

Abstract
The prokaryote-derived CRISPR–Cas genome editing systems have transformed our ability to manipulate, detect, image and annotate specific DNA and RNA sequences in living cells of diverse species. The ease of use and robustness of this technology have revolutionized genome editing for research spanning from fundamental science to translational medicine. Initial successes have inspired efforts to discover new systems for targeting and manipulating nucleic acids, including those from Cas9, Cas12, Cascade and Cas13 orthologs. Genome editing by CRISPR–Cas can utilize non-homologous end joining (NHEJ) and homologous-directed repair (HDR) for DNA repair, as well as single-base editing enzymes. In addition to targeting DNA, CRISPR–Cas-based RNA-targeting tools are being developed for research, medicine and diagnostics. Nuclease-inactive and RNA-targeting Cas proteins have been fused to a plethora of effector proteins to regulate gene expression, epigenetic modifications and chromatin interactions. Collectively, these advances are considerably advancing our understanding of biology and propelling CRISPR–Cas-based tools towards clinical use in gene and cell therapies.
INTRODUCTION
The ability to modulate and edit genetic information is crucial for studying gene function and uncovering biological mechanisms. Since the first demonstration of producing specific DNA fragments with restriction enzymes in 1971, scientists have been harnessing prokaryotic molecules for gene editing1. In addition to restriction enzymes2, classes of DNA-modifying tools include recombinases3 and programmable nucleases such as meganucleases, zinc finger nucleases, transcription activator-like effector nucleases and clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated endonuclease (Cas) systems4. DNA-binding proteins that modify specific loci have tremendously advanced science, biotechnology and medicine. However, the complexity of developing modular DNA-binding proteins to bind at custom targets often requires protein engineering expertise. In the past decade, the CRISPR–Cas9 technology has transformed genome engineering by removing the need for any expertise in engineering custom targeted DNA-binding proteins, because the target specificity of CRISPR–Cas9 relies on base pairing of nucleic acids rather than protein–DNA recognition.
In nature, the CRISPR-Cas system is a prokaryotic adaptive immunity mechanism used to cleave invading nucleic acids5. An assortment of CRISPR-Cas systems exists across diverse species of bacteria and archaea, which differ in their components and mechanisms of action. For example, class 1 CRISPR–Cas systems comprise multi-protein effector complexes whereas class 2 systems have a single effector protein; overall there are 6 CRISPR–Cas types and at least 29 subtypes6-8, and this list of types and subtypes is undergoing rapid expansion. All CRISPR-Cas systems rely on CRISPR RNA (crRNA) or, in experimental CRISPR–Cas9 systems, on the guide RNA (gRNA) for guidance and targeting specificity (Figure 1). Following hybridization of the spacer [G] part of the crRNA to a target sequence that is positioned next to a protospacer adjacent motif (PAM) (or a protospacer flanking sequence (PFS) in type VI systems), the Cas nuclease cleaves the target nucleic acid. Thus, site-specific cleavage at any locus containing a PAM or PFS can be achieved by retargeting CRISPR-Cas systems with designed crRNAs containing appropriate spacer sequences. The discovery and development of type II CRISPR-Cas9 systems and the ease of their use have led to rapid adoption and development of a great range of applications, spanning from fundamental to translational science and medicine9. In turn, the early successes have inspired efforts to discover new CRISPR–Cas systems and develop novel genome engineering applications.
Figure 1. Overview of the main CRISPR–Cas gene editing tools.

a ∣ CRISPR-associated endonuclease 9 (Cas9) proteins rely on RNA guidance for targeting specificity. In engineered CRISPR–Cas9 systems, Cas9 interacts with the backbone of the guide RNA (gRNA). Complementary pairing of the spacer portion of the gRNA to a DNA target sequence positioned next to a 5’ protospacer adjacent motif (PAM) results in generation of a blunt DNA double-strand break by the two Cas9 nuclease domains, RuvC and HNH11-13. b ∣ Cas12a nucleases recognize DNA target sequences with complementarity to the crRNA spacer positioned next to a 3’ PAM. Target recognition results in generation of a staggered DNA double-strand break by a RuvC domain and a putative nuclease (Nuc) domain33. c ∣ Cascade is a multimeric complex that targets DNA that has complementarity to the spacer portion of a crRNA and that is positioned next to a 3’ PAM37-42. Following target recognition, Cascade recruits Cas3 to generate a single-strand nick, which is followed by 3’ to 5’ degradation of the targeted DNA37,39,44,45.
In this review, we discuss recent advances in CRISPR–Cas tools for gene editing and epigenetic modulation, before describing a diverse range of new CRISPR–Cas functions. We discuss next-generation applications such as perturbation of the transcriptome and non-coding genome, single-base editing, genome-wide pooled screens, chromatin reorganization and therapeutic potential moving towards clinical studies.
Advances in genome editing
CRISPR-Cas systems are modular DNA-binding or RNA-binding proteins that can be engineered to bind specific sequences by designing crRNAs or gRNAs containing spacers that complement the target sequence. In addition to binding specific nucleic acid sequences, these proteins also function as nucleases and thus can be used for programmable genome editing.
Broader targeting capacity
By harnessing the unique attributes of various CRISPR-Cas systems, such as PAM specificity, protein size and nuclease activity, a range of CRISPR–Cas-based DNA-targeting tools have been developed for genome editing applications. Additionally, the development of methods for the detection of on-target and off-target interactions has advanced the targeting specificity of CRISPR-Cas tools (Supplementary information Box 1).
CRISPR-Cas9 tools
Cas9 belongs to the class 2 type II CRISPR systems and is the most widely used genome editing tool. Specifically, Streptococcus pyogenes Cas9 (SpCas9) was the first to be used outside of prokaryotic cells10 and reprogrammed for genome editing in mammalian cells11,12; it remains the most commonly used Cas9. Following DNA target recognition, SpCas9 typically generates a blunt double-strand break (DSB) (Figure 1a)13. DNA targeting by SpCas9 relies on the 20-nucleotides long spacer and on the PAM 5’-NGG10,14. Cas9 systems are dual-RNA-guided: a crRNA is responsible for DNA targeting and also hybridizes with the trans-activating crRNA (tracrRNA), which is responsible for forming the complex with Cas915,16. The crRNA and tracrRNA functions can be recapitulated with an engineered single guide RNA (gRNA)10 (Figure 1a).
Recognition of the PAM 5’-NGG (N represents any nucleotide) limits the availability of SpCas9 target sites in the human genome to an average of one target site for every eight base pairs9. To increase the availability of target sites, directed evolution [G] approaches have generated variants with altered PAM specificities (Table 1)17,18. For example, an expanded-PAM SpCas9 variant, xCas9, recognizes 5’-NG, 5’-GAA and 5’-GAT PAM sequences18. Another motivation for engineering Cas9 variants is to increase targeting specificity. In fact, several studies have described mutated Cas9 variants with reduced off-target cleavage following expression of Cas9 and gRNAs from plasmids19-22 or their delivery as ribonucleoprotein (RNP) complexes23. Alternatively, on-target CRISPR-Cas specificity has been increased by engineering secondary structures in the form of RNA hairpins on the spacer region of gRNAs, which increase the thermodynamic barrier to crRNA or gRNA strand invasion at off-target sites while generally maintaining on-target activity24.
Table 1.
Cas9 variants with altered PAM and targeting specificities
| Name | Included mutations | PAM (5’ to 3’) | Notes |
|---|---|---|---|
| SpCas9 | Native Streptococcus pyogenes Cas9 | NGG249 | 1368 amino acids |
| VRER SpCas9 | D1135V, G1218R, R1335E, T1337R | NGCG17 | Altered PAM variant; Bacterial-selection-based screening |
| VQR SpCas9 | D1135V, R1335Q, T1337R | NGAN or NGNG17 | Altered PAM variant; Bacterial-selection-based screening |
| EQR SpCas9 | D1135E, R1335Q, T1337R | NGAG17 | Altered PAM variant; Bacterial-selection-based screening |
| xCas9-3.7 | A262T, R324L, S409I, E480K, E543D, M694I, E1219V | NG, GAA, GAT18 | Altered PAM variant; Phage-assisted continuous evolution |
| eSpCas9 (1.0) | K810A, K1003A, R1060A | NGG | Enhanced specificity; Structure-guided protein engineering19 |
| eSpCas9 (1.1) | K810A, K1003A, R1060A | NGG | Enhanced specificity; Structure-guided protein engineering19 |
| Cas9-HF1 | N497A, R661A, Q695A, Q926A | NGG | Enhanced specificity20 |
| HypaCas9 | N692A, M694A, Q695A, H698A | NGG | Enhanced specificity21 |
| evoCas9 | M495V, Y515N, K526E, R661Q | NGG | Enhanced specificity; yeast-based screening22 |
| HiFi Cas9 | R691A | NGG | Enhanced specificity for ribonucleoprotein delivery23 |
| ScCas9 | Native Streptococcus canis Cas9 | NNG250 | 1375 amino acids |
| StCas9 | Native Streptococcus thermophilus Cas9 | NNAGAAW11,25 | 1121 amino acids |
| NmCas9 | Native Neisseria meningitidis Cas9 | NNNNGATT26-28 | 1082 amino acids |
| SaCas9 | Native Staphylococcus aureus Cas9 | NNGRRT29 | 1053 amino acids |
| CjCas9 | Native Campylobacter jejuni Cas9 | NNNVRYM30 | 984 amino acids |
| CasX | Deltaproteobacteria and Planctomycetes phyla | TTCN32 | 980 amino acids |
AAV, adeno-associated virus
ABA, abscisic acid
ABE, adenine base editor
ABI1, dimerizing ABA-binding protein
ADAR, adenosine deaminase acting on RNA
APEX2, engineered apurinic/apyrimidinic endodeoxyribonuclease 2 peroxidase
APOBEC1, apolipoprotein B mRNA editing enzyme complex-1
BE3, third generation base editor
BLESS, breaks labeling, enrichment on streptavidin and next-generation sequencing
Cas9-HF1, high fidelity Cas9 variant
CIB1, plant-derived cytochrome protein
CIRCLE-seq, circularization for in vitro reporting of cleavage effects by sequencing
CLOuD9, chromatin loop reorganization with CRISPR–dCas9
CORRECT, consecutive re-guide or re-Cas steps to erase CRISPR/Cas-blocked targets
CRISPR, clustered regularly interspaced short palindromic repeats
CRISPRa, CRISPR activation
CRISPRi, CRISPR interference
crRNA, CRISPR RNA
CRY2, plant-derived cytochrome protein
dCas9, catalytically deficient Cas9
Digenome-seq, in vitro Cas9-digested whole-genome sequencing
DNA, deoxyribonucleic acid
DNMT, DNA methyltransferase
DSB, double-strand break
dsDNA, double-stranded DNA
enAsCas12a, enhanced AsCas12a variant
enCHIP, engineered DNA-binding molecule-mediated chromatin immunoprecipitation
EQR, Cas9 variant with triple mutations
eSpCas9, enhanced specificity SpCas9 variant
evoCas9, evolved high fidelity Cas9 variant
FKBP, rapamycin-binding domain
FRB, rapamycin-binding domain
GFP, green fluorescent protein
gRNA, guide RNA
GUIDE-seq, genome-wide, unbiased identification of DSBs enabled by sequencing
HCV, hepatitis C virus
HDR, homologous-directed repair
HEPN, higher eukaryotes and prokaryotes nucleotide binding domain
HiFi Cas9, high fidelity Cas9 variant
HITI, homology-independent targeted integration
HNH, Cas9 nuclease domain
HTGTS, high-throughput, genome-wide, translocation sequencing
HypaCas9, hyper-accurate Cas9 variant
Indels, insertions or deletions
iPSC, induced pluripotent stem cell
lncRNA, long non-coding RNA
KRAB, Krϋppel-associated box
LNP, lipid Nanoparticle
MEMOIR, mutagenesis with optical in situ readout
mRNA, messenger RNA
nCas9, Cas9 nickase
NHEJ, non-homologous end joining
p65, NF-κB transactivating subunit
PAM, protospacer adjacent motif
PAMmer, PAM-presenting oligonucleotide
PFS, protospacer flanking sequence
PYL1, dimerizing ABA-binding protein
REPAIR, RNA editing for programmable A to I replacement
RNA, ribonucleic acid
RNAi, RNA interference
RNP, ribonucleoprotein
RuvC, Cas9 nuclease domain
SHERLOCK, specific high sensitivity enzymatic reporter unlocking
smFISH, single-molecule RNA fluorescence hybridization
ssDNA, single-stranded DNA
ssODN, single-stranded oligodeoxynucleotide
stgRNA, self-targeting gRNA
TET1, ten-eleven translocation methylcytosine dioxygenase 1
tracrRNA, trans-activating crRNA
tRNA, transfer RNA
UGI, uracil glycosylase inhibitor
VIVO, verification of in vivo off-targets
VP64, four repeats of the herpes simplex VP16 activation domain
VQR, Cas9 variant with triple mutations
VRER, Cas9 variant with quadruple mutations
xCas9, expanded PAM SpCas9 variant
AsCas12a – Acidaminococcus spp. Cas12a
CjCas9 – Campylobacter jejuni Cas9
FnCas9 – Francisella novicida Cas9
LbCas12a – Lachnospiraceae spp. Cas12a
LwaCas13a – Leptotrichia wadei Cas13a
NmCas9 – Neisseria meningitidis Cas9
PaCsy4 – Pseudomonas aeruginosa Csy4
PspCas13b – Prevotella sp. P5-125 Cas13b
SaCas9 – Staphylococcus aureus Cas9
ScCas9 – Streptococcus canis Cas9
SpCas9 – Streptococcus pyogenes Cas9
StCas9 – Streptococcus thermophilus Cas9
The discovery and development of additional Cas9 orthologs that recognize different PAM sequences has provided a greater choice of target sites. For example, Streptococcus thermophilus Cas9 recognizes the PAM 5’-NNAGAAW (W represents A or T)11,25 and Neisseria meningitidis Cas9 recognizes 5’-NNNNGATT26-28. These Cas9 orthologs have been repurposed for DNA targeting in bacteria and mammalian cells. Furthermore, the PAM recognized by Staphylococcus aureus Cas9 (SaCas9) is 5’-NNGRRT (R represents A or G)29. Notably, SaCas9 gene editing efficiencies are comparable to SpCas9 and the smaller size of SaCas9 (1,053 amino acids compared to 1,368 amino acids of SpCas9) has enabled its use in size-restricted delivery vectors such as adeno-associated virus (AAV)29. More recently, an even smaller Cas9 ortholog, from Campylobacter jejuni (984 amino acids), was reported to recognize the PAM 5’-NNNVRYM (V represents A, C or G; Y represents C or T)30 and used for targeted genome editing in vivo31. Additional efforts to identify Cas9 orthologs has resulted in the discovery of CasX (980 amino acids), the smallest Cas9 to date32.
CRISPR–Cas12a
Another class II RNA-guided endonuclease that has been reprogrammed for gene editing in human cells is Cas12a (formerly Cpf1)33. As a type V system, Cas12a generates a staggered cut with a 5’ overhang at DNA target sites and does not use a tracrRNA (Figure 1b). In contrast to the generation of blunt ends by Cas9, production of staggered ends by Cas12a may be advantageous for applications such as integrating DNA sequences in a precise orientation. Additionally, Cas12a can cleave crRNA arrays [G] to generate its own crRNAs. This crRNA processing ability facilitates the use of a single customized crRNA array for simplified multiplexed genome editing with multiple crRNAs34.
Cas12a from Acidaminococcus spp. (AsCas12a) and Lachnospiraceae spp. (LbCas12a), the first Cas12a orthologs that were shown to have activity in mammalian cells, recognize the PAM sequence 5’-TTTV upstream of the target sequence. To improve their genome editing activity, an enhanced AsCas12a variant (enAsCas12a) has been engineered35. To increase the targeting range of Cas12a, AsCas12a variants have recently been engineered to recognize the PAMs 5’-TYCV and 5’-TATV36, or PAMs 5’-VTTV, 5’-TTTT, 5’-TTCN and 5’-TATV35. The unique features and cutting mechanism of Cas12a provides a genome editing tool that expands the CRISPR toolbox.
Cascade and Cas3
Type I systems of the class 1 category are the most common type of CRISPR-Cas systems in nature, comprising a multimeric DNA-targeting complex termed Cascade and the endonuclease Cas3 (Figure 1c). Before recruiting Cas3 to a target DNA sequence, Cascade must first bind to DNA through PAM and spacer recognition37-42. Cascade offers greater target site flexibility owing to its promiscuous recognition of PAM sequences43. Recruitment of Cas3 generates a single-stranded nick followed by target DNA degradation through 3’ to 5’ exonuclease activity37,39,44,45. Both the nickase and helicase activities of Cas3 are essential for the degradation of foreign DNA in prokaryotes38. The unique cutting mechanism of Cas3 is being harnessed as an antimicrobial tool by directing native or exogenous type I systems to bacterial genomes for degradation and subsequent cell death46. Exploration to repurpose the nickase, helicase and exonuclease activities of Cas3 may lead to new applications in mammalian cells.
In type I systems, crRNA arrays are processed by the Cascade subunit Csy447. Like Cas12a, this endonuclease activity has been repurposed for directed RNA processing. For example, the Pseudomonas aeruginosa type I-F Csy4 has been used for generating multiple Cas9 gRNAs in human cells48,49.
Mechanisms and uses of gene editing
Gene editing nucleases, including Cas9, function by generating targeted DNA breaks that induce the DNA damage response and stimulate repair by various endogenous mechanisms50. Use of the unique characteristics of the different DNA repair mechanisms has enabled the development of specific genome editing strategies.
NHEJ versus HDR
Eukaryotes predominantly repair DSBs through the error-prone non-homologous end joining (NHEJ) pathway, which leads to accumulation of small insertions or deletions (indels [G]) following repeated cycles of break and repair (Figure 2a). Alternatively, a repair template with homology to the target site can be delivered with Cas9 to stimulate the error-free homology-directed repair (HDR), but typically at a lower efficiency than NHEJ-mediated repair (Figure 2b). NHEJ can be used to produce gene knockouts (deletions) whereas HDR can be used to introduce a specific change in the targeted genomic site, such as a point mutation or insertion of a longer segment of DNA. Increasing the efficiency of HDR following nuclease-mediated DNA breakage is widely pursued to fully harness the power of genome editing to introduce precise genomic alterations51-55.
Figure 2. Genome editing strategies.

Nucleases generate targeted DNA double-strand breaks (DSBs), which can be repaired by different repair pathways. a ∣ Non-homologous end joining (NHEJ)-mediated repair is error-prone and induces small insertion or deletion mutations (indels). Large, targeted deletions can be produced through repair between two DSBs produced by simultaneously targeting nucleases to two genomic sites. Alternatively, homology-independent targeted integrations (HITI) can be directed to a single cut site by providing donor DNA that is independently targeted for cutting65. b ∣ The homology-directed repair (HDR) pathway can be utilized for genome editing by providing either double-strand or single-strand oligodeoxynucleotide (ssODN) donor templates that contain homology arms to the cut target site. Single nucleotide alterations or insertion of larger sequences can be mediated by introducing variations into the donor template, which may also consist of plasmid DNA, viral DNA248 or long single-stranded DNA71. Following HDR, silent mutations — also referred to as blocking mutations (B) — that prevent subsequent target site recognition by the nucleases and formation of NHEJ-mediated indels, can be incorporated into the donor template along with the intended alterations73. c ∣ For single nucleotide C→T (or G→A) conversion, Cas9 nickase has been fused to cytidine deaminases such as APOBEC179. For increased base editing efficiency, two uracil glycosylase inhibitors (UGI) have been fused to a base editor for preventing cellular base excision repair88.
Gene deletions
Following Cas9 cleavage, NHEJ-mediated DNA repair can be harnessed to create gene knockouts. When targeting a coding exon, indel-mediated frameshift mutations, which also typically introduce premature stop codons downstream of the target site, will disrupt gene expression. Alternatively, by simultaneously targeting two sites in a gene, a deletion can be generated between the DSBs11,56-58, including megabase-size deletions59 (Figure 2a). A systematic exploration of Cas9-mediated deletion efficiencies showed an inverse correlation between deletion size and its frequency60. In addition to studies in cells, strategies have been developed for facilitating heritable genomic deletions in organisms such as zebrafish61 and mice62-64. The wide spectrum of possible Cas9-mediated genomic deletions is accelerating the investigation of genes and genetic elements.
Gene insertions
Inserting a DNA sequence encoding an epitope tag or a fluorescent protein into protein-coding genes to monitor endogenously-expressed proteins is a valuable strategy for studying protein function in native cellular settings. Cas9-mediated and NHEJ-mediated gene tagging strategies have been developed based on the integration of linear DNA fragments at nuclease cleavage sites. In homology-independent targeted integration (HITI), a tag is flanked with gRNA target sites, so that Cas9 can simultaneously release it from a plasmid and cleave a recipient genomic target adjacent to the gene of interest65 (Figure 2a). Generic plasmid-based systems to create endogenous amino-terminal66 or carboxy-terminal66,67 gene–tag fusions using non-target-specific universal donor sequences have also been developed. Large-scale gene tagging is now possible due to the simplicity of these modular Cas9-mediated systems. HITI utilizes NHEJ for DSB repair, creating two problems: generation of indels and donor integration in random orientation. To overcome these obstacles, donor sequences can be flanked with homology arms. To circumvent the need for molecular cloning of target-specific donor sequences, single-strand DNA (ssDNA) were used to tag endogenous human genes with GFP-coding sequences68 (Figure 2b). Mice with multiple precise single point mutations were generated using multiplexed HDR in mouse embryonic stem cells69. Cancer modeling in mice can also be achieved by HDR-mediated insertion of missense gain-of-function mutations70. To generate conditional knockout mice at high efficiency by inserting large regulatable cassettes, RNPs were co-delivered with long ssDNA donors containing short homology arms71. Recently, an HDR-dependent strategy termed CORRECT (consecutive re-guide or re-Cas steps to erase CRISPR–Cas-blocked targets) was developed for producing scarless targeted knock-in of disease-relevant mutations72. By making variations in the donor template, edited cell lines, including human pluripotent stem cells, can be generated with pathogenic mutations and with additional, silent mutations that block subsequent target-site recognition by the nucleases and formation of NHEJ-mediated indels73 (Figure 2b).
Translocations
During cancer development, oncogenic fusion genes are frequently created through chromosomal translocations. Translocations can be mediated by illegitimate NHEJ of DSBs located at two non-homologous chromosomes. To generate models for studying the oncogenic properties of fusion proteins, simultaneous Cas9-mediated cleavage at two genomic loci has been used to engineer cancer-relevant translocations in human cells74,75. Cas9-induced chromosomal rearrangements leading to oncogenic gene fusions have been recapitulated also in mice76. These genetically engineered models are important for understanding tumorigenesis and for developing therapeutic strategies against oncogenic fusion proteins.
Single-base editing
The most common genetic variants associated with human disease are point mutations. An ability to edit single nucleotide bases is important for creating genetic disease models and developing corrective therapeutics. Targeted HDR-mediated single-base editing can be achieved by co-delivering Cas9 with a homologous donor sequence that contains the edited nucleotide of choice72. However, such strategies remain inefficient, particularly in post-mitotic cells with decreased HDR activity. Additionally, the need to create DSBs to induce efficient HDR carries the possibility of off-target mutagenesis, and even on-target activation of DNA repair pathways can have adverse consequences on cell viability77,78.
For improved single-base editing, tools have been developed that utilize Cas9 nickase (nCas9) or catalytically deficient Cas9 (dCas9) for site-specific targeting without generating DSBs. For direct conversion of single nucleotides, dCas9 or nCas9 have been fused to cytidine deaminases. Fusion with deaminases such as rat APOBEC1 and lamprey cytidine deaminase 1 can achieve targeted C→T (or G→A) nucleotide conversions within a 5-bp activity window located within the spacer sequence79,80 (Figure 2c). Cellular DNA repair responses can antagonize this process and restore edited bases, therefore a uracil glycosylase inhibitor was also used to prevent base excision repair and increase the efficiency of base editing79-81. A third generation editor (BE3) containing APOBEC1 fused to a 16-residues XTEN linker, nCas9 and a uracil glycosylase inhibitor (APOBEC1–XTEN–dCas9(A840H)-UGI) can achieve permanent conversion of 15–75% of a target nucleotide in mammalian cells79. Furthermore, BE3 has accomplished base editing in vivo through RNP-mediated protein delivery to mouse and zebrafish embryos82,83, AAV-mediated delivery in utero to mice84, and injection of mRNA and gRNA to human embryos85,86. In adult mice, BE3 was used to introduce site-specific nonsense mutations into the Pcsk9 gene, which resulted in lowered cholesterol levels87.
Continued development of BE3 has resulted in improved single-base editing. For example, improved Cas9-mediated targeting specificity has been achieved by combining BE3 with a high-fidelity Cas983. For optimization of base editing, lengthening of the linker between the fused proteins and adding a second copy of the uracil glycosylase inhibitor has led to fourth generation base editors engineered from SpCas9 (BE4) and SaCas9 (SaBE4)88. The base-targeting range has continued to expand following fusion of APOBEC1 to catalytically inactive dLbCas12a, which recognizes a T-rich PAM and has a 6-bp activity window89. Recently, enAsCas12a was used for enhanced base-editing activity35. To narrow the editing window at targets with potential C→T bystander alterations, base editors have been developed with human APOBEC3A for use in human cells90,91 and plants92. Specifically, eA3A-BE3, an engineered APOBEC3A domain (eA3A) fused to BE3, preferentially deaminates cytidines according to a TCR>TCY>VCN hierarchy90. Additionally, APOBEC3A-mediated base editing can be achieved in regions with high DNA methylation levels and CpG dinucleotide content91.
Recently, the base editor toolbox has been expanded to adenine base editors (ABEs), which can perform targeted A→G (or T→C) nucleotide conversions93. A seventh-generation ABE with the highest reported editing efficiencies and on-target activities was developed using directed evolution and protein engineering of a tRNA adenosine deaminase93. Optimized and enhanced cytidine and adenine base editors include BE4max, AncBE4max and ABEmax94.
Unbiased analyses of base-editing specificity is particularly difficult given the prevalence of single-base substitutions in the human genome and the frequency of sequencing errors. An early analysis of base editor specificity revealed off-target sites that are different from what was detected in cells treated with Cas9 alone95. More recently, widespread gRNA-independent off-target activity was reported for cytosine base editors in both plants and mice,96,97 indicating the existence of base-editing activity that is independent of Cas9–DNA interactions. Therefore, future efforts will likely focus on strategies to restrict base editing activity to intended targeted sites.
With the abundance of known point mutations associated with genetic disease, single base editors can be used to make animal models with nonsense mutations or single amino acid substitutions. Moreover, the therapeutic potential of base editors for correction or knockout of clinically relevant human diseases is being explored.98 Base editing may prove particularly useful for multiple-gene targeting, where avoiding the formation of multiple DSBs on different chromosomes that could generate translocations would be particularly desirable.
High-throughput loss-of-function screens
RNA interference (RNAi) has been the primary system for large-scale gene perturbation in mammalian cells, but the limitations of RNAi include incomplete suppression of target genes and frequent off-target effects. These limitations can largely be overcome by Cas9-based methods for gene knockout screening (Box 1). Indeed, Cas9-based high-throughput screens achieve high rates of target validation.99-102 The ease of producing large gRNA libraries coupled with efficient lentiviral delivery platforms — for example, a genomic CRISPR–Cas9 pooled lentivirus library, which can knockout over 18,000 human genes using 3–4 gRNAs per gene99 — has made genomic knockout screens possible in mouse100,101,103 and human cells99,104,105. Such a mouse-genome targeting library was delivered to a mouse model of tumor growth and metastasis, and loss-of-function mutations in known tumor suppressor and novel genes were identified in vivo102. For higher content readout of pooled screens, the modularity of Cas9 has been coupled with single-cell RNA sequencing106-108 By pairing genomic perturbation and transcriptomic analysis within the same cell, higher order interactions can be elucidated including the function of combinatorial interactions. Considerable advances in the optimization gRNA-library design are also improving the quality and throughput of these screens109.
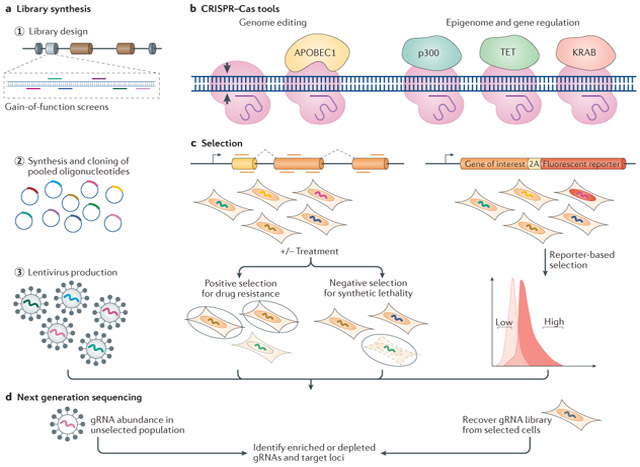
Box 1 ∣. Genome-wide pooled screens using CRISPR–Cas-based tools.
The simplicity of targeting CRISPR–Cas tools to the genome has facilitated high-throughput genetic screening. Genome-scale targeting of Cas9 is possible with synthesis of a guide RNA (gRNA) library. The breadth of the gRNA library can be customized, for example, loss-of-function screens may use saturation mutagenesis and target only exons of human genes99,104,105, and screens to annotate the noncoding genome may target sites of accessible chromatin193 or transcription factor motifs123. The gRNA libraries are generated by synthesizing pools of oligonucleotides, cloning them into plasmids and producing a lentivirus library that encodes the gRNAs (see the figure, part a). A Cas9-expressing cell line can be generated prior to gRNA delivery, or cells can be co-transduced with Cas9 and the gRNA library. Fusion of Cas9 nickase (nCas9) or catalytically deficient Cas9 (dCas9) to different effector proteins can enable genome editing (for example, by the cytidine deaminase APOBEC1) or epigenome and gene regulation (for example, histone acetylation by p300, DNA demethylation by TET dioxygenases or transcription repression by KRAB domains; see the figure, part b). To screen for functional elements, gRNAs that elicit the phenotype of interest must be enriched or depleted. For example, positive selection can identify elements that function in drug resistance237 and negative selection can identify elements involved in synthetic lethality238 (see the figure, part c). Alternatively, gene regulatory elements can be identified by selecting cells with altered gene expression either through direct immunofluorescence staining or through tagging an endogenous gene with a reporter193. By selecting cells with low or high reporter expression, factors that affect gene expression can be identified (see the figure, part c). Following selection, next-generation sequencing and bioinformatics are used to compare the unselected gRNA library with the selected gRNA library and identify enriched and depleted gRNAs and thus specific genomic loci (see the figure, d). A wide range of applications are possible with CRISPR-based screens. Interrogation of gene function can identify genes involved in cell survival and proliferation or cancer genes100,105,238,239; drug targets can be identified based on resistance or sensitivity to drugs, toxins or pathogens238,240. Targeted screens are also mapping the function of the noncoding genome by perturbing enhancer sequences113 or modulating particular sets of genes – for example, targeted activation of all transcription factor genes to identify factors involved in stem cell differentiation241. Although pooled CRISPR-based screens have so far used Cas9-based tools, in the future other Cas proteins could be used for other functions or for orthogonal screening.
Beyond single gene perturbation for therapeutic targeting strategies, combinatorial studies can be used to dissect genetic interactions. For cancer therapy, simultaneous knockout of a synthetic-lethal gene pair can achieve cell killing via multiplexed targeting. Therefore CRISPR–Cas-based double knockout screening has been developed for dissecting genetic interactions and identifying synthetic-lethal drug target pairs of cancer genes110,111.
Excessive DNA damage and cell death resulting from Cas9-induced DSBs may muddle conclusions drawn from knockout screens. Another point raised from recent Cas9 loss-of-function screens is that not all indels result in gene knockout. To address these issues, a DSB-independent knockout method, termed CRISPR-STOP79, was developed using CRISPR base editors to create stop codons by single nucleotide conversion. To expand this induction of stop codons (iSTOP) method, a database of over 3.4 million gRNAs targeting 97-99% of genes in eight eukaryotic species was compiled112. These Cas9-based knockout screens have confirmed known essential genes and mediators of resistance to drugs and toxins and provided novel genetic insights.
Although many initial applications of CRISPR–Cas-based gene editing were directed at studying gene function, a particularly important use of this technology lies in annotating the non-coding genome in ways that have not been previously possible. For example, the BCL11A gene encodes a transcription factor that controls the levels of fetal hemoglobin113; modulation of BCL11A expression by perturbing cell type-specific enhancers could be used as a therapeutic approach for β-haemoglobin disorders. By tiling the 10 kb of the BCL11A enhancer region with a gRNA library, divergence in enhancer–gene interactions was revealed between mice and humans and crucial minimal genetic elements were revealed and validated as targets for fetal hemoglobin reinduction113. This work involved the introduction of indel mutations at noncoding sequences to identify functional gene regulatory elements, which later led to the development of therapeutic strategies to target these sites in preclinical models of sickle cell disease and beta thalassemia114. Alternatively, HDR was used to introduce all possible nucleotide substitutions into a putative gene regulatory element to decipher its function115. Numerous other high-throughput tiling approaches are being used to identify functional elements in regulatory regions116-123. Finally, a genomic screening method that targets splice sites was used to identify long non-coding RNAs (lncRNAs) that are essential for cellular growth124. By using the same library to screen multiple cell lines, cell-type-specific differences in lncRNA essentially were identified.
Molecular recording
To better understand cellular dynamics in response to external (and internal) stimuli, CRISPR–Cas-based tools have been developed to function as molecular recorders by tracking cellular responses in the form of nucleotide alterations. Self-targeting gRNAs (stgRNAs) can be generated so that expression of Cas9 and the stgRNA will result in cleavage and indel mutation accumulation at the stgRNA loci125,126. Thus, a cellular response can be ‘recorded’ by linking cellular responses with the expression of the stgRNA or Cas9. By sequencing the stgRNA locus and determining the level of accumulated mutations, the duration or intensity of the stimulus can be measured. Alternatively, cellular activity can be recorded as individual nucleotide alterations using single-base editors targeted to designated positions on plasmid or genomic DNA127. These CRISPR–Cas-based molecular recording systems have been used to track cellular behavior in response to the presence of small molecules, virus infection, light exposure and multiplexed stimuli in bacteria and human cells125,127.
Cas9-mediated nucleotide alterations are inherited from the founder cell to its descendants, and therefore indels can be used for cell-lineage tracing. To perform whole-organism lineage tracing, accumulation of indel scars over multiple rounds of cell division was recorded following Cas9 and gRNA injection into 1-cell zebrafish embryos containing a compact DNA barcode with multiple Cas9 target sites128. By tracking these scars in hundreds of thousands of cells from individual zebrafish, it was found that most organs derive from relatively few embryonic progenitors128. To increase the number of traceable scars, Cas9 was targeted to its own gRNA spacer sequences. DNA repair mechanisms that form indels within the spacer sequences results in increased scarring complexity, which provides more information for improved phylogenetic annotation126. Another synthetic recording system, termed memory by engineered mutagenesis with optical in situ readout (MEMOIR), was developed to record and subsequently read lineage information out of single cells in situ129. This system combines Cas9-based targeted mutagenesis with multiplexed single-molecule RNA fluorescence hybridization (smFISH) to visualize recorded editing events for studying lineage tracing while maintaining the relative spatial positioning of cells129.
An alternative recording strategy is based on integrating nucleotides into bacterial genomic crRNA arrays as trackable molecular events. This mechanism utilizes the natural adaptation process of prokaryotic CRISPR-Cas systems, in which Cas1 and Cas2 proteins capture short fragments of invading plasmid or phage genetic material and integrate the exogenous sequences as spacers into a crRNA array5,130. Since new spacers are preferentially inserted at the 5’ end of crRNA arrays5, this mechanism can be harnessed for tracking sequential spacer acquisition as a means of recording the temporal order of molecular events. In a population of bacterial cells overexpressing Cas1 and Cas2, synthetic oligonucleotides can be serially electroporated to generate stable genomic recordings of multiple molecular events131. Recently, this technique has been scaled to store synthetic sequences encoding pixel values of black and white images and a short movie into the genomes of living bacteria132. These studies demonstrate the capacity of DNA to encode and store analog data.
CRISPR–Cas TARGETING RNA
Although CRISPR–Cas systems have been valuable for targeting DNA, manipulating RNA is limited by lack of precise and efficient RNA-targeting molecular tools. RNAi and antisense oligonucleotides can inhibit gene expression, but additional tools are needed to expand RNA-targeting applications. Recently, development of CRISPR–Cas technology for binding or cleaving specific RNAs has advanced RNA manipulation in living cells (Figure 3).
Figure 3. RNA targeting tools and their applications.

a ∣ Streptococcus pyogenes Cas9 was repurposed to target RNA (RCas9) by providing it a matching guide RNA (gRNA) and a complementary PAM-presenting oligonucleotide (PAMmer)134. Cas9 orthologs such as Staphylococcus aureus Cas9 and Campylobacter jejuni Cas9, can target RNA in the absence of a PAMmer, thereby demonstrating PAM-independent RNA cleavage138. Cas13 proteins are RNA-guided RNA-targeting nucleases, some requiring recognition of a protospacer flanking sequence (PFS), that generate cuts along target and non-target RNA molecules using two HEPN domains, which are nucleotide-binding domains with RNA cutting activity142. b ∣ Similar to catalytically deficient Cas9 (dCas9), dCas13 maintains the capacity to bind the targeted RNA. For RNA visualization and tracking purposes, a fluorescent protein can be fused to the dCas protein and co-localize with an array of crRNAs or gRNAs135,143. Adenosine deaminase RNA specific (ADAR) enzymes can be fused to dCas for RNA A→I base editing to correct disease-relevant mutations. To promote alternative splicing, dCas13 can be targeted to bind splicing regulating cis elements152. c ∣ Cas13 can be used for targeted RNA degradation in eukaryotic cells for applications such as targeting viral RNA or toxic RNAs that contain microsatellite repeat expansions136. Francisella novicida Cas9 has been repurposed in eukaryotic cells to target the RNA genome of hepatitis C virus141.
RCas9
Cas9 can be made to cleave ssDNA targets by providing a PAM-presenting oligonucleotide (PAMmer) that anneals to ssDNA133. Similarly, a PAMmer can be provided to direct Cas9 to ssRNA targets134 (Figure 3a). To specifically target RNA while avoiding DNA, PAMmers can be designed for RNA sequences that lack PAMs at the corresponding genomic DNA sites. This RNA-targeting Cas9 system, termed RCas9, only requires the design and synthesis of a matching gRNA and complementary PAMmer134. By targeting dCas9 to RNA, RCas9 can be utilized as a programmable RNA-binding protein for RNA recognition (Figure 3b). This modular tool permits detection of endogenous RNA without the need to genetically encode affinity tags on transcripts. RCas9 binding to specific mRNAs has been utilized for their visualization and tracking into stress granules [G] in living cells135. Further development of this technology may provide a useful tool for RNA visualization of mRNAs of low abundance orconcentration.
Catalytically active RCas9 can stimulate site-specific cleavage of ssRNA134. Thus, RCas9 can be used to control cellular processes at the transcript level. Therapeutic strategies to block the expression of toxic RNA can utilize genome editing through DNA targeting, however this involves a risk of causing permanent off-target DNA edits. By contrast, the diagnostic and therapeutic potential for RCas9 has been demonstrated by visualizing and eliminating toxic RNA species associated with microsatellite-repeat expansion[G] diseases136 (Figure 3c). Specific RNA targeting and elimination were observed in patient cells ex vivo, but in vivo efficacy remains to be demonstrated. Although the development of RNA-targeting therapies is hindered by the need for continuously targeting newly synthesized transcripts, AAV delivery is known to support long-term transgene expression137, and truncated versions of RCas9 have been generated that are compatible with the limited AAV packaging capacity136.
Cas9 orthologs
Although in nature Cas9 is thought to preferentially target phage and DNA in bacteria, Cas9 orthologs have the capacity to also target RNA. SaCas9 and Campylobacter jejuni Cas9 (CjCas9) can directly cleave ssRNA in a PAM-independent manner138 (Figure 3a). When targeted to RNA, SaCas9 repressed gene expression in Escherichia coli138 and for CjCas9, crRNA-dependent but PAM-independent binding and cleavage of endogenous RNAs was shown139.
Francisella novicida Cas9 (FnCas9) was originally shown to target bacterial mRNA and alter gene expression140, and has been repurposed to target the RNA genome of hepatitis C virus (HCV) in eukaryotic cells141. This positive-sense ssRNA virus has a cytosolic life cycle and its RNA does not undergo reverse transcription and genomic integration. By targeting the 5’ or 3’ untranslated regions of the HCV genome, FnCas9 inhibited both viral protein production and replication (Figure 3c). Unlike RCas9, RNA targeting by FnCas9 is PAM-independent and thus does not require PAMmers141. In future, FnCas9 could also potentially be used to target negative-sense ssRNA viruses such as those belonging to the filoviridae, paramyxoviridae, or orthomyxoviridae families. Additional studies are needed to clarify the potential physiological consequences of RNA targeting by Cas9 in eukaryotic cells.
Cas13
CRISPR–Cas systems containing naturally RNA-targeting endonucleases have been recently discovered. In bacteria, Cas13a (formerly known as C2c2) is an RNA-guided RNA-targeting nuclease. This class 2 type VI CRISPR protein is activated upon recognition of ssRNA targets142 (Figure 3a). Similar to a PAM sequence, some type VI CRISPR proteins require recognition of a PFS142, however Cas13a from Leptotrichia wadei (LwaCas13a)143 and Cas13b from Prevotella sp. P5-125 (PspCas13b)144 do not. Following target binding, Cas13a cuts at uracil bases anywhere in its vicinity, and this ‘collateral’ cleavage extends also to nearby, untargeted RNAs. Cas13a has been programmed to cleave specific mRNAs in both bacteria and eukaryotic cells142,143. Unexpectedly, collateral cleavage by activated Cas13a was not observed in eukaryotic cells, but the mechanism for this difference remains unknown143. A catalytically inactive Cas13a variant, dCas13a, maintains the ability to bind targeted RNA and was used for live cell imaging of RNA143. Similar to RCas9, dCas13a has been targeted to mRNA to visualize the formation of stress granules135,143.
The collateral cleavage observed following programmed mRNA targeting in bacteria cells has also been demonstrated in vitro with purified Cas13a protein143,145. This promiscuous RNase activity, which is induced upon target recognition has been utilized as a molecular detection platform termed SHERLOCK (specific high sensitivity enzymatic reporter unlocking)146. Following detection of target RNA, Cas13a is activated for collateral-RNA cleavage-mediated release of a reporter signal. On the basis of this method, a diagnostic test was developed to detect viral RNA of specific strains of Zika and Dengue viruses146. Additionally, amplified DNA can be converted to RNA for subsequent Cas13-mediated detection146. Following conversion to RNA, SHERLOCK can be used to detect species-specific bacterial pathogens, discriminate between single-nucleotide polymorphisms in the human genome, and identify cell-free, mutated tumor DNA. Further development has resulted in the improved SHERLOCKV2 molecular detection platform, which can perform quantitative detection, has increased sensitivity, and can be used to detect simultaneously up to four targets147. Recently, Cas12a has also been repurposed as a detection tool. Following targeted activation by dsDNA, Cas12a nonspecifically cleaves ssDNA148. By providing a quenched ssDNA reporter, the collateral cleavage of Cas12a can be used to detect viral DNA in patient samples146,148.
A single-base RNA editing application has been developed by fusing dCas13 to adenosine deaminase acting on RNA (ADAR) enzymes (Figure 3b). This system, termed REPAIR (RNA editing for programmable A to I replacement), can make directed adenosine-to-inosine edits in eukaryotic cells144. In translation and splicing, inosine is functionally equivalent to guanine149,150. To broaden the base conversions achievable by REPAIR, dCas13 could be fused with other RNA editing domains such as that of APOBEC for potential cytidine-to-uridine editing. Additional applications for site-specific binding of dCas13a include studying RNA–protein interactions, visualizing RNA trafficking and localization with fluorescently tagged dCas13a or modulating the function or translation of transcripts with dCas13a fused to different effectors. The application of this RNA editing tool for treating genetic diseases remains to be explored.
Scanning of bacterial genome sequences has led to the identification of a class 2 type VI-D CRISPR effector, termed Cas13d. Similarly to Cas13a, Cas13d-mediated cleavage promotes collateral RNA cleavage in bacteria151 but not when expressed in mammalian cells152. RNA recognition by Cas13d is PFS-independent. dCas13d lacks target-RNA cleavage activity but retains
rRNA array processing activity, and notably, its smaller size makes packaging into vectors like AAV possible for in vivo applications151,152. These characteristics have been utilized to deliver dCas13d and a crRNA array targeting cis elements in pre-mRNAs to manipulate alternative splicing in a neuronal model of frontotemporal dementia152 (Figure 3b).
GENE REGULATION by CRISPR–Cas
Beyond gene editing through the formation of DNA breaks, site-specific gene regulation is possible by engineering Cas9 as a DNA recognition complex rather than a targeted nuclease153. Mutations in the RuvC (D10A) and HNH (H840A) nuclease domains destroy the catalytic activity of Cas9 while maintaining its RNA-guided DNA targeting capacity10,154. The CRISPR–Cas toolbox has been expanded by fusing this dCas9 with diverse effectors such as transcription repressors or activators, epigenetic modifiers, and fluorophores (Figure 4).
Figure 4. Targeted gene regulation and other applications.

a ∣ For transcription repression, catalytically-deficient Cas9 (dCas9) alone or dCas9 fused to effectors such as the transcription repression domain of Krϋppel-associated box domain (KRAB)157 can be targeted to promoters, 5’ untranslated region (5’ UTR) enhancers156,159-161. Transcription activation can be targeted by fusing dCas9 to transcription activation domains such as VP64: VP64–dCas9–VP64 activated the expression of the neuronal transcription-factor genes Brn2, Ascl1 and Myt1l and thus directed the conversion of primary mouse embryonic fibroblasts into neuronal cells172. Similarly, dCas9 was fused to the catalytic domain of methylcytosine dioxygenase TET1 and targeted to the FMR1 gene, to reverse the hypermethylation and silencing of the gene, which is associated with Fragile X syndrome176. b ∣ Inducible Cas9-based systems allow dynamic control of gene targeting. For example, chemical induction by rapamycin of the dimerization of split dCas9 fused to the rapamycin-binding domains of FKBP and FRB activates target-gene expression. Alternatively, light-inducible dimerization of the cytochrome proteins CRY2 and CIBN can be used in photoactivatable systems. Combinations of inducible dCas9-ortholog-based systems can be used for dynamic manipulation of multiple targets simultaneously. For example, dimerization of Streptococcus pyogenes dCas9 (dSpCas9)–KRAB by the addition of abscisic acid (ABA) can repress one gene while dimerization of Staphylococcus aureus dCas9 (dSaCas9)–VP64–p65–Rta (VPR) by the addition of gibberellin can lead to activation of another gene189. c ∣ CRISPR–dCas9 tools can monitor or manipulate chromatin interactions that regulate gene expression. The fusion of dCas9 to the peroxidase APEX2 can be used to biotinylate proteins in the vicinity of a targeted genomic locus; the proteins are then identified using mass spectrometry200. Distal loci can be brought into proximity using chromatin loop reorganization with CRISPR–dCas9 (CLOuD9). In the CLOuD9 system, dSpCas9 and dSaCas9 are fused to the dimerizing, ABA-binding proteins PYL1 and ABI1202. ABA induces targeted protein dimerization and chromatin looping, which can be reversed following its removal to restore the endogenous chromatin conformation.
Transcription regulators
The modularity of dCas9 is exemplified by the ability to tether protein effectors to dCas9 or to the gRNA and still maintain dCas9-mediated DNA targeting. Thus, a versatile DNA-targeting platform can be combined with various protein effectors for a broad range of applications.
CRISPRi
Binding of dCas9 to DNA elements may repress transcription by sterically hindering the RNA polymerase machinery154. dCas9-mediated steric interference, termed CRISPR interference (CRISPRi), works efficiently in prokaryotic cells but is less effective in eukaryotic cells154-156. To enhance the repressive capacity of CRISPR in eukaryotic cells, dCas9 has been tethered to transcription repressor domains such as that of Krϋppel-associated box (KRAB)156, which is found in many natural zinc-finger transcription factors157. KRAB is known to induce heterochromatin formation, and changes in chromatin structure often accompany dCas9–KRAB-targeted transcription repression158. dCas9–KRAB is a robust tool in mammalian cells that can effectively silence single genes and noncoding RNAs by targeting promoter regions, 5’ untranslated regions and proximal and distal enhancer elements156,159-161 (Figure 4a). For improved repressive capabilities, dCas9 was fused to a bipartite repressor consisting of the transcription repression domains of KRAB and of methyl-CpG-binding protein 2162. The versatility of dCas9–KRAB is highlighted by its capacity to repress transcription by targeting both genes and gene-regulatory regions161.
CRISPRa
dCas9 can also be fused to activator effectors for programmed transcription activation, termed CRISPR activation (CRISPRa). In eukaryotes, both reporter genes and endogenous genes can be activated by dCas9 fused to the transcription activation domains of the NF-κB transactivating subunit (p65) or to VP64 (four repeats of the herpes simplex VP16 activation domain)156,163-165. Synergistic gene activation has frequently been observed with these synthetic transcription factors by targeting multiple gRNAs to a promoter region163,164. In addition, synergy can be achieved by combining different activator domains166-170. Multiplexed activation of endogenous genes can also be used for cellular reprogramming171. For example, direct conversion of primary mouse embryonic fibroblasts to induced neuronal cells was achieved following activation of lineage-specific transcription factors by targeting VP64–dCas9–VP64 (dCas9 fused to VP64 at each of its termini) to the endogenous Brn2, Ascl1 and Myt1l genes172 (Figure 4a), and similar approaches have been applied to reprogramming cells into pluripotency173 or to myogenic cells167.
Epigenome editing
Targeted epigenetic modifications, such as acetylation and methylation of histones and methylation of DNA, can be achieved using dCas9-based tools153. For example, the fusion of dCas9 to the catalytic core of the human histone acetyltransferase p300 was targeted to promoters and enhancers for catalyzing the acetylation of histone H3 Lys 27, leading to robust gene activation174. DNA demethylation was achieved using dCas9 fusions with the catalytic domain of methylcytosine dioxygenase TET1. Targeting dCas9–TET1 to the BRCA1 promoter resulted in transcription up-regulation175. As a potential therapy, dCas9–TET1 was used to de-methylate the CGG-expansion mutation in the 5’ untranslated region of the gene FMR1 and reverse its silencing, which is associated with Fragile X syndrome176 (Figure 4a). Importantly, FMR1 expression was maintained following engraftment of edited cells into mouse brains.
For heritable transcriptional silencing, dCas9–KRAB can be used in combination with DNA methyltransferases (DNMTs). Stable silencing of the β2-microglobulin promoter–enhancer was achieved in up to 78% of K562 cells by the transient expression of dCas9 fused to the KRAB domain and to the catalytic domains of DNMT3A and DNMT3L, along with seven gRNAs177. Combined with the robustness of dCas9-mediated targeting, the plethora of potential epigenetic effectors provides many applications for epigenetic studies.
Dynamic control of Cas9 function
Inducible systems function by requiring particular stimuli for gene activation. Based on the type of stimulus, various strategies have been developed for generating inducible Cas9-based systems that permit temporal control of Cas9-mediated gene targeting (Figure 4b).
Chemical induction
Chemical compounds can activate Cas9 expression through inducible promoters. This may be desirable to precisely time gene knockout in certain cell types, rather than use constitutive knockout cell lines. Doxycycline-inducible expression of Cas9 has been used in human pluripotent stem cells178,179 and in adult mice180. However, doxycycline-independent mutagenesis has been observed in the transfected cells, suggesting the expression of Cas9 is leaky in some of these systems180.
Inducible dCas9-based systems also offer versatility in epigenome engineering. A doxycycline-inducible CRISPRi system enabled efficient, tunable and reversible disease modeling in induced pluripotent stem cell (iPSC)-derived cardiomyocytes181. Chemically inducible CRISPRa systems have been developed using conditionally stabilized dCas9–activators182, or split dCas9–activators that dimerize following chemical induction183,184 (Figure 4b). Beneficial uses of inducible split dCas9–activators include minimizing leaky dCas9 expression and targeting multiple genes for multiplexed temporal regulation184.
Optogenetics
Light-inducible dCas9 systems enable precise dynamic regulation of endogenous genes and the possibility of spatial control. For example, light-inducible dimerization of the plant-derived cytochrome proteins CRY2 and CIB1 has been used to create photoactivatable dCas9–p65185 and dCas9–VP64186 (Figure 4b). An anti-CRISPR protein [G] was engineered for light-mediated spatiotemporal control of genome and epigenome editing in human cells by pairing a photosensor from Avena sativa with a SpCas9 inhibitor187. A second-generation optogenetic split-protein system was developed and targeted to upregulate the expression of the gene neurogenic differentiation 1 to induce neuronal differentiation in iPSCs188. For more complex regulation, multiple chemical- and light-inducible systems have been used to dynamically manipulate the activation or repression of multiple genes189 (Figure 4b). These light-inducible systems hold promise for modeling development and disease with reversible and temporal control of gene expression.
Other genomic dCas9 applications
CRISPR-dCas9 gene regulation systems are proving immensely valuable for elucidating the function of transcribed genes. Another important application of these tools lies in understanding the function of the noncoding genome. Cas9 and dCas9-engineered effectors provide an opportunity to explore these genomic regions, for which there are no other tools for direct perturbation.
Annotating the non-coding genome
With the inception of dCas9–effector tools, CRISPRi and CRISPRa methods are also being developed for high-throughput screening to annotate the non-coding genome170,190-194 (Box 1). Epigenome editing with these methods permits efficient perturbation of regulatory elements without mutating the DNA, and CRISPRa-based methods enable gain-of-function studies. CRISPRi and CRISPRa have been combined in parallel screens to target DNaseI hypersensitive sites [G] that surround genes of interest193. This unique approach identified regulatory elements that may be dependent on the direction of dCas9-based transcription perturbation. Collectively, these dCas9-based methods enable the elucidation of the roles of regulatory sequences in their native genomic contexts and allow the screening of lncRNAs whose function might not be altered by introduction of indels with Cas9 nucleases195,196.
Chromatin interactions
Chromatin structure modulates genome function, however elucidating the molecular basis of this modulation has been limited by an inadequate availability of methods to study chromatin–protein interactions. To identify proteins that interact with specific genome loci, the chromatin can be immunoprecipitated using an antibody against a dCas9–tag fusion protein, which is co-expressed with a gRNA that targets the desired DNA sequence. This method, named engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) is then followed by mass spectrometry to identify the locus-associated proteins197. enChIP was used for biochemical analysis of transcription and epigenetic regulation at specific genomic loci in living cells198. Alternatively, dCas9 has been tethered to APEX2, which is an engineered peroxidase that promiscuously labels nearby proteins with biotin199,200. dCas9–APEX2 can be used to biotinylate proteins in the vicinity of a targeted genomic locus; these proteins can then be identified following affinity purification and mass spectrometry (Figure 4c)200.
Regulation of gene expression is also influenced by the formation of long-range chromatin interactions, often referred to as chromatin looping. To better understand the role of chromatin interactions, dCas9-based methods have been developed for precisely modifying chromatin looping. Biotinylated dCas9 has been used to identify chromatin-associated proteins and study long-range chromatin interactions201. Chromatin loop reorganization with CRISPR–dCas9 (CLOuD9) can selectively and reversibly establish chromatin loops and modulate the expression of associated genes202 (Figure 4c). As an alternative to the chemically induced CLOuD9 system, a light-inducible dCas9 system was developed for directing rearrangement of chromatin looping on faster time scales203. A chemically inducible and reversible system termed CRISPR-GO can control spatial genome organization within the cell204. CRISPR-GO enables studying chromatin interactions within nuclear compartments to help elucidate their function. CRISPRi tools such as dCas9–KRAB have also been used to disrupt anchored looping interactions that coordinate changes in gene expression205. These studies have helped to confirm the roles of interactions between loci in the maintenance of gene expression. Chromatin restructuring facilitated by these technologies will be greatly beneficial for studying the dynamic roles of genome architecture in gene regulation.
Imaging loci
Methods to image specific DNA sequences are useful for studying the spatial organization of the genome. Fluorescence in situ hybridization techniques have been valuable for this purpose, however they require cell fixation. For live cell imaging, EGFP-tagged dCas9 and structurally optimized gRNAs have been targeted to repetitive elements and to coding genes206. By targeting a large number of loci, labeling of an entire chromosome was made possible for live cell imaging207. Depending on chromosome length, painting entire human chromosomes could require about 100–800 gRNAs.
To expand these tools to multicolor genome imaging, orthogonal dCas9s have been tagged with different fluorescent proteins208,209. Other dCas9-based multicolor, live cell imaging methods have focused on engineering gRNA scaffolds [G]. By adapting gRNA scaffolds to bind sets of fluorescent proteins, up to 6 targeted chromosomal loci were visualized simultaneously210. Additionally, gRNA aptamer [G] insertions have been engineered that concurrently bind two different fluorescent protein tags211. This dual-color approach is tolerant to photobleaching [G], which makes it useful for long-term imaging of genomic loci.
BIOMEDICAL APPLICATIONS OF CRISPR TOOLS
CRISPR–Cas-based gene editing and epigenome engineering tools have revolutionized our ability to manipulate the genomic functions. Importantly, these tools are now being applied in gene therapy and in enhancing cell therapy.
Pre-clinical gene therapy
Genome editing technologies have transformed the gene therapy paradigm from delivery of an exogenous transgene to editing human genome sequences. The therapeutic potential of making precise, targeted genome modifications includes a wide variety of diseases and disorders, but potential limitations must be overcome as CRISPR–Cas-based technologies advance to the clinic (Box 2). Although the most obvious therapeutic applications of genome editing are correcting mutations that cause genetic diseases, a variety of editing strategies exist that manipulate genes involved in more common, complex disease. For example, by targeting SpCas9 to the mouse cholesterol homeostasis gene Pcsk9 through adenovirus delivery, a reduction in low-density lipoprotein cholesterol was demonstrated following gene disruption and silencing in vivo212. For preclinical assessment of somatic genome editing applications, this work has been expanded to successfully target the human PCSK9 gene in mice engrafted with human hepatocytes213. Similar approaches have been explored using epigenetic silencing of Pcsk9 by viral delivery of dCas9–KRAB.214
Box 2 ∣. Potential limitations of CRISPR–Cas medical applications.
Despite the advances in CRISPR–Cas-based genome engineering technologies, some challenges remain for translating these tools to the clinic:
Adeno-associated virus (AAV), which is the most frequently used gene-therapy delivery vehicle, provides limited packaging capacity of genetic information. This restriction has led to continued development and in vivo testing of smaller Cas9 orthologs such as Staphylococcus aureus Cas9 (SaCas9)29 and Campylobacter jejuni Cas931. Nevertheless, prolonged expression of Cas9 from AAV vectors and integration of AAV vectors into DSBs remain undesirable consequences of AAV delivery.221
Off-target effects, which remain a major concern, can be reduced with preliminary guide RNA selection and optimization. For example, VIVO (verification of in vivo off-targets)242 can be used with CIRCLE-seq (cleavage effects by sequencing)243 to screen off-targets using the genomic DNA from the specific patient or organism. More-sensitive methods are necessary to detect possible off-target editing and to understand the possible implications of any unintended genome changes.
Immunogenicity of Cas proteins is another potential obstacle to their clinical application. Immune responses to Cas9 following its delivery into mouse models is well-documented221,244, but the implications of this for therapeutic approaches are still unclear. Recently, pre-existing adaptive immunity to Streptococcus pyogenes Cas9 and SaCas9 has been detected in human blood samples245-247. In the case of intracellular expression of virus-delivered Cas9, T cell responses may be worrisome if they are reactive to Cas9 peptides displayed by treated cells. More studies are needed to decipher the implications for clinical use of pre-existing immunity. The high prevalence of exposure of the human population to S. pyogenes and S. aureus is an additional motivation for ongoing testing of novel Cas9 orthologs. Other approaches to limit immunogenicity include the reengineering of immunogenic epitopes of Cas proteins, the use of transient immunosuppressive drugs during treatment, or ex vivo cell modification.
A potential limitation is the observation that CRISPR–Cas-mediated gene editing is more efficient in cells that have lost the function of the tumor suppressor p5377,78.
Therapeutic genome editing strategies are currently being explored for ocular diseases such as retinitis pigmentosa, which can result in blindness. Cas9-mediated disruption of the gene Nrl by indel formation preserved the function of cone photoreceptors in three different mouse models of retinal degradation215. A HITI-mediated Cas9 insertion repaired the 1.9kb deletion in the kinase gene Mertk in a retinitis pigmentosa rat model and restored MERTK function65. The gene therapeutics were delivered to the eye using AAV vectors. Importantly, AAV is the most frequently used gene-therapy delivery vehicle due to its effective and safe track record and wide range of tissue targeting. AAV delivery to skeletal and cardiac muscle can be used for treatment of neuromuscular disorders such as Duchenne muscular dystrophy (DMD). In most individuals with DMD, a hotspot of various deletions exists that disturbs the open reading frame of the DMD gene, which encodes dystrophin216. Restoration of the reading frame in vivo has been achieved in several studies following AAV-mediated delivery of CRISPR–Cas9 to excise additional exons through NHEJ around the inherited deletion217-219, including a mutation correction that has been sustained for at least one year after CRISPR–Cas9 administration220,221. These deletion-based editing approaches resulted in the expression of a truncated but partially functional dystrophin. Importantly, progress has been made in advancing these approaches to testing in large-animal models of DMD222. In order to avoid the generation of DSBs, Cas9-mediated single-base editing of splice site donors and acceptors has also been explored in these models223,224.
In addition to viral delivery methods, lipid nanoparticles (LNPs) can be utilized to deliver Cas9 in vivo. Recently, LNPs containing SpCas9 mRNA and a chemically modified gRNA targeting the mouse Ttr gene were delivered to mice225. Following a single administration, a reduction of TTR serum protein levels was observed and levels of in vivo genome editing required for therapeutic benefit were achieved. The clinical significance of CRISPR–Cas-based therapeutics relies on coupling genome editing developments with continued advancements in delivery methods226. In particular, transient, non-viral-mediated delivery strategies may be useful in addressing concerns about long-term expression of immunogenic Cas proteins and integration of DNA vectors into the genome221,227.
Translation to the clinic
The most clinically advanced gene editing strategies rely on ex vivo cell manipulation that provides therapeutic effects following the administration of the cells back to the donor. In particular, engineered autologous T cells have been successful in adoptive T-cell immunotherapy228. Gene editing approaches have been used for enhancing the properties of these engineered cells. For example, the insertion of transgenes encoding programmable chimeric antigen receptors [G] (CARs) into the endogenous T cell receptor alpha constant gene, rather than overexpression of CARs from viral vectors, prevents the exhaustion of T cells from overstimulation229,230. Another important therapeutic application of genome editing is in knocking out components of the human leukocyte antigen system to generate universal cell donors [G],231 which would address the practical and economic challenges of patient-specific autologous cell therapies. Researchers have also targeted programmed cell death protein 1 (PD-1) to block inhibitory signals that prevent T-cell recognition of tumor cells230,232,233. In fact, autologous T cells that were treated ex vivo with Cas9 to knock out PD-1 were infused back into individuals with cancer, in the first use of CRISPR–Cas gene editing in a human clinical trial in the United States or Europe ( NCT03399448)234,235. Also currently underway are the first human trial of CRISPR–Cas to treat a genetic disease, β-thalassemia ( NCT03655678), and the first trial of in vivo genome editing by CRISPR–Cas in retina to treat a rare form of blindness ( NCT03872479).227 Importantly, these CRISPR–Cas-based clinical trials build on a foundation of several genome editing clinical trials using zinc finger nucleases236. Collectively, these clinical trials will establish the therapeutic potential of recently developed genome engineering tools.
CONCLUSIONS AND FUTURE DIRECTIONS
Repurposing CRISPR–Cas systems for use in eukaryotic cells has revolutionized the genome engineering field. Even with the extensive use of type II CRISPR-Cas systems, continued discovery and development of CRISPR systems from prokaryotic species has resulted in new, beneficial technologies, such as Cas13a-based RNA targeting tools. Fusing dCas9 to the plethora of effectors will continue to expand possibilities for targeted epigenetic modulation.
The ease of gRNA-library generation for large-scale Cas9 targeting coupled with advancements in next-generation sequencing has made genome-wide genetic and epigenetic screens readily available. Perturbation at this magnitude will advance our understanding of biological mechanisms and aid the discovery of new therapeutic targets. Additionally, the multiplexed targeting potential of CRISPR–Cas systems will enable more complex and sophisticated manipulation of cellular processes.
As CRISPR–Cas-based therapeutics enter clinical testing, they hold great potential for correcting genetic diseases and enhancing cell therapies. Preclinical results are promising but safety and efficacy need to be monitored closely during these studies. A potential risk of using gene editing methods is the introduction of off-target changes to genome sequence, and thus enhancing methods for detecting rare mutations and quantifying their potential risks will be important for future clinical advancement.
Supplementary Material
Acknowledgements
This work was supported by an Allen Distinguished Investigator Award from the Paul G. Allen Frontiers Group, Open Philanthropy, US National Institutes of Health (NIH) grants R01DA036865, R01AR069085, R21NS103007, R33DA041878, P30AR066527, R41GM119914, R41AI136755, U01HG007900, UM1HG009428, UG3TR002142, and the National Science Foundation grants EFRI-1830957 and DMR-1709527. A.P-O. is supported by a Pfizer-NC Biotech Distinguished Postdoctoral Fellowship.
Glossary
- Spacer
The interchangeable portion of the guide RNA that is complementary to the targeted sequence
- Directed evolution
Method to generate and select for nucleic-acid or protein variants with desirable properties
- crRNA arrays
In bacterial genomes, series of spacers flanked by repeats, which are transcribed as a single pre-crRNA array and subsequently processed into individual crRNAs
- Indels
Small insertions or deletions of nucleotides at repair sites of DNA double-strand breaks
- Stress granules
Denote a type of cytosolic membraneless bodies with high concentrations of RNA and/or proteins, which form in different cell stress conditions
- Microsatellite-repeat expansion
Repetitive DNA sequences that can expand between generations and encode RNAs that are toxic to cells and cause neurological disorders
- Anti-CRISPR protein
A protein that interacts with and inhibits CRISPR–Cas activity
- DNaseI hypersensitive sites
Chromatin regions accessible to the enzyme DNase I; generally denote gene-activity-permissive chromatin
- gRNA scaffolds
The backbone (invariable) portions of gRNAs, which are recognized by Cas proteins
- gRNA aptamer
RNA structures added to the gRNA scaffold, which can bind specific effector molecules
- Photobleaching
Reduction in the intensity of fluorescence emission owing to the imaging of a sample over time
- Seed region
PAM-proximal nucleotides in the target sequence, where spacer mismatches are less tolerated for on-target gRNA binding
- Chimeric antigen receptors
T cell receptors engineered to recognize a specific antigen
- Universal cell donors
Cells engineered to avoid recognition by a recipient immune system
Footnotes
Competing interests
CAG and AKP are inventors on patent applications related to CRISPR technologies. CAG is a scientific advisor to Element Genomics, Locus Biosciences, and Sarepta Therapeutics.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Molecular Cell Biology thanks J. Chen, R. Platt and other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Danna K & Nathans D Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc Natl Acad Sci U S A 68, 2913–2917 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts RJ How restriction enzymes became the workhorses of molecular biology. Proc Natl Acad Sci U S A 102, 5905–5908, doi: 10.1073/pnas.0500923102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olorunniji FJ, Rosser SJ & Stark WM Site-specific recombinases: molecular machines for the Genetic Revolution. Biochem J 473, 673–684, doi: 10.1042/BJ20151112 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Chandrasegaran S & Carroll D Origins of Programmable Nucleases for Genome Engineering. J Mol Biol 428, 963–989, doi: 10.1016/j.jmb.2015.10.014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrangou R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712, doi: 10.1126/science.1138140 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Makarova KS et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13, 722–736, doi: 10.1038/nrmicro3569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shmakov S et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol 15, 169–182, doi: 10.1038/nrmicro.2016.184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koonin EV, Makarova KS & Zhang F Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37, 67–78, doi: 10.1016/j.mib.2017.05.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278, doi: 10.1016/j.cell.2014.05.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821, doi: 10.1126/science.1225829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823, doi: 10.1126/science.1231143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826, doi: 10.1126/science.1232033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garneau JE et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71, doi: 10.1038/nature09523 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J & Almendros C Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740, doi: 10.1099/mic.0.023960-0 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Deltcheva E et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607, doi: 10.1038/nature09886 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasiunas G, Barrangou R, Horvath P & Siksnys V Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109, E2579–2586, doi: 10.1073/pnas.1208507109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleinstiver BP et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485, doi: 10.1038/nature14592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63, doi: 10.1038/nature26155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slaymaker IM et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88, doi: 10.1126/science.aad5227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495, doi: 10.1038/nature16526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JS et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410, doi: 10.1038/nature24268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casini A et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol 36, 265–271, doi: 10.1038/nbt.4066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vakulskas CA et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med 24, 1216–1224, doi: 10.1038/s41591-018-0137-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kocak DD et al. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat Biotechnol, doi: 10.1038/s41587-019-0095-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deveau H et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190, 1390–1400, doi: 10.1128/JB.01412-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esvelt KM et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10, 1116–1121, doi: 10.1038/nmeth.2681 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50, 488–503, doi: 10.1016/j.molcel.2013.05.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou Z et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A 110, 15644–15649, doi: 10.1073/pnas.1313587110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191, doi: 10.1038/nature14299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada M et al. Crystal Structure of the Minimal Cas9 from Campylobacter jejuni Reveals the Molecular Diversity in the CRISPR-Cas9 Systems. Mol Cell 65, 1109–1121.e1103, doi: 10.1016/j.molcel.2017.02.007 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Kim E et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun 8, 14500, doi: 10.1038/ncomms14500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burstein D et al. New CRISPR-Cas systems from uncultivated microbes. Nature 542, 237–241, doi: 10.1038/nature21059 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zetsche B et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771, doi: 10.1016/j.cell.2015.09.038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zetsche B et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 35, 31–34, doi: 10.1038/nbt.3737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleinstiver BP et al. Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat Biotechnol, doi: 10.1038/s41587-018-0011-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao L et al. Engineered Cpf1 variants with altered PAM specificities. Nat Biotechnol 35, 789–792, doi: 10.1038/nbt.3900 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jore MM et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol 18, 529–536, doi: 10.1038/nsmb.2019 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Westra ER et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46, 595–605, doi: 10.1016/j.molcel.2012.03.018 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hochstrasser ML et al. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc Natl Acad Sci U S A 111, 6618–6623, doi: 10.1073/pnas.1405079111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brendel J et al. A complex of Cas proteins 5, 6, and 7 is required for the biogenesis and stability of clustered regularly interspaced short palindromic repeats (crispr)-derived rnas (crrnas) in Haloferax volcanii. J Biol Chem 289, 7164–7177, doi: 10.1074/jbc.M113.508184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richter C, Gristwood T, Clulow JS & Fineran PC In vivo protein interactions and complex formation in the Pectobacterium atrosepticum subtype I-F CRISPR/Cas System. PLoS One 7, e49549, doi: 10.1371/journal.pone.0049549 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiedenheft B et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci U S A 108, 10092–10097, doi: 10.1073/pnas.1102716108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayes RP et al. Structural basis for promiscuous PAM recognition in type I-E Cascade from E. coli. Nature 530, 499–503, doi: 10.1038/nature16995 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinkunas T et al. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J 30, 1335–1342, doi: 10.1038/emboj.2011.41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jackson RN, Lavin M, Carter J & Wiedenheft B Fitting CRISPR-associated Cas3 into the helicase family tree. Curr Opin Struct Biol 24, 106–114, doi: 10.1016/j.sbi.2014.01.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomaa AA et al. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. MBio 5, e00928–00913, doi: 10.1128/mBio.00928-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K & Doudna JA Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329, 1355–1358, doi: 10.1126/science.1192272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nissim L, Perli SD, Fridkin A, Perez-Pinera P & Lu TK Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell 54, 698–710, doi: 10.1016/j.molcel.2014.04.022 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai SQ et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32, 569–576, doi: 10.1038/nbt.2908 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wyman C & Kanaar R DNA double-strand break repair: all's well that ends well. Annu Rev Genet 40, 363–383, doi: 10.1146/annurev.genet.40.110405.090451 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Lin S, Staahl BT, Alla RK & Doudna JA Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766, doi: 10.7554/eLife.04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinder J, Salsman J & Dellaire G Nuclear domain 'knock-in' screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res 43, 9379–9392, doi: 10.1093/nar/gkv993 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song J et al. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat Commun 7, 10548, doi: 10.1038/ncomms10548 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gutschner T, Haemmerle M, Genovese G, Draetta GF & Chin L Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep 14, 1555–1566, doi: 10.1016/j.celrep.2016.01.019 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Aird EJ, Lovendahl KN, St Martin A, Harris RS & Gordon WR Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Communications biology 1, 54, doi: 10.1038/s42003-018-0054-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ran FA et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389, doi: 10.1016/j.cell.2013.08.021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu S et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol 34, 1279–1286, doi: 10.1038/nbt.3715 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Essletzbichler P et al. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res 24, 2059–2065, doi: 10.1101/gr.177220.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Canver MC et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem 289, 21312–21324, doi: 10.1074/jbc.M114.564625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao A et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res 41, e141, doi: 10.1093/nar/gkt464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou J et al. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J 281, 1717–1725, doi: 10.1111/febs.12735 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Yang H et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379, doi: 10.1016/j.cell.2013.08.022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kraft K et al. Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell Rep 10, 833–839 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Suzuki K et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149, doi: 10.1038/nature20565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lackner DH et al. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nat Commun 6, 10237, doi: 10.1038/ncomms10237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmid-Burgk JL, Honing K, Ebert TS & Hornung V CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat Commun 7, 12338, doi: 10.1038/ncomms12338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leonetti MD, Sekine S, Kamiyama D, Weissman JS & Huang B A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci U S A 113, E3501–3508, doi: 10.1073/pnas.1606731113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang H et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918, doi: 10.1016/j.cell.2013.04.025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Platt RJ et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455, doi: 10.1016/j.cell.2014.09.014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Quadros RM et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol 18, 92, doi: 10.1186/s13059-017-1220-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paquet D et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129, doi: 10.1038/nature17664 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Kwart D, Paquet D, Teo S & Tessier-Lavigne M Precise and efficient scarless genome editing in stem cells using CORRECT. Nat Protoc 12, 329–354, doi: 10.1038/nprot.2016.171 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Choi PS & Meyerson M Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun 5, 3728, doi: 10.1038/ncomms4728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Torres R et al. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat Commun 5, 3964, doi: 10.1038/ncomms4964 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Maddalo D et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427, doi: 10.1038/nature13902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haapaniemi E, Botla S, Persson J, Schmierer B & Taipale J CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 24, 927–930, doi: 10.1038/s41591-018-0049-z (2018). [DOI] [PubMed] [Google Scholar]
- 78.Ihry RJ et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 24, 939–946, doi: 10.1038/s41591-018-0050-6 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424, doi: 10.1038/nature17946 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nishida K et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Ma Y et al. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods 13, 1029–1035, doi: 10.1038/nmeth.4027 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Kim K et al. Highly efficient RNA-guided base editing in mouse embryos. Nat Biotechnol 35, 435–437, doi: 10.1038/nbt.3816 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Rees HA et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun 8, 15790, doi: 10.1038/ncomms15790 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rossidis AC et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat Med 24, 1513–1518, doi: 10.1038/s41591-018-0184-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liang P et al. Correction of beta-thalassemia mutant by base editor in human embryos. Protein Cell 8, 811–822, doi: 10.1007/s13238-017-0475-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li G et al. Highly efficient and precise base editing in discarded human tripronuclear embryos. Protein Cell 8, 776–779, doi: 10.1007/s13238-017-0458-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chadwick AC, Wang X & Musunuru K In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler Thromb Vasc Biol 37, 1741–1747, doi: 10.1161/ATVBAHA.117.309881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Komor AC et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Science advances 3, eaao4774, doi: 10.1126/sciadv.aao4774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li X et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat Biotechnol 36, 324–327, doi: 10.1038/nbt.4102 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Gehrke JM et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol 36, 977–982, doi: 10.1038/nbt.4199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang X et al. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat Biotechnol 36, 946–949, doi: 10.1038/nbt.4198 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Zong Y et al. Efficient C-to-T base editing in plants using a fusion of nCas9 and human APOBEC3A. Nat Biotechnol 36, 950–953 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471, doi: 10.1038/nature24644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 36, 843–846, doi: 10.1038/nbt.4172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim D et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol 35, 475–480, doi: 10.1038/nbt.3852 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Jin S et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science, doi: 10.1126/science.aaw7166 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Zuo E et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science, doi: 10.1126/science.aav9973 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song CQ et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat Biomed Eng, doi: 10.1038/s41551-019-0357-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87, doi: 10.1126/science.1247005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hart T et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526, doi: 10.1016/j.cell.2015.11.015 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Parnas O et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686, doi: 10.1016/j.cell.2015.06.059 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen S et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260, doi: 10.1016/j.cell.2015.02.038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C & Yusa K Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32, 267–273, doi: 10.1038/nbt.2800 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Wang T, Wei JJ, Sabatini DM & Lander ES Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84, doi: 10.1126/science.1246981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang T et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101, doi: 10.1126/science.aac7041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dixit A et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866 e1817, doi: 10.1016/j.cell.2016.11.038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jaitin DA et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896 e1815, doi: 10.1016/j.cell.2016.11.039 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 14, 297–301, doi: 10.1038/nmeth.4177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sanson KR et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 9, 5416, doi: 10.1038/s41467-018-07901-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Han K et al. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat Biotechnol 35, 463–474, doi: 10.1038/nbt.3834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shen JP et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods 14, 573–576, doi: 10.1038/nmeth.4225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Billon P et al. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol Cell 67, 1068–1079 e1064, doi: 10.1016/j.molcel.2017.08.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197, doi: 10.1038/nature15521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu Y et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med, doi: 10.1038/s41591-019-0401-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Findlay GM, Boyle EA, Hause RJ, Klein JC & Shendure J Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513, 120–123, doi: 10.1038/nature13695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vierstra J et al. Functional footprinting of regulatory DNA. Nat Methods 12, 927–930, doi: 10.1038/nmeth.3554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Korkmaz G et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat Biotechnol 34, 192–198, doi: 10.1038/nbt.3450 (2016). [DOI] [PubMed] [Google Scholar]
- 118.Diao Y et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat Methods 14, 629–635, doi: 10.1038/nmeth.4264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gasperini M et al. CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am J Hum Genet 101, 192–205, doi: 10.1016/j.ajhg.2017.06.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rajagopal N et al. High-throughput mapping of regulatory DNA. Nat Biotechnol 34, 167–174, doi: 10.1038/nbt.3468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sen DR et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169, doi: 10.1126/science.aae0491 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Canver MC et al. Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat Genet 49, 625–634, doi: 10.1038/ng.3793 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klann TS, Black JB & Gersbach CA CRISPR-based methods for high-throughput annotation of regulatory DNA. Curr Opin Biotechnol 52, 32–41, doi: 10.1016/j.copbio.2018.02.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu Y et al. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat Biotechnol 36, 1203–1210 (2018). [DOI] [PubMed] [Google Scholar]
- 125.Perli SD, Cui CH & Lu TK Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 353, aag0511 (2016). [DOI] [PubMed] [Google Scholar]
- 126.Kalhor R, Mali P & Church GM Rapidly evolving homing CRISPR barcodes. Nat Methods 14, 195–200, doi: 10.1038/nmeth.4108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tang W & Liu DR Rewritable multi-event analog recording in bacterial and mammalian cells. Science 360, eaap8992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McKenna A et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907, doi: 10.1126/science.aaf7907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Frieda KL et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111, doi: 10.1038/nature20777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yosef I, Goren MG & Qimron U Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res 40, 5569–5576, doi: 10.1093/nar/gks216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shipman SL, Nivala J, Macklis JD & Church GM Molecular recordings by directed CRISPR spacer acquisition. Science 353, aaf1175, doi: 10.1126/science.aaf1175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shipman SL, Nivala J, Macklis JD & Church GM CRISPR-Cas encoding of a digital movie into the genomes of a population of living bacteria. Nature 547, 345–349, doi: 10.1038/nature23017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67, doi: 10.1038/nature13011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.O'Connell MR et al. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516, 263–266, doi: 10.1038/nature13769 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nelles DA et al. Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell 165, 488–496, doi: 10.1016/j.cell.2016.02.054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Batra R et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 170, 899–912 e810, doi: 10.1016/j.cell.2017.07.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Naldini L Gene therapy returns to centre stage. Nature 526, 351–360, doi: 10.1038/nature15818 (2015). [DOI] [PubMed] [Google Scholar]
- 138.Strutt SC, Torrez RM, Kaya E, Negrete OA & Doudna JA RNA-dependent RNA targeting by CRISPR-Cas9. Elife 7, e32724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dugar G et al. CRISPR RNA-Dependent Binding and Cleavage of Endogenous RNAs by the Campylobacter jejuni Cas9. Mol Cell 69, 893–905.e897, doi: 10.1016/j.molcel.2018.01.032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sampson TR, Saroj SD, Llewellyn AC, Tzeng YL & Weiss DS A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497, 254–257, doi: 10.1038/nature12048 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Price AA, Sampson TR, Ratner HK, Grakoui A & Weiss DS Cas9-mediated targeting of viral RNA in eukaryotic cells. Proc Natl Acad Sci U S A 112, 6164–6169, doi: 10.1073/pnas.1422340112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abudayyeh OO et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573, doi: 10.1126/science.aaf5573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abudayyeh OO et al. RNA targeting with CRISPR-Cas13. Nature 550, 280–284, doi: 10.1038/nature24049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cox DBT et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027, doi: 10.1126/science.aaq0180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.East-Seletsky A et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273, doi: 10.1038/nature19802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gootenberg JS et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442, doi: 10.1126/science.aam9321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gootenberg JS et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444, doi: 10.1126/science.aaq0179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen JS et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439, doi: 10.1126/science.aar6245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nishikura K Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79, 321–349, doi: 10.1146/annurev-biochem-060208-105251 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tan MH et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254, doi: 10.1038/nature24041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yan WX et al. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol Cell 70, 327–339.e325, doi: 10.1016/j.molcel.2018.02.028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Konermann S et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676.e614, doi: 10.1016/j.cell.2018.02.033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thakore PI, Black JB, Hilton IB & Gersbach CA Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods 13, 127–137, doi: 10.1038/nmeth.3733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Qi LS et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183, doi: 10.1016/j.cell.2013.02.022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Larson MH et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc 8, 2180–2196, doi: 10.1038/nprot.2013.132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451, doi: 10.1016/j.cell.2013.06.044 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Margolin JF et al. Kruppel-associated boxes are potent transcriptional repression domains. Proc Natl Acad Sci U S A 91, 4509–4513 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kearns NA et al. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141, 219–223, doi: 10.1242/dev.103341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gao X et al. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res 42, e155, doi: 10.1093/nar/gku836 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kearns NA et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12, 401–403, doi: 10.1038/nmeth.3325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Thakore PI et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12, 1143–1149, doi: 10.1038/nmeth.3630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yeo NC et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods 15, 611–616, doi: 10.1038/s41592-018-0048-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Perez-Pinera P et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10, 973–976, doi: 10.1038/nmeth.2600 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Maeder ML et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10, 977–979, doi: 10.1038/nmeth.2598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Farzadfard F, Perli SD & Lu TK Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth Biol 2, 604–613, doi: 10.1021/sb400081r (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cheng AW et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23, 1163–1171, doi: 10.1038/cr.2013.122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chakraborty S et al. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Reports 3, 940–947, doi: 10.1016/j.stemcr.2014.09.013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS & Vale RD A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646, doi: 10.1016/j.cell.2014.09.039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chavez A et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12, 326–328, doi: 10.1038/nmeth.3312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Konermann S et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588, doi: 10.1038/nature14136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Black JB & Gersbach CA Synthetic transcription factors for cell fate reprogramming. Curr Opin Genet Dev 52, 13–21, doi: 10.1016/j.gde.2018.05.001 (2018). [DOI] [PubMed] [Google Scholar]
- 172.Black JB et al. Targeted Epigenetic Remodeling of Endogenous Loci by CRISPR/Cas9-Based Transcriptional Activators Directly Converts Fibroblasts to Neuronal Cells. Cell Stem Cell 19, 406–414, doi: 10.1016/j.stem.2016.07.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu P, Chen M, Liu Y, Qi LS & Ding S CRISPR-Based Chromatin Remodeling of the Endogenous Oct4 or Sox2 Locus Enables Reprogramming to Pluripotency. Cell Stem Cell 22, 252–261 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Hilton IB et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33, 510–517, doi: 10.1038/nbt.3199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Choudhury SR, Cui Y, Lubecka K, Stefanska B & Irudayaraj J CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 7, 46545–46556, doi: 10.18632/oncotarget.10234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu XS et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172, 979–992.e976, doi: 10.1016/j.cell.2018.01.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Amabile A et al. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 167, 219–232.e214, doi: 10.1016/j.cell.2016.09.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gonzalez F et al. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 15, 215–226, doi: 10.1016/j.stem.2014.05.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chen Y et al. Engineering Human Stem Cell Lines with Inducible Gene Knockout using CRISPR/Cas9. Cell Stem Cell 17, 233–244, doi: 10.1016/j.stem.2015.06.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dow LE et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol 33, 390–394, doi: 10.1038/nbt.3155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mandegar MA et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18, 541–553, doi: 10.1016/j.stem.2016.01.022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Balboa D et al. Conditionally Stabilized dCas9 Activator for Controlling Gene Expression in Human Cell Reprogramming and Differentiation. Stem Cell Reports 5, 448–459, doi: 10.1016/j.stemcr.2015.08.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zetsche B, Volz SE & Zhang F A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol 33, 139–142, doi: 10.1038/nbt.3149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bao Z, Jain S, Jaroenpuntaruk V & Zhao H Orthogonal Genetic Regulation in Human Cells Using Chemically Induced CRISPR/Cas9 Activators. ACS Synth Biol 6, 686–693, doi: 10.1021/acssynbio.6b00313 (2017). [DOI] [PubMed] [Google Scholar]
- 185.Nihongaki Y, Yamamoto S, Kawano F, Suzuki H & Sato M CRISPR-Cas9-based photoactivatable transcription system. Chem Biol 22, 169–174, doi: 10.1016/j.chembiol.2014.12.011 (2015). [DOI] [PubMed] [Google Scholar]
- 186.Polstein LR & Gersbach CA A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nature chemical biology 11, 198–200, doi: 10.1038/nchembio.1753 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bubeck F et al. Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. Nat Methods 15, 924–927, doi: 10.1038/s41592-018-0178-9 (2018). [DOI] [PubMed] [Google Scholar]
- 188.Nihongaki Y et al. CRISPR-Cas9-based photoactivatable transcription systems to induce neuronal differentiation. Nat Methods 14, 963–966, doi: 10.1038/nmeth.4430 (2017). [DOI] [PubMed] [Google Scholar]
- 189.Gao Y et al. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat Methods 13, 1043–1049, doi: 10.1038/nmeth.4042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fulco CP et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773, doi: 10.1126/science.aag2445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Xie S, Duan J, Li B, Zhou P & Hon GC Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol Cell 66, 285–299 e285, doi: 10.1016/j.molcel.2017.03.007 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Simeonov DR et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115, doi: 10.1038/nature23875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Klann TS et al. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol 35, 561–568, doi: 10.1038/nbt.3853 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Joung J et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12, 828–863, doi: 10.1038/nprot.2017.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu SJ et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, aah7111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Joung J et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 548, 343–346, doi: 10.1038/nature23451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Fujita T & Fujii H Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun 439, 132–136, doi: 10.1016/j.bbrc.2013.08.013 (2013). [DOI] [PubMed] [Google Scholar]
- 198.Fujita T & Fujii H Identification of proteins associated with an IFNgamma-responsive promoter by a retroviral expression system for enChIP using CRISPR. PLoS One 9, e103084, doi: 10.1371/journal.pone.0103084 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lam SS et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54, doi: 10.1038/nmeth.3179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Myers SA et al. Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat Methods 15, 437–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Liu X et al. In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell 170, 1028–1043, doi: 10.1016/j.cell.2017.08.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Morgan SL et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat Commun 8, 15993, doi: 10.1038/ncomms15993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rege M et al. LADL: Light-activated dynamic looping for endogenous gene expression control. bioRxiv, 349340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wang H et al. CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 175, 1405–1417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.D'Ippolito AM et al. Pre-established Chromatin Interactions Mediate the Genomic Response to Glucocorticoids. Cell Syst 7, 146–160 e147, doi: 10.1016/j.cels.2018.06.007 (2018). [DOI] [PubMed] [Google Scholar]
- 206.Chen B et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491, doi: 10.1016/j.cell.2013.12.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhou Y et al. Painting a specific chromosome with CRISPR/Cas9 for live-cell imaging. Cell Res 27, 298–301, doi: 10.1038/cr.2017.9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ma H et al. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci U S A 112, 3002–3007, doi: 10.1073/pnas.1420024112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chen B et al. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci. Nucleic Acids Res 44, e75, doi: 10.1093/nar/gkv1533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ma H et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol 34, 528–530, doi: 10.1038/nbt.3526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Shao S et al. Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system. Nucleic Acids Res 44, e86, doi: 10.1093/nar/gkw066 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ding Q et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res 115, 488–492, doi: 10.1161/CIRCRESAHA.115.304351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wang X et al. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo-Brief Report. Arterioscler Thromb Vasc Biol 36, 783–786, doi: 10.1161/atvbaha.116.307227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Thakore PI et al. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat Commun 9, 1674, doi: 10.1038/s41467-018-04048-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yu W et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat Commun 8, 14716, doi: 10.1038/ncomms14716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Young CS et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 18, 533–540, doi: 10.1016/j.stem.2016.01.021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nelson CE et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407, doi: 10.1126/science.aad5143 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tabebordbar M et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411, doi: 10.1126/science.aad5177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Long C et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403, doi: 10.1126/science.aad5725 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hakim CH et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight 3, doi: 10.1172/jci.insight.124297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Nelson CE et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat Med 25, 427–432, doi: 10.1038/s41591-019-0344-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Amoasii L et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362, 86–91, doi: 10.1126/science.aau1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gapinske M et al. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol 19, 107, doi: 10.1186/s13059-018-1482-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yuan J et al. Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol Cell 72, 380–394 (2018). [DOI] [PubMed] [Google Scholar]
- 225.Finn JD et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell reports 22, 2227–2235, doi: 10.1016/j.celrep.2018.02.014 (2018). [DOI] [PubMed] [Google Scholar]
- 226.Nelson CE & Gersbach CA Engineering Delivery Vehicles for Genome Editing. Annu Rev Chem Biomol Eng 7, 637–662, doi: 10.1146/annurev-chembioeng-080615-034711 (2016). [DOI] [PubMed] [Google Scholar]
- 227.Maeder ML et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 25, 229–233, doi: 10.1038/s41591-018-0327-9 (2019). [DOI] [PubMed] [Google Scholar]
- 228.Miller JF & Sadelain M The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 27, 439–449, doi: 10.1016/j.ccell.2015.03.007 (2015). [DOI] [PubMed] [Google Scholar]
- 229.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117, doi: 10.1038/nature21405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ren J et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 8, 17002–17011, doi: 10.18632/oncotarget.15218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Qasim W et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med 9, doi: 10.1126/scitranslmed.aaj2013 (2017). [DOI] [PubMed] [Google Scholar]
- 232.Schumann K et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A 112, 10437–10442, doi: 10.1073/pnas.1512503112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ren J et al. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research 23, 2255–2266, doi: 10.1158/1078-0432.ccr-16-1300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Torikai H et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 122, 1341–1349, doi: 10.1182/blood-2013-03-478255 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Qasim W et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science translational medicine 9, eaaj2013 (2017). [DOI] [PubMed] [Google Scholar]
- 236.Maeder ML & Gersbach CA Genome-editing Technologies for Gene and Cell Therapy. Mol Ther 24, 430–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhou Y et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491, doi: 10.1038/nature13166 (2014). [DOI] [PubMed] [Google Scholar]
- 238.Wang T et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903 e815, doi: 10.1016/j.cell.2017.01.013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Aguirre AJ et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 6, 914–929, doi: 10.1158/2159-8290.CD-16-0154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Han J et al. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep 23, 596–607, doi: 10.1016/j.celrep.2018.03.045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Liu Y et al. CRISPR Activation Screens Systematically Identify Factors that Drive Neuronal Fate and Reprogramming. Cell Stem Cell 23, 758–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Akcakaya P et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 561, 416–419, doi: 10.1038/s41586-018-0500-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Tsai SQ et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14, 607–614, doi: 10.1038/nmeth.4278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chew WL et al. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods 13, 868–874, doi: 10.1038/nmeth.3993 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Simhadri VL et al. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol Ther Methods Clin Dev 10, 105–112, doi: 10.1016/j.omtm.2018.06.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wagner DL et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat Med 25, 242–248 (2019). [DOI] [PubMed] [Google Scholar]
- 247.Charlesworth CT et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med 25, 249–254, doi: 10.1038/s41591-018-0326-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Wang J et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol 33, 1256–1263, doi: 10.1038/nbt.3408 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832, doi: 10.1038/nbt.2647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Chatterjee P, Jakimo N & Jacobson JM Minimal PAM specificity of a highly similar SpCas9 ortholog. Science advances 4, eaau0766, doi: 10.1126/sciadv.aau0766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.