

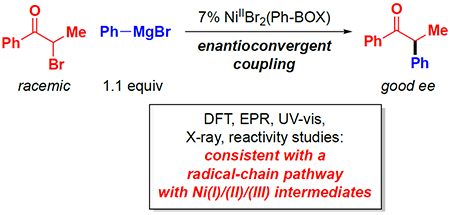
Abstract
In recent years, a wide array of methods for achieving nickel-catalyzed substitution reactions of alkyl electrophiles by organometallic nucleophiles, including enantioconvergent processes, have been described; on the other hand, experiment-focused mechanistic studies of such couplings have been comparatively scarce. The most detailed mechanistic investigations to date have examined catalysts that bear tridentate ligands and, with one exception, processes that are not enantioselective; studies of catalysts based on bidentate ligands could be anticipated to be more challenging, due to difficulty in isolating proposed intermediates as a result of instability arising from coordinative unsaturation. In this investigation, we explore the mechanism of enantioconvergent Kumada reactions of racemic α-bromoketones catalyzed by a nickel complex that bears a bidentate chiral bis(oxazoline) ligand. Utilizing an array of mechanistic tools (including isolation and reactivity studies of three of the four proposed nickel-containing intermediates, as well as interrogation via EPR spectroscopy, UV-vis spectroscopy, radical probes, and DFT calculations), we provide support for a pathway in which carbon–carbon bond formation proceeds via a radical-chain process wherein a nickel(I) complex serves as the chain-carrying radical and an organonickel(II) complex is the predominant resting state of the catalyst. Computations indicate that the coupling of this organonickel(II) complex with an organic radical is the stereochemistry-determining step of the reaction.
Graphical Abstract
INTRODUCTION
Nickel-catalyzed couplings of alkyl electrophiles with organometallic nucleophiles are emerging as a powerful tool for achieving an extensive array of substitution reactions (eq 1).1 Rather than a single universal pathway, it is instead to be expected that the mechanisms of these processes will depend on the identity of the electrophile, the nucleophile, the ligand, and other reaction parameters. Indeed, a wide variety of pathways have been suggested to date, including those illustrated in Figure 1.2,3,4
| (1) |
Figure 1.
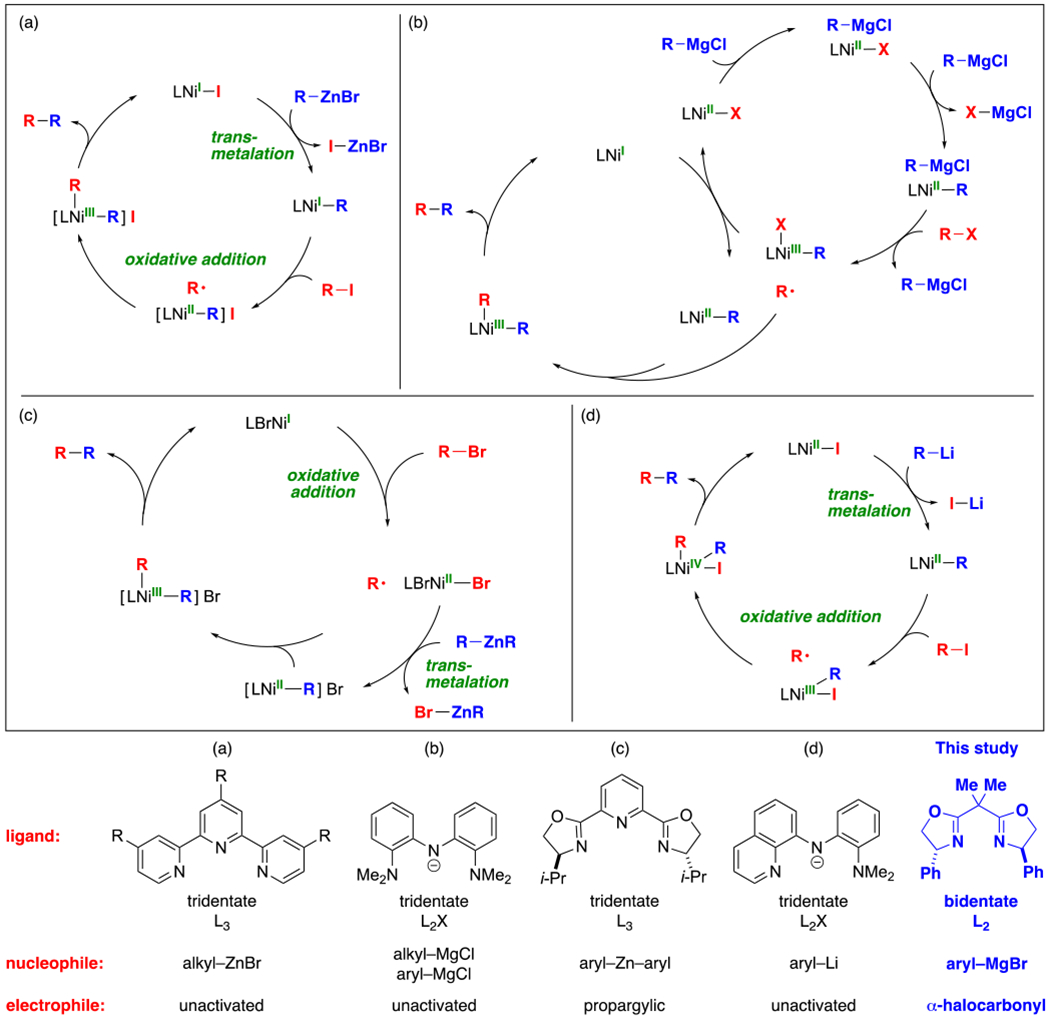
An overview of previous experiment-based mechanistic studies of nickel-catalyzed couplings of alkyl electrophiles with organometallic nucleophiles. For the sake of simplicity, all elementary steps are depicted as being irreversible.
Experiment-based mechanistic investigations of nickel-catalyzed couplings of alkyl electrophiles have focused on catalysts that bear tridentate ligands (Figure 1), despite the fact that bidentate ligands are also useful for many such processes;1 this may be due to the instability of key reactive intermediates in the case of bidentate ligands, due to increased coordinative unsaturation. Furthermore, only one detailed experiment-based mechanistic study has been reported of nickel-catalyzed enantioconvergent reactions (Figure 1c),2c which represent an important subset of nickel-catalyzed couplings of alkyl electrophiles. That investigation examined nickel/pybox-catalyzed Negishi reactions of propargylic halides with organozinc reagents, and it provided support for the radical-chain pathway illustrated in Figure 1c. We have been interested in gaining insight into the generality of this mechanism with regard to other nickel-catalyzed enantioconvergent couplings of alkyl electrophiles.
We therefore decided to investigate enantioconvergent Kumada couplings of α-haloketones effected by a chiral nickel/bis(oxazoline) catalyst (eq 2).5 This process diverges from our previous study (Figure 1c) in several significant ways, including the use of a different family of electrophiles (α-haloketones instead of propargylic halides), a different family of nucleophiles (organomagnesium instead of organozinc reagents), and a different class of ligands (bidentate bis(oxazoline) instead of tridentate pybox).
 |
(2) |
RESULTS AND DISCUSSION
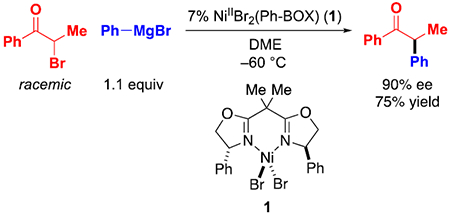
Reaction Conditions.
For this mechanistic study, we have modified the reaction conditions that we described in our original report (eq 2 versus eq 3).5 The most significant change is the use of discrete NiIIBr2(Ph-BOX) (1), rather than a mixture of 7% NiCl2•glyme/9% Ph-BOX, which eliminates any complexity that might arise from the presence of chloride and of excess ligand. An X-ray crystallographic study of NiIIBr2(Ph-BOX) (1) reveals a tetrahedral nickel complex (Figure 2).
 |
(3) |
Figure 2.
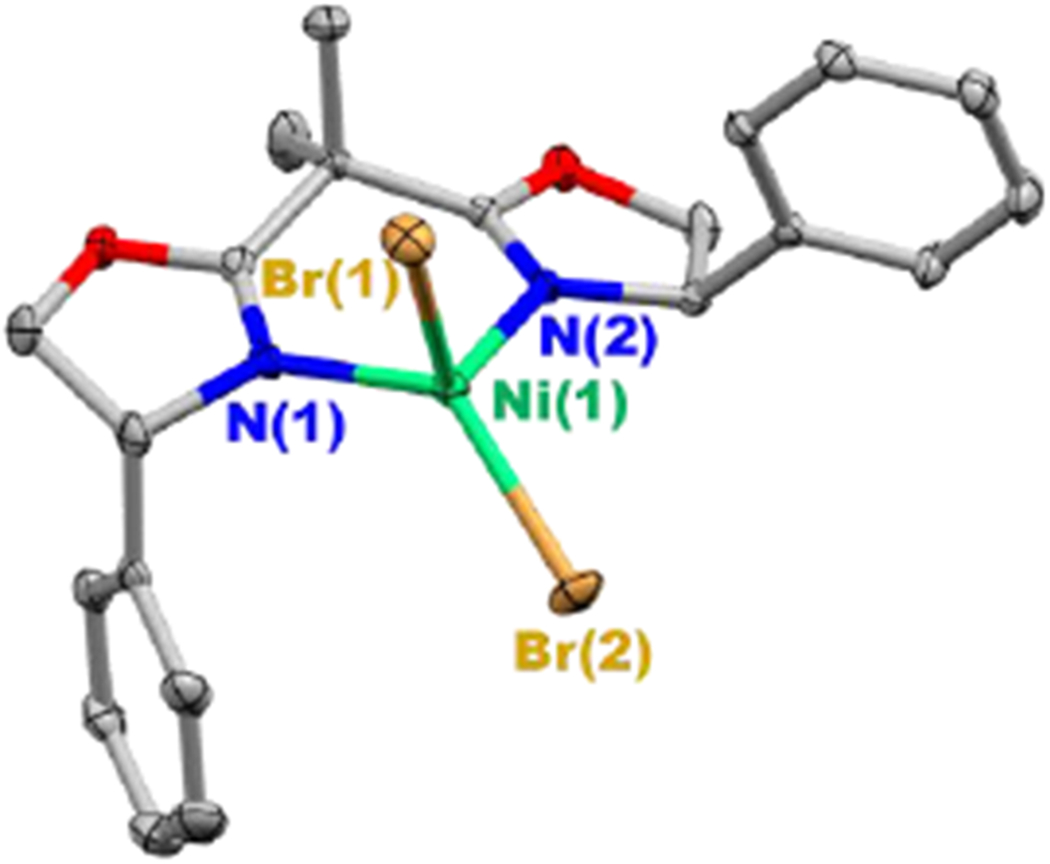
X-ray crystal structure of NiIIBr2(Ph-BOX) (1) (thermal ellipsoids at 30% probability; for clarity, the hydrogen atoms have been omitted).
As illustrated in Figure 3, we have monitored the course of this nickel/bis(oxazoline)-catalyzed Kumada coupling, and we have established that the ee of the product remains ~90% throughout the reaction. Furthermore, the electrophile is essentially racemic throughout the coupling process.
Figure 3.
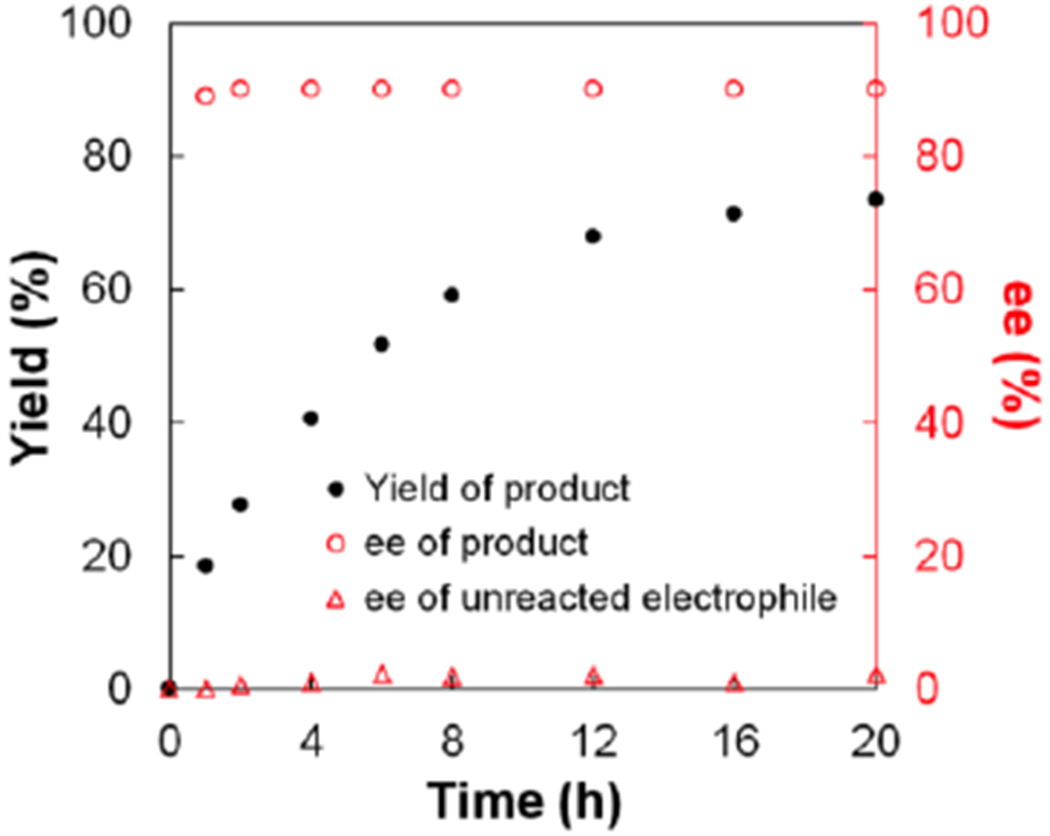
The yield and ee of the coupling product, as well as the ee of the unreacted electrophile, as a function of time (eq 3).
Evidence for the Formation of an Organic Radical from the Electrophile.
When the α-bromoketone illustrated in eq 4 is subjected to the standard coupling conditions, the trans:cis ratio of the cyclized product is 1.3:1 (both diastereomers are racemic). This value is essentially identical to that obtained in a n-Bu3SnH-mediated reductive cyclization of the same α-bromoketone (eq 5; 1.4:1), which is consistent with organic free-radical R serving as a common intermediate in the two processes.6
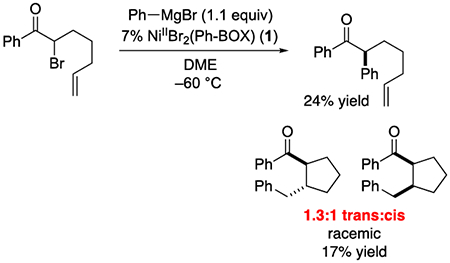 |
(4) |
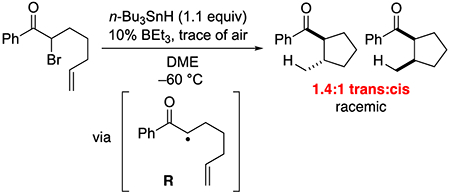 |
(5) |
Identification of the Predominant Resting State of the Catalyst During the Coupling Process: NiIIPhBr(Ph-BOX) (2).
When the standard enantioconvergent Kumada coupling (eq 3) is analyzed via electron paramagnetic resonance (EPR) spectroscopy, no significant quantity of an EPR-active species is detected throughout the course of the reaction, suggesting that NiI (d9) and NiIII (d7) complexes do not accumulate during the coupling process. While the coupling is in progress, the reaction mixture remains light orange-yellow, whereas NiIIBr2(Ph-BOX) (1) is magenta. The d→d absorption for nickel complex 1 (DME at −60 °C) appears at 517 nm, whereas low-temperature UV-vis spectroscopy of a reaction in progress reveals an absorption feature at 474 nm, but not at 517 nm (Figure 4). This rules out NiIIBr2(Ph-BOX) (1) as the resting state of the catalyst during coupling.
Figure 4.
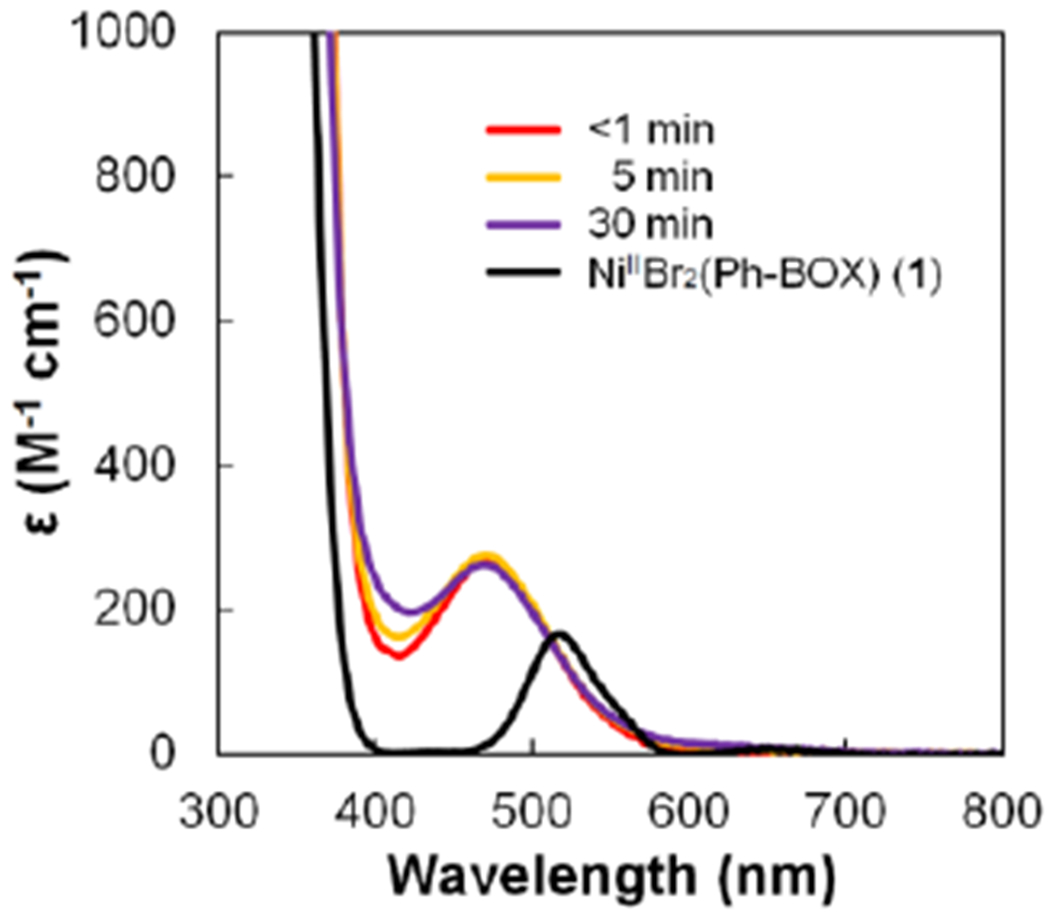
UV-vis spectra of a coupling in progress (eq 3) and of NiIIBr2(Ph-BOX) (1). The absorption from PhMgBr has been subtracted.
On the basis of the UV-vis and the EPR spectroscopic studies, we hypothesized that NiIIPhBr(Ph-BOX) (2) might be the predominant resting state of the catalyst during the Kumada reaction. We therefore sought to independently synthesize nickel complex 2 via the oxidative addition of bromobenzene to Ni(cod)2 in the presence of Ph-BOX. Unfortunately, when attempting to crystallize the desired adduct (2) from the reaction mixture under a variety of conditions, we consistently obtained NiIIBr2(Ph-BOX) (1) instead, along with biphenyl in the supernatant (e.g., eq 6).7
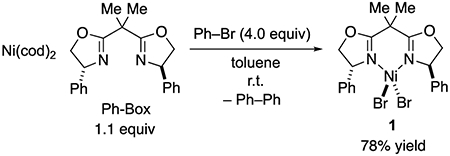 |
(6) |
We conjectured that, under these conditions, NiIIBr2(Ph-BOX) (1) might be generated by the oxidative addition of bromobenzene to nickel(0) to transiently furnish desired NiIIPhBr(Ph-BOX) (2), followed by decomposition to NiIIBr2(Ph-BOX) (1). Consistent with this suggestion, when we monitor a reaction via UV-vis spectroscopy, we observe the formation and then the disappearance of an intermediate, which then affords NiIIBr2(Ph-BOX) (1) (Figure 5). We speculate that the proposed intermediate, NiIIPhBr(Ph-BOX) (2), may correspond to the species that we observe during a coupling reaction (i.e., the absorption maximum at 474 nm in Figure 4; cf. Figure 5b).
Figure 5.
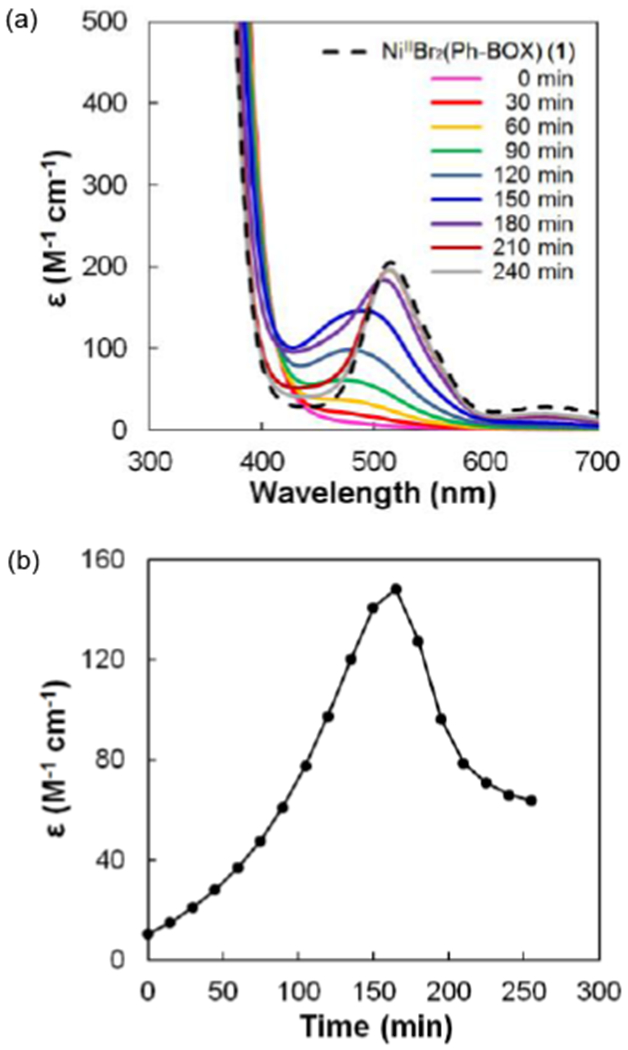
(a) UV-vis spectra of NiIIBr2(Ph-BOX) (1) and of the reaction of Ni(cod)2, Ph-BOX, and Ph-Br (eq 6, except in DME). (b) Absorbance at 474 nm as a function of time.
Because NiIIPhBr(Ph-BOX) (2) appears to be unstable in solution at room temperature, we sought to isolate it via immediate precipitation. Thus, by carrying out the reaction of Ni(cod)2, Ph-BOX, and bromobenzene in n-pentane, we have been able to isolate an orange precipitate, elemental analysis of which is close to that expected for NiIIPhBr(Ph-BOX) (2) (eq 7, R = H).8 Our attempts to obtain X-ray quality crystals have been unsuccessful, leading instead to the formation of NiIIBr2(Ph-BOX) (1) and biphenyl.
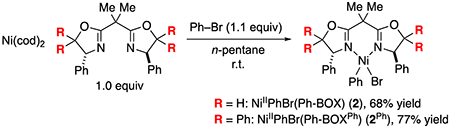 |
(7) |
The decomposition of a related nickel complex, NiIIPhBr(bpy), to generate biphenyl has been observed, and it has been suggested that this occurs via a bimolecular pathway.9 Building on the hypothesis that the decomposition of desired NiIIPhBr(Ph-BOX) (2) might also proceed through a bimolecular reaction, we targeted the synthesis of an analogue of nickel complex 2 that bears a more sterically demanding bis(oxazoline) ligand. Specifically, we have determined that the same conditions that afford complex 2 also provide complex 2Ph in similar yield (eq 7). Complexes 2 and 2Ph, both of which are coordinatively unsaturated, are unstable in solution at room temperature; however, unlike complex 2, complex 2Ph is sufficiently stable in THF/n-pentane at −40 °C that it can be crystallized and structurally characterized, revealing a square-planar nickel complex (Figure 6).
Figure 6.
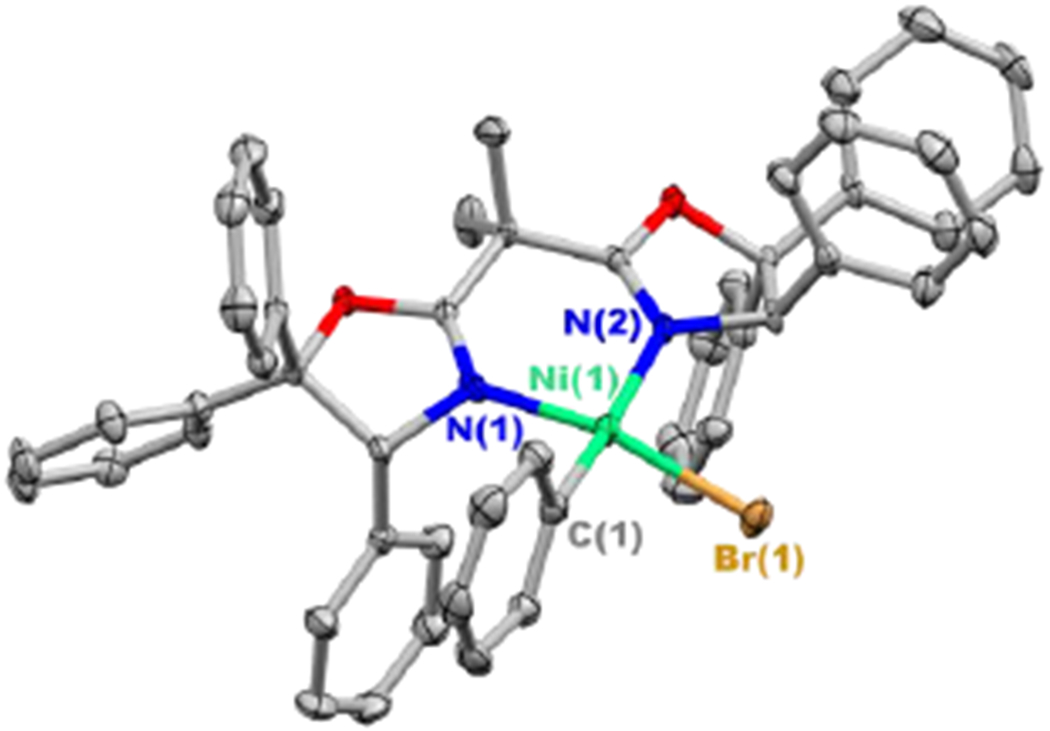
X-ray crystal structure of NiIIPhBr(Ph-BOXPh)•THF (2Ph•THF) (thermal ellipsoids at 30% probability; for clarity, the solvent and the hydrogen atoms have been omitted).
The low-temperature UV-vis spectrum of NiIIPhBr(Ph-BOXPh) (2Ph) is similar to that of the parent complex, NiIIPhBr(Ph-BOX) (2), as well as to that of a coupling reaction in progress (Figure 7a). We hypothesize that the absorption feature at ~470 nm corresponds to the Laporte forbidden d→d transition, consistent with its low absorptivity (ε ~ 250 M−1 cm−1) and with time-dependent density functional theory (DFT) calculations (Figure 7b). Thus, the independent preparation of NiIIPhBr(Ph-BOX) (2) and NiIIPhBr(Ph-BOXPh) (2Ph) support the hypothesis that nickel complex 2 is the predominant resting state of the catalyst during the stereoconvergent Kumada coupling of an α-haloketone catalyzed by a nickel/ bis(oxazoline) catalyst. In our previous study of a Negishi reaction of a propargylic halide catalyzed by a nickel/pybox catalyst (Figure 1c),2c an arylnickel(II) complex was also the principal resting state, and we continued our investigation of the Kumada reaction with this similarity in mind.
Figure 7.
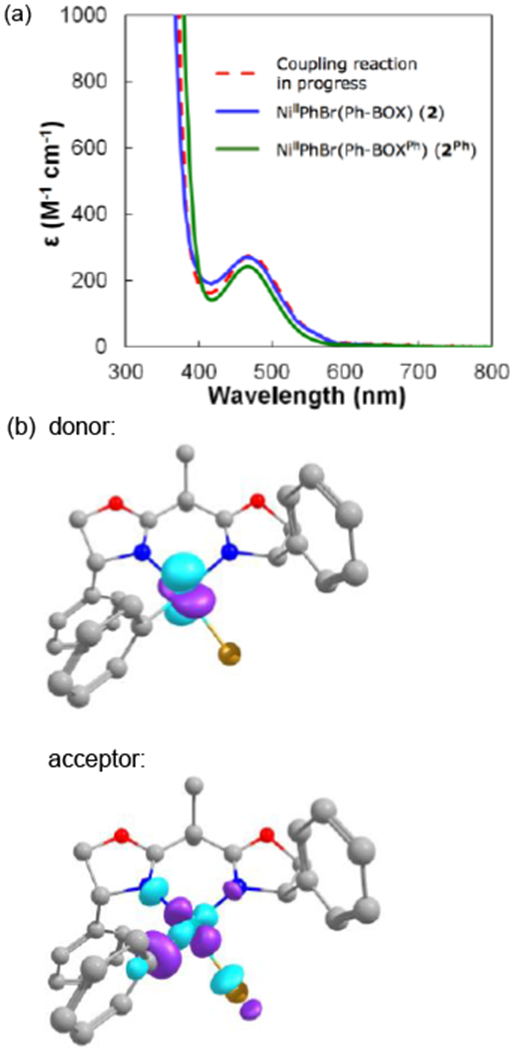
(a) UV-vis spectra of a coupling reaction in progress (eq 3), of NiIIPhBr(Ph-BOX) (2), and of NiIIPhBr(Ph-BOXPh) (2Ph). (b) Natural transition orbital (NTO) representations10 of the computed (B3LYP/6–31G*) lowest energy transition for NiIIPhBr(Ph-BOX) (2).
Formation of NiIBr(Ph-BOX) (3) from NiIIPhBr(Ph-BOX) (2).
Dissolving NiIIPhBr(Ph-BOX) (2) in THF at −40 °C, followed by layering with n-pentane, led to the isolation of NiIBr(Ph-BOX) (3) as yellow crystals, accompanied by biphenyl in the supernatant (eq 8).11 Coordinatively unsaturated nickel complex 3 is unstable in solution at room temperature, undergoing disproportionation to afford NiIIBr2(Ph-BOX) (1) and metallic nickel(0).
 |
(8) |
An X-ray crystallographic study of NiIBr(Ph-BOX) (3) reveals an unsymmetrical trigonal planar geometry at nickel, with N–Ni–Br angles of 148.27(3) and 120.71(3) Å (Figure 8; an unsymmetrical geometry has previously been observed for three-coordinate nickel(I) complexes12). Our DFT calculations indicate that there is a low barrier (~1.4 kcal mol−1) for bending to a symmetrical geometry.
Figure 8.
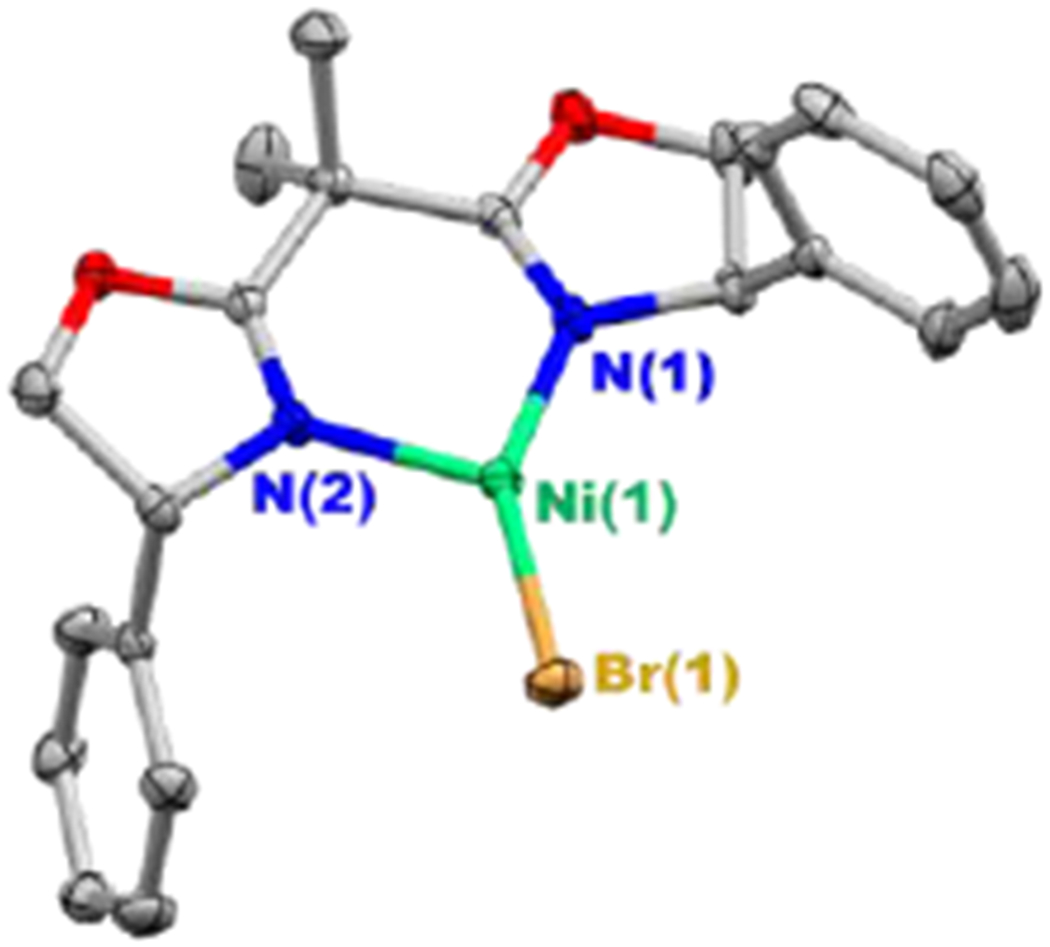
X-ray crystal structure of NiIBr(Ph-BOX) (3) (thermal ellipsoids at 30% probability; for clarity, the hydrogen atoms have been omitted).
The EPR spectrum of NiIBr(Ph-BOX) (3) displays an axial signal with g⊥ = 2.329 and g// = 2.085 (Figure 9a). The observed g anisotropy is consistent with a nickel-centered radical. This assignment is further supported by DFT calculations, which indicate that the majority of the spin density is on nickel (Figure 9b).
Figure 9.
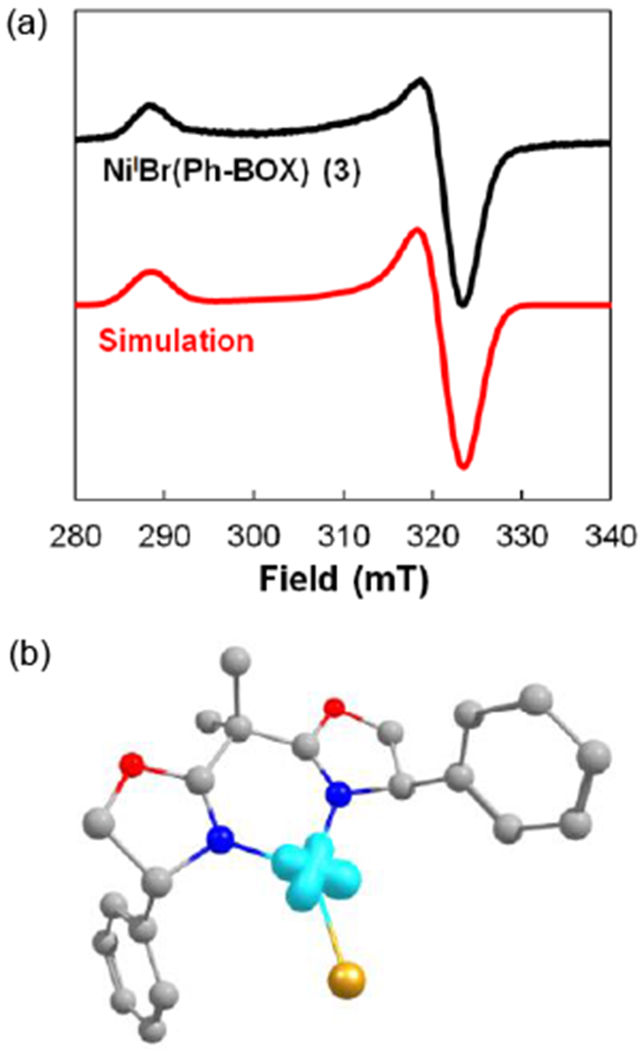
(a) X-band EPR spectrum of NiIBr(Ph-BOX) (3) (collected in toluene glass at 77 K with ν = 9.4 GHz at 2 mW power and a modulation amplitude of 2 G) and simulated spectrum (parameters used for simulation: g1 = 2.084, g2 = 2.087, g3 = 2.329, line width = 5.28); hyperfine couplings from the nitrogen atoms were not resolved. (b) DFT-computed (B3LYP/6-31G*) spin density plot for NiIBr(Ph-BOX) (3).
In DME at −60 °C, NiIBr(Ph-BOX) (3) is yellow, with an intense absorption band observed at ~410 nm in the UV-vis spectrum (Figure 10). The spectrum is distinct from that of a coupling reaction in progress, NiIIBr2(Ph-BOX) (1), and NiIIPhBr(Ph-BOX) (2).
Figure 10.
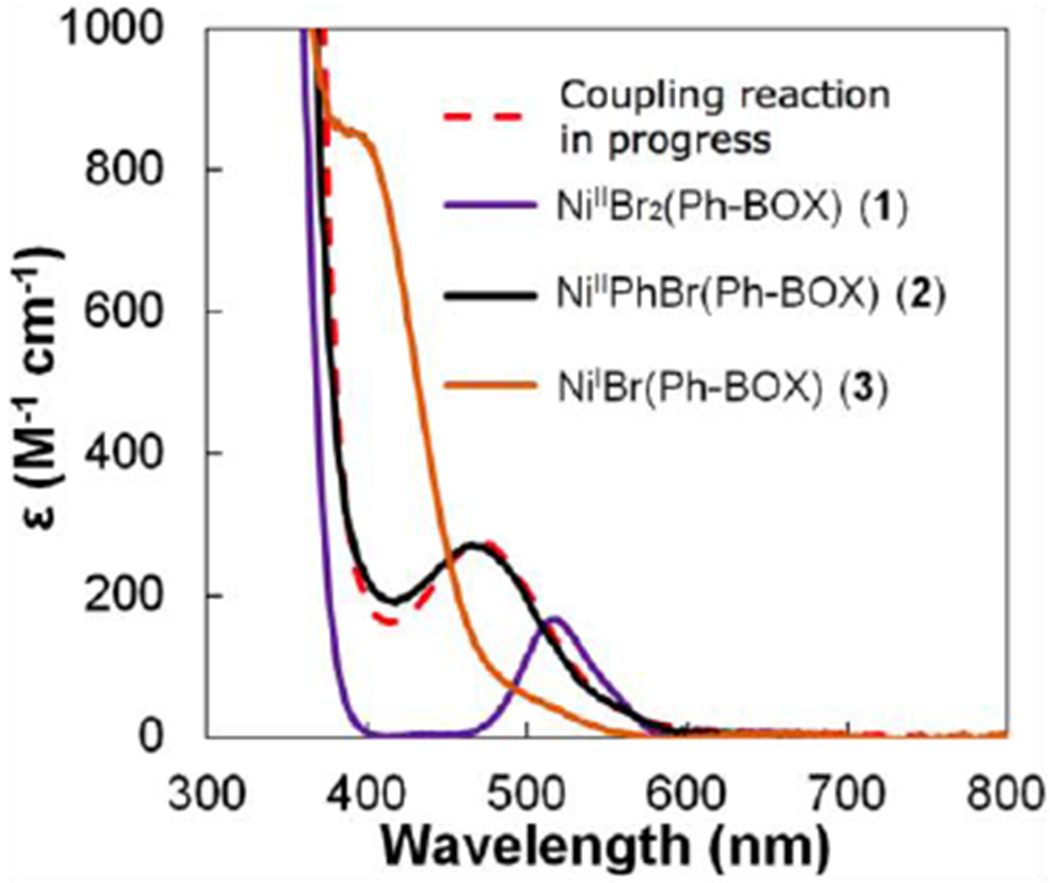
UV-vis spectra of a coupling reaction in progress (eq 3), of NiIIBr2(Ph-BOX) (1), of NiIIPhBr(Ph-BOX) (2), and of NiIBr(Ph-BOX) (3).
Reactivity Studies of Isolated Nickel Complexes: NiIIBr2(Ph-BOX) (1), NiIIPhBr(Ph-BOX) (2), and NiIBr(Ph-BOX) (3).
With several potential intermediates in hand, we turned to investigating the reactivity of the three isolated nickel complexes, vis-a-vis a possible catalytic cycle (Figure 11).2c In step 1, a nickel radical (NiIBr(Ph-BOX), 3) abstracts a halogen atom from the electrophile to generate an organic radical (R•) and NiIIBr2(Ph-BOX) (1). In step 2, the organic radical (R•) reacts with the resting state of nickel (NiIIPhBr(Ph-BOX), 2) to furnish a diorganonickel(III) intermediate (NiIIIRPhBr(Ph-BOX), 4), which reductively eliminates in step 3 to afford the coupling product and to regenerate the chain-carrying radical (NiIBr(Ph-BOX), 3). Meanwhile, in step a, the NiIIBr2(Ph-BOX) (1) that is produced in step 1 reacts with the nucleophile (PhMgBr) to re-form the resting-state nickel complex (NiIIPhBr(Ph-BOX), 2).
Figure 11.
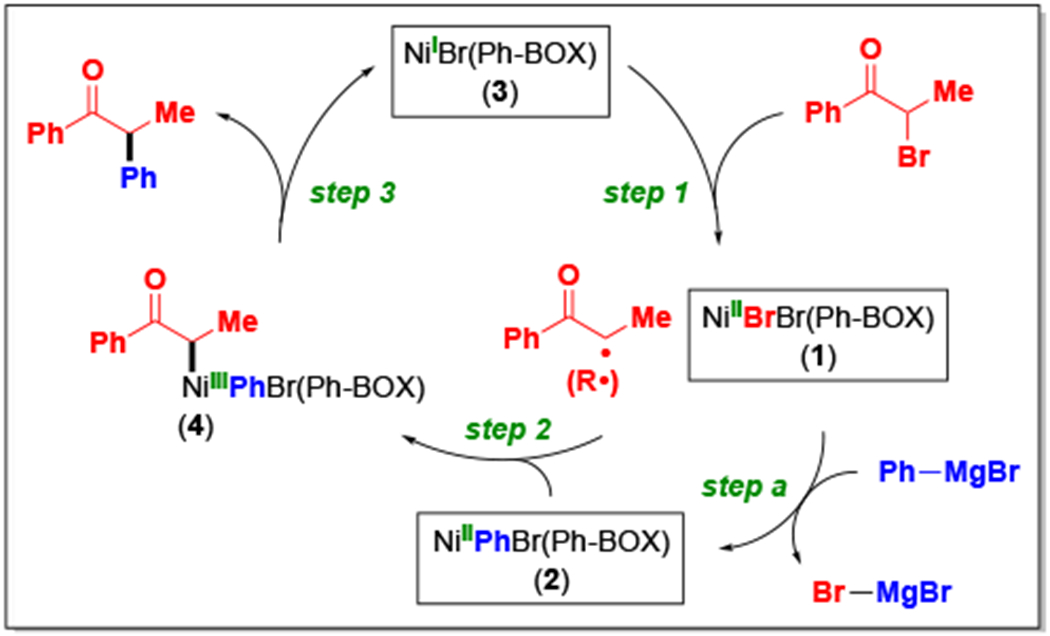
Possible mechanism for the nickel/bis(oxazoline)-catalyzed enantioconvergent Kumada coupling of an α-bromoketone with a Grignard reagent. Intermediates that have been isolated are boxed. For the sake of simplicity, all elementary steps are depicted as being irreversible.
Transmetalation between NiIIBr2(Ph-BOX) (1) and PhMgBr to generate the resting state of nickel (NiIIPhBr(Ph-BOX), 2) (step a in Figure 11).
Treatment of NiIIBr2(Ph-BOX) (1) with excess PhMgBr in DME at −60 °C results in the formation of NiIIPhBr(Ph-BOX) (2) within 30 seconds, as monitored via low-temperature UV-vis spectroscopy (Figure 12). In contrast, nickel complex 1 does not react with the other coupling partner, the α-haloketone, under these conditions.
Figure 12.
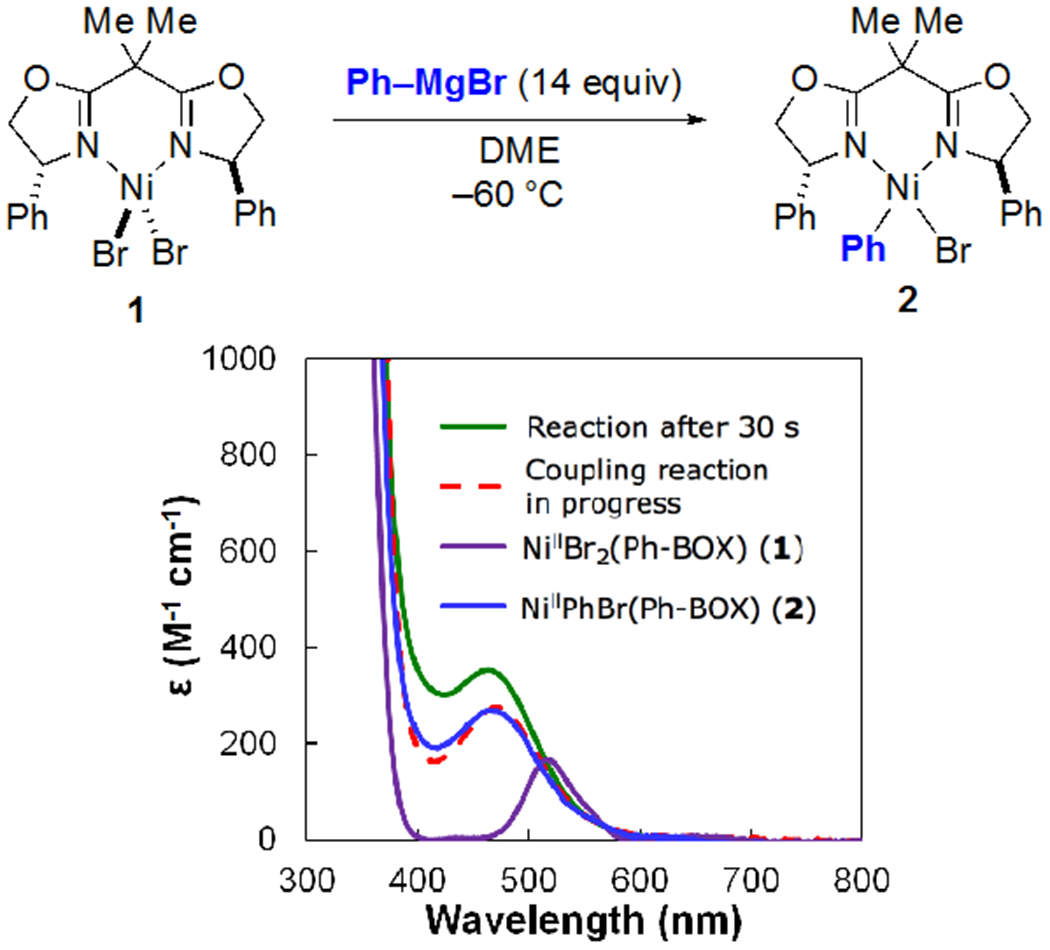
UV-vis spectra of the reaction between NiIIBr2(Ph-BOX) (1) and PhMgBr after 30 seconds, of a separate catalyzed coupling reaction in progress (eq 3), of NiIIBr2(Ph-BOX) (1), and of NiIIPhBr(Ph-BOX) (2).
Abstraction by NiIBr(Ph-BOX) (3) of a halogen atom from the electrophile (step 1 in Figure 11).
Treatment of NiIBr(Ph-BOX) (3) with the α-bromoketone in DME at −60 °C results in an immediate color change from yellow to magenta and in the formation of NiIIBr2(Ph-BOX) (1) (eq 9), as well as a mixture of stereoisomeric products derived from homocoupling of the organic radical generated by bromine abstraction from the electrophile. Analysis of the abstraction reaction by low-temperature UV-vis spectroscopy reveals that the absorption feature at ~410 nm, corresponding to NiIBr(Ph-BOX) (3), disappears in less than 15 seconds (estimated second-order rate constant: >104 M−1 s−1). Nickel complex 3 reacts at least 30 times more rapidly with the α-bromoketone than with PhMgBr under these conditions, which is consistent with the proposed catalytic cycle (Figure 11) and is not supportive of a transmetalation-first pathway (e.g., Figure 1a).
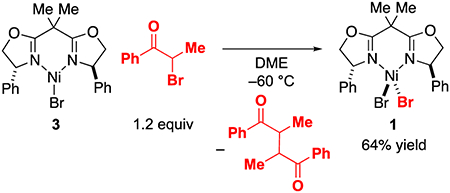 |
(9) |
Coupling of the resting state of nickel (NiIIPhBr(Ph-BOX), 2) with the electrophile.
In a stoichiometric reaction, arylnickel complex NiIIPhBr(Ph-BOX) (2) couples with the α-bromoketone in DME at −60 °C to provide the product in 90% ee and 78% yield (eq 10; comparable to the catalyzed conditions: 90% ee, 75% yield (eq 3)), along with NiIIBr2(Ph-BOX) (1) (Figure 13). When the progress of this stoichiometric reaction is monitored via low-temperature UV-vis and EPR spectroscopy, no significant intermediates are observed, consistent with our observations for the catalyzed coupling (vide supra).
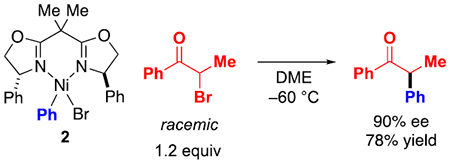 |
(10) |
Figure 13.
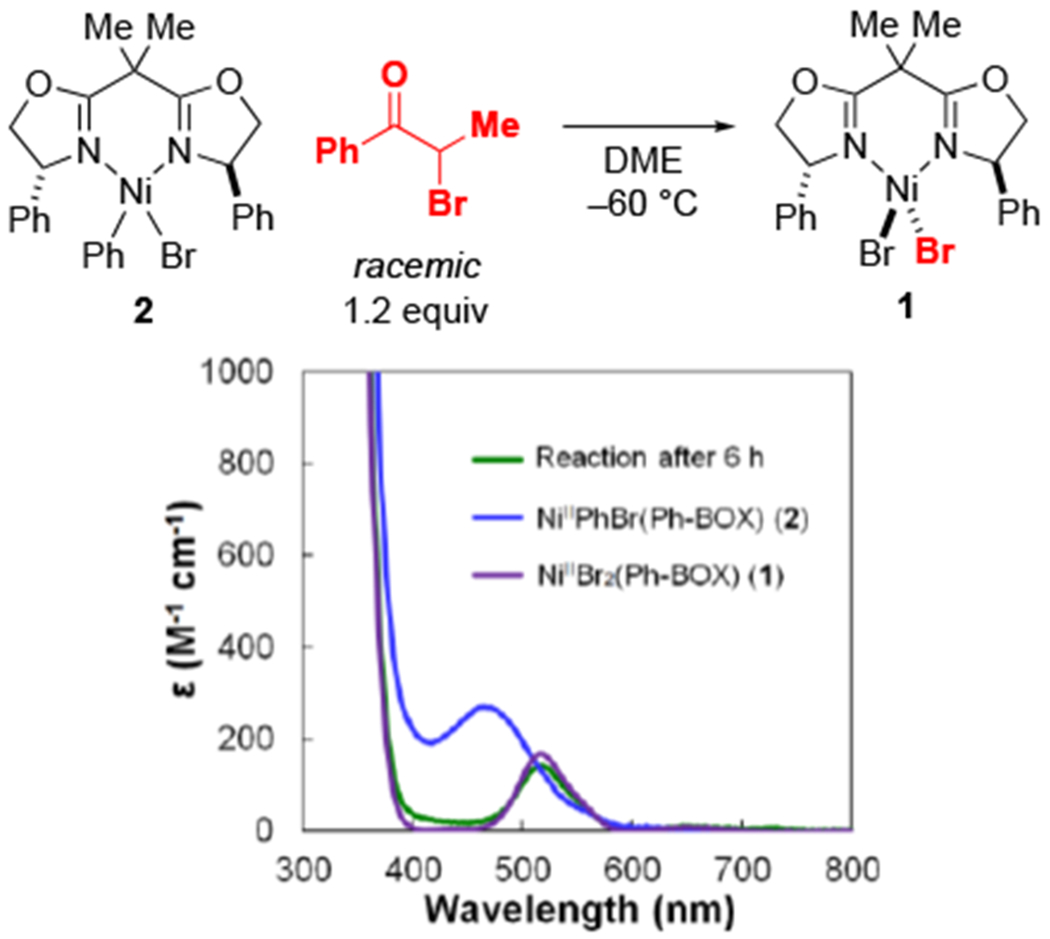
UV-vis spectra of the reaction between NiIIPhBr(Ph-BOX) (2) and an α-bromoketone after 6 hours, of NiIIPhBr(Ph-BOX) (2), and of NiIIBr2(Ph-BOX) (1).
Evidence for Out-of-Cage Nickel–Carbon Bond Formation.
According to the mechanism outlined in Figure 11, coupling of the organic radical with NiIIPhBr(Ph-BOX) (2) occurs through an out-of-cage pathway. 5-Hexenyl radicals that bear a carbonyl group α to the radical have been reported to cyclize with a rate constant of ~105 s−1,13 whereas the rate constant for diffusion is typically >108 s−1.14 Therefore, our observation of cyclized product for the coupling illustrated in eq 4 is consistent with the conclusion that the organic radical has a sufficient lifetime to escape the solvent cage. Furthermore, the ratio of uncyclized (U) to cyclized (C) products increases as the concentration of the nickel catalyst increases (Figure 14), as expected for out-of-cage, but not for in-cage, coupling.
Figure 14.
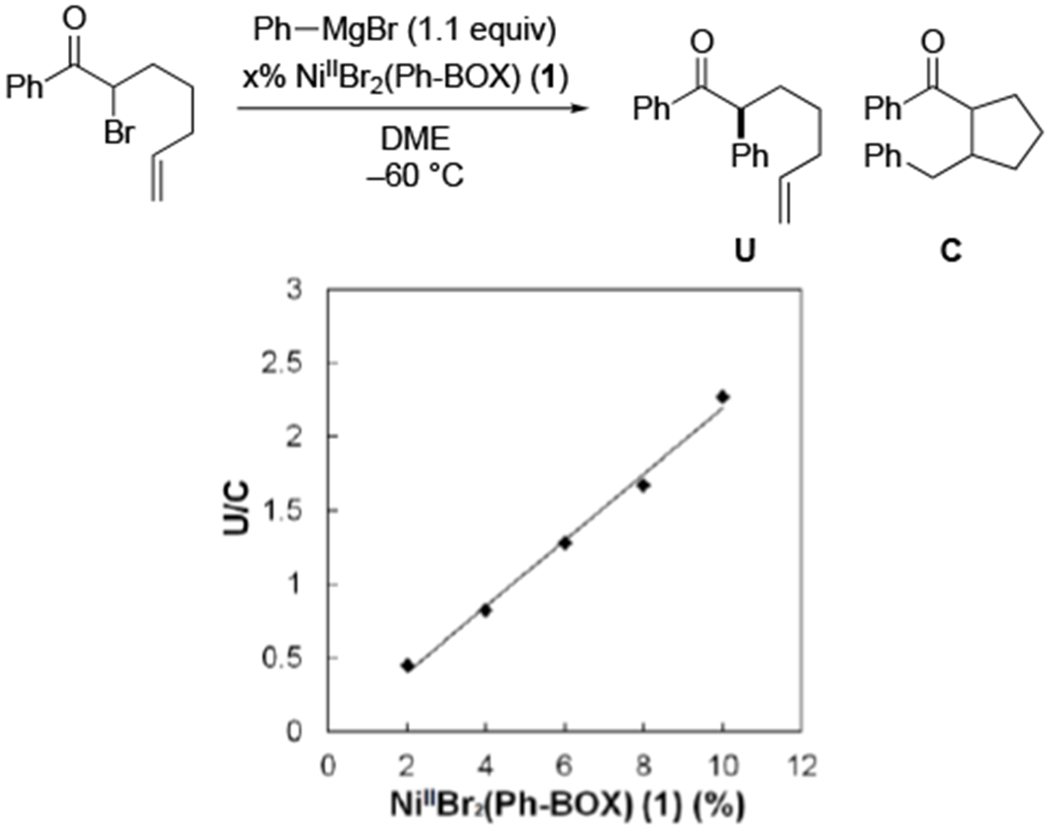
Ratio of uncyclized (U) to cyclized (C) products, as a function of the loading of NiIIBr2(Ph-BOX) (1).
Inhibition by TEMPO and Activation by NiIBr(Ph-BOX) (3).
The pathway outlined in Figure 11 is a radical-chain process wherein NiIBr(Ph-BOX) (3) serves as the chain-carrying radical. Consistent with this mechanism, the addition of TEMPO, a radical trap, inhibits coupling (a TEMPO–electrophile adduct can be identified by ESI–MS15), and the addition of nickel complex 3 accelerates coupling (eq 11).
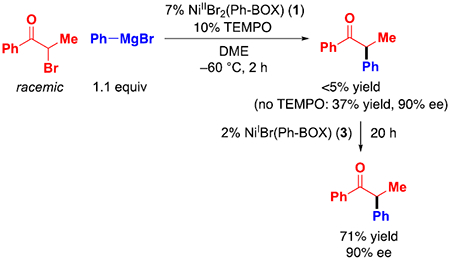 |
(11) |
Mechanistic Summary (Figure 11).
NiIIBr2(Ph-BOX) (1) reacts rapidly (<30 seconds) with PhMgBr in DME at −60 °C to form NiIIPhBr(Ph-BOX) (2) (Figure 12; step a in Figure 11), which is the predominant resting state of nickel during the coupling process (Figure 7a). A small amount of NiIIPhBr(Ph-BOX) (2) decomposes to generate NiIBr(Ph-BOX) (3) (eq 8), the chain-carrying radical. Nickel complex 3 abstracts a bromine from the α-bromoketone to provide an organic radical (R•) and NiIIBr2(Ph-BOX) (1) (eq 9; step 1 in Figure 11). Nickel complex 1 is rapidly converted by PhMgBr to NiIIPhBr(Ph-BOX) (2) (Figure 12; step a in Figure 11), while the organic radical reacts with NiIIPhBr(Ph-BOX) (2) to generate NiIIIRPhBr(Ph-BOX) (4) (step 2 in Figure 11); because NiIIPhBr(Ph-BOX) (2) is the resting state of nickel during catalysis, and therefore at a much higher concentration than the organic radical, radical–radical coupling is avoided. Reductive elimination of NiIIIRPhBr(Ph-BOX) (4) furnishes the coupling product and regenerates NiIBr(Ph-BOX) (3), the chain-carrying radical (step 3 in Figure 11).16
Insight into the Origin of Enantioselectivity: DFT Calculations.
Either step 2 or step 3 (Figure 11) could be stereochemistry-determining in this nickel/bis(oxazoline)-catalyzed enantioconvergent Kumada coupling. Using DFT calculations ((U)M06/6-311+G**,SMD(THF)//(U)B3LYP/6-31G*), we have calculated the relative energies of the two diastereomers of NiIIIRPhBr(Ph-BOX) (4) and the transition states for nickel–carbon bond homolysis (reverse of step 2) and for reductive elimination (step 3) (Figure 15; for more details, see the Supporting Information). The computed energy diagram indicates that nickel–carbon bond formation (step 2) is stereochemistry-determining, with ΔΔG‡ = 1.1 kcal/mol favoring formation of the stereoisomer illustrated in eq 3, corresponding to 86% ee at −60 °C. In contrast, the two previous computational studies of enantioconvergent nickel-catalyzed coupling reactions of alkyl electrophiles suggested that reductive elimination is the stereochemistry-determining step when a chiral pybox and a diamine ligand are used.3c,e
Figure 15.
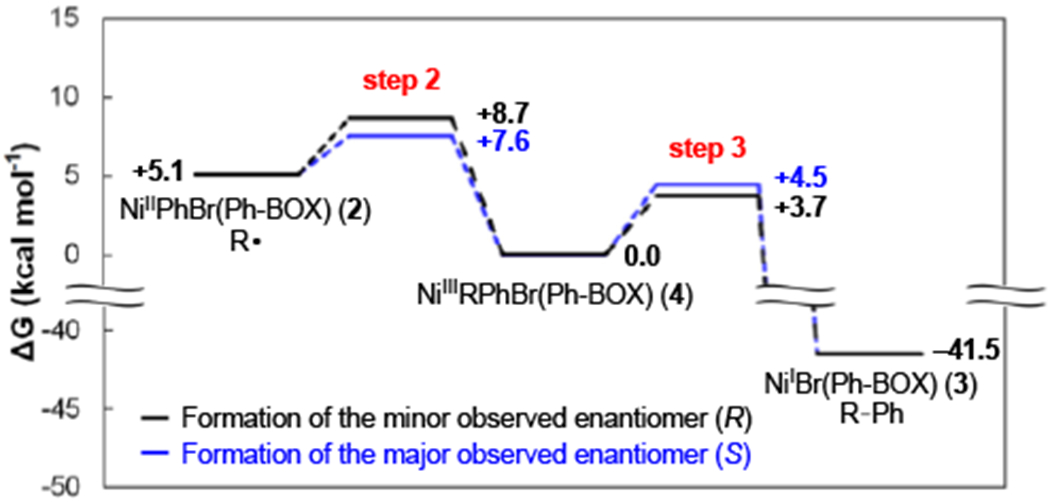
Computed relative free energies for step 2 and for step 3 of the proposed catalytic cycle (Figure 11), calculated at (U)M06/6-311+G**,SMD(THF)//(U)B3LYP/6x-31G*) level of theory.
CONCLUSIONS
There have not been in-depth experiment-focused mechanistic studies of nickel-catalyzed coupling reactions of nucleophiles with alkyl electrophiles wherein bidentate, rather than tridentate, ligands are employed, perhaps due to the difficulty of isolating nickel intermediates that are coordinatively unsaturated. Exploiting a range of tools, we have investigated the mechanism of nickel/bis(oxazoline)-catalyzed enantioconvergent Kumada reactions of racemic α-haloketones, and we have provided further support for a radical-chain pathway, for a coupling that is significantly different from our previous study of nickel/pybox-catalyzed Negishi reactions of propargylic halides. In particular, we have isolated three of the four proposed nickel-containing intermediates (two of which are unstable in solution at room temperature), including an organonickel complex, as well as explored their reactivity. We have identified the likely resting state of nickel during the catalytic cycle, and we have described evidence for a radical pathway that involves out-of-cage nickel–carbon bond construction. Our DFT calculations point to the formation of a diorganonickel(III) complex as the stereochemistry-determining step of the reaction, with the final step in the catalytic cycle (reductive elimination) having a low activation barrier (<5 kcal/mol), which precludes isolation of the diorganonickel(III) complex. Further mechanistic studies of nickel-catalyzed stereoconvergent coupling reactions of alkyl electrophiles are underway.
Supplementary Material
ACKNOWLEDGMENTS
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences; grant R01–GM062871). We thank Lawrence M. Henling, Dr. Paul H. Oyala, Dr. Michael K. Takase, Dr. Scott C. Virgil, Dr. Zhaobin Wang, and the laboratories of Prof. Jonas C. Peters and Prof. Theodor Agapie for assistance and for helpful discussions, and we acknowledge the Dow Next Generation Educator Fund for support of instrumentation at Caltech.
Footnotes
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08185.
Procedures and characterization data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Choi J; Fu GC Transition Metal-Catalyzed Alkyl–Alkyl Bond Formation: Another Dimension in Cross-Coupling Chemistry. Science 2017, 356, eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu GC Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci 2017, 3, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kaga A; Chiba S Engaging Radicals in Transition Metal-Catalyzed Cross-Coupling with Alkyl Electrophiles: Recent Advances. ACS Catal. 2017, 7, 4697–4706. [Google Scholar]; (d) Iwasaki T; Kambe N Ni-Catalyzed C–C Couplings using Alkyl Electrophiles. Top. Curr. Chem 2016, 374, 66. [DOI] [PubMed] [Google Scholar]
- (2).Figure 1 summarizes experiment-focused mechanistic studies wherein at least one proposed organonickel intermediate has been characterized:; (a) Jones GD; Martin JL; McFarland C; Allen OR; Hall RE; Haley AD; Brandon RJ; Konovalova T; Desrochers PJ; Pulay P; Vicic DA Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl-Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc 2006, 128, 13175−13183. [DOI] [PubMed] [Google Scholar]; (b) Breitenfeld J; Ruiz J; Wodrich MD; Hu X Bimetallic Oxidative Addition Involving Radical Intermediates in Nickel-Catalyzed Alkyl–Alkyl Kumada Coupling Reactions. J. Am. Chem. Soc 2013, 135, 12004–12012; Breitenfeld J; Wodrich MD; Hu X Bimetallic Oxidative Addition in Nickel-Catalyzed Alkyl–Aryl Kumada Coupling Reactions. Organometallics 2014, 33, 5708–5715. [DOI] [PubMed] [Google Scholar]; (c) Schley ND; Fu GC Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J. Am. Chem. Soc 2014, 136, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Patel UN; Jain S; Pandey DK; Gonnade RG; Vanka K; Punji B Mechanistic Aspects of Pincer Nickel(II)-Catalyzed C–H Bond Alkylation of Azoles with Alkyl Halides. Organometallics 2018, 37, 1017–1025. [Google Scholar]
- (3).For examples of computation-based mechanistic studies, see:; (a) Lin X; Phillips DL Density Functional Theory Studies of Negishi Alkyl–Alkyl Cross-Coupling Reactions Catalyzed by a Methylterpyridyl-Ni(I) Complex. J. Org. Chem 2008, 73, 3680–3688. [DOI] [PubMed] [Google Scholar]; (b) Chass GA; Kantchev EAB; Fang D-C The Fine Balance Between One Cross-Coupling and Two β-Hydride Elimination Pathways: A DFT Mechanistic Study of Ni(π-allyl)2-Catalyzed Cross-Coupling of Alkyl Halides and Alkyl Grignard Reagents. Chem. Commun 2010, 46, 2727–2729. [DOI] [PubMed] [Google Scholar]; (c) Lin X; Sun J; Xi Y; Lin D How Racemic Secondary Alkyl Electrophiles Proceed to Enantioselective Products in Negishi Cross-Coupling Reactions. Organometallics 2011, 30, 3284–3292. [Google Scholar]; (d) Li Z; Jiang Y-Y; Fu Y Theoretical Study on the Mechanism of Ni-Catalyzed Alkyl–Alkyl Suzuki Cross-Coupling. Chem. Eur. J 2012, 18, 4345–4357. [DOI] [PubMed] [Google Scholar]; (e) Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For an insightful mechanistic study of nickel-catalyzed cross-electrophile coupling, see:; Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lou S; Fu GC Nickel/Bis(oxazoline)-Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic α-Bromoketones. J. Am. Chem. Soc 2010, 132, 1264–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For examples of early suggestions of the formation of a radical from the electrophile in nickel-catalyzed couplings of alkyl electrophiles, see:; (a) Zhou J; Fu GC Suzuki Cross-Couplings of Unactivated Secondary Alkyl Bromides and Iodides. J. Am. Chem. Soc 2004, 126, 1340–1341. [DOI] [PubMed] [Google Scholar]; (b) Anderson TJ; Jones GD; Vicic DA Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl-Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc 2004, 126, 8100–8101. [DOI] [PubMed] [Google Scholar]; (c) Powell DA; Maki T; Fu GC Stille Cross-Couplings of Unactivated Secondary Alkyl Halides using Monoorganotin Reagents. J. Am. Chem. Soc 2005, 127, 510–511. [DOI] [PubMed] [Google Scholar]
- (7).Notes: (a) A reaction in DME proceeded similarly. (b) Attempts to carry out the oxidative addition at low temperature were hampered by solubility issues for Ni(cod)2.
- (8).Elemental analysis calculated for C27H27BrN2NiO2: C, 58.95; H, 4.95; N, 5.09. Found: C, 58.46; H, 5.38; N, 5.17.
- (9).Yamamoto T; Wakabayashi S; Osakada K Mechanism of C–C Coupling Reactions of Aromatic Halides, Promoted by Ni(COD)2, in the Presence of 2,2’-Bipyridine and PPh3, to Give Biaryls. J. Organomet. Chem 1992, 428, 223–237. [Google Scholar]
- (10).Martin RL Natural Transition Orbitals. J. Chem. Phys 2003, 118, 4775–4777. [Google Scholar]
- (11).Using EPR spectroscopy, we have determined that NiIBr(Ph-BOX) (3) also forms slowly from NiIIPhBr(Ph-BOX) (2) in DME at −60 °C.
- (12).For example, see:; (a) Kitiachvili KD; Mindiola DJ; Hillhouse GL Preparation of Stable Alkyl Complexes of Ni(I) and Their One-Electron Oxidation to Ni(II) Complex Cations. J. Am. Chem. Soc 2004, 126, 10554–10555. [DOI] [PubMed] [Google Scholar]; (b) Kogut E; Wiencko HL; Zhang L; Cordeau DE; Warren TH A Terminal Ni(III)-Imide with Diverse Reactivity Pathways. J. Am. Chem. Soc 2005, 127, 11248–11249. [DOI] [PubMed] [Google Scholar]
- (13).For example, see:; Musa OM; Choi S-Y; Horner JH; Newcomb MN Absolute Rate Constants for α-Amide Radical Reactions. J. Org. Chem 1998, 63, 786–793, and references cited therein. [DOI] [PubMed] [Google Scholar]
- (14).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006; p. 156. [Google Scholar]
- (15).Calculated for C18H28NO2 [M+H]+ (coupling of the organic radical with TEMPO): 290.2115, found: 290.2119.
- (16).Our DFT calculations indicate that all of the elementary steps in the proposed catalytic cycle (Figure 11) are exergonic and that O-bound nickel enolates are higher in energy than their C-bound isomers (see the Supporting Informations).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.