, 100 μl of binding buffer (1 mM MnCl2, 1 mM CaCl2, 0.5 M NaCl, and 20 mM Tris–HCl, pH 7.3) and 100 μg of Sambucus nigra (SNA) lectin (VECTOR, Burlingame, CA, USA) were added sequentially in the microtube. To promote the binding of SNA lectin to gangliosides, the samples were incubated at 25°C for 1 h in a Thermomixer (Eppendorf, Hamburg, Germany). Then, 200 μl of washing solution (1 mM MnCl2, 1 mM CaCl2, 0.5 M NaCl, 20 mM Tris–HCl, pH 7.3, and 40 mM NH5CO3) was applied to the filters with 14,000 × g centrifugal force at 25°C for 10 min. This clean‐up step was performed twice to remove non‐glycolipids from the filter. For the elution of gangliosides, 100 μl of an acidic hydrophobic solution (80% acetonitrile, 0.1% formic acid in water) was added twice at 25°C and 14,000 × g for 20 min; the eluents were removed completely by drying and the pellets were resuspended in 10 μl solvent A (20 mM ammonium formate, 0.1% formic acid in acetonitrile/methanol/water mixture (19:19:2)) for further mass spectrometry analysis. Quantification of gangliosides in samples was performed using a 6490 Accurate‐Mass Triple Quadrupole LC‐MS instrument connected to a 1200 series HPLC system with an SB‐C18 2.1 × 50 mm, 1.8 μm column (Agilent Technologies, Wilmington, DE, USA). The mobile phase was pumped at a flow rate of 0.1 ml/min, and 3.5 μl of sample was injected for each run. To determine MRM pair for all ganglioside subclasses, we used data on parent and daughter ions from a previous study (Sturgill et al, 2012). For ganglioside analysis, we optimized MRM parameters with GM3‐d3 as an internal standard. The collision energies for GT1b species were set at 32 and 38 V for GM3‐d3 internal standard (optimized condition). The collision gas and sheath gas temperatures were adjusted to 250 and 350°C, respectively. Finally, we performed a validation study using an internal standard, GM3‐d3. The peak intensity of GT1b species in each sample was normalized to the corresponding level of internal standard and is presented as a percent of the corresponding controls. Data are expressed as mean ± SEM values.
In vivo microdialysis in spinal cord and CSF collection
Interstitial fluid of the spinal cord was collected as previously reported with minor modifications (Jo et al, 2014). In brief, 5 days after L5 SNT, rats were anesthetized using 1.5–3% isoflurane and placed on the stereotaxic frame. After exposing the spinal cord and removing the dura mater, a CMA7 guide cannula (Harvard Apparatus, Holliston, MA, USA) was inserted in the L5 spinal cord dorsal horn (DV: −0.75 mm, degree 60°). The guide cannula was fixed with zinc polycarboxylate dental cement. After fixing the guide cannula thoroughly by dental cement, a CMA7 microdialysis probe (membrane diameter 0.24 mm, length 1 mm, stainless‐steel shaft diameter 0.38 mm) was implanted through the guide cannula. The probe was connected to a CMA402 syringe pump (Harvard Apparatus) with fluorinated ethylene propylene tubing (Harvard Apparatus). Then, the probe was perfused with artificial cerebrospinal fluid (mM: 149 NaCl, 2.8 KCl, 1.2 MgCl2, 1.2 CaCl2, and 5.4 glucose, pH 7.4) into inlet of the probe at a flow rate of 2 μl/min. Perfusates from the outlet of the tubing were collected in the plastic vial on ice. Dialysates were collected at intervals of 1 h for 3 h. Dialysates were stored at −20°C until before use. The CSF sampling was performed as previously described (Rodriguez‐Fanjul et al, 2015). Briefly, 5 days after L5 SNT, mice were anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and the subcutaneous tissue and occipital muscles were carefully separated by dissection with scissors to expose the surface of the dura covering the cisterna magna. The surrounding area was then gently cleaned with cotton swabs to remove any residual blood. After stabbing the dura with a 30‐gauge one‐half‐inch needle, the flowing CSF was collected with a pipette. The CSF from five mice was pooled into one biological replicate and stored at −20°C until before use.
Enzyme‐linked immunosorbent assay (ELISA)
GT1b measurement in ISF, CSF, conditioned medium, and spinal cord tissues was performed as described previously with minor modifications (Holmgren et al, 1980). Briefly, 1 μg/ml of tetanus toxin (Sigma, St. Louis, MO, USA) in coating buffer (BioLegend, San Diego, CA, USA) was directly attached to the 96‐well plate by passive adsorption at 37°C overnight and then the microplate was incubated at 55°C for 3 h to immobilize the toxin firmly. The GT1b ELISA was performed using the tetanus toxin‐coated microplate according to a conventional ELISA protocol. IL‐1β release and BDNF release were analyzed using an IL‐1β ELISA kit (Komabiotech, Seoul, Korea) and BDNF ELISA kit (Boster Biological Technology, Pleasanton, CA, USA) according to the manufacturer's instructions, respectively. Total protein concentrations from each cell lysate were measured using a BCA protein assay kit (Thermo Scientific, Rockford, IL, USA) and used for normalization.
Intrathecal injection
Mice were anesthetized with pentobarbital sodium (25 mg/kg, i.p.) and GT1b (Matreya LLC) in saline solution or vehicle alone was administrated by direct lumbar puncture between the L5 and L6 vertebrae of the spine of WT and Tlr2 −/− mice, using a 10‐μl Hamilton syringe (Hamilton Company, Reno, NV, USA) with a 30‐gauge one‐half‐inch needle as previously described (Hylden & Wilcox, 1980). Polymyxin B (10 μg/ml, Sigma) was used to prevent LPS contamination 30 min before GT1b administration. PDMP (Matreya LLC) was dissolved in 5% Tween 80 (Sigma) in saline and diluted with sterile saline immediately before intrathecal injection. The success of the intrathecal injection was assessed by monitoring the tail‐flick response when the needle penetrated the subarachnoid space.
Immunohistochemistry and live cell imaging
L5 DRG and spinal cord sections were prepared and immunostained as previously described (Lim et al, 2017), with rabbit anti‐Iba‐1 (1:1,000, Wako, Osaka, Japan, 019‐19741), mouse anti‐GT1b (1:1,500, Millipore, Temecula, CA, USA, MAB5608), rabbit anti‐ST3Gal‐II (1:250, Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc‐292044), and FITC‐ or Cy3‐conjugated secondary antibodies (1:200, Jackson ImmunoResearch, West Grove, PA, USA, 715‐095‐151, 715‐165‐150). GT1b immunostaining was performed as previously described (Gong et al, 2002). Briefly, the animals were perfused through the heart with saline solution followed by 10% sucrose. The L5 DRG and dorsal root were collected and snap‐frozen at −80°C. For ganglioside immunostaining, the detergent was omitted from the entire process. For live cell imaging, primary microglia were prepared from WT, Cx3cr1 +/GFP, and Tlr2 −/−/Cx3cr1 +/GFP mice. To measure intracellular reactive oxygen species level, primary microglia cultured from WT mice were treated with 10 μg/ml (4.5 μM) of GT1b for 5.5 h. Then, 10 μM of CM‐H2DCFDA (Life Technologies, Carlsbad, CA, USA) were loaded in the microglia and incubated for 30 min at 37°C. Upon rinsing the microglia with PBS, the fluorescence intensity was measured. The fluorescence intensity of CM‐H2DCFDA was measured and presented as the fold induction. To test the direct interaction between TLR2 and GT1b, primary microglia were incubated with 5 μg/ml (1.6–1.9 μM) of GT1b‐rhodamine for 1 h in the presence or absence of LTA (Sigma) at 37°C in a 5% CO2 incubator. LTA was pre‐treated for 30 min prior to the GT1b‐rhodamine treatment. After incubation, cells were rinsed with glial cell culture medium three times and the rhodamine intensity was measured in whole microglia and expressed as mean ± SEM.
We immunostained 3–5 sequential sections of spinal cord tissue, DRG tissue, and dorsal root tissue at 50‐μm intervals per animal (n = 1) and obtained a representative image under the same condition using a confocal microscope (LSM700, Carl Zeiss, Oberkochen, Germany) in the blind state. For the relative quantification of fluorescence intensity, tissue sections of each group were simultaneously immunostained under the same condition and when shooting a confocal image, laser power and gain values were adjusted to avoid saturation of single pixels. When measuring fluorescent signal intensity, brightness and contrast were adjusted under the same condition in the ZEN software (version ZEN 2010B SP1, Carl Zeiss) to remove the background of each group to be compared. Fluorescent signal intensity was measured in three random areas of a 0.1 × 0.1‐mm per tissue section using the ZEN software, and their mean values were calculated. The fluorescence intensity is presented as the fold induction compared to the corresponding level measured in the control sample. Using differential interference contrast images, we measured ST3Gal‐II and GT1b immunoreactivity in DRG neurons without separate marker staining. Image analysis was performed under blind conditions.
Real‐time RT–PCR
Real‐time RT–PCR experiments were performed as previously described (Lim et al, 2017). The following PCR primer sequences were used: Gapdh forward, 5′‐AGG TCA TCC CAG AGC TGA ACG‐3′; Gapdh reverse, 5′‐CAC CCT GTT GCT GTA GCC GTA‐3′; Il1b forward, 5′‐GTG CTG TCG GAC CCA TAT GA‐3′; Il1b reverse, 5′‐TTG TCG TTG CTT GGT TCT CC‐3′; Tnf forward, 5′‐AGC AAA CCA CCA AGT GGA GGA‐3′; Tnf reverse, 5′‐GCT GGC ACC ACT AGT TGG TTG T‐3′; Bdnf forward, 5′‐TGC AGG GGC ATA GAC AAA AGG‐3′; Bdnf reverse, 5′‐CTT ATG AAT CGC CAG CCA ATT CTC‐3′; St3gal2 forward, 5′‐ATG GCT ACC TTG CCC TAC CT‐3′; St3gal2 reverse, 5′‐GTC CAG ACG GGT GAG ATG TT‐3′; St3gal3 forward, 5′‐AGG CTG CCT TCA CTC TCA TT‐3′; and St3gal3 reverse, 5′‐CAT TGG GTG TGT TCA TGT CA‐3′.
Primary cell and F11 cell culture
Primary mixed glial cell and microglia cultures were prepared according to previously established procedures (Lim et al, 2013). In brief, brain glial cells were cultured from post‐natal days 1–3 for WT, Tlr2 −/−, Cx3cr1 +/GFP, and Tlr2 −/−/Cx3cr1 +/GFP mice. After 2 weeks, the glial cells were trypsinized and seeded into 6‐well plates for Western blot analysis. Primary microglia were harvested from brain mixed with glial cell cultures as previously described (Lim et al, 2013). Microglia were plated in 96‐well plates at a density of 50,000 cells per well and allowed to adhere overnight. Cells were incubated for 6 h in serum‐free glial cell culture medium prior to stimulation. For IL‐1β release, microglia were primed by pre‐treatment of 10 μg/ml GT1b for 2 h prior to stimulation with 5 mM ATP for 1 h. For BDNF release, microglia were stimulated with 10 μg/ml GT1b for 2 h followed by 50 μM ATP for 1 h. Neither the GT1b nor ATP affected cell viability. For nitric oxide production, microglia were stimulated with 10 μg/ml GT1b for 24 or 48 h. F11 cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM l‐glutamine, and 1 × penicillin/streptomycin at 37°C in a 5% CO2 incubator. F11 cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA, HB‐11761). For F11 neuronal differentiation (Ghil et al, 2000), the cells were incubated in the culture media supplemented with 1% fetal bovine serum, 2 mM l‐glutamine, 1 × penicillin/streptomycin, and 0.5 mM of dibutyryl cyclic AMP (Sigma) for 3 days in a 24‐well plate. To depolarize the cells, the KCl concentration in the culture medium was adjusted to 50 mM. The high concentration of potassium did not affect cell viability. Cell supernatants and lysates were collected and stored at −20°C until the day of the assay.
Nitric oxide assay
Nitric oxide production was determined by measuring nitrite level with Griess reagents (Sigma) according to the manufacturer's instructions. In brief, equal volumes of the culture supernatants and the Griess reagents were mixed and then incubated for 15 min at room temperature. Absorbance was measured at 540 nm. The nitrite concentration was calculated using a standard curve, and data are presented as mean ± SEM values.
Western blot analysis
Protein lysates were prepared by lysing glial cells in RIPA buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% NP‐40, 0.5% deoxycholic acid, 0.1% SDS, 1 mM PMSF, and phosphatase inhibitor mixture set IV (Millipore). A total of 20 μg of the cell lysate was separated on a 10% SDS–PAGE by electrophoresis and electrotransferred to a nitrocellulose membrane. Membranes were blocked with 5% bovine serum albumin (BSA) in Tris‐buffered saline containing Tween 20 (20 mM Tris, pH 7.4, 150 mM NaCl, and 0.1% Tween 20) and probed overnight with primary antibody against p‐p38 (Thr180/Tyr182) (1:1,000, Cell Signaling Technology, Danvers, MA, USA, 9211) at 4°C, followed by incubation with horseradish peroxidase‐conjugated secondary antibody at room temperature for 1 h prior to chemiluminescence treatment. For normalization, the membranes were stripped and reprobed with p38 antibody (1:1,000, Cell Signaling Technology, 9212). Immunoreactivity signals were visualized by MicroChemi (DNR Bio‐imaging Systems, Jerusalem, Israel). Three independent experiments were performed, and a representative blot image is shown. The band intensity of p‐p38 was measured and presented as the fold induction compared to the control.
Fluorescent dye‐labeled GT1b
GT1b‐rhodamine was synthesized using labeling probe kit (AAT Bioquest, Sunnyvale, CA, USA) according to the manufacturer's instructions. Briefly, three mg of GT1b (Matreya LLC) were dissolved in chloroform/methanol/distilled water (2:1:0.2). 5(6)‐Carboxy‐X‐rhodamine (16.6 mg) in anhydrous dimethylformamide (DMF, Sigma) was added dropwise to the GT1b solution. A mixture of GT1b and two equivalents of N‐hydroxybenzotriazole (HOBt, Anaspec Inc., San Jose, CA, USA) and 2‐(1H‐benzotriazole‐1‐yl)‐1,1,3,3‐tetramethyluronium (HBTU, Anaspec Inc.), and four equivalents of N,N‐diisopropylethylamine (DIPEA, Sigma) per surface hydroxyl group of the GT1b in anhydrous DMF was stirred for 24 h at room temperature. The crude product was eluted onto a Sephadex LH‐20 (Lipophilic Sephadex, Sigma) column (DMF only) to separate the unreacted excess rhodamine, HOBt, and HBTU. The sample solution was dialyzed (MWCO 1000, Sigma) against pure water for 4 h. After filtration with a 0.22‐μm filter, the sample solution was freeze‐dried. All steps were performed in the absence of light. The synthesis of GT1b‐rhodamine, with the conjugation ester bond, was confirmed using proton NMR spectra (Avance DPX‐300, Bruker, Billerica, MA, USA).
Statistics
The statistical significances of differences were assessed using SigmaPlot software (version 11.0, Systat Software, Inc., San Jose, CA, USA). Statistical analyses were performed using one‐way ANOVA followed by Bonferroni's post‐test for multiple comparisons or Student's t‐test. All data are presented as mean ± SEM, and differences were considered statistically significant when the P‐value was smaller than 0.05.
Author contributions
HL and SJL designed the study, analyzed the data, and wrote the manuscript. JL performed GT1b immunohistochemistry. BY tested effects of gangliosides on primary glial cells. JHO synthesized GT1b‐rhodamine, and J‐SP analyzed the data. HJM and KPK carried out LC‐MRM/MS analysis for gangliosides in spinal cord tissues. YSK and B‐EY performed microdialysis in spinal cord. BGK designed the study. SKB performed L5 SNL and L5 dorsal root rhizotomy. RLS analyzed the data and wrote the manuscript. HL performed the rest of the experiments and analyzed the data. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Acknowledgements
This work was supported by Samsung Science & Technology Foundation (SSTF‐BA1502‐13). HL was supported by the post‐doctoral fellowship of Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1A6A3A01011812). We thank Dr. Allan I. Basbaum, University California at San Francisco, for providing us with the Scn10a‐Cre mice.
The EMBO Journal (2020) 39: e102214
References
- Agarwal N, Offermanns S, Kuner R (2004) Conditional gene deletion in primary nociceptive neurons of trigeminal ganglia and dorsal root ganglia. Genesis 38: 122–129 [DOI] [PubMed] [Google Scholar]
- Ando S (1983) Gangliosides in the nervous system. Neurochem Int 5: 507–537 [DOI] [PubMed] [Google Scholar]
- Beach TG, Woodhurst WB, MacDonald DB, Jones MW (1995) Reactive microglia in hippocampal sclerosis associated with human temporal lobe epilepsy. Neurosci Lett 191: 27–30 [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438: 1017–1021 [DOI] [PubMed] [Google Scholar]
- Ellies LG, Sperandio M, Underhill GH, Yousif J, Smith M, Priatel JJ, Kansas GS, Ley K, Marth JD (2002) Sialyltransferase specificity in selectin ligand formation. Blood 100: 3618–3625 [DOI] [PubMed] [Google Scholar]
- Ghil SH, Kim BJ, Lee YD, Suh‐Kim H (2000) Neurite outgrowth induced by cyclic AMP can be modulated by the alpha subunit of Go. J Neurochem 74: 151–158 [DOI] [PubMed] [Google Scholar]
- Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer‐Lauc M, Li CY, Griffin JW, Schnaar RL, Sheikh KA (2002) Localization of major gangliosides in the PNS: implications for immune neuropathies. Brain 125: 2491–2506 [DOI] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans‐Reinsch Z, Braz J, Devor M et al (2016) Injured sensory neuron‐derived CSF1 induces microglial proliferation and DAP12‐dependent pain. Nat Neurosci 19: 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Zhao W, Appel SH (2009) Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol 4: 389–398 [DOI] [PubMed] [Google Scholar]
- Holmgren J, Elwing H, Fredman P, Svennerholm L (1980) Polystyrene‐adsorbed gangliosides for investigation of the structure of the tetanus‐toxin receptor. Eur J Biochem 106: 371–379 [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL (1980) Intrathecal morphine in mice: a new technique. Eur J Pharmacol 67: 313–316 [DOI] [PubMed] [Google Scholar]
- Inoue K, Tsuda M (2009) Microglia and neuropathic pain. Glia 57: 1469–1479 [DOI] [PubMed] [Google Scholar]
- Jana M, Palencia CA, Pahan K (2008) Fibrillar amyloid‐beta peptides activate microglia via TLR2: implications for Alzheimer's disease. J Immunol 181: 7254–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TS, Finnerup NB (2014) Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol 13: 924–935 [DOI] [PubMed] [Google Scholar]
- Jeong H, Na YJ, Lee K, Kim YH, Lee Y, Kang M, Jiang BC, Yeom YI, Wu LJ, Gao YJ et al (2016) High‐resolution transcriptome analysis reveals neuropathic pain gene‐expression signatures in spinal microglia after nerve injury. Pain 157: 964–976 [DOI] [PubMed] [Google Scholar]
- Ji RR, Suter MR (2007) p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain 3: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MS, Lee JO (2008) Structures of the toll‐like receptor family and its ligand complexes. Immunity 29: 182–191 [DOI] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H et al (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat Med 20: 886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH et al (2008a) Distinct roles of matrix metalloproteases in the early‐ and late‐phase development of neuropathic pain. Nat Med 14: 331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR (2008b) Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin‐1beta, interleukin‐6, and tumor necrosis factor‐alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 28: 5189–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG (2007) Toll‐like receptor (TLR)‐2 and TLR‐4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem 102: 37–50 [DOI] [PubMed] [Google Scholar]
- Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, Choi SY, Park K, Kim JS, Akira S et al (2007) A critical role of toll‐like receptor 2 in nerve injury‐induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem 282: 14975–14983 [DOI] [PubMed] [Google Scholar]
- Kim D, You B, Jo EK, Han SK, Simon MI, Lee SJ (2010) NADPH oxidase 2‐derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury‐induced neuropathic pain. Proc Natl Acad Sci USA 107: 14851–14856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ et al (2013) Neuron‐released oligomeric alpha‐synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4: 1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschning CJ, Schumann RR (2002) TLR2: cellular sensor for microbial and endogenous molecular patterns. Curr Top Microbiol Immunol 270: 121–144 [DOI] [PubMed] [Google Scholar]
- Lim H, Kim D, Lee SJ (2013) Toll‐like receptor 2 mediates peripheral nerve injury‐induced NADPH oxidase 2 expression in spinal cord microglia. J Biol Chem 288: 7572–7579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Lee H, Noh K, Lee SJ (2017) IKK/NF‐kappaB‐dependent satellite glia activation induces spinal cord microglia activation and neuropathic pain after nerve injury. Pain 158: 1666–1677 [DOI] [PubMed] [Google Scholar]
- Lu VB, Biggs JE, Stebbing MJ, Balasubramanyan S, Todd KG, Lai AY, Colmers WF, Dawbarn D, Ballanyi K, Smith PA (2009) Brain‐derived neurotrophic factor drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. J Physiol 587: 1013–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Hayes RL, Lu J, Mayer DJ (1992) Intrathecal GM1 ganglioside and local nerve anesthesia reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res 584: 28–53 [DOI] [PubMed] [Google Scholar]
- Masuda T, Ozono Y, Mikuriya S, Kohro Y, Tozaki‐Saitoh H, Iwatsuki K, Uneyama H, Ichikawa R, Salter MW, Tsuda M et al (2016) Dorsal horn neurons release extracellular ATP in a VNUT‐dependent manner that underlies neuropathic pain. Nat Commun 7: 12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Jayaraj ND, Wilson HM, Ren D, Flood K, Wang XQ, Shum A, Miller RJ, Paller AS (2016) Ganglioside GM3 synthase depletion reverses neuropathic pain and small fiber neuropathy in diet‐induced diabetic mice. Mol Pain 12 10.1177/1744806916666284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander‐Melin M, Blennow K, Bogdanovic N, Dellheden B, Mansson JE, Fredman P (2005) Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent‐resistant membrane domains. J Neurochem 92: 171–182 [DOI] [PubMed] [Google Scholar]
- Okubo M, Yamanaka H, Kobayashi K, Dai Y, Kanda H, Yagi H, Noguchi K (2016) Macrophage‐colony stimulating factor derived from injured primary afferent induces proliferation of spinal microglia and neuropathic pain in rats. PLoS ONE 11: e0153375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ASB, Faergeman NJ (2017) Sphingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol 7: 170069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E (2004) Involvement of toll‐like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279: 7370–7377 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Fanjul J, Fernandez‐Feijoo CD, Camprubi MC (2015) A new technique for collection of cerebrospinal fluid in rat pups. J Exp Neurosci 9: 37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawynok J, Downie JW, Reid AR, Cahill CM, White TD (1993) ATP release from dorsal spinal cord synaptosomes: characterization and neuronal origin. Brain Res 610: 32–38 [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Fromholt SE, Gong YP, Vyas AA, Laroy W, Wayman DM, Heffer‐Lauc M, Ito H, Ishida H, Kiso M et al (2002) Immunoglobulin G‐class mouse monoclonal antibodies to major brain gangliosides. Anal Biochem 302: 276–284 [DOI] [PubMed] [Google Scholar]
- Schnaar RL (2016) Gangliosides of the vertebrate nervous system. J Mol Biol 428: 3325–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL (1999) Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci USA 96: 7532–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JA, Cheung J, Eddinger K, Corr M, Yaksh TL (2013) Toll‐like receptor signaling adapter proteins govern spread of neuropathic pain and recovery following nerve injury in male mice. J Neuroinflammation 10: 148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill ER, Aoki K, Lopez PH, Colacurcio D, Vajn K, Lorenzini I, Majic S, Yang WH, Heffer M, Tiemeyer M et al (2012) Biosynthesis of the major brain gangliosides GD1a and GT1b. Glycobiology 22: 1289–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa K, Iwamori M, Mutai M, Nagai Y (1986) Gangliosides of bovine buttermilk. Isolation and characterization of a novel monosialoganglioside with a new branching structure. J Biol Chem 261: 5625–5630 [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S (1999) Differential roles of TLR2 and TLR4 in recognition of gram‐negative and gram‐positive bacterial cell wall components. Immunity 11: 443–451 [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X et al (2007) Pivotal role for neuronal Toll‐like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA 104: 13798–13803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettamanti G, Bonali F, Marchesini S, Zambotti V (1973) A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim Biophys Acta 296: 160–170 [DOI] [PubMed] [Google Scholar]
- Trbojevic‐Cepe M, Kracun I, Jusic A, Pavlicek I (1991) Gangliosides of human cerebrospinal fluid in various neurologic diseases. J Neurol Sci 105: 192–199 [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto‐Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K (2003) P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424: 778–783 [DOI] [PubMed] [Google Scholar]
- Venkateshwari S, Veluraja K (2012) Conformational analysis of GT1B ganglioside and its interaction with botulinum neurotoxin type B: a study by molecular modeling and molecular dynamics. J Biomol Struct Dyn 30: 255–268 [DOI] [PubMed] [Google Scholar]
- Vunnam RR, Radin NS (1980) Analogs of ceramide that inhibit glucocerebroside synthetase in mouse‐brain. Chem Phys Lipid 26: 265–278 [DOI] [PubMed] [Google Scholar]
- Wang X, Wang C, Zeng J, Xu X, Hwang PY, Yee WC, Ng YK, Wang S (2005) Gene transfer to dorsal root ganglia by intrathecal injection: effects on regeneration of peripheral nerves. Mol Ther 12: 314–320 [DOI] [PubMed] [Google Scholar]
- Watanabe S, Tan‐No K, Tadano T, Higashi H (2011) Intraplantar injection of gangliosides produces nociceptive behavior and hyperalgesia via a glutamate signaling mechanism. Pain 152: 327–334 [DOI] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Kulkarni N, Ledeen RW (2012) Deficiency of ganglioside GM1 correlates with Parkinson's disease in mice and humans. J Neurosci Res 90: 1997–2008 [DOI] [PubMed] [Google Scholar]
- Yang LJ, Zeller CB, Shaper NL, Kiso M, Hasegawa A, Shapiro RE, Schnaar RL (1996) Gangliosides are neuronal ligands for myelin‐associated glycoprotein. Proc Natl Acad Sci USA 93: 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Wang HC, Ding AH, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H (2006) HMGB1 signals through toll‐like receptor (TLR) 4 and TLR2. Shock 26: 174–179 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File