

Abstract
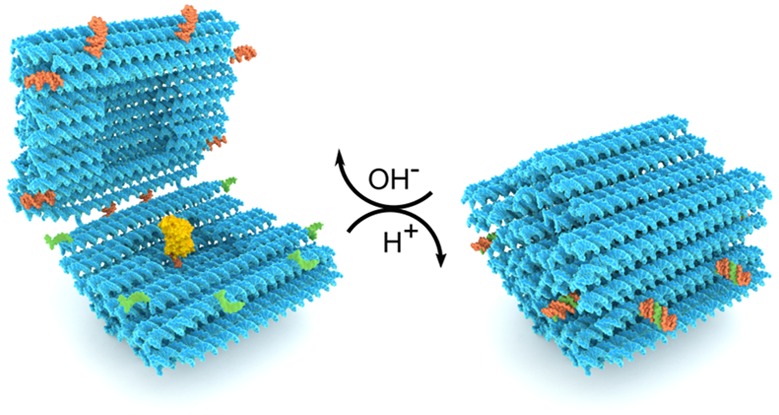
DNA nanotechnology provides a toolbox for creating custom and precise nanostructures with nanometer-level accuracy. These nano-objects are often static by nature and serve as versatile templates for assembling various molecular components in a user-defined way. In addition to the static structures, the intrinsic programmability of DNA nanostructures allows the design of dynamic devices that can perform predefined tasks when triggered with external stimuli, such as drug delivery vehicles whose cargo display or release can be triggered with a specified physical or chemical cue in the biological environment. Here, we present a DNA origami nanocapsule that can be loaded with cargo and reversibly opened and closed by changing the pH of the surrounding solution. Moreover, the threshold pH value for opening/closing can be rationally designed. We characterize the reversible switching and a rapid opening of “pH-latch”-equipped nanocapsules using Förster resonance energy transfer. Furthermore, we demonstrate the full cycle of capsule loading, encapsulation, and displaying the payload using metal nanoparticles and functional enzymes as cargo mimics at physiologically relevant ion concentrations.
Keywords: DNA nanotechnology, DNA origami, pH control, Förster resonance energy transfer, nanoparticles, enzymes, drug delivery
DNA nanotechnology1−3 provides an essential foundation for programmable and innovative nanoscale structures, devices and advanced materials. DNA nanostructures have been successfully used for example in creating static nanophotonic devices,4−7 nanoscopic rulers8,9 and templates for material assembly.10,11 Nevertheless, it is extremely intriguing to explore the possibilities to build dynamic devices12−14 using DNA as a construction material, especially by applying the robust and versatile DNA origami technique.15 Examples of such dynamic DNA origami-based devices include robotics,16−18 molecular-scale precision measurement and diagnostic tools,19 plasmonic/nanophotonic systems,20 reconfigurable metamolecules,21,22 and information relay systems.23 Dynamic DNA nanostructures can also find interesting uses in drug delivery24,25 since these nanoscale devices are readily modifiable for targeted delivery and loading of the specified molecular cargo.26,27 Moreover, they can be programmed to perform user-defined tasks upon external stimuli, for example, encapsulation and subsequent display and release of cargo.28,29
So far, dynamics of signal-responsive prototypes of DNA drug-delivery vehicles have been realized by conformational changes due to a temperature gradient,30 strand displacement reactions,31 DNA–protein interactions,24,25 or by taking advantage of photoactivated properties of the system.29,32 Regulation of the dynamic properties may become extremely challenging in biologically relevant environments, especially with systems relying on the introduction of additional regulating molecules, such as oligonucleotides for strand displacement reactions. For this, pH change of the environment presents an intriguing trigger for the cargo release of drug-delivery vehicles functioning autonomously in biological environments. Various compartments of the cell are characterized by a distinct pH, and deviations from the normal pH can be linked to abnormal cell behavior. For instance, while healthy cells maintain a lower intracellular pH (∼7.2) compared to the extracellular pH (∼7.4), cancer cells are distinguished by an inverted pH gradient with low extracellular pH (∼6.8–7.0) and high intracellular pH (∼7.3–7.6) necessary for the multiple altered characteristics of cancer cells.33,34 This raises an interest to demonstrate controlled cargo release upon similar slight pH increases in physiological conditions. pH-driven actuation also holds the advantage of being simple and easy to implement: DNA motifs such as cytosine-rich i-motifs and Hoogsteen-type polypurine-polypyrimidine triplexes are intrinsically pH responsive,35−37 and incorporating them into larger DNA nanostructures can be used to control the dynamics of the structures, avoiding the need for more laborious or costly chemical modifications or conjugation of external molecules as stimuli-responsive units. Indeed, both DNA origami structures22,38−41 and tile-based DNA polyhedra42−44 have been successfully functionalized with pH-responsive DNA moieties for applications in various fields of research.
In this article, we have designed and assembled a reconfigurable, compact, and cargo-shielding DNA origami nanocapsule (Figure 1A) whose reversible opening/closing cycle can be controlled by a sharp pH change. In other words, the capsule is equipped with multiple rationally designed “pH-latches” that work cooperatively. Each of the latches is comprised of two parts: a double-stranded DNA (dsDNA) and a single-stranded DNA (ssDNA). Depending on the pH of the environment, these counterparts can either form a parallel triplex DNA via Hoogsteen bonding (closed state of the capsule, low pH) or hang freely from the capsule (open state, high pH). The open/close transition (acid dissociation constant, pKa) of the system can be rationally designed by selecting the base contents of the latch strands,22,45 thus adding another level of modularity to the system. We have analyzed the dynamics and functionality of the capsule in detail by Förster resonance energy transfer (FRET) measurements. Moreover, the capsule has a functionalizable cavity for anchoring molecular payloads, and by taking advantage of the reversible opening/closing cycle of the capsule, we have demonstrated successive loading, encapsulation, and display of molecular payloads. We have used gold nanoparticles (AuNPs) as cargo mimics for transmission electron microscopy (TEM)-based verification and horseradish peroxidase (HRP) enzymes for proving the functionality of the encapsulated cargo by characterizing of enzyme activity.
Figure 1.

DNA origami nanocapsule design and function. (A) A hinged DNA origami nanocapsule with a HRP payload can be reversibly opened and closed with the help of eight programmable pH-responsive latches. At high pH, the latches are open (a hairpin (orange) and a ssDNA (green) detached) and the capsule halves can move freely, whereas at low pH, the latches form a triplex DNA thus closing the capsule. (B) A schematic figure series depicts the full cycle of capsule function. The open capsule can be loaded with a cargo (yellow sphere with an attachment strand) via hybridization of the anchoring and attachment strands. The loaded cargo can be encapsulated by lowering pH and further displayed by increasing pH. The dynamics of the opening and closing can be followed by FRET analysis when the capsule is equipped by a FRET pair (green and red dyes). (C) TEM images reveal open nanocapsules with a variety of opening angles from completely open to barely ajar structures. Insets show representative structures with corresponding opening angles depicted at the bottom with the same color codes. The monomer capsule yield after folding is 79 ± 2%. (D) Closed nanocapsules under TEM. Insets show zoomed-in representative structures. Scale bars in subfigures C and D are 50 nm. The width of each inset is 60 nm.
Results and Discussion
Here, we present the design of a dynamic DNA origami nanocapsule and use FRET- and TEM-based analysis to show that the nanocapsule can be opened and closed with solution pH and harnessed for loading and display of cargo molecules. The DNA nanocapsule was folded as a single structure from the 8064-nucleotide (nt) scaffold. The design consists of two capsule halves linked together at one long edge by four ssDNA hinges (Figure 1A), each consisting of 10 nucleotides of nonhybridized scaffold DNA. The outer dimensions of the nanocapsule are ca. 31 nm × 28 nm × 33 nm (w × h × l), making it roughly spherical, and it contains an inner cavity with dimensions of 11 nm × 12 nm × 13 nm. The walls of the nanocapsule consist of two layers of DNA double helices to ensure the required rigidity and stability for shielding the loaded cargo. In order to avoid multimerization or aggregation of the nanocapsules through blunt-end stacking interactions, the outward pointing ends of each DNA helix were passivated with 8-nt long single-stranded poly-T overhangs.
For introducing a closing/opening mechanism of choice, a total of eight latch sites were included in the design at the interface between the capsule halves. The staple strands at these sites can be replaced by strands containing a desired latch functionality. Here, we designed eight pairs of pH latch DNA sequences that were inserted at these sites as staple strand extensions: One part of the latch is a DNA duplex, consisting of complementary polypurine and pyrimidine regions in a single 20 bp hairpin motif. The other part of the latch in the adjacent half is a single-stranded, 20 nt polypyrimidine DNA oligonucleotide that forms a parallel DNA triplex with the duplex. Each latch is designed with a unique base sequence but identical 60% T-A·T base triplex content to ensure that all latches will open at the same pH,22,45 but the triplex formation will only take place between the intended counterparts. The aforementioned T-A·T base content was chosen to yield pKa ∼ 7.2 according to previously reported triplexes with similar lengths.22 This targeted pKa value was selected to demonstrate the capsule function in physiologically relevant conditions (blood pH ∼ 7.4, healthy cells ∼7.2, and cancer cells typically ∼7.4–7.6).33,34,46
In addition to the pH latch (pHL) nanocapsules, we designed and assembled two types of controls: permanently open nanocapsules with no locking mechanism (opC) and permanently closed nanocapsules (clC), in which the pH latches were replaced with complementary ssDNA overhangs. To first confirm the structural integrity and correct folding of all types of nanocapsules, the assembled structures were analyzed with TEM and agarose gel electrophoresis (AGE). The nanocapsules visualized with TEM showed well-defined shapes, negligible aggregation (monomer yield of capsules calculated from the gel electrophoresis was 79 ± 2% (s.d., n = 3)), and clearly distinguishable open and closed conformations (Figure 1C,D). While nanocapsules in a closed state appear structurally homogeneous under TEM, the structural flexibility of open nanocapsules is reflected in a variety of different opening angles. On agarose gels, all folded structures show a well-defined monomer band and low amount of oligomerization, but surprisingly, the open and closed states cannot be differentiated by electrophoretic mobility (Figure S2).
In order to demonstrate that the opening and closing of the pHL nanocapsules can be controlled with solution pH, we labeled the opposing halves of the capsule with an Alexa Fluor 488 (donor)–Alexa Fluor 594 (acceptor) FRET pair using oligonucleotides with fluorophore end modifications. The emission profiles upon donor excitation at 460 nm were measured from samples prepared at various pH values between 6.0 and 8.0 in 1 × TAE (40 mM Tris, ∼20 mM acetic acid, and 1 mM ethylenediaminetetraacetic acid (EDTA)) with 15 mM MgCl2 and 5 mM NaCl. FRET efficiency at each pH was quantified from the acceptor emission intensity increase at 616 nm.
In acidic conditions (pH 6.4–6.8), a donor excitation results in a clearly observable acceptor emission (Figure 2A, inset) and a high FRET efficiency, EFRET = 0.67 ± 0.03 (Figure 2A). This indicates that a significant amount of donor excitation energy is transferred to the acceptor in a close proximity and, subsequently, that a high portion of the capsules are in a closed state. In contrast to the high-FRET state at acidic pH, pHL samples prepared in basic conditions (pH 7.6–8.0) are in a flexible, open state characterized by EFRET ≈ 0. Emission spectra of the open and closed states of the pHL nanocapsule show a high correspondence to opC and clC control samples. Furthermore, emission spectra of opC and clC are not affected by pH (Figure S1), confirming that the observed FRET efficiency differences in pHL nanocapsules result solely from pH-driven conformational changes. Lowering the pH below 6.4 leads to an increased amount of sample aggregation or possibly other forms of loss of structural integrity, observed as a further increase in FRET efficiency unrelated to the pH-dependent conformational change of individual structures (Figure S3). The aggregation was observed to be less significant in opC and clC control samples, indicating that interactions between pH latches of separate nanocapsules may play a role in the aggregation process. At higher pH values, the level of aggregation in all samples is very low, including negligible multimerization via blunt-end stacking interactions due to an effective passivation of the outward-pointing DNA double-helix ends with poly-T overhangs.
Figure 2.

The pH-controlled opening and closing of nanocapsules in solution characterized by FRET. (A) Dependence of FRET efficiency on the solution pH near the pKa = 7.27. Inset figure shows emission spectra of the sample at pH 6.4 and pH 8.0 resulting from the donor excitation at 460 nm. A Hill equation (solid line) has been fitted to the data points to determine values of pKa and Hill coefficient n. FRET efficiency values have been calculated based on average donor and acceptor emission intensity values in three parallel samples. Error limits for each data point have been calculated according to the propagation of error, based on the standard deviation between the initial measurements. (B) Closing and opening kinetics of nanocapsules in the presence of 15, 10, and 5 mM MgCl2. At t = 0, acetic acid is added to the pH 7.7 solution to rapidly decrease the pH to 6.3. At t = 220 min, the NaCl concentration in the samples is increased to a physiological concentration (150 mM). Sodium hydroxide addition at t = 266 min leads to all samples returning to fully open states. FRET efficiency values have been averaged from three parallel (and independent) samples, error bars representing the standard error of the mean. (C) Repeated opening/closing cycles of the nanocapsules equipped with pH latches (pHL). In addition to the pHL nanocapsules, the same pH cycling was performed for permanently closed (clC) and open (opC) control samples. EFRET values in subfigures B and C have both been normalized to the highest efficiency value observed at a 15 mM MgCl2 concentration (in subfigure B).
At pH 7.0–7.5 (within ca. 0.5 pH units) with a pKa of 7.27 ± 0.02, the pHL nanocapsules undergo a steep transition between the closed and open states. The transition is characterized by a high Hill coefficient, 3.9 ± 0.5, consistent with the idea of a two-state system working in a cooperative manner. The pKa value depends on the relative amount of T-A·T base triads in the pH latches (here 60%) and has high agreement to the pKa = 7.2 reported by Kuzyk et al. for plasmonic DNA origami devices actuated with one DNA triplex with identical length (20 base triads) and T-A·T content.22 Due to the good agreement of the results, we propose that the pKa of our system could be adjusted in a similar fashion demonstrated by both Idili et al.45 and Kuzyk et al.22 to a lower pH by decreasing or to a higher pH by increasing the percentage of T-A·T triplets in the pH latch strands, enabling fine-tuning the opening threshold for the desired biological target. Interestingly, we note that the pH sensitivity does not appear to be affected by the number of pH latches, as the shape and steepness of the pH response curves of the pHL nanocapsules and the plasmonic metamolecules presented by Kuzyk et al.15 are also very similar.
In addition to static measurements, we performed series of kinetic measurements following the FRET efficiency in an individual sample after changing the solution pH by addition of small volumes of either acid or base (Figure 2B). We observed that the closed capsules in acidic conditions open extremely rapidly after the addition of sodium hydroxide: Measured FRET efficiency drops to the level of opC control even before the first measurement at 30 s. In contrast, closing the nanocapsules takes place gradually over a time course of several hours after lowering the pH of the sample from 7.7 to 6.3.
When the closing process was initiated in the presence of lowered cation concentrations (10 mM or 5 mM MgCl2), we observed significantly reduced rates of closing as well as a lower amount of closed structures at the end of the incubation period (Figure 2B). Since the positive charge of the divalent cations screens the repulsive electrostatic interactions between the large, negatively charged nanocapsule halves, the result indicates a heavy role of electrostatic effects in the dynamic properties of the system. It is likely that a reduced amount of electrostatic repulsion both increases the probability of formation of the closed state by reducing the energetic penalty of bringing the two capsule halves together as well as stabilizes the formed triplexes in the closed state by decreasing the repulsion both between the large structural units as well as the interacting latch strands. Thus, we found 15 mM MgCl2 to be the required concentration for successfully preparing and closing the nanocapsules. At higher MgCl2 concentrations (e.g., 30 mM), further weakening of the electrostatic repulsion between individual capsules starts to lead to an increased level of aggregation (Figure S3).
The response time and dynamics of stimuli-responsive nanosystems can be heavily dependent on the type of triggering stimulus. In our system, solution pH change and the associated protonation/deprotonation of cytosine N3 atoms in the pH latch oligonucleotides can be expected to take place in a millisecond time scale according to the diffusion rate of ions. For instance, Idili et al.45 reported both folding (associated with intramolecular triplex formation) and unfolding of small pH-responsive triplex nanoswitches in solution taking place within milliseconds, and reconfiguration times of several hundred milliseconds have also been achieved with larger DNA origami devices actuated by ion concentrations.47 In contrast, systems relying on the strand displacement reactions may yield slower reaction rates due to a number of limiting factors: bifurcation in the branch migration (especially for long strands), concentration of the strands in the reaction, and the low diffusion constant of large oligonucleotides. Moreover, each full strand displacement reaction cycle generates byproducts to the solution, which may limit their use in specific applications. The closing kinetics of our system are comparable to similar systems functioning with strand displacement,48 indicating that the closing process is affected more by how the overall design of the DNA origami system promotes the formation of the compact closed state, rather than the initial trigger. The advantage gained by the pH-sensitivity is reflected in the opening phase of the nanocapsules, where the response time is extremely fast and does not rely on the addition of further triggering elements into the system.
After the capsules had been closed at low pH and in the presence of a sufficient level of MgCl2, we tested how their conformational state responds to physiological salt conditions. In the experiment presented in Figure 2B, we showed that closed nanocapsules can be introduced to a physiological NaCl concentration (150 mM) without any change in the FRET efficiency. To be able to carry out the measurements in a kinetic manner, the Mg2+ concentration (5, 10, or 15 mM) was not changed during the measurement. To study the possible effects of a low physiological Mg2+ concentration (∼0.6 mM in blood),49 we also performed a separate experiment where closed pHL capsules (with 15 mM MgCl2) were first diluted to the MgCl2 concentration of 0.6 mM and after that introduced to a 150 mM NaCl concentration (see Supporting Information). Emission spectra collected from these samples showed no decrease of FRET efficiency from the MgCl2 depletion, but in low-Mg2+ conditions, the FRET efficiency was slightly decreased (ca. 22%) after the addition of NaCl (Figure S4A). This observation is supported by the previous observation that Mg2+ ions might be easily replaced by monovalent Na+ in the DNA origami structure at low-Mg2+ conditions (<1 mM), thus causing slightly deformed DNA objects.50 In addition, we also characterized the FRET efficiency of closed pHL nanocapsules in the presence of 1% and 10% blood plasma (solution pH increases through addition of plasma, but nevertheless, it stays below 7.2) (Figure S4B). These results together show that the closed state can be maintained at physiologically relevant conditions, which can be considered an essential property for potential in vivo drug-delivery applications.
We also showed that the opening/closing cycle presented in Figure 2B can be repeated multiple times by the addition of small volumes of acetic acid or sodium hydroxide (Figure 2C). Here, the shorter closing period (90 min vs 300 min in Figure 2B) is not enough to reach the stabilization/plateau of the FRET efficiency in the closing phase. Nevertheless, the shapes of the FRET efficiency curves in each closing phase are very similar to each other, proving the reversibility of the capsule function. This shows that the closing process is not disturbed by repeated adjustment of the solution pH. When the same pH cycling was performed for opC and clC nanocapsules, no effect was observed in the FRET efficiency of opC, but the FRET efficiency of the clC gradually decreased over each sodium hydroxide addition. TEM images of clC samples before and after five rounds of pH cycling indicate that this is linked to the accumulation of structural damage (Figure S5). Furthermore, we tested that the effect is not related to Na+ concentration (increasing up to 65 mM due to increasing volumes of sodium hydroxide) with a control experiment where equal concentration and volume of NaCl was added to the samples (data not shown).
After showing that the nanocapsule opening and closing can be reliably triggered by changing the solution pH, we loaded the nanocapsules in the open state with cargo molecules. We applied both AuNPs and HRP enzymes as two distinct model types of cargo. To visually examine the loading of cargos in the nanocapsule, AuNPs were used because of their high contrast in TEM images, while HRP was chosen for studying the effects of loading and encapsulation on cargo functionality. In the chosen encapsulation strategy, the cargo molecule is first conjugated to one or more DNA oligonucleotides. The conjugate is mixed with nanocapsules that present a complementary, single-stranded oligonucleotide in their inner cavity, so that hybridization of the complementary strands leads to attachment of the cargo inside the cavity (Figure 1B). The AuNP-DNA conjugates were prepared with a modified salting protocol,51 and they were purified and mixed with a nanocapsule, followed by a thermal annealing to increase the loading yield. Analogous HRP-DNA conjugates were produced by covalently linking HRP via its surface lysines to thiol-modified oligonucleotides with the sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) cross-linker.
TEM images of AuNP-loaded nanocapsules show that the pHL nanocapsules can be successfully loaded by anchoring the particles into the nanocapsule cavity at high pH (Figure 3A). By counting the number of open nanocapsule structures that display a AuNP at the expected attachment site, we estimated that the loading yield of the particles is 40–55% (n = 110) (Figure S6). Decreasing the pH leads to cargo encapsulation, and importantly, TEM images confirm that the loaded cargo does not prevent the capsules from closing (Figure 3B). Likewise, the FRET efficiencies of HRP-loaded pHL and opC nanocapsules at pH 6.4 are identical to the corresponding capsule samples without cargo (Figure S7), so that the result obtained from individual structures under TEM is also generalizable to bulk solution.
Figure 3.

High-pH loading (left panel) and low-pH encapsulation (right panel) of AuNPs and HRP. (A) TEM images of nanocapsules in an open state (pH 8.2), displaying loaded AuNP cargo molecules. (B) AuNPs encapsulated within closed pHL nanocapsules at pH 6.4. The width of each frame in subfigures A and B is 50 nm. (C) Catalytic activity of HRP-loaded pHL and opC nanocapsules as well as free HRP at pH 7.8. Product formation (oxidation of ABTS to ABTS•+ by HRP) in the presence of 1 mM ABTS and 4 mM H2O2 is observed with an increase of ABTS•+ absorption at 420 nm. Values at each time point have been calculated as the mean of three parallel samples, and error bars represent the standard error of the mean. (D) Product formation at pH 6.4 (1 mM ABTS, 4 mM H2O2), including an opC control assembled without a cargo anchoring strand (opC-anchor). Product formation curves for all measured ABTS concentrations (0.125–4 mM) at both pH values are presented in Figure S8. (E) Maximum reaction velocities (Vmax) and Michaelis constants (Km) at pH 7.8 from the Michaelis–Menten fit to the initial product formation rates between 0 and 300 s in each sample type. Vmax values have been normalized to the value of free HRP. Values for both Vmax and Km have been calculated as the weighted mean of fit results for three parallel samples, weighted by the fit error. Error bars show the error of the weighted mean. (F) Vmax and Km values at pH 6.4. The values and error bars were calculated as in subfigure E.
In addition to immobilizing cargo molecules within the nanocapsules, we characterized how the functionality of the cargo responds to the loading and encapsulation process. Thus, we compared the enzymatic activity of HRP in three different sample types at both pH 6.4 and 7.8: free HRP without DNA strand(s), HRP inside pHL nanocapsules, and HRP inside opC nanocapsules. The measurements were performed by detecting the rate of ABTS oxidation by HRP in the presence of H2O2. The oxidation of ABTS by HRP leads to the formation of an ABTS•+ radical, which has an absorption maximum at 420 nm, and therefore velocity of the process can be measured spectrometrically by following the increase of A420 after the substrate is mixed to HRP-containing samples.
We first ensured that the cargo attachment within the nanocapsules is specific to the cargo anchoring strand. Since HRP cannot be reliably detected in TEM images, the specificity was assessed with enzyme activity measurements. The opC capsules were assembled either with or without a cargo anchoring strand (additional opC-anchor control), mixed with HRP-DNA conjugates, and purified from unbound enzymes with PEG precipitation.52 After purification, clear HRP activity was detected in the opC samples assembled with the cargo anchoring strand, while the opC-anchor controls showed virtually no activity (Figure 3D). In addition to confirming that HRP binding to the nanocapsules is specific to the anchoring strand, this shows that PEG precipitation efficiently removes unbound enzymes from the samples.
We observed that HRP remains fully functional after loading and encapsulation. Furthermore, the ABTS oxidation rates were higher in the nanocapsule-HRP samples (both pHL and opC) than in the samples containing free HRP at both measured pH values. This can be seen both directly in the product formation curves (Figure 3C,D) as well as in the maximum reaction rates (Vmax) determined for each sample type by a Michaelis–Menten analysis of catalytic rates measured at varying ABTS concentrations (Figure 3E,F) (product formation plots and Michaelis–Menten curves for each sample are presented in Figure S8). All sample types displayed greater reaction rates at pH 6.4 than at pH 7.8, which is a predictable result based on the known fact that the catalytic activity of HRP is pH dependent, increasing with decreasing pH, and a pH optimum at pH 6.0–6.5.53 At pH 6.4, the Vmax of HRP inside both types of nanocapsules at 2 nM nanocapsule concentration was approximately 2 times larger than of free HRP at a 2 nM enzyme concentration (Figure 3F). At pH 7.8, the difference was similar but slightly less prominent (1.5-fold in pH lock capsule sample, and 1.8-fold in the open control) (Figure 3E). Since the quantity Vmax depends on both enzyme concentration and its catalytic activity (Vmax = kcat[E]total, where kcat is the turnover number of the enzyme, and [E]total the enzyme concentration), differences in Vmax can likewise signify either enzyme concentration or activity differences between the compared samples. While the exact HRP concentration in the HRP-loaded nanocapsules is unknown, the DNA origami concentration in the samples sets an effective 2 nM upper limit to the HRP concentration, since this would correspond to a 100% encapsulation yield. In this case, the results would indicate a 2-fold increase in the kcat of HRP upon encapsulation. As with the 40–55% encapsulation yield estimated for the AuNPs, the real encapsulation yield of HRP is also likely smaller than 100%. Thus, the kcat value can likewise be expected to be further enhanced. The activity increase is well in line with previous studies showing that the catalytic activity of enzymes conjugated to DNA nanostructures can differ significantly from free enzymes.53−55 In the case of HRP, this has been observed as an activity increase; Zhao et al.55 reported as high as a 10-fold enhancement of kcat when HRP was confined inside DNA nanocages. While a comprehensive understanding of the mechanisms of such activity effects has not yet been reached,56 one explanation for the higher catalytic rate of DNA origami-conjugated HRP has been suggested to be a lower local pH near the origami surface.53 According to the activity profile reported by Zhang et al.,53 HRP activity is also more sensitive to pH changes around pH 6.4 than pH 7.8, which could in part explain why we also observe a slightly larger activity enhancement at pH 6.4 than at pH 7.8.
Interestingly, at pH 6.4, the formation of ABTS•+ both in pHL and opC nanocapsules is highly similar (Figure 3D). The Michaelis constant Km describes the ability of the substrate to interact with the enzyme, so that higher Km values can indicate either that the enzymes are less accessible to the substrate or that their affinity toward the substrate at the specific conditions is lower. Km values of HRP inside an open or closed nanocapsule, or of free HRP, show no significant differences between each other (Figure 3E,F) at either of the measured pHs. While it could be assumed that at pH 6.4, the closed state of the pHL nanocapsules blocks the diffusion of ABTS to the encapsulated HRP, the results on the contrary suggest that HRP is equally accessible to the substrate in all of the cases. This leads to a conclusion that the nanocapsules are porous enough for the low molecular weight (548.68 g/mol) ABTS to diffuse into the inner cavity. However, encapsulation still provides shielding from larger molecules. The pHL nanocapsules mixed at a closed, low pH state with AuNPs do not contain particles within the capsules (Figure S6), demonstrating that the DNA barrier effectively blocks these larger particles (5 nm gold core) from entering the nanocapsule cavity.
Conclusions
In this work, we have presented the design and characterization of a dynamic DNA origami nanocapsule. We have shown that the nanocapsule can be loaded with various types of molecular cargo, here AuNPs and HRP, and that the cargo can be selectively displayed after exposing the nanocapsule to the specified external cue. Our multilayer DNA origami-based carrier has been designed for high stability and protection of enclosed molecules and for functionalization with multiple stimuli-responsive units for cooperative actuation.
We have shown that when the nanocapsules were equipped with multiple DNA residues that form Hoogsteen triplexes in low pH, reconfiguration between open and closed conformational states could be induced with solution pH changes in a predictable and repeatable manner under physiologically relevant salt conditions. Application of Hoogsteen triplexes for pH-actuation gives an important aspect of programmability for the system. The pKa can be chosen simply by selecting the base content of the latch strands, and the measured pKa was seen to be in close agreement with the predicted value for the latch sequences.22 As determined from FRET analysis, the conformational state of the nanocapsules is also highly sensitive to pH differences. An approximately 0.5 pH unit increase is sufficient to switch the bulk sample from a closed state to an open state with a very rapid response time.
In order to reach full functionality as a drug delivery vehicle, we envision that our system could still be further functionalized with additional stimuli-responsive or targeting groups for specific cell types. It could also be interesting to explore how various protein- or polymer-coating strategies49,57 presented for static objects could be applied for such dynamic systems to increase the in vivo stability and compatibility while preserving the delicate functionality and dynamic properties of the system. All in all, the presented compatibility with physiologically relevant medium, programmability of the pKa value, high pH sensitivity, and fast kinetics of the opening give our system multiple intriguing properties in terms of the development of smart, targeted DNA-based drug delivery systems that are capable of responding to stimuli present in living organisms and that would function independently of additional external triggers.
Methods
Nanocapsule Design and Assembly
The DNA nanocapsule was designed in a honeycomb lattice using caDNAno v 2.2.0,58 and the three-dimensional structure was predicted with CanDo simulations.59,60 Self-assembly of the structures was performed as a single batch reaction containing a 20 nM p8064 scaffold (Tilibit Nanosystems) and 7.5× excess of staple strands (Integrated DNA Technologies) in 1× capsule folding buffer (FOB) (1× capsule FOB: 1× TAE, 15 mM MgCl2, 5 mM NaCl). The reactions were heated in G-storm G1 Thermal Cycler to 65 °C and annealed by cooling to 59 °C with a rate of 1 °C/15 min and then to 12 °C with rate 0.25 °C/45 min. The excess staple strands were removed from the solution using PEG precipitation in the presence of 7.5% (w/v) PEG 8000.52 Folding yield was assessed by integration over the AGE band intensities (Figure S2A).
Fluorescence Measurements
FRET-labeled nanocapsule samples were analyzed with a Biotek Cytation 3 plate reader using black Costar flat bottom 96-well plates. The donor (Alexa Fluor 488) was excited at 460 nm and the acceptor (Alexa Fluor 546) at 560 nm. All FRET measurements were conducted at room temperature. FRET efficiency was calculated from the increase of acceptor emission in the presence of energy transfer:
where IAD is the acceptor emission intensity following donor excitation, IAA is the acceptor emission intensity (at 616 nm) after acceptor excitation, εAA is the acceptor extinction coefficient at the acceptor excitation wavelength (30,817 M–1 cm–1), and εAD and εDD are the acceptor and donor extinction coefficients at the donor excitation wavelength (1985 M–1cm–1 and 19,848 M–1cm–1). Extinction coefficient values at the excitation wavelengths were calculated by combining the measured absorption spectra of the fluorophore-modified oligonucleotides with the fluorophore extinction coefficients at absorption maxima provided by IDT. Calculating the FRET efficiency as described enables using the value of IAA as an internal reference for acceptor emission intensity (and implicitly, quantum yield) in the absence of energy transfer. In addition, the equation includes an assumption that donor and acceptor fluorophores are present in the sample in a 1:1 molar ratio, which can be reasonably assumed for a DNA origami system where both the number and location of the fluorophores are highly controlled.
For the pH titration experiment, nanocapsule samples folded in the normal FOB (pH 8.2) were resuspended to pH-adjusted buffers (pH 6.0–8.0 in 0.2 pH unit intervals) after PEG purification of excess staples. In order to determine the values for the closed state, FRET efficiency EFRET (closed), pKa, and the Hill coefficient n, FRET efficiencies measured at each pH were fitted with the Hill equation:
Kinetic Fluorescence Measurements
FRET efficiency measurements for the kinetic response of the FRET efficiency after solution pH changes were based on single-wavelength recordings of the sample emission intensity at 517 nm (IDD) and 616 nm (IAD) after donor excitation at 460 nm. Changes in capsule conformation were followed by calculating relative FRET efficiency as
where pH of the sample was cycled between 6.1–6.3 and 7.7–8.0 by adding small volumes (2–4 μL) of 0.5 M acetic acid or 0.5 M sodium hydroxide.
TEM Imaging
Nanocapsule samples which were purified with PEG precipitation were absorbed on plasma cleaned (20 s oxygen plasma flash) Formvar carbon-coated copper grids (FCF400-Cu, Electron Microscopy Science) for TEM imaging. A 3 μL droplet of DNA origami solution was applied onto the carbon-coated side of the TEM grid, and the excess sample solution was blotted away with filter paper after an incubation of 2 min. The samples were stained using a 2% aqueous uranyl formate solution with 25 mM NaOH. Excess stain solution was blotted away with filter paper after 40 s. After these procedures, the sample was left to dry under ambient conditions for at least 30 min before imaging. The TEM images were obtained using a FEI Tecnai 12 Bio-Twin instrument operated at an acceleration voltage of 120 kV.
AuNP-DNA Conjugate Preparation and AuNP Encapsulation
The steps of AuNP-DNA conjugation were all carried out at 40 °C with constant shaking, if not stated otherwise. First, 20 μL of AuNPs of 5 nm diameter (Sigma-Aldrich) was incubated with 0.4 μL 1% SDS water solution for 20 min. Then 2 μL of thiolated oligos (100 μM, Integrated DNA Technologies) was added and incubated for 30 min. In the salting process, first 0.2 μL of 2.5 M NaCl was added at 2 min interval for 6 times, followed by additions of 0.4 μL and 0.8 μL of NaCl for 6 times each at the same interval. After the salting, the AuNP-DNA conjugates were mixed with 30 μL of folding buffer (with 0.02% SDS) and incubated for 1 h. Finally, the conjugates were purified from the free oligos by spin-filtration through a 100 kDa MWCO Amicon filter at RT and 14000 rcf for 10 min and repeated for 4 times. In each filtration step, 480 μL of folding buffer at either pH 6 or pH 8 was added in order to match the nanocapsule sample pH. For the AuNP encapsulation, the AuNP-DNA and nanocapsule with complementary cargo strands were mixed in a 10:1 AuNP:origami ratio and thermally annealed from 40 to 20 °C (−0.1 °C/min).
HRP-DNA Conjugation and HRP Encapsulation
HRP (Thermo Scientific) was covalently conjugated to single-stranded 5′-thiol-modified DNA oligonucleotides (Integrated DNA Technologies) by using the sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) cross-linker (Thermo Scientific). To form maleimide-activated HRP, HRP and sulfo-SMCC were individually dissolved in 50 mM phosphate buffer (pH 7.2), mixed in a 1:20 enzyme:cross-linker molar ratio, and incubated at RT for 2 h. The thiol groups in the oligonucleotides were deprotected by incubating the strands dissolved in water with an 100-fold molar excess of tris(2-carboxyethyl)phosphine (TCEP) (Aldrich Chemistry) for 1–2 h at RT. The unreacted cross-linker in the HRP-sulfo-SMCC mixture, as well as excess TCEP in the DNA-TCEP mixture, was removed by spin-filtration with Amicon Ultra 10 kDa cutoff filters (EMD Millipore). One mM EDTA was added in the oligo buffer during spin-filtration. Maleimide-activated HRP was then mixed with the thiol-modified oligos in a 1:2 DNA:enzyme ratio. HRP was used in excess in order to minimize the amount of free oligonucleotides in the product. HRP contains six surface lysines, out of which three have been shown to be accessible to chemical modifications,61 so conjugation with thiol-modified oligonucleotides can be expected to yield a mixture of conjugates with different HRP:DNA ratios. Since this heterogeneity is not relevant for loading the nanocapsule, conjugate composition was not further analyzed. Thus, the conjugates were used without further purification and mixed in a 15–20-fold molar excess with cargo-strand containing nanocapsules in 1× FOB (pH 8.2). To maximize the encapsulation yield, the HRP-nanocapsule mixture was thermally annealed (from 40 to 20 °C, −0.1 °C/40 s). After annealing, the samples were incubated further for 12 h at +4 °C, after which unbound enzymes were removed by PEG precipitation. Enzyme-loaded, purified nanocapsules were resuspended in 1× FOB at either pH 6.4 or 7.8, and the enzymatic activity in the samples was measured after overnight incubation at +4 °C.
HRP Activity Measurements
Enzymatic activity of HRP was determined by measuring the rate of ABTS oxidation in the presence of H2O2 at varying ABTS concentrations. Measurements were performed in 1× FOB adjusted to pH 6.4 or pH 7.8 with acetic acid, at either 2 nM HRP concentration (free HRP samples) or 2 nM DNA origami concentration (HRP-loaded nanocapsule samples) with a total 80 μL sample volume on a clear 96-well microwell plate (Thermo Scientific) at +25 °C. The samples were first diluted with a FOB containing H2O2, in a 4 mM final H2O2 concentration. Immediately after this, an ABTS solution was added to the sample in either 0.125 mM, 0.25 mM, 0.5 mM, 1 mM, 2 mM, or 4 mM final ABTS concentration. Formation of the oxidized product ABTS•+ was monitored by measuring the A420 of the samples in 20 s intervals over 20 min with a Cytation 3 cell imaging multimode reader (BioTek). To determine Vmax and Km for each sample type, the initial oxidation rates for each ABTS concentration were determined by fitting a linear equation to the data points at the first 300 s after starting the reaction, and a Michaelis–Menten equation,
was fitted to the data points. Curve fitting was done with OriginPro version 2019 (OriginLab Corporation, Northampton, MA, USA).
Acknowledgments
We acknowledge the provision of facilities and technical support by Aalto University at OtaNano - Nanomicroscopy Center (Aalto-NMC). The financial support through the Academy of Finland (grant numbers 286845, 308578, 303804, 267497), Jane and Aatos Erkko Foundation, and Sigrid Jusélius Foundation is gratefully acknowledged. This work was carried out under the Academy of Finland Centers of Excellence Programme (2014–2019). We would also like to thank J. Ihalainen for helpful discussions and comments on the manuscript and J. Rumfeldt for help in interpreting the pH titration data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b01857.
Supporting methods, a list of staple strands for pHL, opC, and clC nanocapsules, figures showing the nanocapsule design, additional FRET measurements with opC and clC samples and HRP-loaded nanocapsules, characterization of nanocapsules with AGE, aggregation caused by low pH and high Mg2+ concentrations, Mg2+ depletion and plasma experiments, supplementary TEM images of pH cycling and AuNP loading, and product formation at all ABTS concentrations and Michaelis–Menten curves for HRP activity measurements (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Seeman N. C.; Sleiman H. F. DNA Nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. 10.1038/natrevmats.2017.68. [DOI] [Google Scholar]
- Bathe M.; Rothemund P. W. K. DNA Nanotechnology: A Foundation for Programmable Nanoscale Materials. MRS Bull. 2017, 42, 882–888. 10.1557/mrs.2017.279. [DOI] [Google Scholar]
- Nummelin S.; Kommeri J.; Kostiainen M. A.; Linko V. Evolution of Structural DNA Nanotechnology. Adv. Mater. 2018, 30, 1703721. 10.1002/adma.201703721. [DOI] [PubMed] [Google Scholar]
- Roller E.-M.; Argyropoulos C.; Högele A.; Liedl T.; Pilo-Pais M. Plasmon–Exciton Coupling Using DNA Templates. Nano Lett. 2016, 16, 5962–5966. 10.1021/acs.nanolett.6b03015. [DOI] [PubMed] [Google Scholar]
- Gopinath A.; Miyazono E.; Faraon A.; Rothemund P. W. K. Engineering and Mapping of Nanocavity Emission via Precision Placement of DNA Origami. Nature 2016, 535, 401–405. 10.1038/nature18287. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Jungmann R.; Acuna G. P.; Liu N. DNA Origami Route for Nanophotonics. ACS Photonics 2018, 5, 1151–1163. 10.1021/acsphotonics.7b01580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B.; Kostiainen M. A.; Linko V. DNA Origami Nanophotonics and Plasmonics at Interfaces. Langmuir 2018, 34, 14911–14920. 10.1021/acs.langmuir.8b01843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauer C.; Jungmann R.; Sobey T. L.; Simmel F. C.; Tinnefeld P. DNA Origami as a Nanoscopic Ruler for Super-Resolution Microscopy. Angew. Chem., Int. Ed. 2009, 48, 8870–8873. 10.1002/anie.200903308. [DOI] [PubMed] [Google Scholar]
- Graugnard E.; Hughes W. L.; Jungmann R.; Kostiainen M. A.; Linko V. Nanometrology and Super-Resolution Imaging with DNA. MRS Bull. 2017, 42, 951–959. 10.1557/mrs.2017.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maune H. T.; Han S.-P.; Barish R. D.; Bockrath M.; Goddard W. A. III; Rothemund P. W. K.; Winfree E. Self-Assembly of Carbon Nanotubes into Two-Dimensional Geometries Using DNA Origami Templates. Nat. Nanotechnol. 2010, 5, 61–66. 10.1038/nnano.2009.311. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan S.; Subramaniam S.; Stewart A. F.; Grundmeier G.; Keller A. Regular Nanoscale Protein Patterns via Directed Adsorption through Self-Assembled DNA Origami Masks. ACS Appl. Mater. Interfaces 2016, 8, 31239–31247. 10.1021/acsami.6b10535. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Turberfield A. J.; Mills A. P. Jr.; Simmel F. C.; Neumann J. L. A DNA-Fuelled Machine Made of DNA. Nature 2000, 406, 605–608. 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- Zhang D. Y.; Seelig G. Dynamic DNA Nanotechnology Using Strand-Displacement Reactions. Nat. Chem. 2011, 3, 103–113. 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- Ijäs H.; Nummelin S.; Shen B.; Kostiainen M. A.; Linko V. Dynamic DNA Origami Devices: From Strand-Displacement Reactions to External-Stimuli Responsive Systems. Int. J. Mol. Sci. 2018, 19, 2114. 10.3390/ijms19072114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Marras A. E.; Zhou L.; Su H.-J.; Castro C. E. Programmable Motion of DNA Origami Mechanisms. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 713–718. 10.1073/pnas.1408869112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir Y.; Ben-Ishay E.; Levner D.; Ittah S.; Abu-Horowitz A.; Bachelet I. Universal Computing by DNA Origami Robots in a Living Animal. Nat. Nanotechnol. 2014, 9, 353–357. 10.1038/nnano.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thubagere A. J.; Li W.; Johnson R. F.; Chen Z.; Doroudi S.; Lee Y. L.; Izatt G.; Wittman S.; Srinivas N.; Woods D.; Winfree E.; Qian L. A Cargo-Sorting DNA Robot. Science 2017, 357, eaan6558 10.1126/science.aan6558. [DOI] [PubMed] [Google Scholar]
- Castro C. E.; Dietz H.; Högberg B. DNA Origami Devices for Molecular-Scale Precision Measurements. MRS Bull. 2017, 42, 925–929. 10.1557/mrs.2017.273. [DOI] [Google Scholar]
- Kuzyk A.; Schreiber R.; Fan Z.; Pardatscher G.; Roller E.-M.; Högele A.; Simmel F. C.; Govorov A. O.; Liedl T. DNA-Based Self-Assembly of Chiral Plasmonic Nanostructures with Tailored Optical Response. Nature 2012, 483, 311–314. 10.1038/nature10889. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Schreiber R.; Zhang H.; Govorov A. O.; Liedl T.; Liu N. Reconfigurable 3D Plasmonic Metamolecules. Nat. Mater. 2014, 13, 862–866. 10.1038/nmat4031. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Urban M. J.; Idili A.; Ricci F.; Liu N. Selective Control of Reconfigurable Chiral Plasmonic Metamolecules. Sci. Adv. 2017, 3, e1602803 10.1126/sciadv.1602803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.; Li Z.; Wang P.; Meyer T.; Mao C.; Ke Y. Reconfiguration of DNA Molecular Arrays Driven by Information Relay. Science 2017, 357, eaan3377 10.1126/science.aan3377. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Bachelet I.; Church G. M. A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science 2012, 335, 831–834. 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- Li S.; Jiang Q.; Liu S.; Zhang Y.; Tian Y.; Song C.; Wang J.; Zou Y.; Anderson G. J.; Han J. Y.; Chang Y.; Liu Y.; Zhang C.; Chen L.; Zhou G.; Nie G.; Yan H.; Ding B.; Zhao Y. A DNA Nanorobot Functions as a Cancer Therapeutic in Response to a Molecular Trigger In Vivo. Nat. Biotechnol. 2018, 36, 258–264. 10.1038/nbt.4071. [DOI] [PubMed] [Google Scholar]
- Linko V.; Ora A.; Kostiainen M. A. DNA Nanostructures as Smart Drug-Delivery Vehicles and Molecular Devices. Trends Biotechnol. 2015, 33, 586–594. 10.1016/j.tibtech.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Surana S.; Shenoy A. R.; Krishnan Y. Designing DNA Nanodevices for Compatibility with the Immune System of Higher Organisms. Nat. Nanotechnol. 2015, 10, 741–747. 10.1038/nnano.2015.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A.; Bhatia D.; Saminathan A.; Chakraborty S.; Kar S.; Krishnan Y. Controlled Release of Encapsulated Cargo from a DNA Icosahedron using a Chemical Trigger. Angew. Chem. 2013, 125, 6992–6995. 10.1002/ange.201302759. [DOI] [PubMed] [Google Scholar]
- Takenaka T.; Endo M.; Suzuki Y.; Yang Y.; Emura T.; Hidaka K.; Kato T.; Miyata T.; Namba K.; Sugiyama H. Photoresponsive DNA Nanocapsule Having an Open/Close System for Capture and Release of Nanomaterials. Chem. - Eur. J. 2014, 20, 14951–14954. 10.1002/chem.201404757. [DOI] [PubMed] [Google Scholar]
- Turek V. A.; Chikkaraddy R.; Cormier S.; Stockham B.; Ding T.; Keyser U. F.; Baumberg J. J. Thermo-Responsive Actuation of a DNA Origami Flexor. Adv. Funct. Mater. 2018, 28, 1706410. 10.1002/adfm.201706410. [DOI] [Google Scholar]
- Andersen E. S.; Dong M.; Nielsen M. M.; Jahn K.; Subramani R.; Mamdouh W.; Golas M. M.; Sander B.; Stark H.; Oliveira C. L. P.; Pedersen J. S.; Birkedal V.; Besenbacher F.; Gothelf K. V.; Kjems J. Self-Assembly of a Nanoscale DNA Box with a Controllable Lid. Nature 2009, 459, 73–76. 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- Tohgasaki T.; Shitomi Y.; Feng Y.; Honna S.; Emura T.; Hidaka K.; Sugiyama H.; Endo M. A Photocaged DNA Nanocapsule for Controlled Unlocking and Opening inside the Cell. Bioconjugate Chem. 2019, 10.1021/acs.bioconjchem.9b00040. [DOI] [PubMed] [Google Scholar]
- Webb B. A.; Chimenti M.; Jacobson M. P.; Barber D. L. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- White K. A.; Grillo-Hill B. K.; Barber D. L. Cancer Cell Behaviors Mediated by Dysregulated pH Dynamics at a Glance. J. Cell Sci. 2017, 130, 663–669. 10.1242/jcs.195297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benabou S.; Aviño A.; Eritja R.; González C.; Gargallo R. Fundamental Aspects of the Nucleic Acid I-Motif Structures. RSC Adv. 2014, 4, 26956–26980. 10.1039/C4RA02129K. [DOI] [Google Scholar]
- Hu Y.; Cecconello A.; Idili A.; Ricci F.; Willner I. Triplex DNA Nanostructures: From Basic Properties to Applications. Angew. Chem., Int. Ed. 2017, 56, 15210–15233. 10.1002/anie.201701868. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran A. R.; Rusling D. A. Triplex-Forming Oligonucleotides: A Third Strand for DNA Nanotechnology. Nucleic Acids Res. 2018, 46, 1021–1037. 10.1093/nar/gkx1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuya A.; Watanabe R.; Yamanaka Y.; Tamaki T.; Kaino M.; Ohya Y. Nanomechanical DNA Origami pH Sensors. Sensors 2014, 14, 19329–19335. 10.3390/s141019329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.; Willner I. pH-Stimulated Reconfiguration and Structural Isomerization of Origami Dimer and Trimer Systems. Nano Lett. 2016, 16, 6650–6655. 10.1021/acs.nanolett.6b03418. [DOI] [PubMed] [Google Scholar]
- Majikes J. M.; Ferraz L. C. C.; LaBean T. H. pH-driven Actuation of DNA Origami via Parallel I-Motif Sequences in Solution and on Surfaces. Bioconjugate Chem. 2017, 28, 1821–1825. 10.1021/acs.bioconjchem.7b00288. [DOI] [PubMed] [Google Scholar]
- Burns J. R.; Lamarre B.; Pyne A. L. B.; Noble J. E.; Ryadnov M. G. DNA Origami Inside-Out Viruses. ACS Synth. Biol. 2018, 7, 767–773. 10.1021/acssynbio.7b00278. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Li Y.; Tian C.; Mao C. A Smart DNA Tetrahedron That Isothermally Assembles or Dissociates in Response to the Solution pH Value Changes. Biomacromolecules 2013, 14, 1711–1714. 10.1021/bm400426f. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Kim K. R.; Ahn D. R.; Lee J. E.; Yang E. G.; Kim S. Y. Reversible Regulation of Enzyme Activity by pH-Responsive Encapsulation in DNA Nanocages. ACS Nano 2017, 11, 9352–9359. 10.1021/acsnano.7b04766. [DOI] [PubMed] [Google Scholar]
- Ottaviani A.; Iacovelli F.; Idili A.; Falconi M.; Ricci F.; Desideri A. Engineering a Responsive DNA Triple Helix into an Octahedral DNA Nanostructure for a Reversible Opening/Closing Switching Mechanism: a Computational and Experimental Integrated Study. Nucleic Acids Res. 2018, 46, 9951–9959. 10.1093/nar/gky857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idili A.; Vallée-Bélisle A.; Ricci F. Programmable pH-Triggered DNA Nanoswitches. J. Am. Chem. Soc. 2014, 136, 5836–5839. 10.1021/ja500619w. [DOI] [PubMed] [Google Scholar]
- Kellum J. A. Determinants of Blood pH in Health and Disease. Crit. Care 2000, 4, 6–14. 10.1186/cc644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras A. E.; Shi Z.; Lindell M. G.; Patton R. A.; Huang C.-M.; Zhou L.; Su H.-J.; Arya G.; Castro C. E. Cation-Activated Avidity for Rapid Reconfiguration of DNA Nanodevices. ACS Nano 2018, 12, 9484–9494. 10.1021/acsnano.8b04817. [DOI] [PubMed] [Google Scholar]
- Grossi G.; Jepsen M. D. E.; Kjems J.; Andersen E. S. Control of Enzyme Reactions by a Reconfigurable DNA Nanovault. Nat. Commun. 2017, 8, 992. 10.1038/s41467-017-01072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bila M.; Kurisinkal E. E.; Bastings M. M. C. Engineering a Stable Future for DNA Origami as a Biomaterial. Biomater. Sci. 2019, 7, 532–541. 10.1039/C8BM01249K. [DOI] [PubMed] [Google Scholar]
- Kielar C.; Xin Y.; Shen B.; Kostiainen M. A.; Grundmeier G.; Linko V.; Keller A. On the Stability of DNA Origami Nanostructures in Low-Magnesium Buffers. Angew. Chem., Int. Ed. 2018, 57, 9470–9474. 10.1002/anie.201802890. [DOI] [PubMed] [Google Scholar]
- Heck C.; Kanehira Y.; Kneipp J.; Bald I. Placement of Single Proteins within the SERS Hot Spots of Self-Assembled Silver Nanolenses. Angew. Chem., Int. Ed. 2018, 57, 7444–7447. 10.1002/anie.201801748. [DOI] [PubMed] [Google Scholar]
- Stahl E.; Martin T. G.; Praetorius F.; Dietz H. Facile and Scalable Preparation of Pure and Dense DNA Origami Solutions. Angew. Chem., Int. Ed. 2014, 53, 12735–12740. 10.1002/anie.201405991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Tsitkov S.; Hess H. Proximity Does Not Contribute to Activity Enhancement in the Glucose Oxidase-Horseradish Peroxidase Cascade. Nat. Commun. 2016, 7, 13982. 10.1038/ncomms13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.-L.; Wheeldon I. Kinetic Enhancements in DNA–Enzyme Nanostructures Mimic the Sabatier Principle. ACS Catal. 2013, 3, 560–564. 10.1021/cs300766d. [DOI] [Google Scholar]
- Zhao Z.; Fu J.; Dhakal S.; Johnson-Buck A.; Liu M.; Zhang T.; Woodbury N. W.; Liu Y.; Walter N. G.; Yan H. Nanocaged Enzymes with Enhanced Catalytic Activity and Increased Stability Against Protease Digestion. Nat. Commun. 2016, 7, 10619. 10.1038/ncomms10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küchler A.; Yoshimoto M.; Luginbühl S.; Mavelli F.; Walde P. Enzymatic Reactions in Confined Environments. Nat. Nanotechnol. 2016, 11, 409–420. 10.1038/nnano.2016.54. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan S.; Ijäs H.; Linko V.; Keller A. Structural Stability of DNA Origami Nanostructures Under Application-Specific Conditions. Comput. Struct. Biotechnol. J. 2018, 16, 342–349. 10.1016/j.csbj.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. Rapid Prototyping of 3D DNA-Origami Shapes with caDNAno. Nucleic Acids Res. 2009, 37, 5001–5006. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C. E.; Kilchherr F.; Kim D. N.; Shiao E. L.; Wauer T.; Wortmann P.; Bathe M.; Dietz H. A Primer to Scaffolded DNA Origami. Nat. Methods 2011, 8, 221–229. 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- Kim D.-N.; Kilchherr F.; Dietz H.; Bathe M. Quantitative Prediction of 3D Solution Shape and Flexibility of Nucleic Acid Nanostructures. Nucleic Acids Res. 2012, 40, 2862–2868. 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien A. M.; Ó’Fágáin C.; Nielsen P. F.; Welinder K. G. Location of Crosslinks in Chemically Stabilized Horseradish Peroxidase: Implications for Design of Crosslinks. Biotechnol. Bioeng. 2001, 76, 277–284. 10.1002/bit.1194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.