

Abstract
Joint Species Distribution Modelling (JSDM) is becoming an increasingly popular statistical method for analysing data in community ecology. Hierarchical Modelling of Species Communities (HMSC) is a general and flexible framework for fitting JSDMs. HMSC allows the integration of community ecology data with data on environmental covariates, species traits, phylogenetic relationships and the spatio‐temporal context of the study, providing predictive insights into community assembly processes from non‐manipulative observational data of species communities.
The full range of functionality of HMSC has remained restricted to Matlab users only. To make HMSC accessible to the wider community of ecologists, we introduce Hmsc 3.0, a user‐friendly r implementation.
We illustrate the use of the package by applying Hmsc 3.0 to a range of case studies on real and simulated data. The real data consist of bird counts in a spatio‐temporally structured dataset, environmental covariates, species traits and phylogenetic relationships. Vignettes on simulated data involve single‐species models, models of small communities, models of large species communities and models for large spatial data. We demonstrate the estimation of species responses to environmental covariates and how these depend on species traits, as well as the estimation of residual species associations. We demonstrate how to construct and fit models with different types of random effects, how to examine MCMC convergence, how to examine the explanatory and predictive powers of the models, how to assess parameter estimates and how to make predictions. We further demonstrate how Hmsc 3.0 can be applied to normally distributed data, count data and presence–absence data.
The package, along with the extended vignettes, makes JSDM fitting and post‐processing easily accessible to ecologists familiar with r.
Keywords: community ecology, community modelling, community similarity, hierarchical modelling of species communities, joint species distribution modelling, multivariate data, species distribution modelling
1. INTRODUCTION
One of the main goals of modern community ecology is to identify and disentangle the assembly processes that contribute to the observed variation in the number, abundance, identities and traits of species over space and time. Among the many analytical tools available to community ecologists, species distribution models (SDMs) are becoming increasingly popular (D'Amen, Rahbek, Zimmermann, & Guisan, 2017). To make inferences at the community level, stacked species distribution models (SSDMs) model each species separately and then combine (‘stack’) their predictions to assess community‐level patterns (Calabrese, Certain, Kraan, & Dormann, 2014; Guisan & Rahbek, 2011), whereas joint species distribution models (JSDMs) explicitly acknowledge the multivariate nature of communities by assuming that the species respond jointly to the environment and to each other (Clark, Nemergut, Seyednasrollah, Turner, & Zhang, 2017; Ovaskainen, Tikhonov, Norberg, et al., 2017; Warton et al., 2015).
The Hierarchical Modelling of Species Communities (HMSC) framework belongs to the class of JSDMs, and can be used to interrelate data on species occurrences, environmental covariates, species traits and phylogenetic relationships with community assembly processes (Ovaskainen, Tikhonov, Norberg, et al., 2017). Within HMSC, environmental filtering is modelled at the species level by measuring how the occurrences of each species depend on environmental conditions. These species‐level models are integrated through a hierarchical structure aimed at determining to what extent environmental filtering is structured by species‐specific traits, and/or whether phylogenetically related species exhibit shared environmental responses (Abrego, Norberg, & Ovaskainen, 2017). Biotic interactions and the influence of missing environmental covariates are captured by residual species‐to‐species association matrices, which may be estimated at multiple spatio‐temporal scales (Ovaskainen, Abrego, Halme, & Dunson, 2016; Ovaskainen, Roy, Fox, & Anderson, 2016; Ovaskainen, Tikhonov, Dunson, et al., 2017). HMSC handles common response variable types such as presence–absence data or count data. In a comparison among a large number of single‐species and JSDMs, HMSC ranked first in terms of predictive performance (Norberg et al., 2019).
Compared to Hmsc 2.0 (see Ovaskainen, Tikhonov, Norberg, et al., 2017), Hmsc 3.0 includes several extensions, enabling one to ask how environmental conditions influence species‐to‐species association matrices (Tikhonov, Abrego, Dunson, & Ovaskainen, 2017), to infer species‐to‐species associations from time‐series data of species‐rich communities (Ovaskainen, Tikhonov, Dunson, et al., 2017), and to apply Hmsc to large spatial data (Tikhonov, Duan, et al., 2019). Furthermore, Hmsc 3.0 offers much improved flexibility with respect to the random error structures, model fitting efficiency and greater functionality for post‐processing the results and for making predictions. To make this possible, Hmsc 3.0 has been re‐coded anew rather than upgraded from Hmsc 2.0 and its syntax is not compatible with Hmsc 2.0. The technical specification of the Hmsc 3.0 implementation is given in Appendix S1.
2. HMSC WORKFLOW
Running a typical HMSC analysis includes five main steps: (1) Setting model structure and fitting the model, (2) Examining MCMC convergence, (3) Evaluating model fit, (4) Exploring parameter estimates and (5) Making predictions. Below, we explain each step in turn.
Step 1. Setting model structure and fitting the model. In this step, the user loads the data and makes decisions about model structure, including random effects, environmental covariates and the inclusion or exclusion of species traits and phylogenetic relationships. Model fitting includes running the Markov chain Monte Carlo (MCMC) estimation scheme to sample from the posterior distribution of the model parameters.
Step 2. Examining MCMC convergence. In this step, the user examines whether the MCMC scheme has resulted in a valid approximation of the posterior distribution, in the sense of the chains having reached a stationary distribution and representing a sufficiently large effective number of samples. If not, the results will not be reliable, and thus the user should refit the model with a longer MCMC sampling scheme.
Step 3. Evaluating model fit. Hmsc comes with built‐in functions that can be used to examine different aspects of model fit. Model fit can be evaluated either in terms of explanatory power, that is for the same sampling units that were used to fit the model, or in terms of predictive power through cross‐validation, that is for other sampling units than those used to fit the model. If explanatory power is much higher than predictive power, the specified model is probably too flexible, and the user may wish to re‐consider the model structure and/or the types of environmental variables included.
Step 4. Exploring parameter estimates. In this step, the user can extract numerical summaries of parameter estimates, for example posterior means and quantiles. Hmsc also comes with functions for producing plots that illustrate the posterior distributions of high‐dimensional variables, such as variance partitioning of the explained variation among environmental covariates and random effects, the responses of the species‐to‐environmental covariates, and species‐to‐species associations.
Step 5. Making predictions. Hmsc comes with a generic predict function as well as more specific tools for generating predictions over environmental gradients. The user can evaluate the predictions both at the species level (e.g. occurrence probabilities or abundances of individual species) or at the community level (e.g. species richness or community‐weighted trait means). Similarly, predictions over space can be used to generate maps for distributions of individual species, community‐weighted trait means or regions of common profile. Predictions for focal species can also be made conditional on the known occurrences of other species.
3. REAL DATA EXAMPLE: FINNISH BIRDS
We illustrate the typical Hmsc workflow using spatially explicit bird count data as an example. The full details of this case study are given in Appendix S2.
Step 1. Setting model structure and fitting the model. We fitted HMSC models to count data on the 50 most common Finnish birds surveyed during 914 counts on 200 permanent transect routes during the years 2006–2014. As environmental covariates (the matrix X), we included the categorical variable ‘habitat type’ with the five levels: broadleaved forests, coniferous forests, open habitats, urban habitats and wetlands, and the continuous covariate ‘spring temperature’ (mean in April and May), for which we included also a squared term to allow for intermediate niche optima. Habitat data were based on Corine land cover data from the years 2006 (used for study years 2006–2009) and 2012 (used for study years 2010–2014) and measured within a 300 meters buffer from the census sites. As species traits (the matrix T), we included the categorical variable ‘migration strategy’ with three levels (resident, short‐distance migrant and long‐distance migrant, see Valkama et al., 2014), and the continuous variable ‘body size’ (log‐transformed, according to Cramp et al. 1977–1994). We included in the analyses a phylogenetic tree for the study species, acquired from http://birdtree.org (Jetz et al. 2012). As community‐level random effect, we included the survey route, which we assumed to be spatially structured and hence implemented with the help of spatial latent factors (Ovaskainen, Roy, et al., 2016). We fitted both a probit model to data truncated to presence–absence (Model PA), as well as a lognormal Poisson model for the full count data including zeros and non‐zeros (Model ABU). For both model types, we considered three model variants that included either both environmental covariates and spatial latent variables (XS), only environmental covariates (X), or only spatial latent variables (S). Thus, we fitted in total the six models PA.XS, PA.X, PA.S, ABU.XS, ABU.X and ABU.S. We applied the default priors in Hmsc (see Appendix S1). We sampled the posterior distribution with four MCMC chains, each of which was run for 150,000 iterations, out of which the first 50,000 were removed as burn‐in and the remaining ones were thinned by 100 to yield 1,000 posterior samples per chain, and thus 4,000 posterior samples in total.
Step 2 (Examining MCMC convergence) and Step 3 (Evaluating model fit) are illustrated in Appendix S2.
Step 4. Exploring parameter estimates. While model fit describes how much of the variation in the data the model is able to explain or predict, variance partitioning describes which components of the model that explain the explained variance. For example, in the model PA.XS, the partitioning of the explained variance attributed on average (over the species) 88% to the spatial random effect for route, and only 5% to climatic and 7% to habitat variables. In contrast, in the model PA.X, that did not include spatial random effects, 72% of the explained variance was attributed to climatic and 28% to habitat variables. Environmental filtering can be assessed by examining the β‐parameters (regression slopes) that characterize species niches, that is the influence of environmental variation on species occurrences. In the model PA.X, many species exhibited statistically well‐supported responses to many of the covariates, for example a generally negative response to the squared effect of spring temperature (Figure 1a), suggesting an intermediate optimum. In the model PA.X, the included traits explained 7% of the variation in species occurrence, and the residual variation showed no evidence for a phylogenetic signal, as the posterior median estimate of the phylogenetic signal parameter ρ was 0 (95% credibility interval from 0.00 to 0.22). The data exhibited a clear spatial signal, as for example in the model PA.S the posterior mean estimate of the spatial scale parameter α related to the leading latent variable was 400 km (95% credibility interval from 200 to 1,200 km). The spatial latent variables also indicated a strong co‐occurrence pattern, with a large number of species exhibiting positive associations beyond those explained by the covariates (Figure 1b). The only exception was Phoenicurus phoenicurus, which was typically recorded on routes where few other species were recorded. This species is known to specialize on nutrient‐poor pine forests that have low densities of birds in general (Lehikoinen, Sirkiä, & Tirri, 2017).
Figure 1.
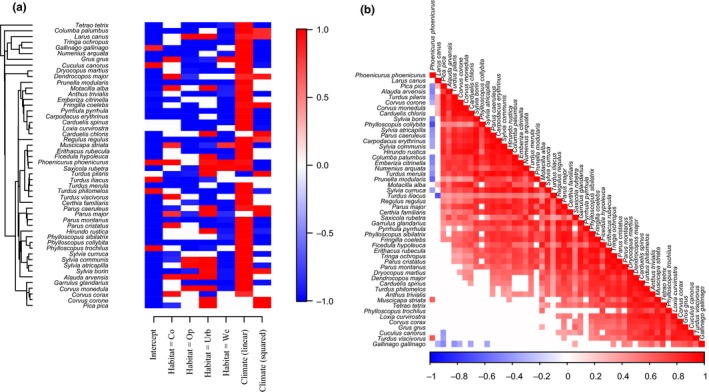
Exploring parameter estimates. Panel (a) shows those β parameters (species responses to environmental covariates) with at least 95% posterior probability of being positive (red) or negative (blue) in model PA.X. The species are ordered according to their phylogeny shown on left. Panel (b) illustrates the residual association structure in model PA.S, with positive associations with high (at least 95% posterior probability) statistical support shown in red and negative associations in blue. The species have been ordered in a way that best illustrates the association structure of the data
Step 5. Making predictions. Hmsc includes functions that make and illustrate predictions over categorical (here the habitat type; Figure 2a–d) as well as continuous (here spring temperature, results shown in Appendix S2) environmental gradients. Concerning the spatial predictions (Figure 2e–h), we have used here a model fitted to data from 200 locations to predict species occurrences in c. 10,000 locations. As we have generated these predictions with model PA.XS, they combine both environmental and spatial information. Predictions can be illustrated at the level of individual species, for example showing that Corvus monedula prefers urban habitats (Figure 2a) and mainly occurs in Southern Finland (Figure 2e). Predictions can also be illustrated at the level of species richness, showing that urban habitats host the most species and wetlands and open habitats the least species (Figure 2b), and a decreasing gradient from south to north (Figure 2f). Predictions can further be illustrated at the level of community‐weighted mean trait values, showing for example that the proportion of resident species is lowest in wetlands (Figure 2c) and decreases towards the north (Figure 2g). One way to visualize variation in community composition is to cluster the predicted communities into a discrete set of communities of common profile. Doing so shows that urban areas have distinct communities (Figure 2d), and that communities are structured along the latitudinal gradient in terms of their species composition (Figure 2h).
Figure 2.
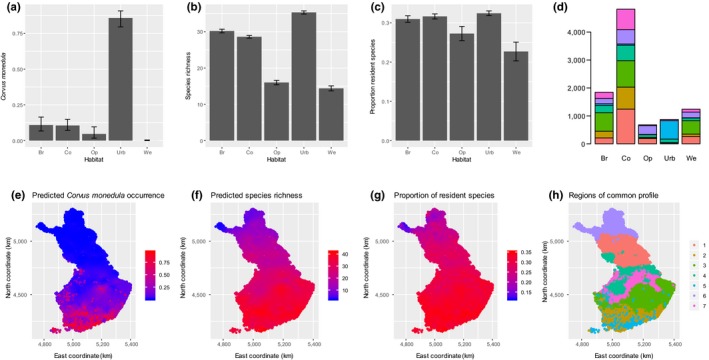
Making predictions. The upper panels exemplify predictions over environmental gradients and the lower panels exemplify spatial predictions, both of which can be used to quantify the influence of covariates on species occurrence (a, e), species richness (b, f), community‐weighted mean traits (c, g) or regions of common profile (d, h). The predictions over environmental gradients are used here to predict species communities in different habitat types, whereas the spatial predictions are used to predict species communities on a grid covering Finland (the training data come from a small subset of 200 locations). The panels (a)–(c) are based on Model PA.X and the remaining panels on Model PA.XS
We further illustrate the workflow of Hmsc with simulated data in four vignettes that are integrated within the package, as well as in Appendix S3, which shows how the type and amount of data influence the mixing properties of the MCMC sampling scheme as well as its ability to recover the true parameter values.
4. CONCLUSION
At present, the most widely applied methods for data analysis in community ecology are based on ordination approaches. While we appreciate the value of ordinations as a fast way for making descriptive analyses, we encourage community ecologists to make even more out of their data by applying also model‐based approaches, especially the newly emerging JSDMs (Warton et al., 2015). We hope that the users find Hmsc 3.0 to provide a highly functional and user‐friendly package for doing so.
AUTHORS' CONTRIBUTIONS
G.T. designed and implemented the main part of the Hmsc 3.0 software. O.O. contributed specific functions to Hmsc, implemented the first versions of the simulated and real data case studies and devised the first drafts of the simulated vignettes. Ø.H.O. devised the first draft of the bird vignette, revised all vignettes and contributed specific functions to Hmsc. N.A. and O.O. wrote the first version of the manuscript. A.L. contributed the data for the bird case study. J.O. revised the technical quality of the package, and M.M.J.d.J. implemented unit tests and the methods for big spatial data. All authors contributed substantially to the writing of the final version of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
This work was funded by Academy of Finland (grants 284601 and 309581 to O.O. and 275606 to A.L.), Jane and Aatos Erkko Foundation (grant to O.O.), and the Research Council of Norway through its Centres of Excellence Funding Scheme (223257) to O.O. via Centre for Biodiversity Dynamics. Finnish Ministry of Environment has supported the census scheme. M.M.J.d.J. was financed by the European Research Council via the project SIZE (647224).
Tikhonov G, Opedal ØH, Abrego N, et al. Joint species distribution modelling with the r‐package Hmsc . Methods Ecol Evol. 2020;11:442–447. 10.1111/2041-210X.13345
DATA AVAILABILITY STATEMENT
The package Hmsc is available at the Comprehensive R Archive Network CRAN (https://CRAN.R-project.org/package=Hmsc (Tikhonov, Oksanen et al., 2019 https://CRAN.R-project.org/package=Hmsc). The development version of Hmsc is available at Github (https://github.com/hmsc-r/HMSC, https://doi.org/10.5281/zenodo.3582925 (Tikhonov, Ovaskainen et al., 2019 https://doi.org/10.5281/zenodo.3582925).
REFERENCES
- Abrego, N. , Norberg, A. , & Ovaskainen, O. (2017). Measuring and predicting the influence of traits on the assembly processes of wood‐inhabiting fungi. Journal of Ecology, 105, 1070–1081. 10.1111/1365-2745.12722 [DOI] [Google Scholar]
- Calabrese, J. M. , Certain, G. , Kraan, C. , & Dormann, C. F. (2014). Stacking species distribution models and adjusting bias by linking them to macroecological models. Global Ecology and Biogeography, 23, 99–112. 10.1111/geb.12102 [DOI] [Google Scholar]
- Clark, J. S. , Nemergut, D. , Seyednasrollah, B. , Turner, P. J. , & Zhang, S. (2017). Generalized joint attribute modeling for biodiversity analysis: Median‐zero, multivariate, multifarious data. Ecological Monographs, 87, 34–56. 10.1002/ecm.1241 [DOI] [Google Scholar]
- D'Amen, M. , Rahbek, C. , Zimmermann, N. E. , & Guisan, A. (2017). Spatial predictions at the community level: From current approaches to future frameworks. Biological Reviews, 92, 169–187. 10.1111/brv.12222 [DOI] [PubMed] [Google Scholar]
- Guisan, A. , & Rahbek, C. (2011). SESAM – A new framework integrating macroecological and species distribution models for predicting spatio‐temporal patterns of species assemblages. Journal of Biogeography, 38, 1433–1444. 10.1111/j.1365-2699.2011.02550.x [DOI] [Google Scholar]
- Jetz, W. , Thomas, G. H. , Joy, J. B. , Hartmann, K. , & Mooers, A. O. (2012). The global diversity of birds in space and time. Nature, 491, 444–448. [DOI] [PubMed] [Google Scholar]
- Lehikoinen, A. , Sirkiä, P. M. , & Tirri, I.‐S. (2017). Yleisten metsälintujen runsaus suhteessa elinympäristöjen piirteisiin. Linnut‐vuosikirja 2016, 54–67. [Google Scholar]
- Norberg, A. , Abrego, N. , Blanchet, F. G. , Adler, F. , Anderson, B. , Anttila, J. , … Ovaskainen, O. (2019). A comprehensive evaluation of predictive performance of 33 species distribution models at species and community levels. Ecological Monographs, 89, e01370 10.1002/ecm.1370 [DOI] [Google Scholar]
- Ovaskainen, O. , Abrego, N. , Halme, P. , & Dunson, D. (2016). Using latent variable models to identify large networks of species‐to‐species associations at different spatial scales. Methods in Ecology and Evolution, 7, 549–555. 10.1111/2041-210X.12501 [DOI] [Google Scholar]
- Ovaskainen, O. , Roy, D. B. , Fox, R. , & Anderson, B. J. (2016). Uncovering hidden spatial structure in species communities with spatially explicit joint species distribution models. Methods in Ecology and Evolution, 7, 428–436. 10.1111/2041-210X.12502 [DOI] [Google Scholar]
- Ovaskainen, O. , Tikhonov, G. , Dunson, D. , Grøtan, V. , Engen, S. , Sæther, B. , & Abrego, N. (2017). How are species interactions structured in species‐rich communities? A new method for analysing time‐series data. Proceedings of the Royal Society B: Biological Sciences, 284(1855), 20170768 10.1098/rspb.2017.0768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovaskainen, O. , Tikhonov, G. , Norberg, A. , Blanchet, F. G. , Duan, L. , Dunson, D. , … Abrego, N. (2017). How to make more out of community data? A conceptual framework and its implementation as models and software. Ecology Letters, 20, 561–576. 10.1111/ele.12757 [DOI] [PubMed] [Google Scholar]
- Tikhonov, G. , Abrego, N. , Dunson, D. , & Ovaskainen, O. (2017). Using joint species distribution models for evaluating how species‐to‐species associations depend on the environmental context. Methods in Ecology and Evolution, 8, 443–452. 10.1111/2041-210X.12723 [DOI] [Google Scholar]
- Tikhonov, G. , Duan, L. , Abrego, N. , Newell, G. , White, M. , Dunson, D. , & Ovaskainen, O. (2019). Computationally efficient joint species distribution modelling of big spatial data. Ecology, in press. 10.1002/ecy.2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonov, G. , Oksanen, J. , deJonge, M. , Opedal, Ø. , & Dallas, T. (2019). Hmsc-r, & ovaskain. hmsc-r/HMSC v3.0-5 (Version v3.0-5). Zenodo. 10.5281/zenodo.3582925 [DOI]
- Tikhonov, G. , Ovaskainen, O. , Oksanen, J. , de Jonge, M. , Opedal, O. , & Dallas, T. (2019). Hmsc: Hierarchical model of species communities. R package version 3.0-4. https://CRAN.Rproject.org/package=Hmsc
- Valkama, J. , Saurola, P. , Lehikoinen, A. , Lehikoinen, E. , Piha, M. , Sola, P. , … Sulonen, H. (2014). The Finnish bird ringing atlas (Vol. 2). Helsinki, Finland: Finnish Museum of Natural History and Finnish Ministry of Environment. [Google Scholar]
- Warton, D. I. , Blanchet, F. G. , O'Hara, R. B. , Ovaskainen, O. , Taskinen, S. , Walker, S. C. , & Hui, F. K. C. (2015). So many variables: Joint modeling in community ecology. Trends in Ecology & Evolution, 30, 766–779. 10.1016/j.tree.2015.09.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The package Hmsc is available at the Comprehensive R Archive Network CRAN (https://CRAN.R-project.org/package=Hmsc (Tikhonov, Oksanen et al., 2019 https://CRAN.R-project.org/package=Hmsc). The development version of Hmsc is available at Github (https://github.com/hmsc-r/HMSC, https://doi.org/10.5281/zenodo.3582925 (Tikhonov, Ovaskainen et al., 2019 https://doi.org/10.5281/zenodo.3582925).