

Abstract
Purpose
Retinoid isomerohydrolase RPE65 has received a tremendous amount of attention due to successful clinical gene therapy for Leber congenital amaurosis (LCA) cases caused by RPE65 mutations. This study aimed to evaluate the frequency of RPE65 mutations and the associated phenotypes based on exome sequencing.
Methods
RPE65 variants were collected from exome sequencing data obtained from 2133 probands with different forms of hereditary retinal degeneration (HRD). Clinical data were collected from probands with homozygous or compound heterozygous variants in RPE65. Associated phenotypes were characterized based on clinical data.
Results
Biallelic RPE65 mutations were detected in 18 families, including eight with LCA, five with early‐onset retinal degeneration, four with fundus albipunctatus‐like (FA‐like) changes and one with high hyperopia. These cases accounted for approximately 3.0% (8/269) of LCA and 0.8% (18/2133) of HRD cases. An almost identical FA‐like change was identified in seven patients from four unrelated families with RPE65 mutations. Classification of mutations suggested that FA‐like changes may be associated with biallelic missense mutations in RPE65.
Conclusion
Fundus albipunctatus‐like (FA‐like) change, a common characteristic fundus sign in RPE65 biallelic mutations, was unexpected but was confirmed by the finding that affected siblings from different families exhibited similar phenotypes. These results enrich our understanding of RPE65 mutation frequencies and their associated phenotypic variants.
Keywords: fundus albipunctatus, mutation frequency, phenotype, RPE65
Introduction
Retinoid isomerohydrolase RPE65, an enzyme encoded by the RPE65 gene (HGNC ID: 10294, OMIM: 180069) (Nicoletti et al. 1995), is responsible for the conversion of all‐trans‐retinyl esters to 11‐cis‐retinol during visual phototransduction in the retinal pigment epithelium (Xue et al. 2004; Moiseyev et al. 2005). Biallelic mutations in RPE65 are an important cause of Leber's congenital amaurosis (LCA) (Marlhens et al. 1997; den Hollander et al. 2008; Cideciyan 2010), the most severe form of hereditary retinal degeneration (HRD) that causes individuals to be born blind. RPE65‐associated LCA has gained a great deal of public attention as a result of a clinical gene therapy (Lee & Lotery 2017; Apte 2018; Kumaran et al. 2018); this new gene therapy targets LCA caused by RPE65 mutations and was recently approved by the Food and Drug Administration as the first directly administered gene therapy of its kind (https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm589467.htm).
The identification of patients with RPE65 mutations is a prerequisite for developing RPE65‐targeted gene therapies. Patients with RPE65 mutations frequently manifest as LCA (Weleber et al. 1993; Gu et al. 1997; Morimura et al. 1998; Cideciyan 2010), occasionally present as early‐onset HRD or retinitis pigmentosa (Gu et al. 1997; Morimura et al. 1998) and rarely present as fundus albipunctatus (FA) (Schatz et al. 2011; Hull et al. 2016; Yang et al. 2017) or cone‐rod dystrophy (Jakobsson et al. 2014). RPE65 mutations are thought to be responsible for approximately 6% of all LCA cases in Caucasians (den Hollander et al. 2008) but for only a few LCA cases in Chinese populations (Xu et al. 2016). However, most of these results are based on analyses of subgroups of patients that consist mostly of those with LCA. Neither the frequency of RPE65 mutations in all forms of HRD nor the variety of associated phenotypes has been well studied. As one disease may mimic another due to phenotypic variation or differences among the stages of the diseases, it is important to determine whether other forms of retinal degeneration are caused by RPE65 mutations. Genotype‐guided phenotype characterization may provide a different view of this than can be achieved by other approaches, such as phenotype‐guided genotype analysis performed in one or a set of candidate genes.
Whole exome sequencing (WES) and targeting exome sequencing (TES) provide genotype information for a group of genes in individuals with different phenotypes. These data provide a practical platform for performing genotype‐guided phenotypic characterization (to determine to what extent the phenotype may vary) of genes in patients with different forms of related diseases have been analysed by WES. In the current study, sequencing variants of RPE65 were selected from our in‐house data obtained from 2133 probands with suspected HRD who underwent WES or TES analysis. Biallelic potential pathogenic mutations were identified in 18 families with phenotypes associated with LCA, early‐onset retinal degeneration (EORD), FA‐like (FA‐like) change or high hyperopia. Two of the 18 families with LCA were previously reported and also included in the current analysis, in which RPE65 mutations were detected by WES or Sanger sequencing. These results provide a brief overview of RPE65 mutation frequencies in addition to data indicating the phenotypic variation that is associated with RPE65 mutations.
Materials and Methods
Probands and family members
Data obtained from probands with different forms of genetic eye diseases and their available family members were collected from our Pediatric and Genetic Clinic, Zhongshan Ophthalmic Center, Guangzhou, China. The clinical data were recorded and the genomic DNA was prepared in our laboratory at the State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center. Written informed consent in accordance with the tenets of the Declaration of Helsinki was obtained from patients’ guardians prior to the collection of clinical data and venous blood samples. This study was approved by the institutional review board of the Zhongshan Ophthalmic Center. Genomic DNA was prepared from leucocytes obtained from venous blood using a previously described method (Wang et al. 2010).
Mutation detection
Our laboratory has so far collected in‐house exome sequencing data from 2133 probands with different forms of HRD, including WES data obtained from 1139 probands, TES data obtained from 994 probands and Sanger sequencing from one proband. The procedure used to perform WES was described in our previous study (Jiang et al. 2015; Li et al. 2015a). In brief, exome as well as their adjacent intronic regions (at least 20 bp) were captured using the Agilent SureSelect Human All Exon Enrichment Kit (50M; Agilent, Santa Clara, CA, USA) array. The exome‐enriched DNA fragments from the sample were sequenced by the Illumina HiSeq system (Illumina, San Diego, CA, USA) with an average sequencing depth of 125‐fold. The reads were aligned with the consensus sequence (UCSC hg19) to detect variants by the Burrows‐Wheeler Aligner (http://bio-bwa.sourceforge.net/). Single nucleotide polymorphisms and small insertions and deletions (Indels) were detected by samtools (http://samtools.sourceforge.net/) on the basis of a Bayesian statistical algorithm. Variant annotations were performed using SnpEff (http://snpeff.sourceforge.net/) and annovar (http://annovar.openbioinformatics.org/en/latest/), whereas the functional prediction of variants was predicted by dbNSFP (http://varianttools.sourceforge.net/Annotation/DbNSFP). Targeting exome sequencing (TES) was performed in our laboratory and targeted 126 genes (including RPE65 and many related genes) frequently mutated in genetic eye diseases in the Chinese population. Sequencing variations in RPE65 were retrieved from the 2133 probands.
Variants detected by WES and TES were initially filtered by multi‐step bioinformatics analyses, as described in our previous study (Jiang et al. 2015; Li et al. 2015a). Candidate variants were also filtered by comparing our data with those available in existing databases, including the Human Genome Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), the 1000 Genomes (http://phase1browser.1000genomes.org/) and the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/). The possible impact of missense changes was predicted using the online tools SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html) (Kumar et al. 2009) or PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) (Flanagan et al. 2010), MutationTaster (http://mutationtaster.org/) and CADD (https://cadd.gs.washington.edu/). Effect on splicing of intronic variants was predicted by the Berkeley Drosophila Genome Project (http://www.fruitfly.org/ [in the public domain]) and Human Splicing Finder (http://umd.be/HSF3/). Candidate disease‐associated variants were confirmed by Sanger‐dideoxy sequencing, and cosegregation in family members was further evaluated.
Probands with biallelic rare variants in RPE65 were selected for further analysis. In rare cases, one heterozygous mutation, c.1430A>G (p.Asp477Gly) (Bowne et al. 2011), has been reported to cause autosomal dominant retinitis pigmentosa, and this mutation was therefore checked.
Phenotype characterization
Routine clinical data obtained from all probands with biallelic RPE65 mutations were reviewed. Additional examinations were carried when necessary, including electroretinogram and fundus photographs. Parents or siblings were examined if available. Phenotypes were classified based on symptoms, visual acuity, fundus changes and electroretinogram data. Two previously reported families with LCA in which RPE65 mutations were detected by either WES or Sanger sequencing were also included in the current analysis.
Results
Biallelic mutations detected in RPE65
In total, biallelic mutations were detected in 18 families (Table 1, Fig 1), with 17 detected by WES or TES and one was detected by Sanger sequencing. These mutations involved 27 different rare nucleotide changes, including missense variants (16), stopgain (four), frameshift insertions (two), frameshift deletion (one), splicing donors (two) and splicing acceptors (two). Twelve mutations were novel while the other 15 have been reported in previous studies (Table 1). Of these 27 variants, 26 were predicted to be damaging by routine SIFT or PolyPhen‐2 tools. The rest one, p.Tyr122His was detected in a proband with high hyperopia and predicted to be benign or tolerated by Polyphen‐2 and SIFT but to be possible disease causing by MutationTaster and CADD. All but one mutation (c.295G>A) was very rare, with no homozygote detected based on the existing database, and the frequencies ranged from none to 0.0002 for 27 mutations). The c.295G>A (p.Val99Ile) mutation had a frequency of 0.0012 for all and 0.0048 for Southern Han, with only one homozygote detected based on the ExAC and 1000 Genomes database. These mutations were confirmed by Sanger sequencing and cosegregated with the disease in the families (Fig 1 and Fig. S1). Of the 18 families with RPE65 mutations, parental analysis was available in 17 families and the results suggested that the two mutations in the 17 families were located in different chromosomes while the parents from one family (F03) were not available. It is difficult to cover the c.713C>G and c.1543C>T mutations in family F03 by the same primers because of a 9738 bp distance between them. However, these two variants are more likely to be biallelic as these two variants were neither observed in the 2132 of the 2133 HRD cases nor in 2871 cases with non‐HRD diseases in our in‐house exome data. None of the probands in the 18 families had mutations in the RDH5 or RLBP1 genes that are known to be associated with FA. The c.1430A>G (p.Asp477Gly) variant associated with autosomal dominant retinitis pigmentosa was not detected in any of the 2133 probands.
Table 1.
Rare variants in biallelic status in 18 probands with genetic eye diseases.
| Variant | Exon | Position at chr01 | Nuleotide change | Effect | PolyPhen‐2 Score | SIFT Score | Mutation taster score | CADD score | Berkeley Drosophila Genome Project | Human Splicing Finder | Exome Aggregation Consortium allele frequency | 1000 genome | Human Genome Mutation Database | rs ID | First reported | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | East Asian | Homo | All | South Han | ||||||||||||||
| 1 | 2 | 68914307 | c.94G>T | p.Gly32Cys | PD (0.985) | D (0.000) | DC (1.000) | 33.0 | / | / | 1/118428 | 0/8592 | 0 | NA | NA | DM | NA | Astuti et al. (2016) |
| 2 | 3 | 68912507 | c.131G>A | p.Arg44Gln | PD (1.000) | D (0.000) | DC (1.000) | 32.0 | / | / | 7/121406 | 0/8654 | 0 | 0.0002 | 0.0000 | DM | rs61751282 | Simovich et al. (2001) |
| 3 | 3 | 68912448 | c.190C>G | p.Gln64Glu | PD (0.996) | D (0.000) | DC (1.000) | 25.8 | / | / | NA | NA | NA | NA | NA | NA | NA | Novel |
| 4 | 3 | 68912438 | c.200T>G | p.Leu67Arg | PD (1.000) | D (0.000) | DC (1.000) | 28.7 | / | / | NA | NA | NA | NA | NA | DM | NA | Xu et al. (2012) |
| 5 | 4 | 68910541 | c.271C>T | p.Arg91Trp | PD (0.999) | D (0.010) | DC (1.000) | 27.4 | / | / | 10/120738 | 1/8620 | 0 | 0.0002 | 0.0000 | DM | rs61752871 | Morimura et al. (1998) |
| 6 | 4 | 68910517 | c.295G>A | p.Val99Ile | B (0.077) | D (0.020) | DC (1.000) | 23.8 | / | / | 36/120964 | 31/8632 | 1 | 0.0012 | 0.0048 | NA | rs143056561 | Li et al. (2011) |
| 7 | 5 | 68910345 | c.364T>C | p.Tyr122His | B (0.046) | T (0.120) | DC (1.000) | 23.0 | / | / | NA | NA | NA | NA | NA | NA | NA | Novel |
| 8 | 5 | 68910279 | c.430T>C | p.Tyr144His | PD (1.000) | D (0.030) | DC (1.000) | 27.4 | / | / | NA | NA | NA | NA | NA | DM | NA | Chen et al. (2013) |
| 9 | 5 | 68910275 | c.434C>A | p.Ala145Asp | PD (0.999) | D (0.000) | DC (1.000) | 26.8 | / | / | NA | NA | NA | NA | NA | DM | NA | Fu et al. (2013) |
| 10 | 5 | 68910216 | c.493C>T | p.Gln165* | / | / | DC (1.000) | 41.0 | / | / | NA | NA | NA | NA | NA | DM | NA | Wang et al. (2015) |
| 11 | 6 | 68906634 | c.545A>G | p.His182Arg | PD (0.993) | D (0.000) | DC (1.000) | 25.4 | / | / | NA | NA | NA | NA | NA | DM | NA | Jacobson et al. (2005) |
| 12 | 6 | 68906540 | c.639dupA | p.Ala214Serfs*20 | / | / | DC (1.000) | / | / | / | NA | NA | NA | NA | NA | DM | NA | Wang et al. (2016) |
| 13 | 7 | 68905256 | c.713C>G | p.Ser238Cys | PD (1.000) | D (0.000) | DC (1.000) | 29.9 | / | / | NA | NA | NA | NA | NA | NA | NA | Novel |
| 14 | 7 | 68905247 | c.722A>G | p.His241Arg | PD (1.000) | D (0.000) | DC (1.000) | 25.9 | / | / | NA | NA | NA | NA | NA | NA | NA | Novel |
| 15 | 8 | 68904907 | c.825C>A | p.Tyr275* | / | / | DC (1.000) | 38.0 | / | / | NA | NA | NA | NA | NA | NA | NA | Novel |
| 16 | 9 | 68904626 | c.997G>C | p.Gly333Arg | PD (1.000) | D (0.010) | DC (1.000) | 34.0 | / | / | NA | NA | NA | NA | NA | NA | NA | Li et al. (2011) |
| 17 | Intron 9 | 68904624 | c.998+1G>A | SD | / | / | DC (1.000) | 33.0 | SSC | SSC | NA | NA | NA | NA | NA | NA | NA | Novel |
| 18 | Intron 9 | 68904000 | c.999‐1G>T | SA | / | / | DC (1.000) | 34.0 | SSC | SSC | NA | NA | NA | NA | NA | NA | NA | Novel |
| 19 | 10 | 68903939 | c.1059dupG | p.Lys354Glufs*11 | / | / | DC (1.000) | 35.0 | / | / | NA | NA | NA | NA | NA | DM | NA | Jacobson et al. (2007) |
| 20 | 10 | 68903931 | c.1067delA | p.Asn356Metfs*17 | / | / | DC (1.000) | 34.0 | / | / | NA | NA | NA | NA | NA | DM | rs281865520 | Marlhens et al. (1997) |
| 21 | 12 | 68897002 | C.1301C>A | p.Ala434Glu | PD (0.999) | T (0.650) | DC (1.000) | 23.7 | / | / | NA | NA | 0 | NA | NA | NA | NA | Novel |
| 22 | 13 | 68896824 | c.1374G>A | p.Trp458* | / | / | DC (1.000) | 43.0 | / | / | NA | NA | NA | NA | NA | DM | NA | Astuti et al. (2016) |
| 23 | 13 | 68896799 | c.1399C>G | p.Pro467Ala | PB (0.918) | D (0.050) | DC (1.000) | 25.7 | / | / | NA | NA | NA | NA | NA | NA | NA | Novel |
| 24 | Intron 13 | 68896747 | c.1450+1delG | SD | / | / | DC (1.000) | / | SSC | SSC | NA | NA | NA | NA | NA | NA | NA | Novel |
| 25 | Intron 13 | 68895611 | c.1451‐1G>A | SA | / | / | DC (1.000) | 34.0 | SSC | SSC | NA | NA | NA | NA | NA | NA | NA | Novel |
| 26 | 14 | 68895558 | c.1503T>G | p.Tyr501* | / | / | DC (1.000) | 36.0 | / | / | NA | NA | NA | NA | NA | NA | NA | Novel |
| 27 | 14 | 68895518 | c.1543C>T | p.Arg515Trp | PD (1.000) | D (0.000) | DC (1.000) | 27.7 | / | / | 2/120986 | 0/8624 | 0 | NA | NA | DM | rs121917745 | Kondo et al. (2004) |
A CADD score over 20 suggests potential damaging.
B = benign, D = damaging, DC = disease causing, PB = possibly damaging, PD = probably damaging, SA = splicing acceptor, SD = splicing donor, SSC = splice site changed, T = tolerated.
Figure 1.
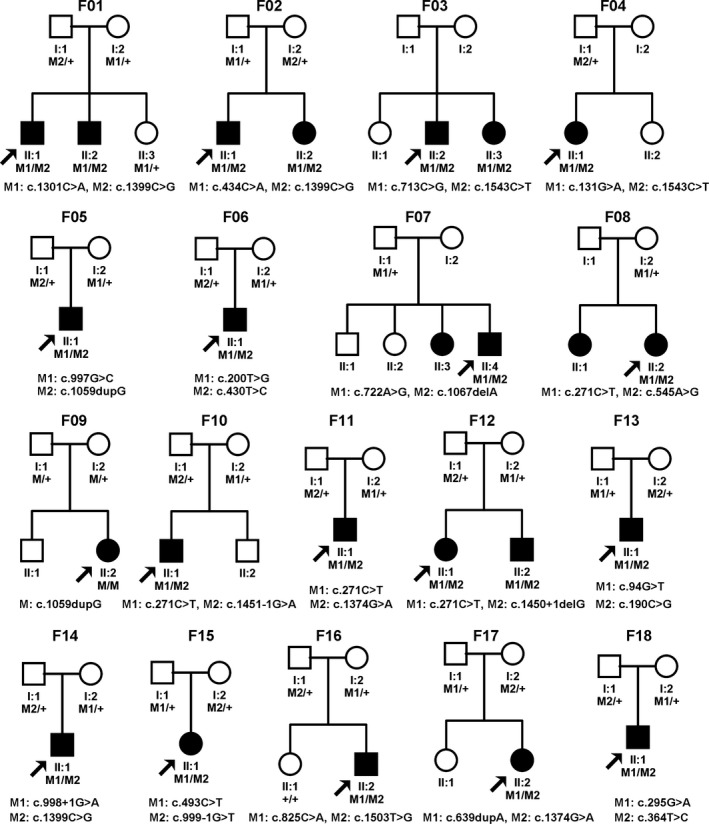
Pedigrees of 18 families with biallelic RPE65 mutations. Squares indicate male individuals while circles indicate females. Shading indicates an affected individual. Proband is indicated by arrows. Family numbers are on top of the pedigrees while mutations are listed under the pedigrees.
Phenotypic characterization
Of the 24 affected individuals in the 18 families, ophthalmological data were available for 21, including all 18 probands and three affected siblings (Table 2). The initial symptoms in the probands were night blindness in seven families, poor vision or no pursuit of objects in six families, nystagmus with poor vision in three families, nystagmus in one family and suspected esotropia in one family. Symptoms appeared as early as 4 months old and no later than 5 years old. Visual acuity ranged from finger counting to normal. Fundus changes ranged from very mild to easily recognizable. Two major types of fundus changes were visualized, including white dot deposits in the mid‐peripheral retina (Figs 2 and 3) and tapetoretinal degeneration (Fig. 4). Macular degeneration was obvious in two probands who also had tapetoretinal degeneration (Fig. 4). Bone‐spicule‐like pigment deposits were not observed in any of the 21 affected individuals. Cone and rod responses on electroretinogram varied from nonrecordable to normal, although typical fundus changes were present. The diseases in the 18 families with RPE65 biallelic mutations could be classified into four groups: LCA in eight families, EORD in five families, FA‐like changes in four families and high hyperopia in one family.
Table 2.
Clinical information of the probands and affected siblings with biallelic RPE65 mutations.
| Family ID | Clinic group | Mutation | Effect | Detection method | Gender | Age (year) at | Axial length or refraction | First symptom | Visual acuity | Fundus changes | ERG recording | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Onset | 1st exam | Last exam | Right | Left | Right | Left | Rods | Cones | ||||||||
| F01‐II:1 | FAP | c.[1301C>A];[1399C>G] | p.[Ala434Glu];[Pro467Ala] | WES | M | 4.7 | 5.7 | 7.9 | 21.49/21.44 mm at 5.7 years | NB | 0.30 | 0.40 | WD | WD | NA | NA |
| F01‐II:2 | FAP | c.[1301C>A];[1399C>G] | p.[Ala434Glu];[Pro467Ala] | SS | M | ECH | 4.3 | 6.4 | +2.75/+2.75 at 3.3 years | NB | 0.40 | 0.40 | WD | WD | Ext | SR |
| F02‐II:1 | FAP | c.[434C>A];[1399C>G] | p.[Ala145Asp];[Pro467Ala] | WES | M | 3.3 | 3.3 | NA | NA | NB | NA | NA | WD | WD | Ext | SR |
| F02‐II:2 | FAP | c.[434C>A];[1399C>G] | p.[Ala145Asp];[Pro467Ala] | SS | F | NA | 8.0 | NA | NA | NA | NA | NA | WD | WD | MiR | N |
| F03‐II:2 | FAP | c.[713C>G];[1543C>T] | p.[Ser238Cys];[Arg515Trp] | WES | M | ECH | 26.0 | NA | −3.25/−3.25 at 26 years | NB | 1.00 | 1.00 | WD | WD | MiR | MiR |
| F03‐II:3 | FAP | c.[713C>G];[1543C>T] | p.[Ser238Cys];[Arg515Trp] | SS | F | ECH | 24.0 | NA | −2.75/−1.75 at 24 years | NB | 0.90 | 0.80 | WD | WD | MoR | MiR |
| F04‐II:1 | FAP | c.[131G>A];[1543C>T] | p.[Arg44Gln];[Arg515Trp] | TES | F | 2.0 | 10.0 | NA | 21.00/20.90 mm at 10 years | NB | 0.40 | 0.40 | WD, TD | WD, TD | SR | SR |
| F05‐II:1 | LCA* | c.[997G>C];[1059dupG] | p.[Gly333Arg];[Lys354Glufs*11] | SS | M | 1.0 | 2.0 | NA | NA | PV | NA | NA | TD, AV | TD, AV | Ext | Ext |
| F06‐II:1 | LCA† | c.[200T>G];[430T>C] | p.[Leu67Arg];[Tyr144His] | WES | M | ECH | 2.1 | 8.8‡ | +6.25/+5.00 at 2.1 years | NYS | 0.10 | 0.10 | TD, AV | TD, AV | Ext | Ext |
| F07‐II:4 | LCA | c.[722A>G];[1067delA] | p.[His241Arg];[Asn356Metfs*17] | WES | M | ECH | 15.0 | NA | −0.75/−1.75 at 15 years | NYS, PV | 0.15 | 0.06 | TD, MD | TD, MD | Ext | SR |
| F08‐II:2 | LCA | c.[271C>T];[545A>G] | p.[Arg91Trp];[His182Arg] | WES | F | ECH | 19.0 | NA | 20.93/20.84 mm at 19 years | NYS, PV | 0.03 | 0.04 | TD, MD, AV | TD, MD, AV | Ext | Ext |
| F09‐II:2 | LCA | c.[1059dupG];[1059dupG] | p.[Lys354Glufs*11];[Lys354Glufs*11] | WES | F | 0.3 | 0.3 | NA | NA | PV | LP | LP | TD, AV | TD, AV | Ext | Ext |
| F10‐II:1 | LCA | c.[271C>T];[1451‐1G>A] | p.[Arg91Trp];[splicing] | WES | M | 0.3 | 4.9 | NA | +5.00/+5.25 at 4.9 years | PV, NYS | 0.10 | 0.15 | TD, AV | TD, AV | NA | NA |
| F11‐II:1 | LCA | c.[271C>T];[1374G>A] | p.[Arg91Trp];[Trp458*] | WES | M | 0.5 | 0.5 | NA | NA | PV | LP | LP | TD, AV | TD, AV | Ext | Ext |
| F12‐II:1 | LCA | c.[271C>T];[1450+1delG] | p.[Arg91Trp];[splicing] | TES | F | ECH | 4.7 | NA | −3.25/−0.50 at 4.7 years | PV, NYS | NA | NA | TD, AV | TD, AV | Ext | Ext |
| F13‐II:1 | EORD | c.[94G>T];[190C>G] | p.[Gly32Cys];[Gln64Glu] | WES | M | 3.0 | 5.3 | NA | 0/−0.25 at 5.3 years | PV | 0.30 | 0.40 | TD | TD | Ext | Ext |
| F14‐II:1 | EORD | c.[998+1G>A];[1399C>G] | [splicing];[p.(Pro467Ala] | TES | M | 2.5 | 2.5 | 5.7‡ | −5.25/−5.75 at 3.5 years | NB | 0.20 | 0.20 | TD | TD | Ext | Ext |
| F15‐II:1 | EORD | c.[493C>T];[999‐1G>T] | p.[Gln165*];[splicing] | TES | F | 2.3 | 3.2 | NA | +2.50/+2.75 at 3.2 years | NB | FC | FC | TD, AV | TD, AV | Ext | Ext |
| F16‐II:2 | EORD | c.[825C>A];[1503T>G] | p.[Tyr275*];[Tyr501*] | TES | M | 1.7 | 7.7 | NA | −2.50/−1.25 at 7.7 years | PV, NB | 0.15 | 0.30 | TD, AV | TD, AV | Ext | Ext |
| F17‐II:1 | EORD | c.[639dupA];[1374G>A] | p.[Ala214Serfs*20];[Trp458*] | TES | F | 2.0 | 5.2 | NA | +3.00/+3.00 at 5.2 years | NYS | 0.10 | 0.10 | TD | TD | NA | NA |
| F18‐II:1 | HH | c.[295G>A];[364T>C] | p.[Val99Ile];[Tyr122His] | WES | M | 2.4 | 2.4 | 6.0‡ | +6.00/+6.50 at 2.4 years | Esotropia | 1.00 | 0.50 | Mild TD§ | Mild TD§ | N | N |
AV = attenuated vessels, ECH = early childhood, EORD = early‐onset retinal degeneration, Ext = Extinguished, F = female, FAP = fundus albipunctatus, FC = finger counting, HH = high hyperopia, LCA = Leber congenital amaurosis, LP = light perception, M = male, MiR = Mildly reduced, MoR = Moderately reduced, N = Normal, NA = Not available, NB = night blindness, NYS = nystagmus, PV = poor vision or no pursuit of objects, SR = severely reduced, SS = Sanger sequencing, TD = tapetoretinal degeneration, TES = targeting exome sequencing, WD = white dot deposits in mid‐peripheral retina, WES = whole exome sequencing.
*RPE65 mutations in this family were detected by Sanger sequencing and reported in our previously study (Li et al. 2011).
†RPE65 mutations in this family were detected by WES and described in our previous study (Chen et al. 2013).
‡Visual acuity for these children was obtained at last examination.
§Normal appearance at posterior fundus but very mild tapetoretinal degeneration in mid‐peripheral retina.
Figure 2.
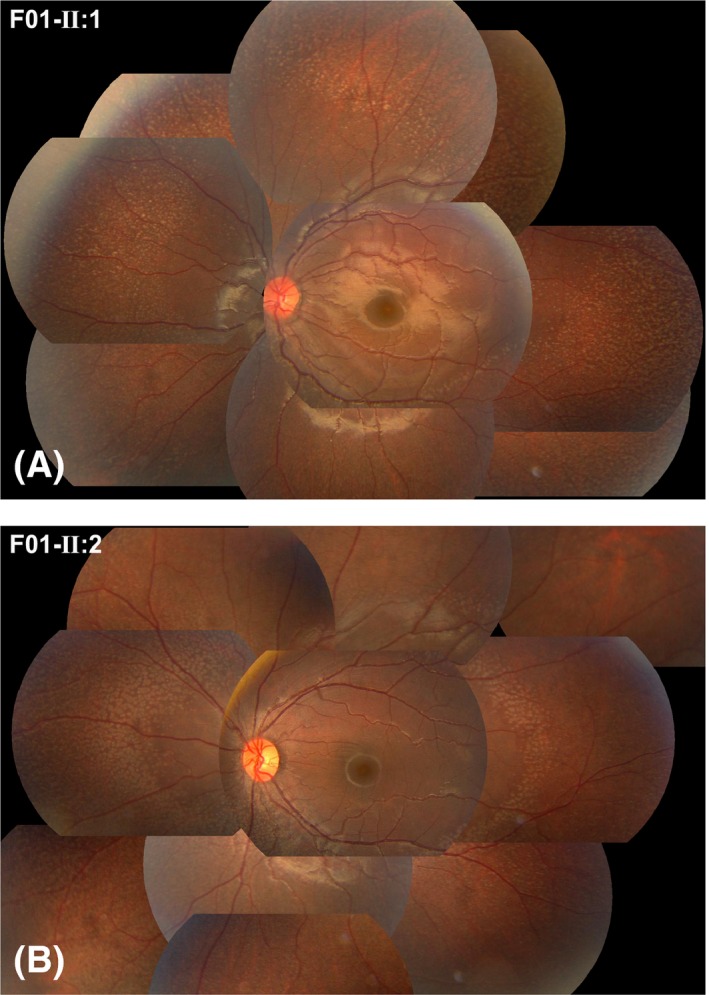
Photos demonstrating the fundus changes typical of fundus albipunctatus‐like changes in two affected brothers in family F01. A number of grey‐white dots were present in the mid‐peripheral retina. The family number and individual ID number that correspond to those shown in Fig. 1 and Table 2 are listed on the top left corner of each photo, as shown in Figs 3, 4, 5.
Figure 3.
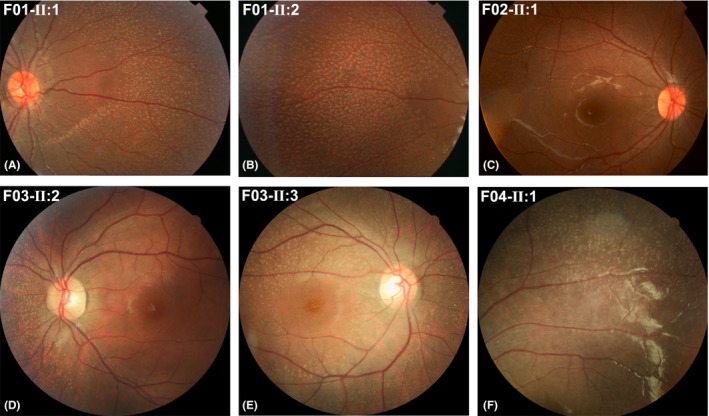
Fundus photos showing fundus albipunctatus‐like changes. A similar feature consisting of grey‐white dots was observed among patients from different families and patients within the same family. These photos also show the varied numbers, varied sizes and varied densities of the grey‐white dots that were observed among different families.
Figure 4.
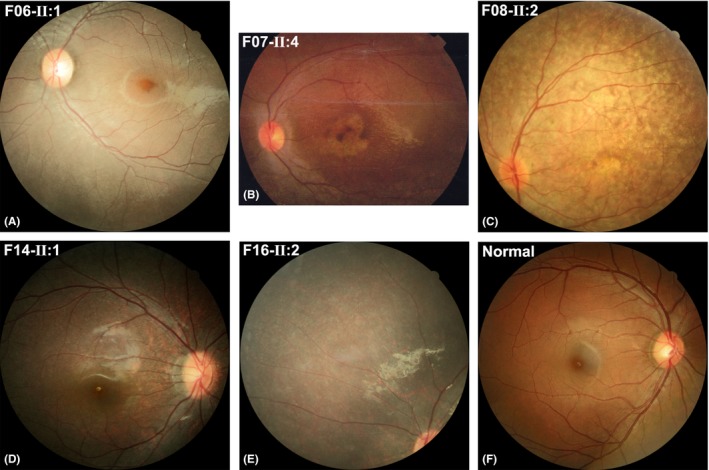
Tapetoretinal degeneration, generalized or located mainly in the mid‐peripheral region, mild or severe, was observed in different patients (A–E). A fundus photo of a normal individual that served as a control (F).
New common and rare phenotypes
Of the four families with FA‐like changes, three had affected siblings (Fig. 1, F01–F03), and one was a singleton case (Fig. 1, F04), suggesting an autosomal recessive trait. Similar fundus changes that were typical for FA‐like changes were documented in the mid‐peripheral region of the retina (Fig. 2 and 3). The type of fundus changes was almost identical between the eyes of each individual, between affected siblings in three families (F01–F03) and similar among all seven patients from unrelated families (F01‐II:1, F01‐II:2, F02‐II:1, F02‐II:2, F03‐II:2, F03‐II:3 and F04‐II:1). The initial symptoms observed in the seven patients with FA‐like changes and RPE65 mutations was night blindness, suggesting a milder phenotype than was found in those in whom poor vision was the initial sign. Five of the seven patients with FA‐like changes had visual acuity better than 0.3 (F01‐II:1, F01‐II:2, F03‐II:2, F03‐II:3 and F04‐II:1), and of these, two had visual acuity close to normal and exhibited mild or moderate changes on electroretinogram, even though they had typical retinal changes of FA‐like changes (F02‐II:2 and F03‐II:2).
One proband in F18 initially visited our clinic due to suspected esotropia. A clinical examination revealed high hyperopia at 2 years and 5 months of age. The boy had a refraction of +6.00/OD and +6.50/OS at that time and a refraction of +5.25/OD and +6.25/OS when he reached 5 years and 2 months old. This patient had two variants (c.295G>A/p.Val99Ile and c.364T>C/p.Tyr122His) that were predicted to be benign by PolyPhen‐2, damaging or tolerated by SIFT, and disease causing by MutationTaster and CADD. A recent examination showed a normal macula on optical coherence tomography, normal cone and rod responses on an electroretinogram performed at 6 years and 3 months of age, and mild tapetoretinal degenerative changes in the mid‐peripheral retina (Fig. 5). High hyperopia or short axial length was recorded in six of the 21 affected individuals (Table 2). Long‐term follow‐up will be performed to document any progress in fundus retinal changes, as progress from a normal‐like retina to typical tapetoretinal degeneration was observed in one proband (F05‐II:1) with LCA and RPE65 mutations after a follow‐up of 6 years (Fig. 5).
Figure 5.
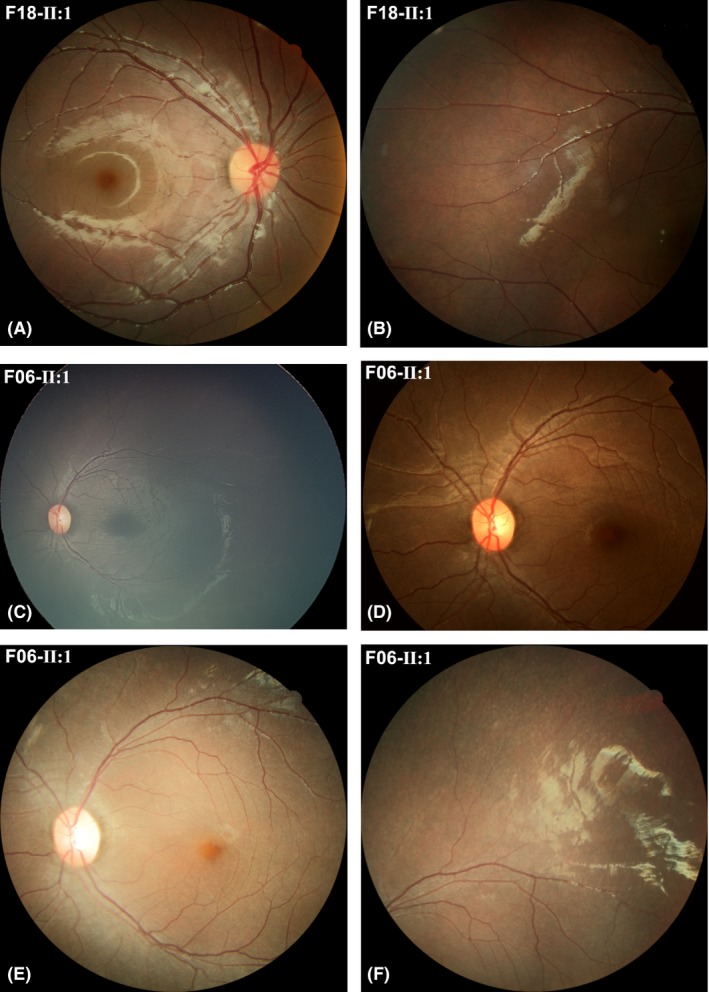
Mild fundus change and progression in retinal degeneration. (A, B) Fundus photos obtained from F18‐II:1, who had high hyperopia. The photos were taken when the patient was 6 years and 3 months old and showed a normal‐like posterior fundus and mild degenerative changes at the mid‐peripheral retina. He had a corrected visual acuity of 1.0 in the right eye and 0.5 in the left eye. (C–F) Fundus photos obtained from F06‐II:1. The photos were taken when the patient was 2 years and 2 months old (C), 5 years old (D) and 8 years and 10 months old (E, F). The fundus changes were insignificant or very minor in the early stage (C, D) but an obvious tapetoretinal degeneration was observed in the posterior (E) and mid‐peripheral retina (F) when the patient was 8 years and 10 months old.
Discussion
Of the 2133 families with HRD, 269 had an initial diagnosis of LCA. Therefore, biallelic RPE65 mutations contributed to approximately 3.0% (8/269) of LCA and 0.8% (18/2133) of HRD cases. Clinically, diseases in 13 of the 18 families could be classified as LCA or EORD, which are common phenotypes previously reported to be associated with RPE65 mutations (Gu et al. 1997; Marlhens et al. 1997; Morimura et al. 1998; Thompson et al. 2000; Simovich et al. 2001; Yzer et al. 2003; Booij et al. 2005; El Matri et al. 2006; Simonelli et al. 2007; Li et al. 2009; Xu et al. 2012; Kabir et al. 2013; Verma et al. 2013; Astuti et al. 2016; Katagiri et al. 2016). In the remaining five families with biallelic RPE65 mutations, we unexpectedly found that the phenotypes were FA‐like changes in four families and high hyperopia in one family.
Fundus albipunctatus‐like (FA‐like) changes were observed in seven patients from four families, suggesting that FA‐like changes are a common phenotype of RPE65‐associated retinopathy. Several lines of evidence support this notion, including the fact that biallelic RPE65 mutations were detected in all seven patients in four families, while mutations in RDH5 and RLBP1 were excluded, and similar fundus changes typical of FA‐like changes were found in different families as well as in different affected siblings. Previously, FA‐like changes had been described in only a few cases with RPE65 mutations; these included an 18‐year‐old woman from Denmark with compound heterozygous mutations (IVS1+5G>A and c.344T>C) (Schatz et al. 2011), a 7‐year‐old British girl with c.433G>A and c.886dupA mutations (Hull et al. 2016), a 3‐year‐old Chinese boy with c.639_640insA and c.982C>T mutations (Yang et al. 2017) and two Japanese patients with c.[1543C>T];[683A>C] or c.[1028T>A];[683A>C] mutations (Katagiri et al. 2018). It remains unknown why FA‐like changes is commonly observed in Chinese patients with biallelic RPE65 mutations. As observed in the current study, patients with FA‐like changes may have relatively good or even normal visual acuity as well as preserved rod‐cone function for many years (Hull et al. 2016). The mutations identified in all four families are missense mutations, and two of the mutations (c.1399C>G and c.1543C>T) were shared by two different families. This may indicate that some missense mutations are associated with this phenotype. RPE65 is not usually screened in patients with FA‐like changes in traditional mutational analysis, especially in phenotype‐guided candidate screening. Another possibility is that FA‐like changes may be seen in only certain periods of the disease course, and longer‐term follow‐up may resolve this issue. Nevertheless, recognizing this new common phenotype may be helpful for identifying patients with RPE65‐associated retinopathy so that they can benefit from potential RPE65 gene therapies.
It is unusual that one proband had high hyperopia and biallelic RPE65 mutations. These mutations may not necessary be causative, but they may have caused a mild phenotype: high hyperopia with mild or late‐onset retinal dystrophy. Currently, no firm evidence demonstrates this causation, but several findings imply a disease‐causing effect: (1) high hyperopia was a common feature as it was recorded in six of the 21 affected individuals examined in this study (Table 2); (2) RPE65 biallelic mutations are very rare in both our in‐house data and existing database; (3) of the two mutant alleles, one (c.364T>C) is novel and absent in the existing database, while the other was a known causative mutation; and (4) a mild phenotype consisting of normal visual acuity or late‐onset retinal dystrophy was previously associated with hypomorphic mutant alleles (Lorenz et al. 2008; Li et al. 2015a,2015b; Hull et al. 2016; Samardzija et al. 2016). Similarly, progress from a normal‐like retina to typical tapetoretinal degeneration was observed in one proband (F05‐II:1) with LCA and RPE65 mutations after 6 years of follow‐up (Fig. 5). We expect that further studies using longer‐term follow‐up will determine whether there is any progress in the fundus retinal changes observed in the proband F18‐II:1.
In summary, genotype‐guided phenotype characterization not only leads to the identification of additional common phenotypes associated with biallelic RPE65 mutations but also reveals the overall frequency of RPE65‐associated eye diseases. Similarly, more information related to many other genes can be obtained by performing genotype‐guided phenotype clarification than when using traditional phenotype‐guided candidate gene analysis.
Supporting information
Fig. S1. Confirmation of mutations and validation in available family members in RPE65 by Sanger sequencing. Pedigrees are shown in the left column. Sequence chromatography of mutations in each family is shown in the middle and right columns with individual number on the left and mutations above sequences.
The authors are grateful to the families for their participation. This work was supported by grants from the Science and Technology Planning Projects of Guangdong (2015A030401032), National Natural Science Foundation of China (81371058), the Key Projects of Guangzhou (201607020013) and the Fundamental Research Funds of the State Key Laboratory of Ophthalmology. Qingjiong Zhang is a recipient of National Science Fund for Distinguished Young Scholars.
References
- Apte RS (2018): Gene therapy for retinal degeneration. Cell 173: 5. [DOI] [PubMed] [Google Scholar]
- Astuti GD, Bertelsen M, Preising MN et al. (2016): Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. Eur J Hum Genet 24: 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booij JC, Florijn RJ, ten Brink JB, Loves W, Meire F, van Schooneveld MJ, de Jong PT & Bergen AA (2005): Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J Med Genet 42: e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Humphries MM, Sullivan LS et al. (2011): A dominant mutation in RPE65 identified by whole‐exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet 19: 1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang Q, Shen T et al. (2013): Comprehensive mutation analysis by whole‐exome sequencing in 41 Chinese families with Leber congenital amaurosis. Invest Ophthalmol Vis Sci 54: 4351–4357. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV (2010): Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res 29: 398–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Matri L, Ambresin A, Schorderet DF et al. (2006): Phenotype of three consanguineous Tunisian families with early‐onset retinal degeneration caused by an R91W homozygous mutation in the RPE65 gene. Graefes Arch Clin Exp Ophthalmol 244: 1104–1112. [DOI] [PubMed] [Google Scholar]
- Flanagan SE, Patch AM & Ellard S (2010): Using SIFT and PolyPhen to predict loss‐of‐function and gain‐of‐function mutations. Genet Test Mol Biomarkers 14: 533–537. [DOI] [PubMed] [Google Scholar]
- Fu Q, Wang F, Wang H et al. (2013): Next‐generation sequencing‐based molecular diagnosis of a Chinese patient cohort with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 54: 4158–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SM, Thompson DA, Srikumari CR et al. (1997): Mutations in RPE65 cause autosomal recessive childhood‐onset severe retinal dystrophy. Nat Genet 17: 194–197. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK & Cremers FP (2008): Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 27: 391–419. [DOI] [PubMed] [Google Scholar]
- Hull S, Holder GE, Robson AG, Mukherjee R, Michaelides M, Webster AR & Moore AT (2016): Preserved visual function in retinal dystrophy due to hypomorphic RPE65 mutations. Br J Ophthalmol 100: 1499–1505. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV et al. (2005): Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc Natl Acad Sci USA 102: 6177–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Aleman TS et al. (2007): RDH12 and RPE65, visual cycle genes causing Leber congenital amaurosis, differ in disease expression. Invest Ophthalmol Vis Sci 48: 332–338. [DOI] [PubMed] [Google Scholar]
- Jakobsson C, Othman IS, Munier FL, Schorderet DF & Abouzeid H (2014): Cone‐rod dystrophy caused by a novel homozygous RPE65 mutation in Leber congenital amaurosis. Klin Monbl Augenheilkd 231: 405–410. [DOI] [PubMed] [Google Scholar]
- Jiang D, Li J, Xiao X, Li S, Jia X, Sun W, Guo X & Zhang Q (2015): Detection of mutations in LRPAP1, CTSH, LEPREL1, ZNF644, SLC39A5, and SCO2 in 298 families with early‐onset high myopia by exome sequencing. Invest Ophthalmol Vis Sci 56: 339–345. [DOI] [PubMed] [Google Scholar]
- Kabir F, Naz S, Riazuddin SA et al. (2013): Novel mutations in RPE65 identified in consanguineous Pakistani families with retinal dystrophy. Mol Vis 19: 1554–1564. [PMC free article] [PubMed] [Google Scholar]
- Katagiri S, Hayashi T, Kondo M et al. (2016): RPE65 mutations in two Japanese families with Leber congenital amaurosis. Ophthalmic Genet 37: 161–169. [DOI] [PubMed] [Google Scholar]
- Katagiri S, Hosono K, Hayashi T et al. (2018): Early onset flecked retinal dystrophy associated with new compound heterozygous RPE65 variants. Mol Vis 24: 286–296. [PMC free article] [PubMed] [Google Scholar]
- Kondo H, Qin M, Mizota A et al. (2004): A homozygosity‐based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Invest Ophthalmol Vis Sci 45: 4433–4439. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S & Ng PC (2009): Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Kumaran N, Michaelides M, Smith AJ, Ali RR & Bainbridge JWB (2018): Retinal gene therapy. Br Med Bull 126: 13–25. [DOI] [PubMed] [Google Scholar]
- Lee H & Lotery A (2017): Gene therapy for RPE65‐mediated inherited retinal dystrophy completes phase 3. Lancet 390: 823–824. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang H, Peng J, Gibbs RA, Lewis RA, Lupski JR, Mardon G & Chen R (2009): Mutation survey of known LCA genes and loci in the Saudi Arabian population. Invest Ophthalmol Vis Sci 50: 1336–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Xiao X, Li S et al. (2011): Detection of variants in 15 genes in 87 unrelated Chinese patients with Leber congenital amaurosis. PLoS ONE 6: e19458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jiang D, Xiao X, Li S, Jia X, Sun W, Guo X & Zhang Q (2015a): Evaluation of 12 myopia‐associated genes in Chinese patients with high myopia. Invest Ophthalmol Vis Sci 56: 722–729. [DOI] [PubMed] [Google Scholar]
- Li Y, Yu S, Duncan T et al. (2015b): Mouse model of human RPE65 P25L hypomorph resembles wild type under normal light rearing but is fully resistant to acute light damage. Hum Mol Genet 24: 4417–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz B, Poliakov E, Schambeck M, Friedburg C, Preising MN & Redmond TM (2008): A comprehensive clinical and biochemical functional study of a novel RPE65 hypomorphic mutation. Invest Ophthalmol Vis Sci 49: 5235–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlhens F, Bareil C, Griffoin JM et al. (1997): Mutations in RPE65 cause Leber's congenital amaurosis. Nat Genet 17: 139–141. [DOI] [PubMed] [Google Scholar]
- Moiseyev G, Chen Y, Takahashi Y, Wu BX & Ma JX (2005): RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci USA 102: 12413–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL & Dryja TP (1998): Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci USA 95: 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti A, Wong DJ, Kawase K, Gibson LH, Yang‐Feng TL, Richards JE & Thompson DA (1995): Molecular characterization of the human gene encoding an abundant 61 kDa protein specific to the retinal pigment epithelium. Hum Mol Genet 4: 641–649. [DOI] [PubMed] [Google Scholar]
- Samardzija M, Barben M, Geiger P & Grimm C (2016): The consequences of hypomorphic RPE65 for rod and cone photoreceptors. Adv Exp Med Biol 854: 341–346. [DOI] [PubMed] [Google Scholar]
- Schatz P, Preising M, Lorenz B, Sander B, Larsen M & Rosenberg T (2011): Fundus albipunctatus associated with compound heterozygous mutations in RPE65. Ophthalmology 118: 888–894. [DOI] [PubMed] [Google Scholar]
- Simonelli F, Ziviello C, Testa F et al. (2007): Clinical and molecular genetics of Leber's congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci 48: 4284–4290. [DOI] [PubMed] [Google Scholar]
- Simovich MJ, Miller B, Ezzeldin H et al. (2001): Four novel mutations in the RPE65 gene in patients with Leber congenital amaurosis. Hum Mutat 18: 164. [DOI] [PubMed] [Google Scholar]
- Thompson DA, Gyurus P, Fleischer LL et al. (2000): Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci 41: 4293–4299. [PubMed] [Google Scholar]
- Verma A, Perumalsamy V, Shetty S, Kulm M & Sundaresan P (2013): Mutational screening of LCA genes emphasizing RPE65 in South Indian cohort of patients. PLoS ONE 8: e73172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Wang P, Li S et al. (2010): Mitochondrial DNA haplogroup distribution in Chaoshanese with and without myopia. Mol Vis 16: 303–309. [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang X, Zou X et al. (2015): Comprehensive molecular diagnosis of a large Chinese leber congenital amaurosis cohort. Invest Ophthalmol Vis Sci 56: 3642–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang Q, Zhang X, Wang Z & Zhao P (2016): Clinical and genetic characteristics of Leber congenital amaurosis with novel mutations in known genes based on a Chinese eastern coast Han population. Graefes Arch Clin Exp Ophthalmol 254: 2227–2238. [DOI] [PubMed] [Google Scholar]
- Weleber RG, Francis PJ, Trzupek KM & Beattie C (1993): Leber congenital amaurosis In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K. & Amemiya A. (eds.). GeneReviews®. Seattle, WA: University of Washington. [Google Scholar]
- Xu F, Dong Q, Liu L, Li H, Liang X, Jiang R, Sui R & Dong F (2012): Novel RPE65 mutations associated with Leber congenital amaurosis in Chinese patients. Mol Vis 18: 744–750. [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Xiao X, Li S et al. (2016): Molecular genetics of Leber congenital amaurosis in Chinese: new data from 66 probands and mutation overview of 159 probands. Exp Eye Res 149: 93–99. [DOI] [PubMed] [Google Scholar]
- Xue L, Gollapalli DR, Maiti P, Jahng WJ & Rando RR (2004): A palmitoylation switch mechanism in the regulation of the visual cycle. Cell 117: 761–771. [DOI] [PubMed] [Google Scholar]
- Yang G, Liu Z, Xie S, Li C, Lv L, Zhang M & Zhao J (2017): Genetic and phenotypic characteristics of four Chinese families with fundus albipunctatus. Sci Rep 7: 46285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yzer S, van den Born LI, Schuil J et al. (2003): A Tyr368His RPE65 founder mutation is associated with variable expression and progression of early onset retinal dystrophy in 10 families of a genetically isolated population. J Med Genet 40: 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Confirmation of mutations and validation in available family members in RPE65 by Sanger sequencing. Pedigrees are shown in the left column. Sequence chromatography of mutations in each family is shown in the middle and right columns with individual number on the left and mutations above sequences.