

Zusammenfassung
Die Hämophagozytische Lymphohistiozytose (HLH), in der Literatur auch Hämophagozytosesyndrom oder Makrophagenaktivierungssyndrom im Rahmen einer rheumatologischen Vorerkrankung, wird auf Intensivstationen aufgrund des sepsisähnlichen Krankheitsbildes in über 70 % der Fälle nicht erkannt. Es wird von einem 30-jährigen, zuvor gesunden Patienten berichtet, der nach einer 3‑monatigen Asien- und Südamerikareise mit unklarem Fieber zunächst auf die infektiologische Normalstation der Charité – Universitätsmedizin Berlin aufgenommen und nach 3 Tagen bei respiratorischer Insuffizienz auf die Intensivstation verlegt wurde. Durch die sofortige HLH-Diagnostik mit Initiierung der spezifischen immunsupprimierenden Therapie mittels Dexamethason, Immunglobulinen und Anakinra konnte der Patient nach insgesamt 14-tägigem Krankenhausaufenthalt vollständig genesen in die Häuslichkeit entlassen werden. Es werden weiterhin aktuelle diagnostische und therapeutische Möglichkeiten diskutiert. Ferritin als entscheidender diagnostischer Marker sollte bei jedem Patienten mit unklarem Organversagen bestimmt werden.
Schlüsselwörter: Hämophagozytische Lymphohistiozytose, Hämophagozytosesyndrom, Makrophagenaktivierungssyndrom, Sepsis, Ferritin
Abstract
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome or macrophage activation syndrome within a pre-existing rheumatological disease, remains undiagnosed in over 70% of all cases in intensive care units (ICU) due to the sepsis-like clinical presentation. This report describes the case of a 30-year-old previously healthy male patient who was admitted to the normal infectiology ward of the Charité – Universitätsmedizin Berlin with unclear fever after a 3‑month journey around Asian and South America. The patient was transferred to the ICU after 3 days because of respiratory failure. Due to the immediate diagnostics of HLH and initiation of specific immunosuppressive treatment with dexamethasone, immunoglobulins and anakinra, the patient completely recovered and could finally be discharged after a 2‑week stay in hospital. Furthermore, the current diagnostic and therapeutic options are discussed. Ferritin is a decisive diagnostic marker that should be determined in every patient with unclear organ failure.
Keywords: Hemophagocytic lymphohistiocytosis, Hemophagocytic syndrome, Macrophage activation syndrome, Sepsis, Ferritin
Falldarstellung
Anamnese
Der 30-jährige, zuvor gesunde Patient wurde initial nach 3‑monatiger Asien- und Südamerikareise aufgrund von Fieber bis 39,0 °C, stärksten holozephalen Kopfschmerzen, Übelkeit ohne begleitendes Erbrechen und ohne Diarrhöe in deutlich reduziertem Allgemeinzustand in der Rettungsstelle der Charité – Universitätsmedizin Berlin vorstellig. Die Freundin des Patienten war während der letzten Woche der Reise an einem unspezifischen grippalen Infekt erkrankt. Der Impfpass des Patienten enthielt die aktuellen Impfungen gegen Typhus, Gelbfieber, Mumps/Masern/Röteln, Meningokokken und Hepatitis A/B.
Aufgrund der starken Zephalgien in Verbindung mit hohem Fieber wurde der Patient unter dem Verdacht einer bestehenden Meningitis/Enzephalitis auf die infektiologische Normalstation aufgenommen. Eine Therapie mit Ceftriaxon und Ampicillin wurde initiiert. Bis zum Folgetag konnte eine Meningitis/Enzephalitis mittels cCT, cMRT sowie liquordiagnostisch ausgeschlossen werden. Der Eiweißgehalt des Liquors war dezent erhöht und im cMRT zeigte sich eine diskret akzentuierte, leptomeningeale Kontrastmittelaufnahme, was beides mit einem unspezifischen entzündlichen Geschehen zu vereinbaren war. Der HIV-Test war negativ. Das Röntgenbild des Thorax bei Aufnahme war ebenfalls unauffällig (Abb. 1a).

Im Labor zeigten sich in den nächsten 48 h eine Leukopenie, eine Thrombozytopenie, ein weiter steigendes CRP, eine Hyponatriämie, erhöhte Hämolyseparameter (relevant erhöhtes freies Hb und bis unter die Nachweisgrenze erniedrigtes Haptoglobin), eine milde Erhöhung von Bilirubin, AST, ALT und eine Verlängerung der aPTT. Es entwickelten sich ein stammbetontes makulopapulöses Exanthem und schließlich eine akute respiratorische Insuffizienz. Als Korrelat zeigten sich in der Thoraxröntgenaufnahme bihiläre Infiltrate (Abb. 1b) und auskultatorisch bilateral basal ein abgeschwächtes Atemgeräusch sowie feuchte feinblasige Rasselgeräusche, sodass die Verlegung auf die infektiologisch-pneumologische Intensivstation erfolgte.
Klinischer Befund
Bei Übernahme auf die Intensivstation war der Patient zu allen Qualitäten orientiert, kreislaufstabil bei tachykardem Sinusrhythmus (HF 116/min, RR 105/60 mmHg), jedoch respiratorisch deutlich insuffizient. Der Patient war tachypnoisch bei einer AF von 30–35/min, unter 6 l/min Sauerstoff via Nasenbrille lag der paO2 bei 53,6 mmHg. Der paCO2 lag bei 30,4 mmHg, sodass bei einem Oxygenierungsindex von 122 mmHg von einem moderaten ARDS und einer kurzfristig drohenden ventilatorischen Erschöpfung des Patienten auszugehen war. Die Nierenfunktion war normal, es lag eine schwere Hyponatriämie von 119 mmol/l vor (Tab. 1). Es bestanden keine peripheren Ödeme, keine obere Einflussstauung, der Volumenstatus war als euvolämisch zu bewerten, das bereits oben beschriebene Exanthem persistierte. Auf Nachfragen offenbarte der Patient ein starkes Durstgefühl und beschrieb, große Mengen Leitungswasser zu sich genommen zu haben. Seit einem Nierensteinleiden in der Vergangenheit berichtete er über eine Trinkmenge von 4–5 l Wasser/Tag. Im thorakalen CT zeigten sich bilateral im Mittel- und basalen Oberlappen dominierend eine zentrilobuläre Milchglastrübung sowie bilaterale Pleuraergüsse (Abb. 1c). Im abdominellen CT fielen eine Hepatosplenomegalie und geringe Mengen Aszites auf. Die cCT sowie eine transthorakale Echokardiographie waren unauffällig. Der extravaskuläre Lungenwasserindex lag erhöht bei 11 ml/kgKG (Ref: 3–7 ml/kgKG).
| Kh, Aufnahme | ITS, Aufnahme | ITS, Tag 2 | ITS, Tag 3 | ITS, Tag 4 | ITS, Tag 5 | ITS, Tag 6 | ITS, Entlassung | Kh, Entlassung | |
|---|---|---|---|---|---|---|---|---|---|
|
Hämoglobin (g/dl) Ref: 13,5–17,0 |
16,1 | 12,2 | 12,6 | 11,9 | 9,8 | 9,2 | – | 10,4 | 13,6 |
|
Leukozyten (/nl) Ref: 3,9–10,5 |
2,68 | 2,82 | 4,97 | 5,91 | 11,6 | 11,9 | – | 11,2 | 9,46 |
|
Myelozyten (%) Ref: 0,0–0,0 |
– | – | – | 1,9 | – | – | – | 1,0 | – |
|
Stabkernige (%) Ref: 0,5–10,0 |
– | 32 | – | 8,5 | – | – | – | 2,1 | – |
|
Segmentkernige (%) Ref: 40,0–70,0 |
– | 54 | – | 83,0 | – | – | – | 67,7 | – |
|
Lymphozyten (%) Ref: 20,0–44,0 |
10,8 | 6,0 | – | 1,9 | – | – | – | 15,6 | 31,8 |
|
Monozyten (%) Ref: 2,0–9,5 |
4,9 | 3,0 | – | 1,9 | – | – | – | 6,3 | 7,1 |
|
Eosinophile (%) Ref: 0,5–5,5 |
0,0 | 0,0 | – | 0,0 | – | – | – | 0,0 | 0,0 |
|
Basophile (%) Ref: 0,0–1,8 |
0,7 | 2,0 | – | 0,9 | – | – | – | 0,0 | 0,3 |
|
Thrombozyten (/nl) Ref: 150–370 |
64 | 49 | 53 | 90 | 157 | 254 | – | 443 | 394 |
|
ALAT (U/l) Ref: <41 |
82 | 152 | 243 | 263 | 227 | 224 | – | 220 | 200 |
|
ASAT (U/l) Ref: <05 |
128 | 232 | 389 | 380 | 241 | 185 | – | 117 | 84 |
|
Bilirubin (mg/dl) Ref: <1,2 |
1,7 | 2,06 | 2,71 | 1,78 | 1,11 | 0,89 | – | 0,91 | 1,00 |
|
Triglyzeride (mg/dl) Ref: <200 |
– | – | 395 | 307 | 321 | 281 | – | 240 | – |
|
Kreatinin (mg/dl) Ref: 0,7–1,2 |
0,88 | 0,78 | 0,63 | 0,56 | 0,5 | 0,33 | – | 0,54 | 0,65 |
|
TPZ (%) Ref: 70–130 |
75 | 66 | 75 | 91 | 77 | 66 | – | 70 | 95 |
|
aPTT (s) Ref: 26,0–40,0 |
50,1 | 59,7 | 48,3 | 51,6 | 46,9 | 44,6 | – | 41,6 | 42,2 |
|
Fibrinogen (g/l) Ref: 1,6–4,0 |
– | – | 1,32 | – | – | – | – | – | – |
|
CRP (mg/l) Ref: <5,0 |
98,1 | 179,9 | 167,0 | 133 | 54,4 | 28,9 | – | 10,9 | 1,3 |
|
PCT (µg/l) Ref: <0,5 |
– | – | 1,08 | 0,72 | 0,37 | 0,07 | – | – | – |
|
Ferritin (µg/l) Ref: 13,0–150,0 |
– | – | 7692 | 7183 | 6057 | 4264 | – | 2736 | 2878 |
|
sIL-2R (U/ml) Ref: <710,0 |
– | – | 9964 | – | – | – | – | – | – |
| Max. Kerntemperatur (°C) | 36,5 | 37,8 | 39,0 | 37,2 | 36,9 | 37,0 | 36,4 | 36,4 | – |
|
pH Ref: 7,2–7,5 |
– | 7,478 | 7,446 | 7,428 | 7,492 | 7,504 | – | – | – |
|
pO2 (mmHg) Ref: 60–300 |
– | 53,6 | 65,3 | 87,7 | 110 | 99,1 | – | – | – |
|
pCO2 (mm Hg) Ref: 32–60 |
– | 30,4 | 33,2 | 38,1 | 32,8 | 30,3 | – | – | – |
|
HCO3− (mmol/l) Ref: 15–30 |
– | 22,3 | 22,5 | 24,7 | 24,9 | 23,6 | – | – | – |
|
sBE (mmol/l) Ref: −10 bis +10 |
– | −0,8 | −1,0 | 0,8 | 1,8 | 0,8 | – | – | – |
|
O2Hb (%) Ref: >90 |
– | 87,1 | 91,6 | 95,4 | 97,1 | 96,7 | – | – | – |
|
sO2 (%) Ref: >90 |
– | 89,3 | 93,7 | 97,5 | 99,2 | 98,8 | – | – | – |
|
Hb (g/dl) Ref: 7,0–18,0 |
– | 12,3 | 12,4 | 11,7 | 9,9 | 9,3 | – | – | – |
|
K (mmol/l) Ref: 3,0–6,0 |
– | 4,0 | 3,8 | 4,1 | 3,8 | 3,9 | – | – | – |
|
Na (mmol/l) Ref: 130–160 |
– | 119 | 123 | 130 | 134 | 134 | – | – | – |
|
Ca (mmol/l) Ref: 0,8–1,5 |
– | 1,12 | 1,12 | 1,19 | 1,19 | 1,16 | – | – | – |
|
Cl (mmol/l) Ref: 90–130 |
– | 96 | 97 | 102 | 104 | 107 | – | – | – |
|
Glucose (mg/dl) Ref: 70–150 |
– | 92 | 103 | 139 | 135 | 158 | – | – | – |
|
Lactat (mg/dl) Ref: ≤20 |
– | 11 | 10 | 10 | 7 | 14 | – | – | – |
| SOFA-Score | – | 8 | 8 | 4 | 2 | 4 | 0 | 0 | – |
ALAT Alanin-Aminotransferase, APACHE2 Acute Physiology and Chronic Health Evaluation 2, aPTT aktivierte partielle Thromboplastinzeit, ASAT Aspartat-Aminotransferase, CRP C-reaktives Protein, Hb Hämoglobin, PCT Prokalzitonin, sIL-2R löslicher Interleukin-2-Rezeptor, SOFA Sequential Organ Failure Assessment, TPZ Thromboplastinzeit
Laborchemisch fielen eine Thrombozyto- und Leukopenie mit Lymphopenie sowie eine deutliche Linksverschiebung auf; das CRP war erhöht. Die Gerinnungsdiagnostik deutete auf eine disseminierte intravasale Koagulopathie (DIC) hin. Die hepatischen Laborparameter (erhöhte Transaminasen, Bilirubin) zeigten eine dezente, funktionell nichtrelevante Leberschädigung (Tab. 1). In der Urindiagnostik zeigten sich ein Natrium von 62 mmol/l sowie eine Osmolalität von 394 mosmol/kg bei zu diesem Zeitpunkt im Serum vorliegender Osmolalität von 250 mosmol/kg und einem Natrium von 125 mmol/l.
Bei Aufnahme und im Verlauf waren weder ein Infektionserreger noch eine serologische Reaktion als Hinwies auf einen spezifischen Erreger nachweisbar (Multiplex-Virus-PCR aus Rachenspülwasser und Liquor, Lues, HIV/HBV/HCV/HEV/HSV/CMV/EBV/RSV/VZV, Masern/Mumps/Röteln/Adenovirus/Coronavirus/Metapneumovirus/Rhinovirus/Enterovirus/Influenza/Parainfluenza/Parechovirus, Rickettsien, Leptospira, Cryptococcus, Malaria, Dengue-Virus, Legionellen). Repetitive Blut‑, Liquor- und Urinkulturen blieben ohne Wachstum. In der autoimmunologischen Diagnostik ergab sich ebenfalls kein Hinweis auf eine zugrunde liegende Autoimmunerkrankung und dadurch vermittelte Entzündungsreaktion: RF, ACPA, cANCA, pANCA, ANA, Anti-dsDNA, Anti-GBM waren nicht pathologisch verändert; es lag kein Komplementverbrauch vor.
Nachdem die beschriebene Diagnostik keine Erklärung für die systemische Entzündungsreaktion und damit verbundenen Organschädigungen erbrachte, wurde bei prävalenter LDH-Erhöhung sowie einem erhöhten β2-Mikroglobulin zum Ausschluss einer Neoplasie eine Knochenmarkpunktion durchgeführt. Es erfolgten außerdem die Bestimmungen von Ferritin sowie Triglyzeriden zur Abklärung der seltenen Differenzialdiagnose Hämophagozytische Lymphohistiozytose (HLH).
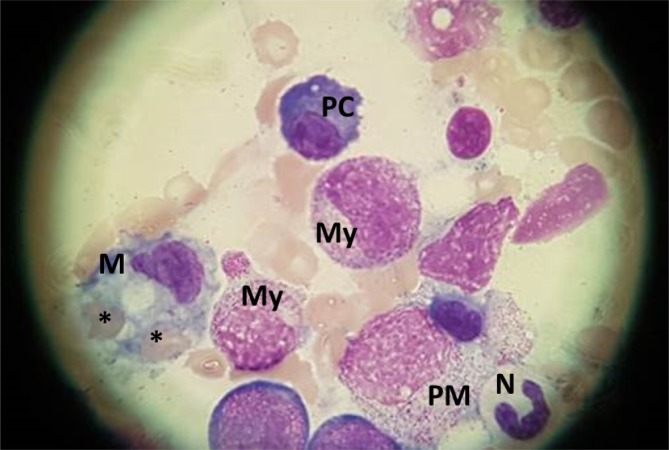
Einen Hinweis für eine akut behandlungsbedürftige Neoplasie erbrachte die Diagnostik nicht. Ferritin sowie Triglyceride im Plasma waren relevant erhöht, und im Ausstrich des Knochenmarks zeigten sich einzelne Monozyten mit nachzuweisender Hämophagozytose (Abb. 2). Weitergehende immunologische Analysen zeigten eine verminderte basale Hochregulation der Expression von CD107a auf NK-Zellen bei jedoch normwertigem Befund nach Stimulation sowie einen relevant erhöhten löslichen IL-2-Rezeptor (sIL-2R) (Tab. 1). Aufgrund der Splenomegalie von 14 cm wurde im Rahmen der Knochenmarkdiagnostik eine molekulargenetische Stufendiagnostik auf myeloproliferative Erkrankungen veranlasst. Dabei wurde eine JAK2-V617F-Mutation (Exon 12) in niedriger Allelfrequenz unter 1 % gefunden, die eine myeloproliferative Erkrankung sehr unwahrscheinlich erscheinen lässt.
Diagnose
In Zusammenschau aller vorliegenden Befunde der Anamnese wurde in Übereinstimmung mit den aktuell dafür geltenden HLH-2004-Diagnosekriterien (Tab. 2) eine HLH diagnostiziert. Im Rahmen dieses Hyperinflammationssyndroms kam es zum akuten Lungenversagen, zur Hämolyse, Hepatitis, einer milden DIC, Fieber und einer die ausgeprägten Zephalgien erklärenden zerebralen Affektion. Die Hyponatriämie wurde der Polydipsie zugeordnet, die allerdings durch die zentralnervöse inflammatorische Affektion aggraviert worden sein kann. Als Auslöser der HLH wurde eine unspezifische Virusinfektion angesehen.
| Fieber |
| Splenomegalie |
| Zytopenie in mind. 2 Zellreihen (Hb <9 g/dl, Thrombozyten <100/nl, neutrophile Granulozyten <1,0 /nl) |
| Hypertriglyzeridämie und/oder Hypofibrinogenämie (Triglyzeride [nüchtern] ≥3 mmol/l, Fibrinogen <1,5 g/l) |
| Ferritin ≥500 µg/l |
| sIL-2R ≥2400 U/ml |
| Erniedrigte NK-Zellaktivität |
| Hämophagozytose in Knochenmark, Leber, Milz oder Lymphknoten |
Hb Hämoglobin, sIL-2R löslicher Interleukin-2-Rezeptor, NK natürliche Killerzelle
Therapie und Verlauf
Zu Beginn der Intensivtherapie wurde die antiinfektive Behandlung bei Verdacht auf eine schwere ambulant erworbene Pneumonie auf Meropenem und Clarithromycin zur Abdeckung der atypischen Erreger umgestellt und für 7 Tage fortgeführt. Aufgrund der vorliegenden respiratorischen und drohenden ventilatorischen Insuffizienz wurde der Patient intermittierend nichtinvasiv beatmet; im Wechsel erfolgte eine „High-flow“-Sauerstofftherapie. In Zusammenschau aller erhobenen Befunde wurde die Diagnose HLH bereits am Folgetag der ITS-Aufnahme gestellt. Bei Diagnosestellung waren 5 der HLH-2004-Kriterien (Tab. 2) erfüllt, nach Vorliegen der Befunde des Knochenmarkausstrichs sogar 6 Kriterien. Die spezifische immunsuppressive Therapie wurde am Tag der Diagnosestellung mit Dexamethason (10 mg/m2KOF und Tag), Immunglobulinen (0,5 g/kgKG über 3 Tage) und dem IL-1-Rezeptor-Antagonist Anakinra 100 mg/Tag begonnen. Bereits innerhalb von 12 h trat eine deutliche klinische Verbesserung ein: Der Patient entfieberte, die respiratorische Insuffizienz war rasch regredient, der radiologisch pulmonale Befund normalisierte sich (Abb. 1d), und das initial erhöhte extravaskuläre Lungenwasser befand sich wieder im Normbereich. Die pathologisch veränderten Laborparameter waren regredient; die Zephalgien sistierten.
Die schwere Hyponatriämie wurde initial mit NaCl behandelt und damit die Natriumkonzentration um 4 mmol/l angehoben. Die Hyponatriämie werteten wir nicht als ursächlich für die neurologische Symptomatik, da diese bereits vor der Entwicklung der Hyponatriämie bestand, und die Zephalgien erst mit dem Beginn der immunsuppressiven Therapie sistierten. Im Verlauf war das Ferritin ebenfalls deutlich regredient (Abb. 3), sodass der Patient nach 7 Tagen intensivstationärer Behandlung auf die Normalstation verlegt werden und nach weiteren 5 Tagen klinisch vollständig erholt in die Häuslichkeit entlassen werden konnte.

Der Patient wurde ambulant weiterbetreut. Im Verlauf traten Schlafstörungen aufgrund der Steroidtherapie auf; sonst war er klinisch nicht beeinträchtigt. Dexamethason wurde in der Dosierung von 7,5 mg/Tag bis Tag 27 fortgeführt. Anakinra wurde noch bis Tag 41 fortgesetzt. Das Ferritin sank unter dieser Therapie bis zum Tag 27 in den Referenzbereich ab. Auch nach Absetzen der Medikation sank das Ferritin weiter. Es gab im Verlauf keinen Anhalt für eine Reaktivierung der HLH (Abb. 3).
Die abschließende Einordnung der in niedriger Allelfrequenz unter 1 % festgestellten JAK2-V617F-Mutation erfolgt in längerfristigen Verlaufskontrollen in der hämatologischen Ambulanz. Dabei ergaben sich bis 4 Monate nach Krankenhausentlassung keine Hinweise auf eine myeloproliferative Erkrankung, sodass eine Wiederholung der molekulargenetischen Testung geplant ist. Im Follow-up nach 4 Monaten lag bei dem Patienten ein unauffälliges Differenzialblutbild vor.
Diskussion
Die HLH ist ein lebensbedrohliches Hyperinflammationssyndrom, bei dem es durch eine Fehlfunktion natürlicher Killerzellen oder zytotoxischer T‑Zellen zu einer exzessiven Immunaktivierung mit Zytokinsturm, Hämophagozytose und Multiorganversagen kommt. Unterschieden wird eine angeborene Form bei Kindern von einer adulten Form bei Erwachsenen, die in der Regel sekundär durch Infektionen, Malignome oder Autoimmunerkrankungen ausgelöst wird [2]. Der Patient zeigte das Vollbild der HLH mit dem Vorliegen von 6 der 8 HLH-2004-Kriterien, obwohl die Diagnose bereits ab 5 positiven Kriterien gestellt werden kann. Die Hochregulation der CD107a-Expression auf NK-Zellen nach Stimulation in den definierten Normbereich macht eine genetische Komponente als Auslöser unwahrscheinlich. Obwohl ein Nachweis der Virusinfektion trotz breiter Diagnostik nicht gelang, ist eine unspezifische Virusinfektion als Trigger der HLH sehr wahrscheinlich, zumal die Freundin des Patienten ebenfalls erkrankt war und die Symptome, v. a. das begleitende Exanthem, die meningeale Reizung sowie die Hepatosplenomegalie der Virusinfektion zugeordnet werden können.
Das klinische Bild der HLH ist der Sepsis ähnlich, weshalb >70 % aller Patienten mit HLH auf Intensivstationen nicht erkannt und als Sepsis fehldiagnostiziert werden [3]. Dabei kann die HLH sowohl eine Sepsis imitieren als auch durch eine Sepsis selbst ausgelöst werden [4]. Gleichzeitig sind eine frühzeitige Diagnose und Therapie für das Überleben entscheidend, allerdings liegt die Letalität auch mit immunsuppressiver Therapie bei über 50 % [5]. Bei hochgradigem Verdacht auf HLH darf der Therapiebeginn nicht verzögert werden, auch wenn erst 4 Kriterien erfüllt sind [6]. Auf Intensivstationen besteht außerdem bei häufigem Einsatz von Antipyretika, Hämodialyse, ECMO und Transfusionen die Herausforderung, die Kriterien Zytopenie, Fieber, Ferritin, sIL-2R und Fibrinogen zu beurteilen. Wichtig ist, dass der Nachweis der Hämophagozytose im Knochenmark bei vorliegender HLH oft sehr schwierig ist und in über 35 % der Fälle nicht gelingt [7].
Im Rahmen der Hyperinflammation mit konsekutiver pulmonalvaskulärer Schrankenstörung kam es zum Lungenversagen mit moderatem ARDS; es entwickelten sich eine Polyserositis mit Pleuraergüssen und Aszites sowie eine milde Hepatitis und eine Verbrauchskoagulopathie. Auch die Zephalgien können der Hyperinflammation zugeordnet werden. Dank einer hohen Vigilanz beim Erkennen der HLH wurde der Patient umgehend diagnostiziert, behandelt und konnte nach insgesamt 14-tägigem Krankenhausaufenthalt vollständig genesen entlassen werden. Durch die zeitnahe Einleitung der spezifischen immunsuppressiven Therapie ist anzunehmen, dass ein weiteres Voranschreiten der HLH zu einem schweren ARDS und ausgeprägtem Multiorganversagen verhindert wurde.
Ferritin als wichtiger Hinweisgeber zum Vorliegen der HLH wies ab einem Wert von 3951 µg/l eine Sensitivität von 88 % und eine Spezifität von 82 % auf Intensivstationen auf [8]. Im Fall eines fortgeschrittenen Leberversagens und laufender Dialyse sowie Massentransfusion ist der einmalig gemessene Ferritinwert allerdings nicht als HLH-spezifisch einzuschätzen. Die zusätzliche Bestimmung von sIL-2R ist in diesen Fällen sehr sinnvoll und zeigte ab Werten von 2,515 U/ml eine Sensitivität von 100 % und eine Spezifität von 72,5 % [9]. Bei Zweifelsfällen zur Diagnose kann der H‑Score (http://saintantoine.aphp.fr/score/) berechnet oder das deutsche HLH-Register (http://www.hlh-registry.org/hlh-register-fuer-erwachsene/) für Therapieentscheidungen konsiliarisch kontaktiert werden.
Die spezifische immunsuppressive Therapie des Patienten wurde durch Dexamethason, Immunglobuline und Anakinra initiiert und war effektiv. Die Immunsuppression durch Glukokortikoide ist neben Etoposid elementarer Bestandteil des HLH-1994-Protokolls; zusätzlich werden Immunglobuline empfohlen [10, 11]. Etoposid als wirksamstes Element des HLH-1994-Protokolls wurde aufgrund des raschen Ansprechens der primären Immunsuppression nicht eingesetzt. Stattdessen wurde entsprechend den Erfahrungen von Kumar et al. [12] neben der empfohlenen Therapie mit Dexamethason und Immunglobulinen der IL-1-Rezeptor-Antagonist Anakinra eingesetzt. Anakinra reduzierte in einer Post-hoc-Subgruppenanalyse einer Sepsistherapiestudie bei Patienten mit Sepsis und Zeichen der HLH (hepatobiliäre Dysfunktion und DIC) die Mortalität um 47 % [13]. Ob jedoch im vorliegenden Fall Anakinra den Krankheitsverlauf signifikant beeinflusst hat oder allein Dexamethason mit einer Immunglobulintherapie ähnliche Wirksamkeit gezeigt hätte, lässt sich retrospektiv nicht beurteilen. Während im vorliegenden Fall die immunsupprimierende Therapie ohne genaue Kenntnis der Trigger-Erkrankung rasch zur kompletten Remission führte, sind in vielen Fällen die spezifische Therapie der Trigger-Erkrankung (Infektion, Malignom, Autoimmunopathie) und die konsequent fortgesetzte Diagnostik bei Nichtansprechen der Primärtherapie von herausragender Bedeutung für den Therapieerfolg [6].
Fazit für die Praxis
Bei Patienten mit Sepsis, die unzureichend auf eine antiinfektive Therapie ansprechen oder eine ausgeprägte Inflammation zeigen, muss eine Hämophagozytische Lymphohistiozytose (HLH) als Differenzialdiagnose ausgeschlossen werden.
Ferritin und löslicher Interleukin-2-Rezeptor (sIL-2R) sind entscheidende diagnostische Marker, um im Kontext des Gesamtbildes die HLH von der Sepsis abzugrenzen bzw. eine Überlappung aufzuzeigen.
Eine frühzeitige Diagnose ist prognoserelevant und bedarf umgehender immunsuppressiver Therapie mittels Hochdosisglukokortikoiden, Immunglobulinen und ggf. Anakinra bzw. Etoposid in therapierefraktären Fällen.
Einhaltung ethischer Richtlinien
Interessenkonflikt
G. Lachmann, C. Knaak, P. La Rosée, C. Spies, P. Nyvlt, C. Oberender, L.E. Sander, N. Suttorp und H. Müller-Redetzky geben an, dass kein Interessenkonflikt besteht.
Dieser Beitrag beinhaltet keine von den Autoren durchgeführten Studien an Menschen oder Tieren. Die Ethikkommission der Charité – Universitätsmedizin Berlin hat den vorliegenden Fallbericht genehmigt (EA1/176/16). Der Patient hat der Veröffentlichung zugestimmt.
Literatur
- 1.Henter JI, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 2.Lachmann G, et al. Hemophagocytic lymphohistiocytosis : a diagnostic challenge on the ICU. Anaesthesist. 2016;65(10):776–786. doi: 10.1007/s00101-016-0216-x. [DOI] [PubMed] [Google Scholar]
- 3.Lachmann G, et al. Hemophagocytic Lymphohistiocytosis: potentially Underdiagnosed in intensive care units. Shock. 2018;50(2):149–155. doi: 10.1097/SHK.0000000000001048. [DOI] [PubMed] [Google Scholar]
- 4.Machowicz R, Janka G, Wiktor-Jedrzejczak W. Similar but not the same: differential diagnosis of HLH and sepsis. Crit Rev Oncol Hematol. 2017;114:1–12. doi: 10.1016/j.critrevonc.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 5.Barba T, et al. Hemophagocytic Lymphohistiocytosis in intensive care unit: a 71-case strobe-compliant retrospective study. Medicine (Baltimore) 2015;94(51):e2318. doi: 10.1097/MD.0000000000002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.La Rosee P. Treatment of hemophagocytic lymphohistiocytosis in adults. Hematology Am Soc Hematol Educ Program. 2015;2015:190–196. doi: 10.1182/asheducation-2015.1.190. [DOI] [PubMed] [Google Scholar]
- 7.Ramos-Casals M, et al. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–1516. doi: 10.1016/S0140-6736(13)61048-X. [DOI] [PubMed] [Google Scholar]
- 8.Saeed H, et al. Evaluating the optimal serum ferritin level to identify hemophagocytic lymphohistiocytosis in the critical care setting. Int J Hematol. 2015;102(2):195–199. doi: 10.1007/s12185-015-1813-1. [DOI] [PubMed] [Google Scholar]
- 9.Hayden A, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529–2534. doi: 10.1182/bloodadvances.2017012310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henter JI, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol. 1997;28(5):342–347. doi: 10.1002/(SICI)1096-911X(199705)28:5<342::AID-MPO3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 11.La Rosee P, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465–2477. doi: 10.1182/blood.2018894618. [DOI] [PubMed] [Google Scholar]
- 12.Kumar B, et al. A personalized diagnostic and treatment approach for Macrophage activation syndrome and secondary Hemophagocytic Lymphohistiocytosis in adults. J Clin Immunol. 2017;37(7):638–643. doi: 10.1007/s10875-017-0439-x. [DOI] [PubMed] [Google Scholar]
- 13.Shakoory B, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of Macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. 2016;44(2):275–281. doi: 10.1097/CCM.0000000000001402. [DOI] [PMC free article] [PubMed] [Google Scholar]