

Two-component signal transduction systems (TCSs) are broadly conserved in the bacterial kingdom and generally contain two molecular components, a sensor histidine kinase and a receiver protein. Sensor histidine kinases alter their phosphorylation state in direct response to a physical or chemical cue, whereas receiver proteins “receive” the phosphoryl group from the kinase to regulate a change in cell physiology. We have discovered that a single-domain receiver protein, FixT, binds an Fe-S cluster and controls Caulobacter heme homeostasis though its function as a negative-feedback regulator of the oxygen sensor kinase FixL. We provide evidence that the Fe-S cluster protects FixT from Lon-dependent proteolysis in the cell and endows FixT with the ability to function as a second, autonomous oxygen/redox sensor in the FixL-FixJ signaling pathway. This study introduces a novel mechanism of regulated TCS feedback control by an Fe-S-binding receiver domain.
KEYWORDS: Caulobacter crescentus, Fe-S, FixL, FixT, feedback inhibition, hypoxia, kinase inhibitor, lon, proteases, receiver domain, two-component regulatory systems
ABSTRACT
Two-component signaling systems (TCSs) function to detect environmental cues and transduce this information into a change in transcription. In its simplest form, TCS-dependent regulation of transcription entails phosphoryl-transfer from a sensory histidine kinase to its cognate DNA-binding receiver protein. However, in certain cases, auxiliary proteins may modulate TCSs in response to secondary environmental cues. Caulobacter crescentus FixT is one such auxiliary regulator. FixT is composed of a single receiver domain and functions as a feedback inhibitor of the FixL-FixJ (FixLJ) TCS, which regulates the transcription of genes involved in adaptation to microaerobiosis. We sought to define the impact of fixT on Caulobacter cell physiology and to understand the molecular mechanism by which FixT represses FixLJ signaling. fixT deletion results in excess production of porphyrins and premature entry into stationary phase, demonstrating the importance of feedback inhibition of the FixLJ signaling system. Although FixT is a receiver domain, it does not affect dephosphorylation of the oxygen sensor kinase FixL or phosphoryl-transfer from FixL to its cognate receiver FixJ. Rather, FixT represses FixLJ signaling by inhibiting the FixL autophosphorylation reaction. We have further identified a 4-cysteine motif in Caulobacter FixT that binds an Fe-S cluster and protects the protein from degradation by the Lon protease. Our data support a model in which the oxidation of this Fe-S cluster promotes the degradation of FixT in vivo. This proteolytic mechanism facilitates clearance of the FixT feedback inhibitor from the cell under normoxia and resets the FixLJ system for a future microaerobic signaling event.
INTRODUCTION
Environmental sensing in bacteria is often mediated by two-component signaling systems (TCSs), consisting of a sensory histidine kinase and a response regulator (1). In canonical TCSs, the sensory histidine kinase autophosphorylates a single conserved histidine residue in response to an environmental cue. This phosphoryl group is then transferred from the histidine to a conserved aspartate residue on the receiver (REC) domain of its cognate response regulator. The phosphorylated response regulator elicits a cellular response, generally via an effector domain at its C terminus (1). In many cases, this effector domain directly binds DNA and regulates transcription. However, a subclass of response regulators, known as the single-domain response regulators (SDRRs), consist of only a REC domain and induce a cellular response via protein-protein interactions (2–6). Although the general model of TCSs is relatively simple, these so-called two-component systems can in fact be part of complex phosphorelays involving multiple proteins, including sensory histidine kinases, histidine phosphotransfer proteins, and SDRRs (7–11).
FixL-FixJ (FixLJ) is an oxygen-responsive TCS that incorporates a regulatory SDRR, FixT, to control system output (12–14). FixL is a histidine kinase whose autophosphorylation activity is regulated by oxygen binding to a heme iron center on its sensory Per-Arnt-Sim (PAS) domain (15–17). FixL has higher kinase activity under microaerobic conditions, which results in increased phosphorylation of its cognate response regulator, FixJ. Phosphorylated FixJ induces a microaerobiosis-specific transcriptional program both directly via DNA binding and indirectly via upregulation of the transcription factor fixK (18, 19). FixL and FixJ were first identified in Sinorhizobium meliloti and have been most extensively characterized in diazotrophic Rhizobiales (20, 21). In this group of bacteria, FixLJ primarily regulates microaerobic respiration and nitrogen fixation, a process that is extremely sensitive to oxygen levels. In addition, some Rhizobiales employ FixLJ to regulate heme biosynthesis, anaerobic nitrate respiration, and the expression of hydrogenases (22). In select organisms, FixLJ also upregulates the SDRR fixT, which functions as a feedback inhibitor of the FixL-FixJ system (12, 13).
Initial biochemical work demonstrated that S. meliloti FixT directly inhibits the accumulation of autophosphorylated FixL (14). This inhibition may arise via effects on the histidine autophosphorylation reaction and/or the reverse reaction with ADP. In addition, in vitro experiments did not detect FixT phosphorylation or the inhibition of phosphotransfer from FixL to FixJ (14). Thus, despite having the primary structure of an SDRR, FixT does not apparently act as a competing receiver domain in S. meliloti. Interestingly, a subsequent genetic screen determined that S. meliloti FixT activity requires the presence of an additional gene, asnO, that is related to glutamine amidotransferase proteins and involved in N-acetylglutaminylglutamine synthesis (23, 24). This finding raises the possibility that FixT may sense metabolic signals and integrate information into the FixLJ circuit. However, the mechanism by which AsnO affects FixT activity is not understood.
fixL, fixJ, and fixT are prevalent in the order Rhizobiales, but they have also been identified in Caulobacter crescentus, a free-living bacterium originally isolated from freshwater (12). C. crescentus is an obligate aerobe that does not fix nitrogen, and its FixLJ TCS primarily serves to activate the expression of high-affinity terminal oxidases and select metabolic pathways, including heme biosynthesis, under microaerobic conditions (12). Similar to S. meliloti, C. crescentus FixLJ strongly induces expression of the SDRR feedback inhibitor fixT (21.5% sequence identity to S. meliloti FixT). In this study, we use the C. crescentus FixLJ system as a model to investigate the biological importance of TCS feedback control. Unlike S. meliloti, we observe distinct defects in a C. crescentus strain lacking fixT, highlighting the importance of negative-feedback regulation of the FixLJ system. In addition, we conduct experiments aimed at defining the molecular mechanism by which FixT represses FixLJ signaling. Our results demonstrate that C. crescentus FixT preferentially inhibits the forward FixL autophosphorylation reaction, which is the biochemical basis of its function as an inhibitor of FixLJ-dependent transcription. Most notably, we identify a 4-cysteine (4-Cys) motif unique to Caulobacter FixT. This feature of primary structure supports binding of an Fe-S cluster and influences the function of FixT as an inhibitor of FixLJ. Mutational analysis implicates the Fe-S cluster in stabilizing FixT against degradation by the Lon protease, and we provide evidence that oxidation of the Fe-S cluster destabilizes FixT in the cell. We conclude that FixT is a novel Fe-S-binding SDRR capable of autonomously detecting changes in oxygen or the cellular redox state, and transducing those signals, via proteolytic susceptibility, into a change in FixLJ signaling.
RESULTS
The loss of fixT affects porphyrin metabolism and growth.
Feedback inhibition is a common mechanism to restrain biological signaling circuits (25, 26). Besides derepression of FixLJ-dependent transcription, no phenotypic consequences for loss of the fixT feedback inhibitor have been reported to date. We postulated that the physiological importance of FixT might be more evident in C. crescentus than in S. meliloti. Using a β-galactosidase transcriptional fusion assay, we found that the FixLJ system was active in cultures grown to stationary phase with limited aeration, and, importantly, the deletion of fixT resulted in strong derepression of FixLJ-dependent transcription (Fig. 1A). Under these same conditions, ΔfixT mutant cells exhibited a red pigmentation compared with wild-type cells (Fig. 1B). This color change was genetically complemented by ectopic fixT expression. To further characterize this color phenotype, we compared the absorbance spectra of lysates from wild-type and ΔfixT mutant cells. These lysates exhibited a characteristic porphyrin absorbance spectrum, with a Soret maximum at 414 nm and four Q bands at 506 nm, 542 nm, 580 nm, and 632 nm (Fig. 1C) (27). Importantly, the magnitude of these peaks correlated with the degree of pigmentation observed in cell pellets (Fig. 1B and C). Previous work found that the FixLJ TCS upregulates the transcription of several genes involved in heme biosynthesis, and thus, it is likely that the porphyrin(s) we observe derive from this metabolic pathway (12). Moreover, the presence of 4 Q bands, rather than 2, indicates an accumulation of free-base porphyrins, such as the heme precursor protoporphyrin IX (see Fig. S1A in the supplemental material) (27). In agreement with these data, excitation of wild-type and ΔfixT mutant lysates with 404-nm light generated fluorescence maxima at 588 nm, 634 nm, and 698 nm (Fig. S1B). The latter two maxima suggest the presence of free-base porphyrin(s), whereas the peak at 588 nm may reflect porphyrin(s) bound to a closed-shell metal like Zn (28). This accumulation of free-base porphyrins in the ΔfixT mutant strain suggests that heme biosynthesis may outpace iron availability. To test this hypothesis, we grew wild-type and ΔfixT mutant cells in PYE medium supplemented with 10 μM FeSO4. Lysates from these cells lost the fluorescence characteristics of free-base porphyrins (Fig. S1B). In addition, the absorbance spectra of lysates from Fe-treated cells exhibited only 2 Q bands (Fig. S1C). These results provide evidence that, relative to wild-type cells, ΔfixT mutant cells strongly upregulate heme biosynthesis and accumulate free-base porphyrin precursors due to insufficient iron availability.
FIG 1.

Loss of fixT leads to derepression of FixLJ-dependent transcription, protoporphyrin accumulation, and a stationary-phase growth defect. (A) Activity of a PfixK-lacZ transcriptional reporter in wild-type (WT) or ΔfixT mutant cells grown to high density with limited aeration (average ± SD, n = 3 biological replicates, each with 3 averaged technical replicates). *, P < 0.0005, Student’s t test. PfixK is directly activated by FixLJ (12, 18, 19). (B) Cell pellets harvested from cultures grown as in panel A. Plasmid-free WT and ΔfixT mutant pellets are on the left. Strains carrying either empty pMT805 (EV) or xylose-inducible pMT805-fixT (fixT++) for complementation are on the right. (C) Top, absorption spectra of cell lysates showing prominent Soret peaks (414 nm) and 4 Q bands. Strain genotypes are annotated as in panel B. Bottom, quantification of lysate absorbance at 414 nm (average ± SD, n = 3). *, P < 0.005, Student’s t test; **, P < 0.0001, one-way ANOVA followed by Dunnett’s posttest comparison to ΔfixT mutant EV. (D) Growth curve, measured by the optical density at 660 nm (OD660), following entry into stationary phase for WT and ΔfixT mutant cells (top) and plasmid-bearing complementation strains (bottom). Points are the averages ± SD of the results from 3 biological replicates.
Comparison of porphyrin signatures to protoporphyrin IX and effects of Fe supplementation. (A) Absorption spectrum of ΔfixT mutant lysate compared with protoporphyrin IX (Sigma Aldrich) in dimethyl sulfoxide (DMSO). (B) Fluorescence emission scan of wild-type (WT), ΔfixT mutant, and Fe-treated ΔfixT mutant lysates (excitation [Ex.], 404 nm). (C) Absorption spectra of lysates from ΔfixT mutant cells grown with (black) or without (gray) 10 μM FeSO4. Inset highlights Q bands in the 500- to 650-nm region. (D) Optical density at 660 nm (OD660) of cultures grown for 24 h with (black bars) or without (gray bars) the addition of 10 μM FeSO4 (average ± SD, n > 3). *, P < 0.001, one-way ANOVA; P < 0.05, Dunnett’s posttest comparison to wild type (WT) untreated. Download FIG S1, TIF file, 0.8 MB (832.7KB, tif) .
Copyright © 2020 Stein et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In addition to exhibiting a color change consistent with the accumulation of heme precursors, ΔfixT mutant cultures also displayed growth defects at high culture density. Specifically, the terminal optical density of ΔfixT mutant cultures was lower than that of the wild type when grown with limited aeration (Fig. 1D). We observed this difference despite the fact that the exponential growth rates of wild-type and ΔfixT mutant cultures were indistinguishable (Fig. S2). Ectopic overexpression of fixT complemented this stationary-phase defect (Fig. 1D). We surmised that the inability of ΔfixT mutant cells to grow to high density might be due to the accumulation of heme precursors observed in these cells, as high levels of free porphyrins are generally toxic (29). Alternatively, upregulation of heme biosynthesis might indirectly lead to a growth defect at high cell density by contributing to iron starvation. In agreement with either of these hypotheses, supplementation of iron in the growth medium largely mitigated the high-density growth defect in ΔfixT mutant cells (Fig. S1D). We conclude that under microaerobic conditions with limited iron, the loss of fixT leads to a detrimental accumulation of porphyrins in C. crescentus.
Growth of wild-type and ΔfixT mutant strains. Growth curves during exponential phase of wild-type (WT, blue) and ΔfixT mutant (orange) strains measured by the optical density at 660 nm (OD660). The points are the averages ± SD of the results from 3 biological replicates. Strains were grown under oxygen-limited conditions as in Fig. 1. Download FIG S2, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2020 Stein et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
FixT acts as a direct inhibitor of the FixL kinase.
Biochemical studies of S. meliloti FixT indicated that it inhibits the accumulation of autophosphorylated FixL (14). To examine whether C. crescentus FixT functions in a similar manner, we tested its inhibitory activity in vitro using purified FixT, soluble FixL (FixL118–495, missing the transmembrane domain), and FixJ(D55A) (a nonphosphorylatable mutant). Previous work noted that FixL autokinase activity is enhanced by FixJ, indicating that FixL and FixJ likely work as a complex in vivo (30, 31). Using [γ-32P]ATP, we observed that FixL robustly autophosphorylates over a 30-minute time course in the presence of FixJ(D55A). However, the addition of excess FixT inhibited the accumulation of autophosphorylated FixL by ∼3-fold (Fig. 2A). This inhibitory activity did not require FixL stimulation by FixJ(D55A), indicating that FixT does not simply compete with FixJ(D55A) for binding to FixL (Fig. 2B). Moreover, FixT completely blocked the activation of FixL by FixJ(D55A), even at equimolar excess FixJ(D55A) and FixT. Given the low-micromolar apparent affinities of FixL for both FixT and FixJ(D55A), these data suggest that the biochemical effects of FixT are dominant over those of FixJ (Fig. 2B and 3B). Thus, similar to S. meliloti FixT, C. crescentus FixT inhibits the accumulation of phosphorylated FixL in the presence or absence of FixJ. This inhibition may arise from effects on FixL autophosphorylation and/or FixL dephosphorylation.
FIG 2.

FixT inhibits FixL autophosphorylation in vitro. (A) Autophosphorylation of FixL (5 μM) in the presence or absence of FixT (80 μM). Reactions were carried out in the presence of FixJ(D55A) (20 μM). (B) FixL (5 μM) autophosphorylation with increasing FixJ(D55A) concentrations in the presence or absence of FixT (80 μM). Reactions were quenched after 15 min and mixtures separated by SDS-PAGE, and phosphorylated-FixL was quantified by phosphorimaging. Values were normalized to the reaction without FixT and FixJ, shown as the average ± SD, n = 3. (C) Disappearance of phosphorylated FixL (5 μM), purified from nucleotides, in the presence of excess FixT (80 μM) and/or ADP (0.5 mM). (D) The disappearance of phosphorylated FixL (5 μM) 30 s after the addition of FixJ (2.5 μM) in the presence or absence of excess FixT (80 μM).
FIG 3.
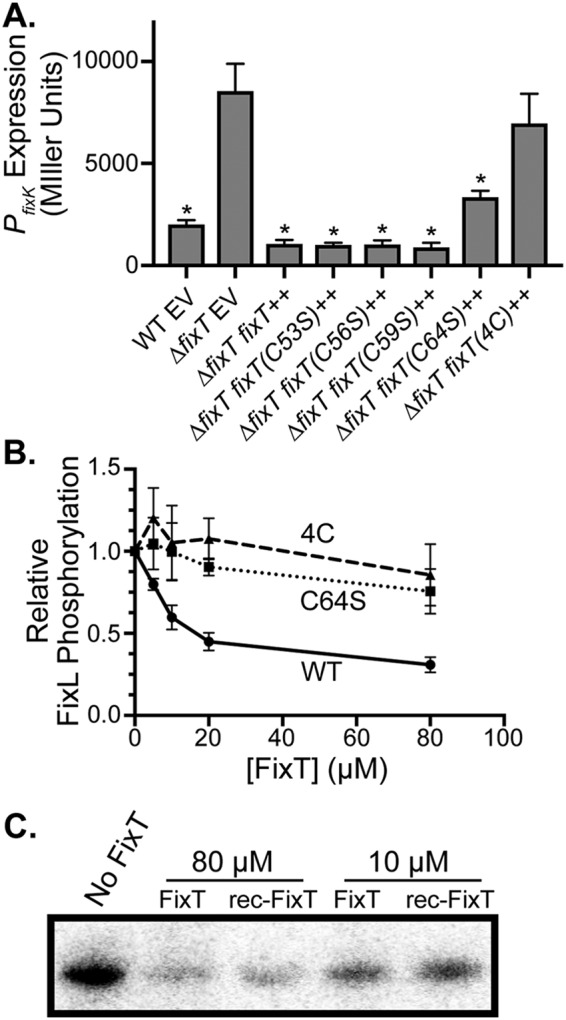
The 4-Cys motif, but not the Fe-S cluster, is necessary for FixL inhibition in vitro. (A) PfixK-lacZ transcriptional reporter activity in strains carrying an empty vector (EV) or a plasmid overexpressing wild-type or mutant fixT alleles (average ± SD, n = 3 biological replicates, each with 3 averaged technical replicates). *, P < 0.0001, one-way ANOVA followed by Dunnett’s posttest comparison to ΔfixT mutant EV. (B) Relative FixL (5 μM) autophosphorylation, in the presence of FixJ(D55A) (20 μM), with increasing concentrations of wild-type (WT) or mutant FixT. Phosphorylation was measured as in Fig. 2B and normalized to the reaction without FixT, shown as the average ± SD, n = 3. (C) Autophosphorylation of FixL (5 μM) in the presence of FixT or rec-FixT. Phosphorylation reactions were performed under anaerobic conditions.
To address the effect of FixT on FixL dephosphorylation (either hydrolysis or a reverse reaction with ADP), we examined the disappearance of phosphorylated FixL after removing nucleotides by desalting. The addition of excess FixT had no effect on the slow loss of FixL-phosphate observed over 15 min, indicating that FixT does not act as a phosphatase (Fig. 2C). The addition of excess ADP led to a rapid loss of the phosphoryl group from FixL, perhaps by a reverse reaction to form ATP (32). The presence of excess FixT did not affect this phosphoryl loss (Fig. 2C). Taken together, these results indicate that FixT primarily reduces the accumulation of phosphorylated FixL by inhibiting the FixL autophosphorylation reaction, rather than by stimulating the loss of FixL-phosphate.
We also examined whether FixT could interfere with phosphoryl-transfer from FixL to FixJ. To test this possibility, we monitored the dephosphorylation of FixL after the addition of FixJ. The addition of FixJ alone induced a rapid (within 30 s) loss of FixL-phosphate. Moreover, a 32-fold molar excess of FixT did not inhibit dephosphorylation of FixL by FixJ (Fig. 2D). Thus, in agreement with our results with FixJ(D55A), these data indicate that FixT does not interfere with the interaction between FixL and FixJ. This observation, paired with the fact that we never observed phosphorylation of FixT in our experiments (data not shown), provides evidence that FixT does not act as a competitive phosphoryl sink.
FixT binds an Fe-S cluster in vitro.
Our biochemical dissection of FixT activity indicated that it functions similarly to S. meliloti FixT (14). However, we observed that C. crescentus FixT, unlike S. meliloti FixT, contains a 4-Cys motif (C53, C56, C59, and C64) reminiscent of iron-sulfur (Fe-S)-binding motifs in other proteins (Fig. 4A) (33, 34). We also noted that C. crescentus FixT was light brown at the early stages of purification, indicating the potential presence of bound iron. Given these observations, we hypothesized that FixT may bind an Fe-S cluster. To test this possibility, we attempted to chemically reconstitute an Fe-S cluster on aerobically purified FixT in an anaerobic chamber. Upon reconstitution, FixT adopted a dark-brown color reminiscent of many Fe-S proteins (35–37). The absorption spectra of this reconstituted sample (rec-FixT) exhibited a shoulder in the blue region of the visible spectrum (λ ∼420 nm), similar to previously reported spectra for [4Fe-4S] cluster-binding proteins (Fig. 4B) (33). Importantly, these spectral features were not evident in nonreconstituted FixT (Fig. 4B). Fe measurements using ferrozine detected an average ± standard deviation (SD) of 3.8 ± 0.3 Fe atoms per rec-FixT monomer (n = 3). These data indicate that FixT is capable of binding an Fe-S cluster, most likely in the form of a [4Fe-4S] cluster. Additionally, we mutated each position in the 4-Cys motif to Ser [FixT(4C)] and tested the ability of this mutant protein to bind a reconstituted Fe-S cluster. Reconstitution reactions with FixT(4C) produced granular black material indicating the formation of iron sulfides instead of Fe-S clusters. rec-FixT(4C) also exhibited relatively strong absorbance beyond 650 nm, which was further evidence of significant iron-sulfide formation (Fig. 4B) (33). We conclude that the FixT 4-Cys motif is important for productive reconstitution of an Fe-S cluster.
FIG 4.

FixT binds an Fe-S cluster. (A) Sequence alignment of FixT proteins from several Caulobacter species and S. meliloti. The position of the first amino acid residue of each sequence is on the left. Cys residues aligning with the C. crescentus 4-Cys motif are in red. Additional nearby Cys residues are in blue. The position corresponding to C. crescentus C64 is marked by an arrow. (B) Absorption spectra of aerobically purified FixT (FixT), anaerobically reconstituted wild-type FixT (rec-FixT), and anaerobically reconstituted FixT(4C) [rec-FixT(4C)]. (C) Absorption spectra of rec-FixT collected at 5-min intervals after exposure to ambient air. (D) Absorption spectra of anaerobically purified H6-Sumo-FixT and H6-Sumo-FixT(4C) at 400 μM. The 10-min decay curve of FixT with a reconstituted Fe-S cluster (rec-FixT) from panel C is shown for comparison. (E) Absorption spectra of anaerobically purified H6-Sumo-FixT collected at 5-min intervals after exposure to ambient air.
We surmised that the FixT Fe-S cluster is likely sensitive to oxygen exposure, as (i) we observe little pigmentation and absorbance in aerobically purified FixT, and (ii) most solvent-accessible [4Fe-4S] clusters react with molecular oxygen (33, 38, 39). To test this hypothesis, we measured absorption spectra after exposing rec-FixT to the ambient atmosphere. We observed a spectral transition within the first 5 min characterized by a red shift of the major absorbance peak, a shoulder at 465 nm, and increased absorbance at ∼540 nm. This initial transition was followed by a slow overall loss in absorbance (Fig. 4C). These changes are consistent with decay to a [2Fe-2S] cluster, followed by a slower loss of remaining iron and sulfide (40–42). Thus, like many Fe-S clusters, the FixT cluster is sensitive to atmospheric concentrations of molecular oxygen, raising the possibility that it acts as a sensor of cytoplasmic oxygen levels.
To examine whether FixT can acquire this oxygen-sensitive Fe-S cluster in cells, we anaerobically purified FixT and FixT(4C) (retaining their H6-Sumo tags), heterologously expressed in Escherichia coli, and examined their absorption spectra. The absorption spectrum of anaerobically purified FixT was most similar to the decay spectra for rec-FixT, with an absorption peak at 420 nm and a shoulder at 465 nm (Fig. 4D). In contrast, anaerobically purified FixT(4C) exhibited no significant absorbance at 420 nm, indicating that the 4-Cys motif is indeed required for Fe-S cluster binding. Consistent with these spectra, we detected Fe in the FixT sample (0.21 ± 0.07 Fe atoms per FixT monomer, n = 2) but not in the FixT(4C) sample [−0.01 ± 0.02 Fe atoms per FixT(4C) monomer, n = 2]. Similar to rec-FixT, exposure to ambient atmosphere led to a cluster decay in anaerobically purified FixT, with an early transition marked by an increase in absorbance at ∼540 nm (Fig. 4E). Thus, a subfraction of FixT purified from E. coli contains an Fe-S cluster similar to that seen in our in vitro reconstitution experiments.
The FixT 4-Cys motif plays an important role in feedback inhibition.
We next sought to test the importance of the 4-Cys motif for the regulatory function of FixT in vivo. Using the PfixK-lacZ fusion (a reporter of FixLJ-dependent transcription) under microaerobic growth conditions, we measured the effect of individual and combined Cys-to-Ser mutations on the ability of FixT to repress FixLJ output. Ectopic overexpression of fixT strongly repressed transcription from the PfixK-lacZ reporter plasmid (Fig. 3A). Overexpression of FixT C53S, C56S, and C59S single mutants repressed transcription similarly to wild-type FixT. However, the C64S single mutant only partially repressed PfixK-lacZ activity. We note that this cysteine is broadly conserved in FixT proteins, unlike other residues in the 4-Cys motif (Fig. 4A). Importantly, the mutation of all four Cys residues [FixT(4C)] abolished repression entirely (Fig. 3A). Thus, the 4-Cys motif is critical for FixT function in vivo.
To examine the importance of these Cys residues for function in vitro, we purified the C64S and 4C mutant proteins and tested their inhibitory activities against FixL. These FixT mutants were both defective at inhibiting FixL autophosphorylation, even at 32-fold molar excess (Fig. 3B). At these concentrations, we did not observe significant differences between the mutant proteins, as might be expected from the in vivo β-galactosidase reporter assay (Fig. 3A). It is possible that the dynamic range of our in vitro assay is not adequate to robustly detect modest inhibition by the C64S mutant protein. Nevertheless, our data clearly demonstrate that the 4-Cys motif, and C64 in particular, is critical for FixT inhibitory activity.
Given that the 4-Cys motif is important for forming an Fe-S cluster, we next tested whether this Fe-S cluster might affect the ability of FixT to inhibit FixL autophosphorylation in vitro. Interestingly, aerobically purified FixT (FixT) and Fe-S-bound FixT (rec-FixT) similarly inhibited FixL autophosphorylation under anaerobic conditions (Fig. 3C). Thus, FixT does not require the Fe-S cluster to inhibit FixL autokinase activity in vitro.
FixT protein levels are regulated by Lon proteolysis.
Although the Fe-S cluster per se does not affect the activity of FixT as a FixL inhibitor, it may play an important structural role. In fact, many Fe-S clusters affect protein folding, thermodynamic stability, and/or proteolytic stability (43, 44). We also hypothesized that proteolysis may play a significant role in regulating FixT as a feedback inhibitor, because FixT should be cleared from the cell under aerobic conditions to allow future microaerobic transcriptional responses. To probe FixT proteolytic stability, we first examined steady-state FixT levels in strains carrying deletions of energy-dependent protease genes. To avoid complications that might arise from endogenous transcriptional regulation of fixT, we deleted the native fixT gene and monitored FixT expressed from a xylose-inducible promoter. Western blots of gel-resolved lysates from these strains detected low levels of FixT in all but the ΔfixT Δlon mutant, where FixT levels were dramatically higher (Fig. 5A). We conclude that the Lon protease plays an important role in the degradation of FixT in C. crescentus. To ensure that this result was not an artifact of fixT overexpression, we also examined natively expressed FixT levels in wild-type (WT) and Δlon mutant cultures grown to saturation. Natively expressed FixT was below the limit of detection in wild-type cells but observable in Δlon mutant cells (Fig. 5B). As a control, we also demonstrated that FixT signal was absent from ΔfixT Δlon mutant cells. Together, these results indicate that Lon regulates FixT steady-state levels in vivo. To more directly assess proteolysis by Lon, we examined the half-life of FixT, in lon+ and Δlon mutant backgrounds, after inhibiting translation with oxytetracycline. Due to low protein levels at stationary phase (presumably as a result of reduced translation rates), we conducted these experiments using mid-exponential-phase cells. Upon the addition of oxytetracycline, we observed a loss of FixT in the lon+ background with a half-life of 42 ± 4.9 min (Fig. 5C). Importantly, the loss of FixT was slower in Δlon mutant cells (half-life not determined but >60 min). We also assayed the in vivo stability of FixT(4C) and found that it was degraded more rapidly than was the wild-type protein, with a half-life of 13 ± 3.9 min (Fig. 5C). As expected, this degradation was also lon dependent. Taken together, our results support a model in which FixT is degraded by Lon in vivo and the loss of the 4-Cys motif enhances degradation. Given our in vitro reconstitution experiments, these data also suggest that the Fe-S cluster may protect FixT against Lon proteolysis. To test this hypothesis, we measured the half-lives of FixT and FixT(4C) in cultures with enhanced aeration, as we have shown that the FixT Fe-S cluster is sensitive to oxygen in vitro. To enhance aeration, we reduced the culture volume from 10 ml to 4 ml to increase headspace and increased the tilt angle during shaking. FixT was degraded more quickly under these conditions (compared with FixT under limited aeration), with an ∼2-fold shorter half-life (24 min versus 42 min, P < 0.05, Student's t test). In contrast, FixT(4C) degradation was not significantly affected by increased aeration. Thus, FixT proteolytic stability is sensitive to the degree of culture aeration if the 4-Cys motif is present.
FIG 5.

The Lon protease degrades FixT in vivo. (A) Western blot of lysates from strains lacking ATP-dependent protease systems probed with anti-FixT antibody. Strains lack chromosomal fixT and carry either an empty vector (EV) or a fixT overexpression plasmid (fixT++). Coomassie stain of the membrane is shown as a loading control. (B) Western blot using an anti-FixT antibody to assess steady-state levels of endogenously expressed fixT in wild-type (WT), Δlon mutant, and ΔfixT Δlon mutant cell lysates under saturating microaerobic conditions. The Coomassie stain of the membrane is shown as a loading control. (C) In vivo degradation experiments using exponential-phase cells grown under limited aeration (determined by culture volume and tilt depicted in cartoon). Left, representative anti-FixT Western blots of ΔfixT (lon+) mutant and ΔfixT Δlon (Δlon) mutant cells overexpressing fixT or fixT(4C). Cells were sampled at the indicated intervals after arresting translation with oxytetracycline. Coomassie stains of membranes are shown as a loading control. Right, quantification of FixT levels from replicates of the experiment shown to the left (average ± SD, n = 3). The half-life (t1/2; average ± SD, n = 3) of FixT in each strain is displayed to the right of the graph. The difference between FixT and FixT(4C) half-lives in the lon+ background is significant (P < 0.005, Student’s t test). (D) In vivo degradation experiments as in panel C using exponential-phase cells grown under enhanced aeration (smaller volume and greater tilt angle, as shown in cartoon). Left, representative anti-FixT Western blot of ΔfixT (lon+) mutant cells overexpressing either fixT or fixT(4C). Right, quantification of FixT levels from replicates of the experiment shown to the left. Points and half-lives are shown as the averages ± SD, n = 3.
DISCUSSION
Importance of feedback inhibition in the FixLJ system.
The deletion of fixT in C. crescentus leads to the accumulation of porphyrins and premature entry into stationary phase (Fig. 1 and S1). These are the first reported phenotypes for the loss of FixT-dependent feedback inhibition in any organism. These phenotypes are also apparently linked, as supplementation of the growth medium with iron alters the absorbance of accumulated porphyrins and alleviates the growth defect. Why are ΔfixT phenotypes apparent in C. crescentus, whereas none have been reported in S. meliloti? Previous studies found that FixLJ upregulates hemA, hemB, hemE, and hemN in C. crescentus (12). In contrast, S. meliloti FixLJ only significantly upregulates hemN (45). We suggest that multilevel regulation of heme biosynthesis by C. crescentus FixLJ underlies the porphyrin accumulation defect. This link between unchecked FixLJ-dependent transcription and porphyrin accumulation highlights an important physiological role for FixT in C. crescentus. Overinduction of heme biosynthesis is likely metabolically taxing, particularly in oligotrophic environments where iron levels may be limiting. Moreover, in low-iron environments, heme overinduction leads to an accumulation of potentially toxic porphyrin intermediates (29). Thus, in C. crescentus, FixT may function largely to restrain the activation of heme biosynthesis by FixLJ, balancing heme production with the levels of available nutrients, including iron.
On the mechanism of FixT inhibition.
Although the physiologic importance of FixT is more readily apparent in C. crescentus than in S. meliloti, its mechanism of action is similar in the two organisms. Our results provide evidence that C. crescentus FixT primarily inhibits the FixL autophosphorylation reaction, as we observed no effect of FixT on FixL-phosphate hydrolysis or the reverse reaction with ADP (Fig. 2). FixT may inhibit FixL autophosphorylation by interfering with ATP binding. However, our inhibition experiments were conducted using a large excess of ATP (1 mM), making this model unlikely. Moreover, FixT does not appear to interfere with the binding of ADP to FixL. Therefore, the data are most consistent with a model in which FixT affects the catalytic activity of the FixL kinase domain.
These observations are largely in agreement with those for S. meliloti FixT. Although Garnerone et al. did not examine the reverse reaction with ADP, they observed that FixT inhibits the accumulation of autophosphorylated FixL (14). We note that the degree of inhibition observed in their experiments was greater than that in ours. This discrepancy may reflect inherent differences in inhibitory activity or FixL affinity between the two homologous systems and thus evolutionary tuning of the degree of feedback in different organisms. Alternatively, this discrepancy may simply be a result of technical differences in the assay conditions or the particular constructs used in each in vitro system.
Our characterization of FixT activity places it in contrast to other well-studied histidine kinase inhibitors, such as KipI and Sda, which block both autophosphorylation and the reverse reaction with ADP (46, 47). These inhibitors appear to sterically interfere with communication between the ATP binding site on the catalytic domain and the histidine phosphorylation site, thus affecting the phosphorylation reaction in both directions (47, 48). In contrast, our data suggest that FixT preferentially inhibits the forward autophosphorylation reaction. Perhaps FixT preferentially binds to and stabilizes unphosphorylated FixL, thereby largely affecting only the autophosphorylation reaction. Further biochemical and structural studies are necessary to characterize the interaction between FixL and FixT and reveal the molecular basis of inhibition.
FixT 4-Cys motif, Fe-S cluster, and proteolysis.
Despite sharing a common mechanism of kinase inhibition, C. crescentus and S. meliloti FixT differ in the presence of a 4-Cys motif. In fact, the 4-Cys motif is largely conserved within the Caulobacter genus but is absent in non-Caulobacter FixT proteins (Fig. 4A). Our in vivo and in vitro data provide evidence that this 4-Cys motif is important for both Fe-S cluster binding and FixT function as a FixL inhibitor (Fig. 3 and 4). To our knowledge, C. crescentus FixT represents the first REC domain found to bind an Fe-S cluster. Thus, FixT may be added to a growing list of SDRR proteins with noncanonical ligands and/or modifications (6, 49, 50). The stoichiometry of this Fe-S cluster remains an open question; spectroscopic techniques such as electron paramagnetic resonance (EPR) or Mössbauer spectroscopy are necessary to definitively determine cluster type (41). However, our initial spectroscopic data and Fe measurements are consistent with FixT binding a [4Fe-4S] cluster.
Although our data indicate that FixT is capable of binding an Fe-S cluster, we did not detect a significant difference in the abilities of FixT and Fe-S-reconstituted FixT (rec-FixT) to inhibit FixL autophosphorylation in vitro. The 4-Cys mutant deficient in Fe-S cluster binding [FixT(4C)] is also defective in FixL inhibition; thus, we cannot discern whether the Fe-S cluster per se contributes to inhibitory activity in vivo. It is possible that our Fe-S-reconstituted FixT does not fully reflect the form found in vivo and therefore does not capture relevant effects of the Fe-S cluster on inhibitory activity. That said, our data do provide evidence that the Fe-S cluster plays a significant role in controlling FixT degradation by the Lon protease.
Lon is most widely known for its role in degrading damaged, unfolded, and misfolded proteins in the cytoplasm (51, 52). However, Lon is also known to degrade a variety of specific, folded substrates, including SulA and DnaA (53–55). Our work demonstrates that FixT is also a substrate of Lon in vivo. As yet, we do not know the signal(s) that target FixT for degradation. Lon may recognize folded FixT via specific degron(s) or may simply degrade FixT upon full or partial unfolding. We can infer, however, that this degradation is likely to be physiologically relevant, as tuning FixT steady-state levels by deletion or overexpression has observable effects on FixLJ-dependent transcription (Fig. 1 and 3). Thus, FixT levels are positioned such that changes in fixT expression and/or the rate of Lon degradation will affect FixLJ signaling.
Notably, the assembly states of Fe-S ligands have been shown to influence the degradation rates of several proteins (44, 56, 57). For example, E. coli fumarate and nitrate reductase regulator (FNR) is a [4Fe-4S]-binding transcription factor that is active under anaerobic conditions. Upon oxygen exposure, the FNR cluster degrades, leading to monomerization and degradation by the ClpXP protease (40, 44). We propose that the FixT Fe-S cluster may play a similar role in impeding degradation by the Lon protease. Our in vivo results clearly show that the FixT(4C) mutant, which does not efficiently reconstitute an Fe-S cluster in vitro, is degraded 3- to 4-fold faster than is the wild-type protein (Fig. 5). Moreover, enhancing culture aeration, and thus presumably dissolved oxygen levels, reduces the half-life of wild-type FixT. Given the oxygen sensitivity of the FixT Fe-S cluster, these results strongly suggest that Fe-S-bound FixT resists Lon proteolysis. We hypothesize that the Fe-S cluster may increase FixT thermodynamic stability or induce conformational or oligomeric changes that block degron recognition.
Mounting evidence in bacteria and eukaryotes points to an ancient role for Lon in degrading Fe-S proteins and regulating Fe-S assembly pathways. In Salmonella enterica, Lon selectively degrades the Fe-S protein FeoC under aerobic conditions, thereby destabilizing FeoB and reducing ferrous iron transport (56). A recent proteomic study in E. coli identified several putative Lon substrates that bind Fe-S cofactors, as well as the Fe-S assembly protein IscS (58). In eukaryotes, multiple substrates of the mitochondrial Lon homologue have been identified, many of which are Fe-S-binding metabolic enzymes or Fe-S cluster assembly proteins (57, 59–64). Moreover, disrupting mitochondrial Fe-S clusters, either genetically or via oxidative stress, enhances the degradation of several Fe-S-binding Lon substrates (57, 59). Lon family proteases may be particularly suited to detecting damage in Fe-S-binding proteins, as a loss of Fe-S clusters can induce conformational changes that reveal hydrophobic regions, which are canonical Lon recognition sites (43, 65).
Importantly, the regulation of FixT levels by proteolysis provides an additional node for signal integration into the FixLJ TCS (Fig. 6). We expect that the FixT Fe-S cluster will respond to physiochemical changes in the cytoplasm. We have shown that the FixT Fe-S cluster is sensitive to ambient air, and thus, perhaps oxygen (or other oxidizing agents) triggers rapid FixT degradation, allowing reset of the FixLJ system upon return to aerobic growth. Moreover, differing affinities of FixL and the FixT Fe-S cluster for oxygen could significantly alter the “on” and “off” kinetics of the FixLJ system. Yet, Fe-S clusters are also known to respond to other inputs, such as nitric oxide stress and the status of iron-sulfur assembly pathways (which can, in turn, reflect iron availability or oxidative stress) (39). Further work is required to more comprehensively determine the signals sensed in vivo by the FixT Fe-S cluster. Finally, Lon itself may relay environmental information into the FixLJ system, as factors like cell cycle stage, adaptor proteins, and/or substrate competition might affect its activity toward FixT (66, 67).
FIG 6.
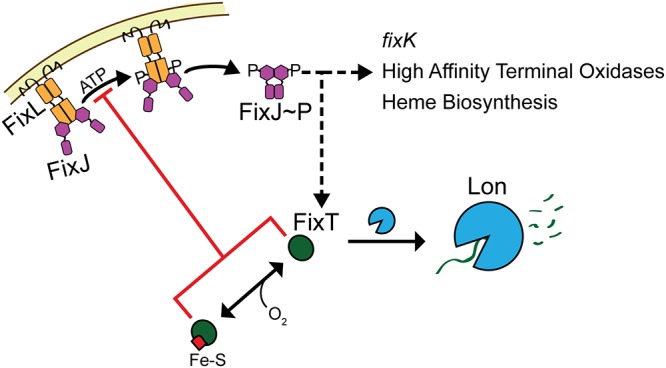
Proteolytic regulation of FixT tunes feedback inhibition of the FixLJ-mediated microoxic response. Shown is a model of FixLJ signaling incorporating posttranslational regulation of FixT. As a feedback inhibitor, FixT is capable of integrating additional information into the FixLJ-signaling system. Low oxygen levels increase FixL autokinase activity, leading to increased FixJ phosphorylation and transcriptional activation (dashed black arrow) of the microaerobic regulon, which includes the fixK transcription factor, high-affinity terminal oxidases, heme biosynthesis genes, and fixT. FixT directly inhibits the autophosphorylation activity of FixL (red line). In addition, FixT can bind an Fe-S cluster that does not affect inhibition of FixL in vitro. Instead, our data support a model in which loss of the Fe-S cluster accelerates the degradation of FixT by the Lon protease, thereby reducing FixT-dependent feedback inhibition. Cellular cues, such as oxygen (or potentially other signals), can affect the state of the FixT Fe-S cluster, and thus, FixT protein levels. In addition, Lon-dependent degradation of FixT may be sensitive to additional factors, such as adaptor proteins.
Although FixT Fe-S-binding appears to be restricted to Caulobacter species, sensory input via proteolysis may be a more universal regulatory theme. A previous study reported an intriguing genetic interaction between fixT and asnO in S. meliloti (23). As yet, no mechanism has been published explaining why asnO is required for FixT activity in vivo. Given our results, perhaps AsnO directly or indirectly protects FixT from proteolysis. We look forward to future studies that will test whether S. meliloti and other species also employ Lon (or other proteases) to regulate FixT levels, and thus FixLJ signaling output, in the cell.
MATERIALS AND METHODS
Strains and plasmids.
All plasmids were cloned using standard molecular biology techniques. The primers used are available upon request. See Table S1 for strain and plasmid information. For in vivo overexpression, FixT constructs were inserted into pMT805, a replicating plasmid for xylose-inducible expression (68). ftsH and hslU in-frame knockout plasmids were generated by cloning homologous upstream and downstream regions into pNTPS138. Plasmids were transformed into C. crescentus CB15 strains by electroporation or triparental mating. For protein expression, FixT, FixL118–495, and FixJ were inserted into an H6-Sumo pET23b expression vector. We note that the FixT sequence referenced in this work is the original open reading frame (ORF) analyzed by Crosson et al. (12). This sequence predicts an earlier start codon, and therefore 3 additional N-terminal amino acids (MRR), than that by the sequence currently annotated in the NCBI. Knockout strains were generated in a CB15 ΔfixT background by a double-recombination strategy (69). ftsH knockouts were recovered as described previously (70).
Strains and plasmids used in this work. Download Table S1, DOCX file, 0.1 MB (24.5KB, docx) .
Copyright © 2020 Stein et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Protein expression and purification.
All constructs were expressed as H6-Sumo fusions in BL21(DE3) cells (Rosetta or BL21 Star) and grown in LB medium. For FixT (for aerobic purification) and FixJ expression, cultures were grown at 37°C, induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density at 600 nm (OD600) of 0.5 to 0.6, and harvested after 1.5 to 3 h. For FixT expression prior to anaerobic purification, cultures were grown at 37°C, transferred to a sealed glass bottle at OD600 of 0.6, and induced with 0.5 mM IPTG for 3.5 h. To express FixL, cultures were grown at 37°C to an OD600 of 0.6, 5-aminolevulinic acid was added to 250 μM, and cells were grown for 1 additional hour before adding 0.5 mM IPTG. Cultures were then grown overnight at 18°C.
To purify FixT, cell pellets were lysed, using an LV1 microfluidizer, in TL buffer (20 mM Tris [pH 8], 150 mM NaCl, 10 mM imidazole, 1 mM dithiothreitol [DTT]), with the addition of 1 mM phenylmethylsulfonyl fluoride (PMSF) and 5 μg/ml DNase I. Lysates were clarified and subjected to Ni-nitrilotriacetic acid (NTA) affinity chromatography (Ni-NTA Superflow resin; Sigma-Aldrich) before and after cleavage with Ulp1 protease. Samples were incubated with 10 mM EDTA for 2 h, followed by dialysis for 2 h at room temperature into T buffer (20 mM Tris [pH 8], 150 mM NaCl, 1 mM DTT). Protein was further purified by gel filtration chromatography (HiLoad 16/600 Superdex, 200 pg) in T buffer. FixL was purified in a similar manner as FixT, but pellets were lysed in LL buffer (20 mM Tris [pH 8], 150 mM NaCl, 10 mM imidazole, 10% glycerol) with the addition of 1 mM PMSF and 5 μg/ml DNase I. Lysates were purified twice by Ni-NTA, followed by gel filtration chromatography in L buffer (20 mM Tris [pH 8], 150 mM NaCl, 10% glycerol). FixJ was purified by double Ni-NTA chromatography in the same manner as FixL and stored in L buffer. All purified proteins were flash frozen in liquid nitrogen and stored at –80°C.
For anaerobic FixT purifications, pellets were lysed in an anaerobic chamber (Coy) by sonication. Ni-NTA purification was carried out as described above but under anaerobic conditions and without Ulp1 cleavage (proteins retained their H6-Sumo tag). Absorption spectra were collected as described for reconstituted FixT samples.
Phenotypic analysis and β-galactosidase assays.
Strains were grown overnight in 2 ml PYE plus 0.15% xylose (chloramphenicol and oxytetracycline added to 1 μg/ml when appropriate) to saturation at 30°C. Cultures were then diluted to an OD660 of 0.025 in 10 ml PYE plus 0.15% xylose and grown at 30°C in 20-mm tubes. Growth curves were made using this dilution as time zero. Phenotypic analyses and β-galactosidase assays were carried out after 24 h of growth.
For phenotypic analyses, equal numbers of cells (2.25 ml·OD660; i.e., volume [ml] = 2.25/OD600 of culture) were centrifuged, lysed in 300 μl BugBuster master mix (Millipore), and clarified before analyzing supernatants by absorption and fluorescence spectroscopy in a Tecan 20M plate reader.
β-Galactosidase assays were performed as described previously after diluting cultures 1:10 in PYE (71).
Phosphorylation assays with FixL.
All phosphorylation assays were carried out at room temperature in kinase buffer (25 mM Tris [pH 7.6], 1 mM MgCl2, 1 mM MnCl2, 1 mM CaCl2, 50 mM KCl). Assays were performed aerobically using the met form of FixL118–495, except for those using Fe-S-reconstituted FixT (rec-FixT) (performed anaerobically with met FixL118–495). For autophosphorylation, FixL (5 μM), FixJ(D55A) (20 μM), and FixT (80 μM) were incubated 5 min before the addition of ATP (1 mM cold ATP, 30 nM [γ-32P]ATP). Samples were quenched with an equal volume of 5× SDS loading buffer (250 mM Tris [pH 6.8], 50% glycerol, 10% SDS, 10 mM DTT, 0.02% bromophenol blue), electrophoresed on Mini-Protean TGX stain-free 12% gels (Bio-Rad), and exposed to a phosphor screen for 1.5 to 3 h before imaging with a Bio-Rad PMI system.
To assay the effect of increasing FixJ(D55A), FixL (5 μM) was incubated with or without FixT (80 μM) and various FixJ(D55A) concentrations. Autophosphorylation was carried out as described above, and 15-min time points were removed. After imaging, bands were quantified using ImageJ and normalized to the reaction without FixT and FixJ. Assays with increasing concentrations of FixT were carried out similarly in the presence of 5 μM FixL and 20 μM FixJ(D55A).
To observe the loss of phosphorylated FixL, FixL (10 μM) was allowed to autophosphorylate in the presence of 40 μM FixJ D55A for 30 min before the removal of nucleotides using Zeba Spin desalting columns (Thermo Fisher). FixL was then added 1:1 to mixes with or without 160 μM FixT and/or 1 mM ADP.
For phosphotransfer assays, FixL (10 μM) was allowed to autophosphorylate for 30 min before mixing 1:1 with 5 μM FixJ and/or 160 μM FixT. Samples were taken after 30 s and analyzed as described above.
Reconstitution of the FixT Fe-S cluster.
Forty-microliter aliquots of FixT were degassed under a vacuum for 2 h and brought into an anaerobic chamber (Coy) at room temperature. Samples were reduced for 15 min with 10 mM DTT and brought to 30 to 40 μM in R buffer (50 mM Tris [pH 8.3], 150 mM NaCl, 5 mM DTT). FeCl3 was added to a 5- to 7-fold excess of FixT and allowed to incubate for 10 min. Na2S was then added equimolar to FeCl3, and the reaction was allowed to proceed for 1.5 to 2 h. For nonreconstituted FixT controls, FeCl3 and Na2S were omitted from the reactions. The reaction mixtures were then desalted using PD-10 desalting columns (GE Healthcare) and concentrated using centrifugal filters. Following concentration, samples were removed from the anaerobic chamber and analyzed by Pierce Coomassie Plus (Bradford) reagent (Thermo Fisher) and a ferrozine (Thermo Fisher) assay, as described previously (72). For absorption spectroscopy, proteins were diluted to 50 μM in 96-well clear-bottom plates (Thermo Scientific), covered with crystal-clear sealing film (Hampton Research), removed from the anaerobic chamber, and immediately analyzed in a Tecan 20M plate reader. The sealing film was then removed, and decay spectra were recorded. Buffer-only spectra were also recorded and subtracted as background.
Western blotting and in vivo protein degradation experiments.
For steady-state overexpression experiments, cultures were grown as described for growth curves and β-galactosidase assays. Cells were collected (1.5 ml·OD660; i.e., volume [ml] = 1.5/OD660 of culture), spun for 2 min at 13,000 × g, and lysed in 200 μl BugBuster master mix plus 2 mM PMSF. Lysates were cleared and denatured in 1× SDS loading buffer. To assay native steady-state levels, primary overnight cultures were diluted to an OD660 of 0.025 in 100 ml of PYE plus 0.15% xylose in a 250-ml flask and grown for 24 h. Samples were spun down, and cell pellets were resuspended in T buffer (2.5 ml/OD660; i.e., volume [ml] = 2.5 × OD660 of culture). Three milliliters of each sample was lysed using an LV1 microfluidizer, cleared by centrifugation, and denatured with 1× SDS loading buffer. For in vivo degradation experiments, overnight cultures were diluted to an OD660 of 0.1 in 10 or 4 ml (20-mm tubes) and grown to an OD660 of ∼0.4. The cells were then treated with 25 μg/ml oxytetracycline to stop translation. Samples were removed at the indicated times (Fig. 5C and D) after oxytetracycline addition (0.3 ml·OD660; i.e., volume [ml] = 0.3/OD660 of culture), spun for 2 min at 13,000 × g, resuspended with 40 μl of 2.5× loading buffer, and frozen at −80°C. After thawing, samples were treated with 250 U Benzonase (Sigma-Aldrich) for 15 min before boiling for 3 min at 95°C.
All Western blot samples were electrophoresed on Mini-Protean TGX stain-free 4 to 20% (wt/vol) precast gels (Bio-Rad). Proteins were transferred to Immobilon-P membranes (EMD Millipore) or Immun-Blot low-fluorescence (LF) polyvinylidene difluoride (PVDF; Bio-Rad) in a wet transfer apparatus. Membranes were probed with polyclonal antibodies (produced by Josman, LLC) raised against FixT (1:4,000 dilution), incubated with either goat anti-rabbit IgG-horseradish peroxidase (IgG-HRP) conjugate (1:5,000 dilution; Thermo Fisher) or goat anti-rabbit IgG cross-adsorbed secondary antibody, Alexa Fluor 488 (1:1,000 dilution; Invitrogen), and, when appropriate, developed with SuperSignal West Femto maximum sensitivity substrate (Thermo Fisher). The blots were imaged using a Bio-Rad ChemiDoc MP Imager. Bands were quantified using the ImageJ software and normalized to the values at time zero for each blot. For each lon+ degradation experiment, curves were fit to a single exponential decay (R2 > 0.95) in Prism (GraphPad) to determine a half-life, which was then averaged across three independent experiments.
ACKNOWLEDGMENTS
We thank Tao Pan and Eric L. Hegg for the use of equipment, and we thank John Anderson, Peter Chien, and members of the Crosson laboratory for helpful discussion.
The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under awards F32 GM128283 (to B.J.S.) and R01 GM087353 and R35 GM131762 (to S.C.).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We declare no conflicts of interest.
Footnotes
Citation Stein BJ, Fiebig A, Crosson S. 2020. Feedback control of a two-component signaling system by an Fe-S-binding receiver domain. mBio 11:e03383-19. https://doi.org/10.1128/mBio.03383-19.
Contributor Information
Patricia J. Kiley, University of Wisconsin—Madison.
Dianne K. Newman, California Institute of Technology.
REFERENCES
- 1.Stock AM, Robinson VL, Goudreau PN. 2000. Two-component signal transduction. Annu Rev Biochem 69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 2.Rood KL, Clark NE, Stoddard PR, Garman SC, Chien P. 2012. Adaptor-dependent degradation of a cell-cycle regulator uses a unique substrate architecture. Structure 20:1223–1232. doi: 10.1016/j.str.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iniesta AA, McGrath PT, Reisenauer A, McAdams HH, Shapiro L. 2006. A phospho-signaling pathway controls the localization and activity of a protease complex critical for bacterial cell cycle progression. Proc Natl Acad Sci U S A 103:10935–10940. doi: 10.1073/pnas.0604554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paul R, Jaeger T, Abel S, Wiederkehr I, Folcher M, Biondi EG, Laub MT, Jenal U. 2008. Allosteric regulation of histidine kinases by their cognate response regulator determines cell fate. Cell 133:452–461. doi: 10.1016/j.cell.2008.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenbach M. 1996. Control of bacterial chemotaxis. Mol Microbiol 20:903–910. doi: 10.1111/j.1365-2958.1996.tb02531.x. [DOI] [PubMed] [Google Scholar]
- 6.Nesper J, Hug I, Kato S, Hee C-S, Habazettl JM, Manfredi P, Grzesiek S, Schirmer T, Emonet T, Jenal U. 2017. Cyclic di-GMP differentially tunes a bacterial flagellar motor through a novel class of CheY-like regulators. Elife 6:e28842. doi: 10.7554/eLife.28842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaczmarczyk A, Hochstrasser R, Vorholt JA, Francez-Charlot A. 2014. Complex two-component signaling regulates the general stress response in Alphaproteobacteria. Proc Natl Acad Sci U S A 111:E5196–E5204. doi: 10.1073/pnas.1410095111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lori C, Kaczmarczyk A, de Jong I, Jenal U. 2018. A single-domain response regulator functions as an integrating hub to coordinate general stress response and development in alphaproteobacteria. mBio 9:e00809-18. doi: 10.1128/mBio.01534-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reyes Ruiz LM, Fiebig A, Crosson S. 2019. Regulation of bacterial surface attachment by a network of sensory transduction proteins. PLoS Genet 15:e1008022. doi: 10.1371/journal.pgen.1008022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perego M, Hanstein C, Welsh KM, Djavakhishvili T, Glaser P, Hoch JA. 1994. Multiple protein-aspartate phosphatases provide a mechanism for the integration of diverse signals in the control of development in B. subtilis. Cell 79:1047–1055. doi: 10.1016/0092-8674(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 11.Gottschlich L, Bortfeld-Miller M, Gäbelein C, Dintner S, Vorholt JA. 2018. Phosphorelay through the bifunctional phosphotransferase PhyT controls the general stress response in an alphaproteobacterium. PLoS Genet 14:e1007294. doi: 10.1371/journal.pgen.1007294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crosson S, McGrath PT, Stephens C, McAdams HH, Shapiro L. 2005. Conserved modular design of an oxygen sensory/signaling network with species-specific output. Proc Natl Acad Sci U S A 102:8018–8023. doi: 10.1073/pnas.0503022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foussard M, Garnerone AM, Ni F, Soupène E, Boistard P, Batut J. 1997. Negative autoregulation of the Rhizobium meliloti fixK gene is indirect and requires a newly identified regulator, FixT. Mol Microbiol 25:27–37. doi: 10.1046/j.1365-2958.1997.4501814.x. [DOI] [PubMed] [Google Scholar]
- 14.Garnerone AM, Cabanes D, Foussard M, Boistard P, Batut J. 1999. Inhibition of the FixL sensor kinase by the FixT protein in Sinorhizobium meliloti. J Biol Chem 274:32500–32506. doi: 10.1074/jbc.274.45.32500. [DOI] [PubMed] [Google Scholar]
- 15.Gilles-Gonzalez MA, Ditta GS, Helinski DR. 1991. A haemoprotein with kinase activity encoded by the oxygen sensor of Rhizobium meliloti. Nature 350:170–172. doi: 10.1038/350170a0. [DOI] [PubMed] [Google Scholar]
- 16.Monson EK, Weinstein M, Ditta GS, Helinski DR. 1992. The FixL protein of Rhizobium meliloti can be separated into the heme-binding oxygen-sensing domain and a functional C-terminal kinase domain. Proc Natl Acad Sci U S A 89:4280–4284. doi: 10.1073/pnas.89.10.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lois AF, Weinstein M, Ditta GS, Helinski DR. 1993. Autophosphorylation and phosphatase activities of the oxygen-sensing protein FixL of Rhizobium meliloti are coordinately regulated by oxygen. J Biol Chem 268:4370–4375. [PubMed] [Google Scholar]
- 18.Reyrat JM, David M, Blonski C, Boistard P, Batut J. 1993. Oxygen-regulated in vitro transcription of Rhizobium meliloti nifA and fixK genes. J Bacteriol 175:6867–6872. doi: 10.1128/jb.175.21.6867-6872.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batut J, Daveran-Mingot ML, David M, Jacobs J, Garnerone AM, Kahn D. 1989. fixK, a gene homologous with fnr and crp from Escherichia coli, regulates nitrogen fixation genes both positively and negatively in Rhizobium meliloti. EMBO J 8:1279–1286. doi: 10.1002/j.1460-2075.1989.tb03502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon R, Kahn D. 2004. Genetic regulation of biological nitrogen fixation. Nat Rev Microbiol 2:621–631. doi: 10.1038/nrmicro954. [DOI] [PubMed] [Google Scholar]
- 21.David M, Daveran ML, Batut J, Dedieu A, Domergue O, Ghai J, Hertig C, Boistard P, Kahn D. 1988. Cascade regulation of nif gene expression in Rhizobium meliloti. Cell 54:671–683. doi: 10.1016/s0092-8674(88)80012-6. [DOI] [PubMed] [Google Scholar]
- 22.Fischer HM. 1994. Genetic regulation of nitrogen fixation in rhizobia. Microbiol Rev 58:352–386. doi: 10.1128/MMBR.58.3.352-386.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergès H, Checroun C, Guiral S, Garnerone AM, Boistard P, Batut J. 2001. A glutamine-amidotransferase-like protein modulates FixT anti-kinase activity in Sinorhizobium meliloti. BMC Microbiol 1:6. doi: 10.1186/1471-2180-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sagot B, Gaysinski M, Mehiri M, Guigonis J-M, Le Rudulier D, Alloing G. 2010. Osmotically induced synthesis of the dipeptide N-acetylglutaminylglutamine amide is mediated by a new pathway conserved among bacteria. Proc Natl Acad Sci U S A 107:12652–12657. doi: 10.1073/pnas.1003063107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groisman EA. 2016. Feedback control of two-component regulatory systems. Annu Rev Microbiol 70:103–124. doi: 10.1146/annurev-micro-102215-095331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cinquin O, Demongeot J. 2002. Roles of positive and negative feedback in biological systems. C R Biol 325:1085–1095. doi: 10.1016/s1631-0691(02)01533-0. [DOI] [PubMed] [Google Scholar]
- 27.Gouterman M. 1961. Spectra of porphyrins. J Mol Spectrosc 6:138–163. doi: 10.1016/0022-2852(61)90236-3. [DOI] [Google Scholar]
- 28.Seybold PG, Gouterman M. 1969. Porphyrins XIII: fluorescence spectra and quantum yields. J Mol Spectrosc 31:1–13. doi: 10.1016/0022-2852(69)90335-X. [DOI] [Google Scholar]
- 29.Dailey HA, Dailey TA, Gerdes S, Jahn D, Jahn M, O’Brian MR, Warren MJ. 2017. Prokaryotic heme biosynthesis: multiple pathways to a common essential product. Microbiol Mol Biol Rev 81:e00048-16. doi: 10.1128/MMBR.00048-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilles-Gonzalez M-A, Gonzalez G, Sousa EHS, Tuckerman J. 2008. Oxygen-sensing histidine-protein kinases: assays of ligand binding and turnover of response-regulator substrates. Methods Enzymol 437:173–189. doi: 10.1016/S0076-6879(07)37010-9. [DOI] [PubMed] [Google Scholar]
- 31.Sousa EHS, Gonzalez G, Gilles-Gonzalez M-A. 2005. Oxygen blocks the reaction of the FixL-FixJ complex with ATP but does not influence binding of FixJ or ATP to FixL. Biochemistry 44:15359–15365. doi: 10.1021/bi051661h. [DOI] [PubMed] [Google Scholar]
- 32.Gilles-Gonzalez MA, Gonzalez G. 1993. Regulation of the kinase activity of heme protein FixL from the two-component system FixL/FixJ of Rhizobium meliloti. J Biol Chem 268:16293–16297. [PubMed] [Google Scholar]
- 33.Freibert S-A, Weiler BD, Bill E, Pierik AJ, Mühlenhoff U, Lill R. 2018. Biochemical reconstitution and spectroscopic analysis of iron-sulfur proteins. Methods Enzymol 599:197–226. doi: 10.1016/bs.mie.2017.11.034. [DOI] [PubMed] [Google Scholar]
- 34.Mulholland SE, Gibney BR, Rabanal F, Dutton PL. 1999. Determination of nonligand amino acids critical to [4Fe-4S]2+/+ assembly in ferredoxin maquettes. Biochemistry 38:10442–10448. doi: 10.1021/bi9908742. [DOI] [PubMed] [Google Scholar]
- 35.Nakamaru-Ogiso E, Yano T, Ohnishi T, Yagi T. 2002. Characterization of the iron-sulfur cluster coordinated by a cysteine cluster motif (CXXCXXXCX27C) in the Nqo3 subunit in the proton-translocating NADH-quinone oxidoreductase (NDH-1) of Thermus thermophilus HB-8. J Biol Chem 277:1680–1688. doi: 10.1074/jbc.M108796200. [DOI] [PubMed] [Google Scholar]
- 36.Huang Q, Hong X, Hao Q. 2008. SNAP-25 is also an iron-sulfur protein. FEBS Lett 582:1431–1436. doi: 10.1016/j.febslet.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoch DC, Arnon DI, Sweeney WV. 1975. Characterization of two soluble ferredoxins as distinct from bound iron-sulfur proteins in the photosynthetic bacterium Rhodospirillum rubrum. J Biol Chem 250:8330–8336. [PubMed] [Google Scholar]
- 38.Crack JC, Green J, Thomson AJ, Le Brun NE. 2014. Iron-sulfur clusters as biological sensors: the chemistry of reactions with molecular oxygen and nitric oxide. Acc Chem Res 47:3196–3205. doi: 10.1021/ar5002507. [DOI] [PubMed] [Google Scholar]
- 39.Crack JC, Green J, Thomson AJ, Le Brun NE. 2012. Iron–sulfur cluster sensor-regulators. Curr Opin Chem Biol 16:35–44. doi: 10.1016/j.cbpa.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 40.Khoroshilova N, Popescu C, Münck E, Beinert H, Kiley PJ. 1997. Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc Natl Acad Sci U S A 94:6087–6092. doi: 10.1073/pnas.94.12.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orme-Johnson WH, Orme-Johnson NR. 1982. Iron-sulfur proteins: the problem of determining cluster type, p 68–96. In Spiro TG. (ed), Iron-sulfur proteins, 1st ed. Wiley-Interscience, New York, NY. [Google Scholar]
- 42.Jervis AJ, Crack JC, White G, Artymiuk PJ, Cheesman MR, Thomson AJ, Le Brun NE, Green J. 2009. The O2 sensitivity of the transcription factor FNR is controlled by Ser24 modulating the kinetics of [4Fe-4S] to [2Fe-2S] conversion. Proc Natl Acad Sci U S A 106:4659–4664. doi: 10.1073/pnas.0804943106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leal SS, Gomes CM. 2010. Iron-sulfur clusters, protein folds, and ferredoxin stability, p 81–96. In Gomes CM, Wittung-Stafshede P (ed), Protein folding and metal ions: mechanisms, biology and disease. CRC Press, Boca Raton, FL. [Google Scholar]
- 44.Mettert EL, Kiley PJ. 2005. ClpXP-dependent proteolysis of FNR upon loss of its O2-sensing [4Fe–4S]. J Mol Biol 354:220–232. doi: 10.1016/j.jmb.2005.09.066. [DOI] [PubMed] [Google Scholar]
- 45.Bobik C, Meilhoc E, Batut J. 2006. FixJ: a major regulator of the oxygen limitation response and late symbiotic functions of Sinorhizobium meliloti. J Bacteriol 188:4890–4902. doi: 10.1128/JB.00251-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Grau R, Perego M, Hoch JA. 1997. A novel histidine kinase inhibitor regulating development in Bacillus subtilis. Genes Dev 11:2569–2579. doi: 10.1101/gad.11.19.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowland SL, Burkholder WF, Cunningham KA, Maciejewski MW, Grossman AD, King GF. 2004. Structure and mechanism of action of Sda, an inhibitor of the histidine kinases that regulate initiation of sporulation in Bacillus subtilis. Mol Cell 13:689–701. doi: 10.1016/s1097-2765(04)00084-x. [DOI] [PubMed] [Google Scholar]
- 48.Cunningham KA, Burkholder WF. 2009. The histidine kinase inhibitor Sda binds near the site of autophosphorylation and may sterically hinder autophosphorylation and phosphotransfer to Spo0F. Mol Microbiol 71:659–677. doi: 10.1111/j.1365-2958.2008.06554.x. [DOI] [PubMed] [Google Scholar]
- 49.Barak R, Welch M, Yanovsky A, Oosawa K, Eisenbach M. 1992. Acetyladenylate or its derivative acetylates the chemotaxis protein CheY in vitro and increases its activity at the flagellar switch. Biochemistry 31:10099–10107. doi: 10.1021/bi00156a033. [DOI] [PubMed] [Google Scholar]
- 50.Desai SK, Kenney LJ. 2017. To ∼P or not to ∼P? Non-canonical activation by two-component response regulators. Mol Microbiol 103:203–213. doi: 10.1111/mmi.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 52.Gur E. 2013. The Lon AAA+ protease, p 35–51. In Dougan DA. (ed), Regulated proteolysis in microorganisms. Springer, Dordrecht, the Netherlands. [Google Scholar]
- 53.Mizusawa S, Gottesman S. 1983. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc Natl Acad Sci U S A 80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jonas K, Liu J, Chien P, Laub MT. 2013. Proteotoxic stress induces a cell-cycle arrest by stimulating Lon to degrade the replication initiator DnaA. Cell 154:623–636. doi: 10.1016/j.cell.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J, Zeinert R, Francis L, Chien P. 2019. Lon recognition of the replication initiator DnaA requires a bipartite degron. Mol Microbiol 111:176–186. doi: 10.1111/mmi.14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H, Lee H, Shin D. 2015. Lon-mediated proteolysis of the FeoC protein prevents Salmonella enterica from accumulating the Fe(II) transporter FeoB under high-oxygen conditions. J Bacteriol 197:92–98. doi: 10.1128/JB.01826-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Major T, Janowsky von B, Ruppert T, Mogk A, Voos W. 2006. Proteomic analysis of mitochondrial protein turnover: identification of novel substrate proteins of the matrix protease Pim1. Mol Cell Biol 26:762–776. doi: 10.1128/MCB.26.3.762-776.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arends J, Griego M, Thomanek N, Lindemann C, Kutscher B, Meyer HE, Narberhaus F. 2018. An integrated proteomic approach uncovers novel substrates and functions of the Lon protease in Escherichia coli. Proteomics 18:e1800080. doi: 10.1002/pmic.201800080. [DOI] [PubMed] [Google Scholar]
- 59.Bota DA, Davies K. 2002. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol 4:674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- 60.Bender T, Lewrenz I, Franken S, Baitzel C, Voos W. 2011. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol Biol Cell 22:541–554. doi: 10.1091/mbc.E10-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ngo JK, Davies K. 2009. Mitochondrial Lon protease is a human stress protein. Free Radic Biol Med 46:1042–1048. doi: 10.1016/j.freeradbiomed.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guillon B, Bulteau AL, Donzé MW, Schmucker S, Friguet B, Puccio H, Drapier JC, Bouton C. 2009. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe–S proteins. FEBS J 276:1036–1047. doi: 10.1111/j.1742-4658.2008.06847.x. [DOI] [PubMed] [Google Scholar]
- 63.Song J-Y, Marszalek J, Craig EA. 2012. Cysteine desulfurase Nfs1 and Pim1 protease control levels of Isu, the Fe-S cluster biogenesis scaffold. Proc Natl Acad Sci U S A 109:10370–10375. doi: 10.1073/pnas.1206945109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ciesielski SJ, Schilke B, Marszalek J, Craig EA. 2016. Protection of scaffold protein Isu from degradation by the Lon protease Pim1 as a component of Fe–S cluster biogenesis regulation. Mol Biol Cell 27:1060–1068. doi: 10.1091/mbc.E15-12-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gur E, Sauer RT. 2008. Recognition of misfolded proteins by Lon, a AAA+ protease. Genes Dev 22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gora KG, Cantin A, Wohlever M, Joshi KK, Perchuk BS, Chien P, Laub MT. 2013. Regulated proteolysis of a transcription factor complex is critical to cell cycle progression in Caulobacter crescentus. Mol Microbiol 87:1277–1289. doi: 10.1111/mmi.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vass RH, Zeinert RD, Chien P. 2016. Protease regulation and capacity during Caulobacter growth. Curr Opin Microbiol 34:75–81. doi: 10.1016/j.mib.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thanbichler M, Iniesta AA, Shapiro L. 2007. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res 35:e137. doi: 10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ried JL, Collmer A. 1987. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in Gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 57:239–246. doi: 10.1016/0378-1119(87)90127-2. [DOI] [PubMed] [Google Scholar]
- 70.Fischer B, Rummel G, Aldridge P, Jenal U. 2002. The FtsH protease is involved in development, stress response and heat shock control in Caulobacter crescentus. Mol Microbiol 44:461–478. doi: 10.1046/j.1365-2958.2002.02887.x. [DOI] [PubMed] [Google Scholar]
- 71.Foreman R, Fiebig A, Crosson S. 2012. The LovK-LovR two-component system is a regulator of the general stress pathway in Caulobacter crescentus. J Bacteriol 194:3038–3049. doi: 10.1128/JB.00182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams HL, Johnson DJ, Haut MJ. 1977. Simultaneous spectrophotometry of Fe2+ and Cu2+ in serum denatured with guanidine hydrochloride. Clin Chem 23:237–240. doi: 10.1093/clinchem/23.2.237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of porphyrin signatures to protoporphyrin IX and effects of Fe supplementation. (A) Absorption spectrum of ΔfixT mutant lysate compared with protoporphyrin IX (Sigma Aldrich) in dimethyl sulfoxide (DMSO). (B) Fluorescence emission scan of wild-type (WT), ΔfixT mutant, and Fe-treated ΔfixT mutant lysates (excitation [Ex.], 404 nm). (C) Absorption spectra of lysates from ΔfixT mutant cells grown with (black) or without (gray) 10 μM FeSO4. Inset highlights Q bands in the 500- to 650-nm region. (D) Optical density at 660 nm (OD660) of cultures grown for 24 h with (black bars) or without (gray bars) the addition of 10 μM FeSO4 (average ± SD, n > 3). *, P < 0.001, one-way ANOVA; P < 0.05, Dunnett’s posttest comparison to wild type (WT) untreated. Download FIG S1, TIF file, 0.8 MB (832.7KB, tif) .
Copyright © 2020 Stein et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Growth of wild-type and ΔfixT mutant strains. Growth curves during exponential phase of wild-type (WT, blue) and ΔfixT mutant (orange) strains measured by the optical density at 660 nm (OD660). The points are the averages ± SD of the results from 3 biological replicates. Strains were grown under oxygen-limited conditions as in Fig. 1. Download FIG S2, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2020 Stein et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains and plasmids used in this work. Download Table S1, DOCX file, 0.1 MB (24.5KB, docx) .
Copyright © 2020 Stein et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.