

Abstract
Cancer cell lines are not homogeneous nor are they static in their genetic state and biological properties. Genetic, transcriptional and phenotypic diversity within cell lines contributes to the lack of experimental reproducibility frequently observed in tissue-culture-based studies. While cancer cell line heterogeneity has been generally recognized, there are no studies which quantify the number of clones that coexist within cell lines and their distinguishing characteristics. We used a single-cell DNA sequencing approach to characterize the cellular diversity within nine gastric cancer cell lines and integrated this information with single-cell RNA sequencing. Overall, we sequenced the genomes of 8824 cells, identifying between 2 and 12 clones per cell line. Using the transcriptomes of more than 28 000 single cells from the same cell lines, we independently corroborated 88% of the clonal structure determined from single cell DNA analysis. For one of these cell lines, we identified cell surface markers that distinguished two subpopulations and used flow cytometry to sort these two clones. We identified substantial proportions of replicating cells in each cell line, assigned these cells to subclones detected among the G0/G1 population and used the proportion of replicating cells per subclone as a surrogate of each subclone's growth rate.
INTRODUCTION
Cancer cell lines are used to study tumor growth, evaluate the biology underlying metastasis and determine drug sensitivities. However, it is increasingly recognized that cancer cell lines have subpopulations with extensive fitness diversity (1,2). This may lead to different drug responses within the same cell line (2). The characterization of cancer cell lines and their intrinsic clonal complexity has generally been qualitative. In contrast, for most cancer cell lines, there is very little known about the total number of coexisting subclones and their genomic features. Addressing this issue, new genomics methods such as single cell DNA-sequencing (scDNA-seq) and single cell RNA sequencing (scRNA-seq) can be used to quantitatively determine the cellular diversity within any given cancer tissue sample or cell line. ScDNA-seq identifies somatic genetic alterations, such as somatic copy number variations (CNVs). Likewise, scRNA-seq data can be used to infer CNVs albeit with limited resolution. Single cell CNVs provide a unique perspective on intratumoral heterogeneity and subclonal structure (3–6).
There are only a limited number of studies which combine both scDNA-seq and scRNA-seq for the analysis of cancer (5,7–10). Integrating these two methods provides granular information about the clonal membership and the transcriptional state of a given cell. Along these lines, we developed a new analysis framework for joint single cell genomics, whereby clones are defined via scDNA-seq and scRNA-seq, allowing for independent evaluation for the accuracy of clone size and characteristics. We analyzed the CNVs from both whole genomes and transcriptomes of thousands of cells originating from nine gastric cancer cell lines. We used our analytical framework to perform a joint analysis that delineated both a cell line's subclonal composition as well as the transcriptional states of these specific cell populations (Figure 1A). Demonstrating the feasibility of isolating clonal cells, we used our method to discover subclone-specific cell surface markers in the cell line NUGC-4. Then, we used these markers to isolate cells from each of two distinct NUGC-4 subclones. Moreover, we estimate the proportion of replicating cells per clone and propose it can serve as a surrogate metric to distinguish clonal stasis from ongoing in-vitro evolution. Overall, our study brings new insights into what drives and maintains genetic cell diversity in-vitro.
Figure 1.

Single cell DNA-seq and RNA-seq delineate cell cycle state heterogeneity of gastric cancer cell lines. (A) Overview of the study using joint single cell DNA and RNA sequencing on nine gastric cancer cell lines. Integrating scDNA- and scRNA-seq data informs a cell's clone membership, pathway activities and cell cycle state in tandem. (B) Using single cell DNA sequencing, 829 cells from the NUGC-4 gastric cancer line were classified according to three features: ploidy (x-axis); the number of breakpoints in their genome (y-axis); the breakpoints’ proximity to the origin of replication (ORI) per chromosome as denoted by a color bar. Low frequency breakpoints (≤1% across all cells) were counted for each chromosome. For cells in S-phase, these counts are correlated to the number of ORIs per chromosome. In contracts to cells in S-phase, cells in G0/G1 had fewer breakpoints which did not correlate to chromosomal ORI counts. (C) The percentages of G0/G1 cells estimated from scDNA-seq and scRNA-seq (y-axis), were positively correlated with the cell lines’ doubling times (x-axis). (D) Cell-cycle phase assignment based on scRNA-seq. 3797 NUGC-4 cells (columns) were clustered according to the activity of 39 pathways involved in cell-cycle progression (rows). Clusters were classified as to whether they had cells in G0/G1 (black), cells in S phase (cyan) or cells in G2M (red).
MATERIALS AND METHODS
Gastric cancer cell lines
Gastric cancer cell lines were purchased from ATCC (KATOIII, NCI-N87, SNU-16), KCLB (SNU-668, SNU-601, SNU-638), JCRB (MKN-45, NUGC-4) and ECACC (HGC-27). Microsatellite Instability (MSI) status was previously assessed for all cell lines and found negative for all but one cell line (SNU-638). Identity of each cell line was determined through independent karyotyping. Cells were checked for mycoplasma contamination. Cells were cultured in their recommended media conditions at 37°C. Afterward, the cells were processed into suspensions with standard procedures. Briefly, this process involved trypsinizing the cells, followed by inactivation by fetal bovine serum (FBS). We performed washes by centrifugation at 400 g in 1× phosphate-buffered saline with 0.04% bovine serum albumin. To remove cellular debris and cellular aggregates, we filtered cells through a Flowmi cell strainer (Wayne, NJ, USA) before proceeding to single-cell DNA and RNA sequencing.
Library preparation protocol for scDNA-seq
Single-cell DNA libraries were generated using a high-throughput, droplet-based reagent delivery system using a two-stage microfluidic procedure. First, cells were encapsulated in a hydrogel matrix and treated to lyse and unpackage DNA. Second, a gel bead (GB) was functionalized with copies of a unique droplet-identifying barcode (sampled from a pool of ∼737 000) and co-encapsulated with the hydrogel cell bead in a second microfluidic stage to separately index the genomic DNA (gDNA) of each individual cell. Unless otherwise stated, all reagents were part of a beta version of the Gel Bead and Library Kit for single cell CNV analysis (10× Genomics Inc., Pleasanton, CA, USA). In the first microfluidic chip, cell beads (CBs) were generated (Supplementary Methods). Cell bead-gel beads (CBGBs) were generated by loading CBs, barcoded gel beads, enzymatic reaction mix and partitioning oil in a second microfluidic chip (Supplementary Methods). A two-step isothermal incubation yielded genomic DNA fragments tagged with an Illumina read 1 adapter followed by a partition-identifying 16-bp barcode sequence. The library preparation was completed per the manufacturer's protocol. Polymerase chain reaction (PCR) was performed using the Illumina P5 sequence and a sample barcode with the following conditions: 98°C for 45 s, followed by 12–14 cycles (dependent on cell loading) of 98°C for 20 s, 54°C for 30 s and 72°C for 30 s. An incubation step at 72°C was performed for 1 min before holding at 4°C. Libraries were purified with SPRIselect beads (Beckman Coulter, Brea, CA, USA) and size-selected to ∼550 bp. At last, sequencing libraries were quantified by qPCR before sequencing on the Illumina platform using NovaSeq S2 chemistry with 2 × 100 paired-end reads.
ScDNA-seq data processing and CNV calling
Sequencing data were processed with the Cellranger-DNA pipeline, which automates sample demultiplexing, read alignment, CNV calling and report generation. In this study, we used a beta version for all analyses (6002.16.0). Paired-end FASTQ files and a reference genome (GRCh38) were used as input. Cellranger-DNA output includes copy number calls for each cell. Cellranger-DNA is freely available at https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/algorithms/overview and details of the pipeline are described in Supplementary Methods.
ScRNA-seq data processing
Cellranger software suite 1.2.1 was used to process scRNA data, including sample demultiplexing, barcode processing and single cell 3′ gene counting. The cDNA insert, which is contained in the read 2, was aligned to the GRCh38 human reference genome. The reference GTF contained 33 694 entries, including 20 237 genes, 2337 pseudogenes and 5560 Antisense (non-coding DNA). Cellranger provided a gene-by-cell matrix, containing the read count distribution of each gene for each cell.
Calling CNVs from scRNA-seq with LIAYSON
The algorithm, linking single-cell genomes among contemporary subclone transcriptomes (LIAYSON), is an approach we developed to profile the CNV landscape of each scRNA-sequenced single cell of a given sample. The algorithm relies on two assumptions: (a) a cell's average copy number state for a given genomic segment influences the mean expression of genes within that segment across the same set of cells; and (b) the copy number variance of a given genomic segment across cells reflects the cells’ expression heterogeneity for genes within that same segment (Supplementary Figure S3A and B). Let 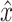 be the measured copy number of a given cell-segment pair, and x its corresponding true copy number state. The probability of assigning copy number x to a cell i at locus j depends on: (i) cell i’s read count at locus j and (ii) cell i’s read count at other loci, i.e. how similar the cell is to other cells that have copy number x at locus j. For (i), we fit a Gaussian kernel on the read counts at locus j across cells to identify the major and the minor copy number states of j as the highest and second highest peak of the fit respectively (Supplementary Methods). For (ii), we use Apriori (11)—an algorithm for association rule mining—to find groups of loci that tend to have correlated copy number states across cells (Supplementary Methods). LIAYSON is implemented in R and is available on CRAN at the following URL https://cran.r-project.org/web/packages/liayson.
be the measured copy number of a given cell-segment pair, and x its corresponding true copy number state. The probability of assigning copy number x to a cell i at locus j depends on: (i) cell i’s read count at locus j and (ii) cell i’s read count at other loci, i.e. how similar the cell is to other cells that have copy number x at locus j. For (i), we fit a Gaussian kernel on the read counts at locus j across cells to identify the major and the minor copy number states of j as the highest and second highest peak of the fit respectively (Supplementary Methods). For (ii), we use Apriori (11)—an algorithm for association rule mining—to find groups of loci that tend to have correlated copy number states across cells (Supplementary Methods). LIAYSON is implemented in R and is available on CRAN at the following URL https://cran.r-project.org/web/packages/liayson.
Identification of coexisting clones from scDNA-seq or scRNA-seq
Let M be the matrix of copy number states per non-private segment per G0/G1 cell, derived either from scRNA- or from scDNA-seq, with entries (i, j) pointing to the copy number state of cell i for segment j. Pairwise distances between cells were calculated in Hamming space (12) of their segmental copy number profiles (rows in M), weighted by segment length (Supplementary Figure S6). We used the BIONJ algorithm (13) to reconstruct a tree of G0/G1 cells from the distance matrix. A subtree was designated as a clone if the maximum distance between its cell members was less than 20% of the genome. At last, we used the Pearson Correlation Coefficient calculated across segments to assign S and G2M cells to the clones detected among the G0/G1 population. The copy number profile of each detected subclone was calculated as the average profile of single cells assigned to that subclone.
Integration of scRNA-seq- and scDNA-seq derived clones
Let R and D be the scRNA- and scDNA-seq derived clone-by-segment matrices of copy number states. Furthermore, let S: = SR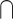 SD, where SR and SD are the segments defining the columns of R and D respectively. We defined X: = RS U DS as the union of scRNA-seq and scDNA-seq derived clones at overlapping genomic locations. We used the same hierarchical clustering procedure as above, only this time clones rather than cells were arranged into the resulting tree T. We iterated through all binary subtrees t
SD, where SR and SD are the segments defining the columns of R and D respectively. We defined X: = RS U DS as the union of scRNA-seq and scDNA-seq derived clones at overlapping genomic locations. We used the same hierarchical clustering procedure as above, only this time clones rather than cells were arranged into the resulting tree T. We iterated through all binary subtrees t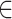 T and assigned clones within t as:
T and assigned clones within t as:
True positives (TPs) – t contains both, an scRNA- and an scDNA-clone
False positives (FPs) – t contains two scRNA-clones
False negatives (FNs) – t contains two scDNA-clones.
To validate scDNA-seq derived clone detection, we used the same procedure, except the roles of FPs and FNs were flipped. Clones comprising >4% cells, which were not confirmed by both techniques, were excluded from further analysis.
Flow cytometry sorting of NUGC-4
We used the scRNA-seq data to identify cell surface markers that are differentially expressed between co-existing clones, in order to physically separate them via flow cytometry. For fluorescence-activated cell sorting (FACS), cells were incubated for 30 min on ice with antibodies at dilutions determined by titration experiments. Antibodies used in this study include: anti-human CD13 (ANPEP) PeCy7 (clone WM15; BioLegend) 1:10, anti-human CD184 (CXCR4) BV421 (clone 12G, BioLegend) 1:10, anti-human TM4SF4 APC (R&D) 1:10, anti-human ITM2C (clone 2E8G11, Proteintech) 1:10. For the detection of the unconjugated anti-human ITM2C antibody cells were subsequently washed and stained for 30 min on ice with anti-mouse IgG2A FITC (Biolegend) 1:100. Corresponding isotype immunoglobulin served as controls. Flow cytometric sorting was performed using a FACSAriaFusion instrument (BD Biosciences, San Jose, CA, USA) and analyses were performed using the FlowJo software (Tree Star, Ashland, OR, USA).
Intra-cell line differential gene expression
We used Seurat version 2.3.4 to identify differentially expressed genes between members of a given clone and members of any other clone detected in the same cell line. Only groups of cells with identical cell line origin were compared. For each comparison we used the Wilcox rank-sum test implemented in Seurat (function 'FindMarkers'), while accounting for variability in gene coverage across cells. To visualize the population structure within a cell line we used the UMAP (14) (Uniform Manifold Approximation and Projection) or tSNE (t-Distributed Stochastic Neighbor Embedding) dimension reduction techniques.
Karyotyping of sorted NUGC-4 subpopulations
Cell cultures were harvested by standard cytogenetic methodologies using Colcemid® mitotic arrest (0.05 μg/ml, 20 °C, variable time), hypotonic shock (0.075 M KCl, 20 °C, 15 min) and fixation (3:1 methanol/acetic acid). Metaphase slide preparations were stained using the GTW banding method and mitotic chromosomes imaged, analyzed and karyotyped with a Leica DM6000 microscope equipped with a 100x oil immersion objective and CytoVision® imaging software (Leica).
RESULTS
Single cell sequencing identifies variable fractions of replicating cells across cancer cell lines
We used a droplet-based partitioning technology to conduct scDNA-seq for CNVs (‘Materials and Methods’ section). Using this approach, we sequenced 8824 single cells from nine gastric cancer cell lines and determined copy number status across the genome of each cell. Overall, for each cell line we sequenced between 0.5 and 2.2 million unique reads per cell, translating to a median effective coverage of 279 reads per 1 Mb per cell (Supplementary Table S1). Sequencing data was of high quality, with mapping rates of at least 97% across all cell lines, and an average duplication rate of 12% across all cell lines. We identified an average of 2198 CNVs per sample that were present in more than 1% of cells per cell line. The majority of these CNVs (95%) were associated with ongoing DNA replication in a cell, further referred to as replication-specific CNVs (Supplementary Tables S1 and 2). The remaining CNVs were a consequence of aneuploidy and segregated the cells into multiple clones. All cell lines demonstrated clonal diversity (Figure 1A). CNVs and aneuploidy status were confirmed by both SNP array analysis and karyotyping of these same cell lines (Supplementary Figure S1A–G). The average ploidy across cells was consistent with that as determined with karyotyping (Supplementary Figure S1F).
The overall strategy of the analysis is show in Figure 1A. CNVs that distinguish any two cells of a population either have a stable representation in the genome or they are a transient consequence of DNA replication during mitosis. To distinguish G0/G1 cells from cells in the S phase of the cell cycle, we used three features (Figure 1B and Supplementary Figure S2B–J): (i) the cell's ploidy, (ii) its number of CNVs and (iii) the distance of CNVs to replication origins (15). The proportion of G0/G1 cells ranged from 58% in SNU-16 to 82% in SNU-668 (Figure 1B and Supplementary Table S1). For a subset of the cell lines, we used flow cytometry to generate comparison data of DNA content (Supplementary Figure S2A). The percentage of replicating cells per scDNA-seq was positively correlated to the percentage of replicating cells per flow cytometry (r = 0.86, P = 0.063; Supplementary Figure S5A). The percentage of G0/G1 cells per scDNA-seq was also proportional to the doubling time of the cell line (r = 0.76, P = 0.017; Figure 1C). Specifically, a smaller proportion of cells in S-phase was an indicator of slower cell growth and showed an association with fewer years since the cell line was established (Supplementary Figure S5B).
We used scRNA-seq to validate our scDNA-seq's cell cycle assignment. For this comparison, we conducted scRNA-seq of 28 209 single cells for the same nine gastric cancer cell lines (Supplementary Table S3). Differences in the passage number between the two single cell sequencing assays were below two for 78% of the cell lines and the extent of confluence was typically at 80–90% (Supplementary Table S2). Activity profiles of multiple cell cycle pathways have been shown to provide robust cell cycle status classification across different cell types (16). For each individual cell, we quantified the activity of 39 cell-cycle pathways from the REACTOME database (17) and used these results to determine cell cycle state (Supplementary Table S4 and Figure S8A–D). Pathways were classified into three groups depending on their main activation timing during G0/G1, S and G2M. We performed hierarchical clustering of cells and classified clusters based on their cells’ pathway activity (Figure 1D). The percentages of G0/G1 cells, assigned with scDNA-seq versus scRNA-seq, were correlated (r = 0.73, P = 0.026, Figure 1C).
Subclonal signatures of genomic instability and ongoing selection
We used scDNA-seq to characterize the underlying subclonal structure of the cell lines. Approximately 95% of the CNVs identified by scDNA-seq were found in less than 1% of the G0/G1 population. We ascribed these events to variance related to DNA replication and not representing cancer genome CNVs (Supplementary Table S1). The remaining CNV segments distinguished the subclones within the G0/G1 population. We calculated the pairwise distances between cells in Hamming space and applied a neighbor joining algorithm (13), to build a tree of G0/G1 cells (‘Materials and Methods’ section). We defined a clone as the largest subtree within which the maximum distance between its cell members was <20% of the genome (Figure 2A). The term subclone size refers to the relative fraction of cells assigned to a specific clone. To assign S-phase cells to the subclones detected among the G0/G1 population, we determined the cellular similarity with a Pearson correlation. For example, this approach identified four clones within the G0/G1 population of NCI-N87 (Figure 2A–C). The percentage S cells assigned to each of these four subclones were proportional to their respective G0/G1 representation (Figure 2A and B).
Figure 2.

Single cell DNA-seq characterizes clonal composition and evolution in gastric cancer cell lines. (A) Copy number landscape of G0/G1 cells (left) is shown alongside S cells (right) for each clone detected in NCI-N87 (left color bars). (B) Copy number segmentation profile shown for an G0/G1- and an S representative of the two largest clones in (A) (cyan and purple). Arrows indicate genomic regions where the two clones diverge. (C) Genetic events leading to the divergence of the four co-existing clones in NCI-N87. (D) The percentage of replicating cells per clone increased with percentage of G0/G1 cells per clone in NCI-N87 as well as in the other eight cell lines, indicating predominant clonal stasis (Pearson r = 0.88; P < 2e-16). Selection of clones (color-coded) was calculated as probability of sampling the % replicating cells observed for a given clone, conditional on the G0/G1 representation of that same clone using the hypergeometric distribution. Clones were assigned to three groups: positive selection (n = 17), no selection (n = 34) and negative selection (n = 6). (E) Number of selected clones per cell line. [CN: copy number].
There were two to 12 subclones present for any given gastric cancer cell line (Supplementary Table S5). Of all the genomic regions affected by subclonal CNVs in the SNU-668 cell line, 14% of these regions had at least three copy number states—each state represented by a significant number of cells (Supplementary Figure S9A). That is, for these CNV affected regions, two copy number states alone were insufficient to represent the majority of the cell population (≥90% cells). The same applied for 8% of subclonal CNV regions in SNU-16 (Supplementary Figure S9B). This is significant because algorithms that quantify genetic intra-tumor heterogeneity by deconvoluting bulk sequencing data typically assume that two copy number states suffice to represent a given genomic region (18,19). Clones whose genomes diverge in these genomic regions would thus remain undetectable with bulk sequencing. Along the same lines, an average of 12% subclones large enough to be detectable with bulk sequencing had a cellular frequency that was in close proximity to at least one other co-existing subclone (Supplementary Figure S9C). Because deconvolution algorithms rely on cellular frequencies alone to define subclones, these algorithms would coalesce such subclone pairs of similar size.
Approximately half of the variation in subclone counts across cell lines was attributed to differences in ploidy and/or the duration since the cell line was first established in culture (adjusted R2 = 0.53; P = 0.044; Supplementary Table S6). Longer time in culture was predictive of fewer clones (P = 0.025; coefficient = −0.29), while higher ploidy predicted more clones, albeit at borderline significance (P = 0.054; coefficient = 3.09). The former observation is consistent with a recent finding showing that in vitro CNV acquisition rate decreases over time, while signatures of proliferation increase, in line with clonal selection of fitter clones (1).
Shifts in a cancer cell line's subclonal composition have been shown to frequently result from in vitro selection, rather than a stochastic process (1,2). As a surrogate of in vitro selection among the cancer cell subclones, we calculated the difference between percentage replicating cells and G0/G1 cells per subclone, which we refer to as a selection coefficient. To estimate the statistical significance associated with a given selection coefficient, we used a hypergeometric distribution—we determined if the subclone's number of replicating cells was within a range consistent with its G0/G1 representation (Supplemental Methods). The two cell-cycle states had similar proportions for a given subclone, indicating predominance of clonal stasis (Pearson r = 0.88; P < 2e-16; Figure 2D). Seventeen subclones (30%) had a higher percentage of replicating cells than expected from their G0/G1 population size (FDR adjusted P < = 0.05; Figure 2D and E). We interpreted this result to be a potential indication of positive selection. Conversely, six subclones (11%) had fewer replicating cells than expected from their G0/G1 representation, suggesting they were under negative selection (FDR adjusted P < = 0.05; Figure 2D and E). The overrepresentation of positively selected clones compared to negatively selected ones was a result consistent with a recent study showing that in vitro evolution is primarily driven by positive selection (2). Quantifying the growth rate of an individual subclone at the time of sample collection may prove useful in predicting the clonal composition of future cell line populations.
Consilience of scDNA- and scRNA-seq on G0/G1 subclonal architectures
We used the scRNA-seq results to determine whether subclones derived from scDNA-seq can be confirmed by an independent ascertainment method. For this comparison, we inferred CNVs from scRNA-seq. Gene expression has been shown to be proportional to the gene's copy number state for the majority of genes (20), suggesting that scRNA-seq derived expression features can inform CNV status. However, other mechanisms of gene regulation alter expression, thus confounding the influence of segmental copy number. One algorithm for calling CNVs from scRNA-seq data demonstrated good performance, particularly for large segments, above 10 Mb, and for large subclones (21). However, this method's precision fell below 50% for smaller subclones, making up 20% or less of the total cells (21). To address this limitation, we developed and applied an algorithm called LIAYSON, which uses scRNA-seq to deconvolute bulk CNV profiles into single cell-specific copy numbers (Supplemental Methods). This approach relies on gene expression to estimate the variance in copy number, but not the mean copy number across cells (Supplementary Figure S3) and is less influenced by regulators of expression levels other than CNVs. Genomic segments spanning 10 Mb contain on average sufficient genes (at least 20 genes with expression results), to facilitate CNV calling. But the locus-specific resolution on CNVs is a function of the architecture of the human genome and the tissue-specificity of gene expression. Between 25 and 80% of segments per cell line passed these metrics for this analysis.
With the CNV results from scRNA-seq, we identified a range of three to 11 subclonal populations across the nine gastric cancer cell lines (Supplementary Table S5). The number of scRNA-seq and scDNA-seq derived clones were highly correlated (r = 0.93. P = 3E-4; Figure 3A). As another validation of concordance between the two techniques, we performed hierarchical clustering of subclone-specific CNV profiles (Figure 3B–D). We defined true positives as clusters containing subclones identified by both techniques and false positives and -negatives as clusters containing subclones identified by just one single technique (Supplemental Methods). To determine the concordance between scDNA-seq and scRNA-seq, we calculated the F1 score which considers both the precisions and the recall of a test. The F1 score was 0.47 for clones below 4% abundance, but increased to ≥0.7 for clones above 4% (Supplementary Figure S3C). Based on this result, we excluded any subclones smaller than 4% and not confirmed by both single cell methods. Posterior saturation curves of scDNA-seq library sizes were calculated for each cell line as previously described (4) and indicated that we had statistical power to detect these subclones (Supplementary Table S7).
Figure 3.

Consilience of scDNA- and scRNA-seq on G0/G1 clonal architectures. (A) Correlation between number of clones inferred by scRNA- and scDNA-seq across all nine cell lines. (B–E) Integrated analysis of cell line NCI-N87. ScRNA-seq derived copy number landscape of 2334 G0/G1 cells detected in NCI-N87 (B), independently distinguishes the same four clones as scDNA-seq of 742 G0/G1 cells (C). Clone membership is color coded on the left. (D) Each CNV profile found by scRNA-seq had an equivalent CNV profile in the scDNA-seq data, applying to a similar % of cells. (E) A UMAP map of NCI-N87 cells shown in panel C based solely on their expression signatures. Clones defined by copy number alterations, are enriched in specific areas of the transcriptionally defined UMAP. (F and G) Differences in passage number between scDNA- and scRNA-seq experiments for SNU-668 (F) and SNU-16 (G) accompany differences in clonal composition (39) observed between the two techniques. [CN: copy number].
Citing an example, our scDNA-seq and scRNA-seq results identified four subclones in NCI-N87 with similar proportional sizes (Figure 3B–D). On closer examination, we observed that the copy number states of several smaller segments (<10 Mb; Supplementary Figure S7), were not assigned for any clone by scRNA-seq, but were identified by scDNA-seq. For these genomic regions, the number of genes with adequate expression levels was too low to allow assignment by scRNA-seq. This result is in line with the expectation that scDNA-seq provides a higher resolution of subclonal CNVs than scRNA-seq.
Results for the remaining gastric cancer cell lines were similar. Among the nine cell lines, the subclonal size, as determined by scRNA-seq, correlated with the scDNA-seq results (Pearson r = 0.93, P < 2e-16; Supplementary Figure S3D). An average of 88% cells per cell line were assigned to subclones confirmed independently by both scDNA-seq and scRNA-seq (Supplementary Table S5). Concordance between the two techniques was dependent on sequence depth, subclone size (Supplementary Figure S3C–F) and the number of subclones per a given cell line (Supplementary Table S5). Higher differences in passage number between scDNA- and scRNA-seq experiments were correlated with a significant divergence between clonal compositions measured by the two methods (r = 0.71, P = 0.032; Supplementary Figure S3G). The magnitude of this divergence is likely a function of the extent of genome instability in a growing cell line. For example, the clonal composition of SNU-668 remained more stable between the two measurements than that of SNU-16, even though differences between passage numbers were slightly smaller for the latter (five for SNU-16 versus seven for SNU-668; Figure 3F and G). Overall, both single-cell sequencing assays independently identified clonal architectures with similar features, increasing our confidence in their biological significance.
Sorting cells from subclones with distinct CNV characteristics
The integration of scDNA- and scRNA-seq results proved useful for isolating cells from subclones based on their expression characteristics. Clones with specific CNV alterations were enriched in separate areas of the transcriptionally defined UMAP (Figure 3E). This result suggested that we could use this transcriptome information to prioritize cell surface markers for physical separation of genetically defined cells for a given clonal population.
We examined two distinct clonal populations detected in NUGC-4. An extra copy of a large genomic region on chromosome 2q distinguished these two clones, further referred to as cloneamplif2q and clonegain2q (Figure 4A and B). Based on the scRNA-seq results, we identified four cell surface marker genes with expression profiles that differentiated themselves among the two clones (‘Materials and Methods’ section). Subsequently, we identified specific antibodies for the corresponding marker proteins and used them for flow sorting two subpopulations, further referred to as subpopulationamplif2q and subpopulationgain2q (Figure 4C and Supplementary Figure S5C). Afterward, we conducted a karyotyping test of these sorted cells from the two subpopulations. Our results showed that subpopulationamplif2q contained exclusively cells with a chromosome 2 rearrangement. In comparison, 65% of the cells from subpopulationgain2q lacked this rearrangement (Figure 4D), suggesting the two subpopulations were indeed enriched for cloneamplif2q and clonegain2q, respectively. Subpopulationgain2q was almost ten times larger than subpopulationamplif2q and the variance of its expression profile was higher, likely accounting for the lower purity of sorting.
Figure 4.

Integrated single cell sequencing informs live-cell-sorting of NUGC-4 clones. (A) Phylogenetic tree of the four clones detected by scDNA-seq in NUGC-4. (B) G0/G1 cells are shown in tSNE space—every dot is a cell, and two clones of interest are highlighted in purple and green. The purple clone has three copies of a large genomic region on chromosome 2q (2q gain), whereas the green clone has an extra copy of that same segment (2q amplification). Cells are colored based on their CNVs, whereas their location in tSNE space is based on their expression signature. (C) The top four cell surface markers informing separation of the green and purple clone in tSNE space were: ANPEP, CXCR4, TM4SF4 and ITM2C. We used flow cytometry with these four markers to enrich for the respective clone. (D) Cytogenetic analysis of chromosome 2 identified three karyotypes among the two isolated subpopulations (top row), including a rearrangement on the q-arm (red arrow). All of the cells (100%) from the green subpopulation had a chromosome 2q rearrangement, whereas the purple subpopulation contained only 35% cells with the rearrangement.
To estimate how many subclones can be isolated in each cell line with this strategy, we quantified how well a given set of cell surface markers can separate a clone of interest from the remaining clones in a cell line. We clustered all cells based on the expression of the corresponding cell surface markers (up to four genes). Across all G0/G1 cells we then calculated the Pearson correlation coefficient between clone membership and cluster membership (Supplementary Table S8). Out of 41 confirmed clones, for 16 clones the correlation between cell surface cluster membership and clone membership was at least as high as for the two isolated NUGC-4 clones, suggesting it may be possible to sort them at a similar accuracy as the two NUGC-4 clones. These experiments underscored the utility of transcriptome and genomic CNV data integration for genotype-phenotype comparisons.
Identification of pathways associated with accumulation of copy number alterations
The rate of accumulation of CNVs is of interest because it contributes to the speed at which a population evolves. CNVs contribute considerably to intra-tumor heterogeneity (22,23). Most somatic CNVs accumulate during DNA replication (24). Alternative end-joining causes insertion deletions (indels) and template switching events can lead to short amplifications (25). The mechanisms responsible for generating large CNVs are complex in their biology. To determine if there were specific pathways associated with CNV accumulation, we focused on CNV events that were present in a small proportion of cells. The rationale behind this strategy is that the number of cells carrying a given CNV is correlated to the time at which the CNV event happened (18). The more recent a CNV the less time selection had to act upon it, rendering CNVs of low cellular frequency a better proxy of CNV accumulation rate than high-frequency-CNVs. This analysis was possible because of the high resolution of scDNA-seq in identifying these cancer CNVs in a small number of cells.
We defined rare CNVs as genomic segments above 1 Mb long that have an altered copy number status in less than 10% of the cells for a given clonal branch. We clustered cells based on their rare CNVs and calculated the Simpson diversity index (26) of cell-clusters found within a given clone (Supplementary Figure S4A). This intra-clone diversity (ICD) index was used as surrogate metric for CNV accumulation rate per clone (Figure 5A) and was not confounded by clone size (Supplementary Figure S4B). There was a high variability of CNV accumulation rates across co-existing clones within a cell line, with clones from the same cell line sometimes residing at opposite sides of the CNV accumulation rate spectrum (Figure 5A and B). Cell cycle state classification was conservative for the G0/G1 state, to minimize a potential contribution of false positive S cells to this result. But given the complexity of the cell cycle and the fact that cell cycle state is a continuous rather than discrete variable, we cannot fully exclude the possibility of its contribution as confounding factor.
Figure 5.

Identification of pathways associated with accumulation of CNVs. (A) Diversification of two clones found in KATOIII based on rare CNVs, i.e. CNVs found in <10% of clone members. Phylogenies were used to calculate a clone's Simpson-index as surrogate of its further diversification. The two clones were on opposite ends of the ICD spectrum. (B) Thirty-nine pathways (y-axis) associated with ICD across 41 confirmed clones (x-axis). Clones were sorted according to their ICD index. (C–E) The correlation between ICD (x-axis) and pathway activity per clone (y-axis) is shown for three pathways. A positive correlation coefficient is often observed within as well as across cell lines. Solid lines depict linear regression fits. Dotted lines indicate a simple connection between two data points—this was applied to the cell line with only two confirmed clones: MKN-45.
Using the overlaid scRNA-seq data from these same lines, we then compared the pathway activity of a clone with its CNV accumulation rate. For each clone and each pathway from the REACTOME database (17), we quantified the average pathway activity among clone members (ranging from 31 to 1441 G0/G1 cells) with GSVA (27). Overall, we identified 39 pathways that were either positively or negatively correlated with CNV accumulation rate per clone (Figure 5B): within at least three individual cell lines (|Pearson r | ≥0.7 for each cell line) and across all nine cell lines (|r|≥0.25).
Pathways positively correlated with CNV accumulation rate included non-homologous end-joining; Packaging Of Telomere Ends; DNA Damage/Telomere Stress Induced Senescence and DNA double strand break repair activity (Figure 5C and D). These pathways are established in their contributory role in the acquisition of CNVs (24), thus validating our choice of the ICD index as surrogate measure of CNV accumulation. Interestingly, there was an overrepresentation of metabolic functions among pathways anticorrelated with CNV accumulation rate. These included the Nicotinate metabolism (Figure 5E), previously shown to contribute to the DNA damage repair process in response to chemotherapy (28). Metabolic pathways have been proposed as features that enable one to classify different types of DNA damage (29). Our results indicate that clones within the same cell line may differ in their DNA-damage response. Other functions negatively associated with CNV accumulation were toll-like receptor (TLR) signaling pathways (Figure 5B).
We conclude that quantification of both, genomic instability and transcriptional activity, allows us to confirm previously known pathways involved in genome integrity and identify new candidate features that may contribute to genome maintenance.
DISCUSSION
For this study, we demonstrated a new scDNA-seq technology that enabled the interrogation of intratumoral heterogeneity from thousands of cells per sample. Adding transcriptomes at the resolution of single cells to single cell genomes, showed that gastric cancer cell lines have substantial genetic and transcriptional diversity. This result is consistent with other studies showing that cancer evolution continues in-vitro (1,30). It suggests that using these in-vitro systems to study specific drug sensitivities should be limited to a narrow time window, where the clonal composition of the cell line is expected to stay within a predefined range. Our results suggest that the width of this window depends on the cell line's ploidy and the number of years since it has been in culture.
In contrast to a prior study, that joined scDNA-seq with scRNA-seq to identify subclones (5), we chose to compare subclones identified independently by each single cell technology, trading a higher clone detection power for the opportunity of validating clone detection accuracy. Co-clustering clones identified independently by either single cell technique intrinsically controlled for false positives: whether two clones co-cluster does not only depend on their own genetic content, but also on the content of other clones identified in the sample (Figure 3D). Integrating the transcriptome and genome features improved our clone detection resolution to identify clones down to 2% cellular fraction. This high sensitivity is likely to have broad applicability as the dominant subclones of resistant tumors (31,32), metastases (33,34), patient-derived xenografts and cell lines (1) often originate from minor subclones in the primary tumor. Single cell data integration enabled us to discover cell surface markers that we used for flow cytometry sorting of clones (Figure 4). In the future this approach can be leveraged to test subclone-specific drug sensitivities without having to re-sequence the population each time after drug exposure to see which clone survived.
Cellular diversity in cancer cell lines can be the result of stochastic drift or of ongoing selection in culture. Several approaches have been developed to quantify selection using either time-resolved sequence data from longitudinal studies (35,36) or by observing differences in the statistical structure and shape of genealogies reconstructed from a fitness diverse asexual population (37,38). Our integrated sequencing approach may facilitate prediction of selective forces imposed on a clone at the time of cell harvest, by simply comparing its S and G0/G1 representations (Figure 2A and D). To dissect the effects of selection and mutation on intratumor heterogeneity and estimate the rate at which CNVs accumulate in a given clone, we quantified how often clone members carry unique/rare CNVs. We identified pathways whose expression was associated with CNV accumulation rate, including DNA repair mechanisms, metabolic pathways and TLR signaling pathways (Figure 5B). We speculate that repair of single- and double-strand breaks may be less error-prone in the presence of TLR signaling, in line with previous reports of a link between activation of TLRs and increased functional DNA repair (28).
In the future, the coexistence of multiple clones within the same cell line can be leveraged to learn generally applicable strategies that differentiate between the sensitivities of co-existing clones and that characterize clonal competition, cooperation and adaptation to changing environments.
DATA AVAILABILITY
The datasets generated for this study are available in the National Institute of Health's SRA repository; accession number PRJNA498809 and in the Gene Expression Omnibus (GEO accession number GSE142750). Code to reproduce parts of this analysis is available at the following URL https://github.com/noemiandor/cloneid. The repository contains various functions used throughout the manuscript, such as identification of differentially expressed cell surface markers (function ‘findCloneMarkers’) and integration of scRNA- and scDNA-seq perspectives on clones (function ‘mergePerspectives’).
A Jupyter notebook to demonstrate its utility is under https://github.com/noemiandor/cloneid/blob/master/CLONEID_applicationExample_CLs.ipynb.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
Supplementary Material
FUNDING
NIH[NHGRI P01HG000205 to B.T.L., M.A.K., S.M.G., H.P.J.]; NCI [R00CA215256 to N.A., NHGRI R01HG006137 to S.M.G., H.P.J.; NCI [U01CA217875 to A.S., H.P.J.]; American Cancer Society (to J.S., H.P.J.); Research Scholar Grant [RSG-13–297-01-TBG]; Clayville Foundation (to H.P.J.); Gastric Cancer Foundation (to H.P.J.); Seiler Family Foundation (to H.P.J.).
Conflict of interest statement. C.C. is a former employee of 10× genomics.
REFERENCES
- 1. Ben-David U., Ha G., Tseng Y.Y., Greenwald N.F., Oh C., Shih J., McFarland J.M., Wong B., Boehm J.S., Beroukhim R. et al.. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017; 49:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ben-David U., Siranosian B., Ha G., Tang H., Oren Y., Hinohara K., Strathdee C.A., Dempster J., Lyons N.J., Burns R. et al.. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018; 560:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casasent A.K., Schalck A., Gao R., Sei E., Long A., Pangburn W., Casasent T., Meric-Bernstam F., Edgerton M.E., Navin N.E.. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell. 2018; 172:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gao R., Davis A., McDonald T.O., Sei E., Shi X., Wang Y., Tsai P.C., Casasent A., Waters J., Zhang H. et al.. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016; 48:1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim C., Gao R., Sei E., Brandt R., Hartman J., Hatschek T., Crosetto N., Foukakis T., Navin N.E.. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018; 173:879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Y., Waters J., Leung M.L., Unruh A., Roh W., Shi X., Chen K., Scheet P., Vattathil S., Liang H. et al.. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014; 512:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dey S.S., Kester L., Spanjaard B., Bienko M., van Oudenaarden A.. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015; 33:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Macaulay I.C., Haerty W., Kumar P., Li Y.I., Hu T.X., Teng M.J., Goolam M., Saurat N., Coupland P., Shirley L.M. et al.. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods. 2015; 12:519–522. [DOI] [PubMed] [Google Scholar]
- 9. Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L. et al.. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014; 344:1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L.Y., Guo J., Cao W., Zhang M., He J., Li Z.. Integrated sequencing of exome and mRNA of large-sized single cells. Sci. Rep. 2018; 8:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Borgelt C., Kruse R.. Härdle W, Rönz B. Induction of Association Rules: Apriori Implementation. Compstat. 2002; HD: Physica-Verlag; 395–400. [Google Scholar]
- 12. Molparia B., Nichani E., Torkamani A.. Assessment of circulating copy number variant detection for cancer screening. PLoS One. 2017; 12:e0180647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997; 14:685–695. [DOI] [PubMed] [Google Scholar]
- 14. Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W.. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018; 37:38–44. [DOI] [PubMed] [Google Scholar]
- 15. Langley A.R., Gräf S., Smith J.C., Krude T.. Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res. 2016; 44:10230–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scialdone A., Natarajan K.N., Saraiva L.R., Proserpio V., Teichmann S.A., Stegle O., Marioni J.C., Buettner F.. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods. 2015; 85:54–61. [DOI] [PubMed] [Google Scholar]
- 17. Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R. et al.. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014; 42:D472–D477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andor N., Harness J.V., Muller S., Mewes H.W., Petritsch C.. EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics. 2014; 30:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roth A., Khattra J., Yap D., Wan A., Laks E., Biele J., Ha G., Aparicio S., Bouchard-Cote A., Shah S.P.. PyClone: statistical inference of clonal population structure in cancer. Nat. Methods. 2014; 11:396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fehrmann R.S., Karjalainen J.M., Krajewska M., Westra H.J., Maloney D., Simeonov A., Pers T.H., Hirschhorn J.N., Jansen R.C., Schultes E.A. et al.. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015; 47:115–125. [DOI] [PubMed] [Google Scholar]
- 21. Fan J., Lee H.O., Lee S., Ryu D.E., Lee S., Xue C., Kim S.J., Kim K., Barkas N., Park P.J. et al.. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 2018; 28:1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen G., Mulla W.A., Kucharavy A., Tsai H.J., Rubinstein B., Conkright J., McCroskey S., Bradford W.D., Weems L., Haug J.S. et al.. Targeting the adaptability of heterogeneous aneuploids. Cell. 2015; 160:771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mroz E.A., Tward A.D., Hammon R.J., Ren Y., Rocco J.W.. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med. 2015; 12:e1001786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G.. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009; 10:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hartlerode A.J., Willis N.A., Rajendran A., Manis J.P., Scully R.. Complex breakpoints and template switching associated with non-canonical termination of homologous recombination in mammalian cells. PLos Genet. 2016; 12:e1006410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Merlo L.M., Shah N.A., Li X., Blount P.L., Vaughan T.L., Reid B.J., Maley C.C.. A comprehensive survey of clonal diversity measures in Barrett's esophagus as biomarkers of progression to esophageal adenocarcinoma. Cancer Prev. Res. (Phila.). 2010; 3:1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanzelmann S., Castelo R., Guinney J.. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piacente F., Caffa I., Ravera S., Sociali G., Passalacqua M., Vellone V.G., Becherini P., Reverberi D., Monacelli F., Ballestrero A. et al.. Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT inhibitors, and DNA repair. Cancer Res. 2017; 77:3857–3869. [DOI] [PubMed] [Google Scholar]
- 29. Bhute V.J., Palecek S.P.. Metabolic responses induced by DNA damage and poly (ADP-ribose) polymerase (PARP) inhibition in MCF-7 cells. Metabolomics. 2015; 11:1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roschke A.V., Tonon G., Gehlhaus K.S., McTyre N., Bussey K.J., Lababidi S., Scudiero D.A., Weinstein J.N., Kirsch I.R.. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 2003; 63:8634–8647. [PubMed] [Google Scholar]
- 31. Johnson B.E., Mazor T., Hong C., Barnes M., Aihara K., McLean C.Y., Fouse S.D., Yamamoto S., Ueda H., Tatsuno K. et al.. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014; 343:189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morrissy A.S., Garzia L., Shih D.J., Zuyderduyn S., Huang X., Skowron P., Remke M., Cavalli F.M., Ramaswamy V., Lindsay P.E. et al.. Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016; 529:351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gerlinger M. Metastasis seeding cells: lone invaders or mass migrators. Clin. Cancer Res. 2018; 24:2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turajlic S., Xu H., Litchfield K., Rowan A., Chambers T., Lopez J.I., Nicol D., O’Brien T., Larkin J., Horswell S. et al.. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell. 2018; 173:581–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khatri B.S. Quantifying evolutionary dynamics from variant-frequency time series. Sci. Rep. 2016; 6:32497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nené N.R., Dunham A.S., Illingworth C.J.R.. Inferring fitness effects from time-resolved sequence data with a delay-deterministic model. Genetics. 2018; 209:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dayarian A., Shraiman B.I.. How to infer relative fitness from a sample of genomic sequences. Genetics. 2014; 197:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O’Fallon B.D., Seger J., Adler F.R.. A continuous-state coalescent and the impact of weak selection on the structure of gene genealogies. Mol. Biol. Evol. 2010; 27:1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith M.A., Nielsen C.B., Chan F.C., McPherson A., Roth A., Farahani H., Machev D., Steif A., Shah S.P.. E-scape: interactive visualization of single-cell phylogenetics and cancer evolution. Nat. Methods. 2017; 14:549–550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated for this study are available in the National Institute of Health's SRA repository; accession number PRJNA498809 and in the Gene Expression Omnibus (GEO accession number GSE142750). Code to reproduce parts of this analysis is available at the following URL https://github.com/noemiandor/cloneid. The repository contains various functions used throughout the manuscript, such as identification of differentially expressed cell surface markers (function ‘findCloneMarkers’) and integration of scRNA- and scDNA-seq perspectives on clones (function ‘mergePerspectives’).
A Jupyter notebook to demonstrate its utility is under https://github.com/noemiandor/cloneid/blob/master/CLONEID_applicationExample_CLs.ipynb.