

Abstract
Detection of multiple viruses is important for global analysis of gene or protein content and expression, opening up new prospects in terms of molecular and physiological systems for pathogenic diagnosis. Early diagnosis is crucial for disease treatment and control as it reduces inappropriate use of antiviral therapy and focuses surveillance activity. This requires the ability to detect and accurately diagnose infection at or close to the source/outbreak with minimum delay and the need for specific, accessible point-of-care diagnosis able to distinguish causative viruses and their subtypes. None of the available viral diagnostic assays combine a point-of-care format with the complex capability to identify a large range of human and animal viruses. Microarray detection provides a useful, labor-saving tool for detection of multiple viruses with several advantages, such as convenience and prevention of cross-contamination of polymerase chain reaction (PCR) products, which is of foremost importance in such applications. Recently, real-time PCR assays with the ability to confirm the amplification product and quantitate the target concentration have been developed. Furthermore, nucleotide sequence analysis of amplification products has facilitated epidemiological studies of infectious disease outbreaks and monitoring of treatment outcomes for infections, in particular for viruses that mutate at high frequency. This review discusses applications of microarray technology as a potential new tool for detection and identification of acute encephalitis-causing viruses in human serum, plasma, and cell cultures.
Keywords: Respiratory Syncytial Virus, West Nile Virus, Infectious Agent, Severe Acute Respiratory Syndrome, Microarray Technology
Introduction
Microarray technology is a promising technology used to study the expression level of various genes simultaneously or to genotype multiple regions of a genome in a particular cell type of an organism, at a particular time and conditions. This allows comparison of gene expression between normal and infected cells. The method involves placing thousands of gene sequences in known locations on a glass slide called a gene chip. Based on such easy implementation of a huge number of tests for identification of viral agents affecting humans, these tests are helpful for blood screening; they can also be used as a diagnostic tool, albeit imperfect as they provide only an indirect measure of infection but cannot advise clinicians regarding whether the infection is recent or in progress, or regarding response to therapy [52, 102, 104]. Antibody-based testing can also fail to detect current infection, because it usually takes a few days to weeks for the immune system to raise an antibody response to an infectious agent [56, 79, 125]. In the past, tests have been developed based on direct quantification of the infectious agent in a sample taken from the patient [41]. Such tests detect the presence of nucleic acids (the genetic material) of infectious agents in blood or other samples. The most common method is the polymerase chain reaction (PCR), which can detect 100 copies or more of an infectious agent in a single sample [29, 79]. PCR employs an enzymatic reaction to amplify specific nucleic acid sequences of infectious agents that may be present in the sample [59, 114]. There are several problems with this technique [78]: viral agents can mutate very quickly, and PCR primers may therefore not recognize the infectious agent, producing false or weak results [22]. The subsequent process is based on direct hybridization of the infectious agent’s nucleic acid to a synthetic nucleic acid probe [23, 62]. The hybridized infectious agent is then detected by extension using nonenzymatic methods. The main disadvantage of this technique is required an additional quantity of the patient’s sample (minimum 1 ml blood compared with 0.1 ml for PCR) [51, 70, 100]. Virus culture generally results in good specificity, but not all viruses can be cultured, and technical expertise is required to understand the cytopathic effect (CPE) and to read stained preparations. Also, this method is time consuming and labor intensive due to the long incubation period of some viruses, and it is very difficult to culture a variety of cell types at once [104, 110].
During the last two decades and today, viral diseases remain the main cause of mortality in humans. In recent times, the appearance of infectious diseases has become more serious, as represented by new pathogens such as re-emerging viruses causing acquired immune-deficiency syndrome, acute encephalitis syndrome, Hendra, Nipah, severe acute respiratory syndrome (SARS), and avian influenza [27, 28, 73]. Many factors contribute to the emergence of viral infections, potentially including genetic exchange or mutation, adaptation to new hosts or vectors, rapid transport, trade, migration of people, and changing values or lifestyles [8]. The resulting rapid epidemiological changes in the community may lead to both new and old viruses that can emerge and cause outbreaks at unexpected times and locations. The fight against such emergent viral infections requires the development of a comprehensive strategy.
Using conventional techniques to analyze gene expression, researchers are able to survey a relatively small number of genes at once [24]. Microarray profiling offers many potential advances in terms of diagnostic and therapeutic interventions in human diseases because of its unparalleled ability for high-throughput gene expression analysis. This technology provides powerful tools for the scientific community [15], and scientists are using microarray technology to try to understand fundamental aspects of growth and development as well as to explore the underlying genetic causes of many human diseases [48, 49]. However, the limitations of this technique are related in part to issues regarding the various methodologies and experimental designs, as well as difficulties in the interpretation of results. Despite these limitations, microarray technology has been used efficiently in disease diagnosis.
Acute Encephalitis Syndrome
Encephalitis means inflammation of brain matter. More than 100 different infectious pathogens and several toxins have been identified as causative agents of encephalitis, although in many cases no pathogen can be detected. Accurate etiological diagnosis is required to increase the usefulness of surveillance of acute encephalitis, especially in view of concerns about new and re-emerging infections. Viruses that infect the central nervous system (CNS) may selectively involve the spinal cord (myelitis), brain stem (e.g., rhomb encephalitis), cerebellum (cerebellitis), or cerebrum (encephalitis). Almost every acute viral CNS infection results in meningeal as well as parenchymal inflammation to varying degrees [12]. Fundamental clinical and laboratory findings are mainly similar despite the different causative agents, consisting of fever and headache in addition to distorted cerebral position, frequently accompanied by seizures and central neurologic abnormality. Cerebrospinal fluid (CSF) is abnormal in >90 % of cases, characteristically showing lymphocytic pleocytosis, slightly elevated protein level, and normal glucose [113]. In unusual diseases, such as West Nile virus (WNV) meningoencephalitis or cytomegalovirus (CMV) radiculomyelitis, polymorphonuclear cells rather than lymphocytes could be the principal cell type, providing useful diagnostic evidence. Nevertheless, in spite of these variations, standard CSF study rarely leads to exact identification of the etiologic agent [11].
A clinician may be faced with a patient of any age, at any time of year, with acute-onset fever and altered mental status including symptoms such as confusion, disorientation, coma, and inability to talk, or new-onset seizures (excluding simple febrile seizures) [61]. Other symptoms may include an increase in irritability, abnormal behavior greater than that seen with usual febrile illness, and altered mental status, enabling initial differentiation of encephalitis from noninfectious causes of brain dysfunction (encephalopathy) [11, 41, 97].
A huge number of tests to detect viral pathogens are available, but they have not been validated for identification of divergent viruses using traditional methods of assay gene expression; researchers can therefore identify only a relatively small number of genes at once [24]. Cell cultures using the traditional tube method can be used for isolation and detection of a wide variety of viruses, including unanticipated agents, mixed-culture antiviral susceptibility testing, serotyping, and epidemiologic studies. They offer increased sensitivity over rapid antigen tests, but require a long incubation period for some viruses as well as acquisition and maintenance of a variety of cell culture types in-house. Shell vials with centrifugation can also be used, but reading of pre-CPE stained preparations is both time consuming and labor intensive. Also, unanticipated agents may be missed when pre-CPE staining targets only one or a few viruses, and isolates from fixed/stained vials are not available. Nonculture antigen detection using Immunofluorescence (IF) monoclonal antibodies (mAbs) takes 40 min per sample, generally offering good sensitivity (which varies with the virus detected) and excellent specificity. CMV antigenemia testing is more sensitive than traditional or shell-vial culture for CMV in blood, but generally not as sensitive as cell culture. Also, it requires expertise for reading and is not useful for all viruses; flavivirus sensitivity is especially poor. Non-IF antigen detection requires 30 min per sample, offering generally good specificity for respiratory syncytial virus (RSV) and influenza A and B viruses. No special technical expertise is required, and results are available very rapidly, enabling application in point-of-care testing. However, it generally offers poor sensitivity compared with cell culture and is currently only available for RSV and influenza A and B viruses. Additional testing of negative samples by cell culture is recommended, the most common method being by PCR, which can detect 100 copies or more of an infectious agent in a single sample [79]. PCR uses an enzymatic reaction to amplify specific nucleic acid sequences from the infectious agent if they are present in the sample [114]. There are several problems with this method [78]. PCR uses specific nucleic acid sequences (primers) from a known sequence of the infectious agent [55, 88]. Therefore, if the infectious agent has not been sequenced, PCR cannot be used. Similarly, if the infectious agent mutates very rapidly, the primers may not recognize the infectious agent and a false-negative test will result [21, 22]. This is a major problem with detection of viruses, which undergo very rapid mutation, especially in response to drug treatment [14, 27]. Since PCR uses an enzymatic reaction, the enzyme can be inhibited by impurities in the patient sample, also leading to false-negative results [7]. In addition, the short sequences of primers are only specific to an infectious agent at definite temperatures, making the test reliant on very strict conditions [117]. The specificity of the primers also makes it difficult to detect more than one agent simultaneously in a single PCR reaction [19, 46]. Multiplex PCR reactions exist, but generally they are not quantitative and can only detect two or a maximum of three agents concurrently [6].
Detection of multiple viruses causing acute encephalitis syndrome (AES) is important for global analysis of gene content and expression, opening prospects for new molecular and physiological systems to control pathogenic diagnosis. Early diagnosis is crucial for disease treatment and control, as it reduces inappropriate use of antiviral therapy and focuses surveillance activity [85, 88]. This requires the ability to detect and accurately diagnose infection at or close to the source/outbreak with minimum delay and the need for specific, accessible point-of-care diagnosis able to distinguish causative viruses and their subtypes. None of the available viral diagnostic assays combines a point-of-care format with the complex capability to identify a large range of viruses causing AES. Biomedical research evolves and advances not only through the compilation of knowledge but also through the development of new technologies [9]. Microarray detection provides a useful, labor-saving tool for detection of multiple viruses, offering several advantages such as convenience and prevention of cross-contamination. Microarray technology aims to monitor the whole genome on a single chip, providing researchers with a clearer picture of the interactions among thousands of genes simultaneously [25]. This represents a major methodological advance and illustrates how the advent of a new technology can provide powerful tools for research [15].
Microarrays can help answer gradually more multifaceted questions and execute additional complicated experiments. Researchers may be able to infer possible functions of new genes based on similarities in expression profile compared with known genes [32, 115]. Finally, such studies increase the number of accessible gene families, providing a novel guide for synchronized gene expression across gene families as well as for completely new groups of genes. In addition, since discovery of any gene usually interrelates with that of many others, knowledge of how these genes are organized can be improved through such analyses, and exact information of these interrelationships will emerge. Use of microarrays may also accelerate identification of genes involved in the progress of various diseases by enabling scientists to examine a much larger number of genes [104]. This technology will also assist with assessment of gene expression and function at cellular level, illuminating how manifold gene products work mutually to construct substantial chemical responses to both static and changing cellular needs. Scientists may use microarray technology to try to understand fundamental aspects of growth and development as well as to explore the underlying genetic causes of many human diseases [114, 122].
Principles of Microarray Technology
Microarray technology is based on hybridization of two DNA strands formed of complementary nucleic acids linked by tight noncovalent bonds. In the hybridization technique, after washing, nonspecific bonding sequences are removed while only strongly paired strands will remain hybridized. Consequently, fluorescently labeled target sequences designed to bind to a particular probe sequence create a signal which depends on the potency of the hybridization and the number of paired bases, the hybridization conditions (such as temperature), and washing after hybridization. The overall potency of the signal from a spot depends on the amount of test sample bound to the corresponding probes at that location [84]. Hybridized targets can then be detected using one of many reporter-molecule systems. Microarrays use relative quantization in which the intensity of a feature is compared to the intensity of the same feature under a different condition, and the identity of the feature is known by its position [127].
Types of Microarray
Two types of DNA microarray are widely available for data analysis: complementary DNA (cDNA) arrays and oligonucleotide arrays (Fig. 1).
Fig. 1.
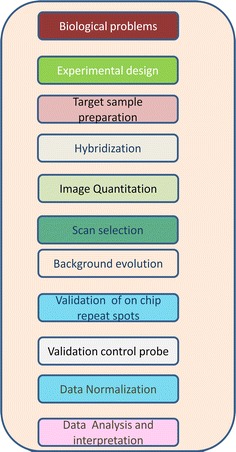
Microarray assignment workflow
cDNA Arrays
This type of chip offers a high-density microarray, most often being derived from cDNA (hence the name). Such chips are usually made by robotically spotting onto a large glass surface [76, 77]. Hybridization is carried out using fluorescently labeled messenger RNA (mRNA) corresponding to the cDNA, and hybridized duplexes are identified by the color fluorescence detection method. Thus, they can be used to study gene expression patterns in time and space. If a gene is overexpressed in a particular disease state, then more sample cDNA, as compared with control cDNA, will hybridize to the spot representing that gene. In turn, the red fluorescence of this spot will have greater intensity than its green fluorescence [22]. Once the expression profile of different genes involved in a disease has been distinguished, cDNA derived from a diseased sample taken from any person can be hybridized to conclude whether the expression profile of the gene from the individual corresponds to the expression profile of a known disease [113].
Oligonucleotide Arrays
In these arrays, the expression strength of a gene is assessed by means of a probe set consisting of 11–20 individual probe pairs. In most recent gene chips, the number of probe pairs has stabilized and is now 10–13 [27, 28, 96 ]. Every probe pair includes an ideal-match 22–25-mer oligonucleotide probe, which is planned to hybridize exclusively to a unique gene transcript, and a variance probe of the same length, which varies from the ideal match probe by a single base in the core of the sequence [46, 55, 118]. The aim of the variance probe is to calculate unfocused hybridization. Probe position algorithms created by Affymetrix read the signals from each 22–25 oligonucleotide probe set to obtain the particular values as a hybridization blueprint of the 22–25 probes. Each ideal match probe has a corresponding mismatch probe which contain the same 22–25 bases long sequences as the ideal match probe, except for the fact that the middle base (11–13) in the chain is substituted for the compliment of the 11–13th base of its consequent ideal match probe. This is meant to give an estimate of non-specific binding, which occurs when m-RNA that not targeted binds to ideal match [60, 94, 95]. In addition it has been exposed to the sign perverse by the ideal match probes not completely unfocused signal. In particular when a transcript is there at far above the ground levels, the label point strength also hybridizes to the variance probe [113, 114]. However, observed expression levels also include variation that is introduced during the process of carrying out the experiment, which could be classified as obscuring variation [57]. Affymetrix has approached the normalization problem by proposing that intensities should be scaled so that each array has the same average value. The distribution of probe intensities is the same across a set of arrays [80, 89, 96]. Propose arametric and non-parametric methods to achieve this. All these approaches depend on the choice of a baseline array. Currently it is up to the researcher to decide on the most significant result for their particular intention for high sensitivity and low variability, or a low false-positive rate [66].
Methods
Briefly, a limited sequence is immobilized on the microarray surface and binds to the target RNA during hybridization. The captured target is labeled with an additional fluorophore-conjugated DNA oligonucleotide (specifically, the label sequence). Positive control spots, in which a capture sequence hybridizes directly to a complementary label sequence, are included to aid visual analysis. After hybridization and precise washing, the microarray is scanned in a laser-based fluorescence scanner with 5-μm resolution [8].
Sequence Selection and AES Chip Microarray Design
AES virus-specific capture and label sequences may be selected [97]. The possibility of false-positive signals resulting from direct hybridization of label sequences to capture sequences is examined by incubation of the label sequences in the absence of any other nucleic acids at room temperature for 2 h in standard hybridization buffer. Capture sequences found to exhibit cross-reactivity with label sequences are removed from the array layout, along with the corresponding label sequence, and the arrays are reprinted [33]. This process is repeated until the microarray demonstrates no false-positive signals in the absence of viral RNA. The resulting array contains capture sequences and the corresponding label sequences. Each capture sequence is spotted in triplicate, and a single limited sequence with a complementary fluorescence-labeled sequence in solution is used as a positive control on each array. The positive control serves as a direct indication of whether the hybridization conditions are adequate and also as a spatial marker for ease of presentation [40, 53, 60, 67].
Microarray Slide Preparation
The substrate used for these studies comprises an OmniGrid microarray spotter with solid core pins with 550 μm pitch between spots.
Samples
Viral samples can be purified from whole blood, plasma, serum, throat swabs, cerebral spinal fluid, virus-infected supernatants, and other cell–free body fluids [101].
Chip Processing
Figure 1 shows a schematic of the dispensing protocol. The details of each processing step are described below.
Nucleic Acid Extraction
Nucleic acids may be extracted from clinical samples by using a nucleic acid purification kit, omitting RNase digestion, or manually by triazle methods according to the manufacturer’s recommended protocols. RNA is bound to an advanced silica gel membrane under optimal buffering conditions [39, 97]. A simple two-step washing protocol ensures that PCR inhibitors such as proteins or divalent cations are completely removed, leaving high-quality RNA to be eluted in Milli-Q water [12]. RNA purification processes are generally performed by TRIzol or using a kit method for degradation of the omnipresent RNases and DNA contamination from genomic DNA in the source material [79]. Purification from a viral source carries with it the possible additional challenge of low or varying viral titers. In addition to looking for a kit that can handle low titers as well as overcome the other traditional challenges, users must find a method that is easy to use and that ensures that the typical concentration of extracted RNA meets the requirements of downstream applications [61].
Primer Design, Probes, and Arrays to Confirm the Identity of Viral Strains
The most critical aspect of successful PCR is primer design. All things being equal, a poorly designed primer can result in a PCR reaction that will not work [47, 48]. The primer sequence determines numerous parameters such as the length of the product, its melting temperature, and ultimately the yield [4, 42]. A poorly designed primer can result in little or no product due to nonspecific amplification and/or primer–dimer formation, which can become competitive enough to suppress product formation [13, 19, 111]. This subsection provides rules that should be considered when designing PCR primers. More comprehensive coverage of this subject can be found elsewhere [23, 26]. Several variables must be taken into account when designing PCR primers [126, 127]. Among the most critical are: primer length, melting temperature (T m), specificity, complementary primer sequences, G/C content and polypyrimidine (T, C) or polypurine (A, G) stretches, and 3′-end sequence; each of these critical elements is discussed in turn [121, 127, 129].
Practically all viral strains carry a unique DNA sequence that differentiates it from other strains, and this sequence can be used for probe design [1, 35]. The probe DNA binds specifically to the target gene corresponding to the viral strain prepared from a clinical sample [31, 53, 120]. Using a variety of such probes, various pathogens in a clinical sample can be determined in a single trial. The DNA probe representing the selected target should be designed considering various aspects such as probe length, GC content, molar concentrations, self-hybridization possibilities, and a limit on the number of single-nucleotide repeats [60, 63]. The melting temperature, secondary structure, and binding position in the target DNA are factors that can affect the signal intensity, specificity, and sensitivity [66, 71]. Typical parameters for such DNA probes include the following: minimum length of 35 and maximum up to 40 bases, melting temperature minimum (T min) of 70 °C and maximum (T max) of 75 °C, and GC content of 45–50 % [86, 92, 93].
Exceptional to the limits of DNA-DNA hybridization models, determining the array equal to the most favorable DNA-DNA duplex on a microarray is sturdy [69, 116]. Computationally, the best possible arrangement among two DNA sequences should be clearly described in terms of a generalized, accurate distance algorithm [19, 94, 123]. Enormously set, the accurate impassiveness between two sequences correspond to the whole number of insertions, deletions, and substitutions that are needed to transform one sequence into the other. From the standpoint of DNA cross-hybridization, a substitution corresponds to a mismatched pair of nucleotides whereas insertions/deletions correspond to gaps in the duplex DNA–DNA [23, 26]. The lower the number of mismatches and gaps in the alignment, the smaller the edit distance. On the other hand, exact area does not make available enough information with regards to the effectiveness of hybridization [46, 127].
Oligonucleotide probes, are usually much shorter (9–24-mer) and are often customized to integrate an amine or thiol linker that allow covalent attachment of the oligonucleotide to a covered glass face [71, 86, 120, 121]. The alterations of probes put in substantial expense to array construction [86]. For illustration, unchanged oligonucleotide probes are able to be balanced in alkaline buffer (pH 12) and put down instinctively on to acid-washed slides; they stick on to the slide face via hydrogen bonds and electrostatic attraction and are then accessible to form duplexes with corresponding strands of target DNA [93]. This addition scheme is vigorous transversely a broad range of temperature (4–95 °C) pH (1–10), and ionic buffers (e.g., 0–4 M NaCl) [116]. The compassion of recognition can be improved if acid-washed slides are coated with epoxy-silane before probe deposition [19, 112]. Underprivileged eminence slides have rough float up and may well auto-fluorescence, thereby producing background signal that interferes with spot finding and quantification [23, 124]. Auto-fluorescence can be predominantly challenging at what time signal strength is short, since is the case with expression arrays. To keep away from these harms, premium ready slides are available [19, 66].
RNA Quantification
The RNA concentration can be used to determine the amount of sample lost while cleaning using a kit. Transcribed viral nucleic acid may be purified by using a kit or manually, and quantified by measurement of the optical absorbance at 260 nm [50, 61, 97]. The concentration of RNA in the crude transcription product is calculated beforehand [128, 129].
Many post-PCR applications require removal of unincorporated primers, primer–dimers, and other reaction components from the PCR product. Traditional purification methods such as ethanol precipitation are difficult to automate. The clean-up system can be automated to purify 96 or 384 reactions in less time. PCR clean-up is especially amenable to automation because no vacuum or centrifugation steps are required, in contrast to many filter-based methods [5, 11, 26, 31].
Sample Amplification and Labeling
Reverse-transcription (RT) reactions may be prepared using a reasonable quantity (40–200 ng) of total RNA or random hexamers/gene-specific primers (Fig. 2). The mixture is heated and immediately cooled on ice before addition of dithiothreitol, dNTPs/aa-dUTP (a mixture of dGTP, dATP, dCTP, dTTP, and aminoallyl-dUTP), Superscript III reverse transcriptase, and first-strand buffer. The mixture can then be incubated, followed by the application of specific reaction conditions [3, 20, 24].
Fig. 2.
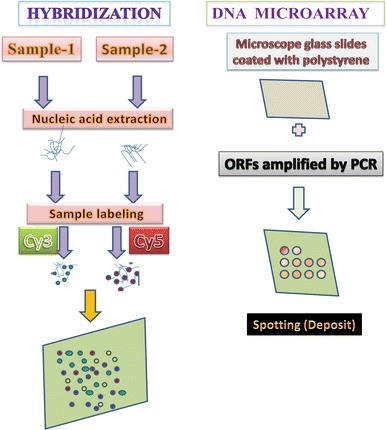
Principle of microarray assay for gene expression. ORFs open reading frames
cDNA from cells under two different conditions is extracted and labeled with two different fluorescent labels, for example, a green dye (cyanine 3) for cells for condition 1 and a red dye (cyanine 5) for cells at condition 2 (to be more accurate, the labeling is typically done by synthesizing single-strand DNAs that are complementary to the extracted mRNA using an enzyme called reverse transcriptase) [34, 41, 48]. Both extracts are washed over the microarray. Labeled gene products from the extracts hybridize to their complementary sequences at the spots due to their preferential binding; complementary single-strand nucleic acid sequences tend to attract each other, and the longer the complementary parts, the stronger the attraction [61, 75, 79] .
Hybridization
Samples for determination probably include DNA from a number of species, at diverse concentrations depending on the relative species abundances. A microarray protocol for quantitative evaluation of species diversity has not yet been developed. However, a universal significance of microarray hybridization in area of genomic transcription levels take on vigorous hybridization of two differentially labeled samples of cDNA to the same microarray slide [82, 114]. The main principle behind microarray technology is hybridization between two DNA strands, i.e., the property of complementary nucleic acid sequences to specifically pair with each other by forming hydrogen bonds between complementary nucleotide base pairs [79]. A greater number of complementary base pairs in a nucleotide sequence means tighter noncovalent bonding between the two strands. After washing off nonspecific bonding sequences, only strongly paired strands will remain hybridized [82, 83]. So, fluorescently labeled target sequences that bind to a probe sequence generate a signal that depends on the strength of the hybridization as determined by the number of paired bases, hybridization conditions (such as temperature), and washing after hybridization. The total strength of the signal from a spot (feature) depends on the amount of target sample bound to the probes present at that spot [114, 115]. Microarrays use relative quantization in which the intensity of a feature is compared with the intensity of the same feature under a different condition, while the identity of the feature is determined by its position [43]. An alternative to microarrays is serial analysis of gene expression, where the transcriptome is sequenced, allowing absolute measurements [54]. This allows quantitative discrimination between transcription levels in two samples when the relative variation is greater than about twofold [2, 20]. Using the same technique, it should be possible to quantitate >2-fold differences in species abundance between two samples, allowing rapid and sensitive examination of differences in species abundance [8, 10, 22].
Microarray Imaging
Microarray images are acquired by a laser scanner that executes a regional scan of the slide and creates a digital map or image for each dye, showing the fluorescent intensities for each pixel [24, 45]. For a particular microarray examination, the scanner generates two 16-bit tagged image file (TIFF) layouts, one for each fluorescent dye [57, 125]. Dissimilar dyes absorb and emit light at different wavelengths [115, 116]. To quantify the amount of each of the two fluorescent dyes at each spot, the scanner applies light excitation at the different wavelengths and measures at the different emission wavelengths [44, 48]. The dyes used usually are Cy3 and Cy5, with emission in the ranges of 510–550 nm and 630–660 nm, respectively [51, 54]. These dyes enable measurement of the amount of sample bound to a spot based on the level of fluorescence emitted when excited by the laser. If the RNA from the sample in condition 1 is abundant, the spot will be green, whereas if the RNA from the sample in condition 2 is abundant, the spot will be red. If both are equal, the spot will be yellow, while if neither is present it will not fluoresce and will appear black [58, 75]. Thus, from the fluorescence intensities and colors for each spot, the relative expression levels of the genes in both samples can be estimated.
A number of conditions [e.g., scan rate, laser power, photomultiplier tube (PMT) voltage] can be adjusted by the user at the time of scanning [24, 80, 103]. New, higher-power lasers provide additional photons for excitation and generate higher signal and noise [58, 75]. Higher PMT voltage results in greater amplification of photons to electrons and generates more detector noise and signal [81, 129].
It is preferable to employ higher laser fluence rather than higher PMT voltage, as this stimulates more signal photons rather than producing more signal per photon [87, 127]. However, elevated laser power can destroy hybridized samples through photobleaching, and depending on the number of scans to be performed on each sample, the laser power needs to be adjusted accordingly [63, 64].
Image quantification limitations (e.g., adaptive, fixed sphere, spot distance) should be carefully assessed and determined for each project as a whole, also depending on the array design, slide type, and spot morphology [8, 89, 106]. It should be noted that the image quantification method should be identical for all slides in a project, whereas the image acquisition parameters, for instance, laser power and/or photomultiplier parameters, can be optimized from slide to slide [9, 10].
Commonly designed for each slide a secure alignment of laser power and PMT is chosen though scanning [23]. The choice of these two parameters is finally determined so that almost all expression on the chip can be captured [37, 90]. However, it has been observed that not all genes spotted onto a chip can be measured accurately for a single scanner setting [44], as there may be genes with expression of 50,000 or more to genes with expression as low as 200 or even less [78]. The choice of these two parameters is finally determined so that almost all expression on the chip can be captured [105]. However, it has been observed that not all the genes spotted on the chip can be measured accurately for a single scanner setting [107]. There are genes with expression ranging from 50,000 or more to genes with expression as low as 200 or even less [115]. Such a wide range of expression is impossible to capture accurately in a single scan with a fixed setting [116]. However, a single scan with particular PMT and laser settings is certainly suitable for most, but certainly not all, of the intensity range [8–10]. Thus, there is a need to capture various ranges of gene expression values and then combine the information from all scans before further analysis is carried out [37, 44, 78, 107].
Data Analysis
The probes represent (partial) genomic sequences of a gene, positioned or fixed onto a glass slide, whereas the aim is the surveys of gene expression, in a highly parallel and comprehensive manner [16, 18, 128]. Probe variants also provide discrete, dissimilar nucleotide sequences corresponding to the same gene [10]. Signal potency is commonly sequence dependent. For this reason, averaging of the indicated intensities is not appropriate, and probe variants should be investigated separately in the final data evaluation steps. Probe copies exhibit similar response for various instances on the chip [23]. In theory, they should exhibit the same expression, being included for signal amplification. Confidence in the reliability of gene investigation is based on authentication research [17, 25]. Probe replicates are mainly developed based on two important approaches, as described below [74].
Our preferred option to specify a sufficient number of biological samples per condition is to determine the core value for each replicated set of genetic material probes on a chip and use this as the “true value” for that gene probe [66]. The median provides a robust measure and its determination inherently ignores outlier values within a replicate set, in contrast to the arithmetic mean [30, 58].
An alternative is to consider all replica values in the considered dataset, e.g., to apply analysis of variance (ANOVA)-type methods afterwards [53, 54]. Investigational inaccuracy is then pooled with genetic variation. Microarray data may require further processing aimed at reducing the dimensionality of the data to aid comprehension and more focused analysis for each gene [123, 124]. Other methods permit analysis of data consisting of a low number of biological or technical replicates [24, 37].
Data Normalization
Normalization is an important process in the investigation of DNA microarrays when evaluating data from diverse arrays or color controls [36, 38]. Analysis of microarrays can be scientifically influenced by various effects such as nucleic acid extraction, cDNA preparation, sample labeling, assimilation, imaging, and spot detection. In addition, there are effects that are exceptional to individual arrays, such as special effects of various probes, spotting effects, region effects, and pin effects. Normalization endeavors to compensate for such effects through use of internal controls [51–53].
The statistical analysis begins with the scanning file itself. Various parameters for the distribution of the pixels in a particular spot are given likes mean, mode, median, standard deviation and the main part correct reserved to elucidate the potency assessment of a précised spot in both channels [10, 109]. The scanned files provide the position values for the center intensity of both channels and their background [20]. The background noise measures the intensity of the mRNA for the slide even if no material was spotted [23]. Using all this information go to the next step, those spots with disgusting excellence that should not be used for advance analysis [34, 37]. The different incorporation properties of the dyes and their different physical characteristics make this the most important source of systematic error in two-color microarrays [38, 42]. The difference in overall intensity between different arrays can be due to real biological variation from one condition to another or just experimental noise [84]. The different expression level of a particular gene in a particular array can also be due to biological variability of the gene or to some noise [37]. If the overall intensity of the hybridized samples is different, this may also be due to some experimental error or to real biological activity [48]. This factor is therefore important in the choice of the within-array normalization method. Besides these factors, it must also be considered that some part of the probe will attach to the slide even when there is no spotted material, thereby contributing to the foreground intensity [44]. However, a reliable estimator for background intensity has not yet been provided. The data should be normalized sequentially to eliminate all nonbiological variation introduced by the investigational procedure and to facilitate assessment of the intensity values contained across slides [24].
Analysis
A brief outline of the initial data analysis of the absolute expression levels within an experiment is shown in Fig. 3. For microarray projects that are designed to study defined gene pathways and interactions, a maximum of annotation and statistical reliability is required [40, 65]. We suggest that the minimum result set for each gene should include: fold-changes of mean expression level per condition and P values from significance testing [24].
Fig. 3.
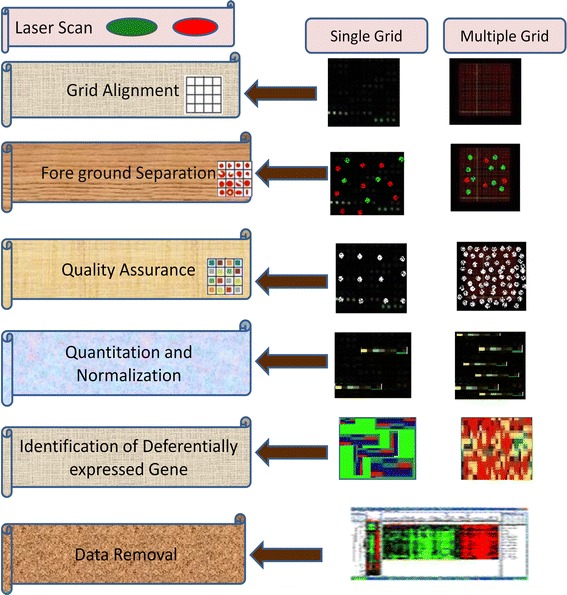
Schematic flow diagram of bioinformatics in microarray development
Data Visualization
Data points for biological chip replicates are usually the mean values of each gene for a given condition. Limited subsets of interesting genes can obviously be plotted by means of simple vertical bar charts of log(ratio) or absolute values [53]. This can be complemented by custom, project-dependent graphs, often integrating annotation data with gene expression results. Expression profiles of genes within one condition or of each gene across a number of conditions can be subjected to cluster analysis [99].
Condition Means and Confidence Intervals
These parameters are required to present the expression level of a gene, and enable better interpretation of fold-changes or variation. In most dual-dye experiments, confidence intervals can be calculated for the fold-change itself [48, 51, 53].
Calculation of 95 % confidence intervals (CI) for means can be done in various ways. If there are sufficient numbers of observations for each condition, the common formula based on a t-distribution can be used [91]. Broadly speaking, the values making up both groups of observations are randomly reassigned to each group a large number of times, with the desired statistic (mean, CI95, t, etc. [97, 98]) being calculated for each such similar run [107]. The accumulated set of newly generated statistics is then used to “estimate” the corresponding parameter for the original data [125].
Significance Tests
A simple two-sample t test or Welch t test is often the first tool of choice for statistical inference. Adjustment for multiple testing changes the obtained P values but not the order of sorted significance values [108]. If two conditions can be assumed to be dependent (e.g., cell lines) then paired t tests can increase the statistical power [119]. Nonparametric testing (e.g., using the Mann–Whitney U test) is an alternative with less power that nonetheless works better under the assumption that the underlying distributions are nonsymmetrical. However, for the very small numbers of observations (i.e., 5–7) typically available in microarray studies, the resulting P values can be less useful as a filtering tool [72]. Statistical power can generally be improved by employing bootstrap versions of significance tests. For most microarray studies, P values resulting from significance testing must be interpreted with care in cases with few biological replicates per group [8, 10, 98].
ANOVA-type methods [53] are somewhat more involved, being appropriate where there is more than one experimental factor under investigation (e.g., treatment and dose, or biological replicates and hybridization replicates) [84]. It is important to note that the expression of individual genes of interest is usually backed up by verification using other techniques such as RT-PCR, in situ hybridization, and Northern blotting [31, 48, 66].
Explorative Methods
Explorative methods can be used to identify genes or samples with similar expression profiles, indicating coregulation or sample type, respectively [84, 91]. If coregulation or time effects are of interest, (graphical) principal component analysis can be used to assess the number of clusters that may be contained within the data, which can then serve as the input parameter for the number of expected clusters in a K-means or self-organizing map (SOM) clustering approach [127]. Because of the nature of explorative methods, we recommend using several combinations of algorithms and distance measures (SOM and hierarchical, both with Pearson correlation and Euclidean distance as a minimum) to highlight different features in the data [68].
Conclusions
Available techniques to screen a broad range of viruses are intrinsically biased and thereby constrained to detection of a restricted number of candidate viruses. To overcome this difficulty, an approach to widen viral recognition based on a combination of viral genomics and long-oligonucleotide microarray technology is required. To accomplish this objective, extremely conserved nucleotide sequences within a viral family can be selected for presentation on the microarray. By using these most conserved sequences, it is hoped to maximize the possibility of detecting all members of each viral family, as well as unsequenced, unknown, or newly evolved family members. A secondary, but corresponding, ambition is to take advantage of the high resolution of microarray hybridization to distinguish among viral subtypes, which is a complex and difficult task with conventional methods.
References
- 1.Afrakhte M, Schultheiss TM. Construction and analysis of a subtracted library and microarray of cDNAs expressed specifically in chicken heart progenitor cells. Dev Dynam. 2004;230:290–298. doi: 10.1002/dvdy.20059. [DOI] [PubMed] [Google Scholar]
- 2.Alizadeh A, Eisen M, Davis RE, Ma C, Sabet H, Tran T, Powell JI, Yang L. The lymphochip: a specialized cDNA microarray for the genomic-scale analysis of gene expression in normal and malignant lymphocytes. Cold Spring Harb Symp Quant Biol. 1999;64:71–78. doi: 10.1101/sqb.1999.64.71. [DOI] [PubMed] [Google Scholar]
- 3.Alizadeh AA, Eisen MB, Davis R, Ma EC, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 4.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 5.Anonymous (March 31, 2006) Posting date. Biological diagnostics manufacturing: request for information. U.S. Department of Health and Human Services Solicitation 2006-Q-08478. http://www.fbo.gov/spg/HHS/CDCP/PGOA/2006%2DQ%2D08478/SynopsisR.html. Accessed 2 Dec 2011
- 6.Bellau-Pujol S, Vabret A, Legrand L, Dina J, Gouarin S, Petitjean- Lecherbonnier J, Pozzetto B, Ginevra C, Freymuth F. Development of three multiplex RT-PCR assays for the detection of 12 respiratory RNA viruses. J Virol Methods. 2005;126:53–63. doi: 10.1016/j.jviromet.2005.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 8.Berthet N, Leclercq I, Dublineau A, Shigematsu S, Burguiere AM, et al. High-density resequencing DNA microarrays in public health emergencies. Nat Biotechnol. 2010;28:25–27. doi: 10.1038/nbt0110-25. [DOI] [PubMed] [Google Scholar]
- 9.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, et al. Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 10.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert CJ, Aach Ansorge W, et al. Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 11.Briese T, Palacios G, Kokoris M, Jabado O, Liu Z, Renwick N, Kapoor V, Casas I, Pozo F, Limberger R, Perez-Brena P, Ju J, Lipkin WI. Diagnostic system for rapid and sensitive differential detection of pathogens. Emerg Infect Dis. 2005;11:310–313. doi: 10.3201/eid1102.040492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Briese T, Paweska JT, McMullan LK, Hutchison SK, Street C, Palacios G, Khristova ML, Weyer J, Swanepoel R, Egholm M, Nichol ST, Lipkin WI. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brownie J, Shawcross S, Theaker J, Whitcombe D, Ferrie R, Newton C, Little S. The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res. 1997;25:3235–3241. doi: 10.1093/nar/25.16.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brust S, Duttmann H, Feldner J, Gurtler L, Thorstensson R, Simon F. Shortening of the diagnostic window with a new combined HIV p24 antigen and anti-HIV-1/2/O screening test. J Virol Methods. 2000;90:153–165. doi: 10.1016/S0166-0934(00)00229-9. [DOI] [PubMed] [Google Scholar]
- 15.Bryant PA, Venter D, Robins-Browne R, Curtis N. Chips with everything: DNA microarrays in infectious diseases. Lancet Infect Dis. 2004;4:100–111. doi: 10.1016/S1473-3099(04)00930-2. [DOI] [PubMed] [Google Scholar]
- 16.Buckley MJ (2000) Spot user’s guide. CSIRO Mathematical and Information Sciences, Sydney, Australia. http://www.cmis.csiro.au/iap/Spot/spotmanual.htm. Accessed 5 Jan 2012
- 17.Call DR, Bakko MK, Krug MJ, Roberts MC. Identifying antimicrobial resistance genes with DNA microarrays. Antimicrob Agents Chemother. 2003;47:3290–3295. doi: 10.1128/AAC.47.10.3290-3295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Call DR, Borucki MK, Loge FJ. Detection of bacterial pathogens in environmental samples using DNA microarrays. J Microbiol Methods. 2003;53:235–243. doi: 10.1016/S0167-7012(03)00027-7. [DOI] [PubMed] [Google Scholar]
- 19.Callow MJ, Dudoit S, Gong EL, Speed TP, Rubin EM. Microarray detection and characterization of Plum pox virus: serological methods. EPPO Bull. 2000;36:254–261. [Google Scholar]
- 20.Candotti D, Richetin A, Cant B, Temple J, Sims C, Reeves I, Barbara JAJ, Allain JP. Evaluation of a transcription-mediated amplification-based HCV and HIV-1 RNA duplex assay for screening individual blood donations: a comparison with a minipool testing system. Transfusion. 2003;43:215–225. doi: 10.1046/j.1537-2995.2003.00308.x. [DOI] [PubMed] [Google Scholar]
- 21.Chen SH, Lin CY, Cho CS, Lo CZ, Hsiung CA. Primer design assistant (PDA): a web-based primer design tool. Nucleic Acids Res. 2003;31:3751–3754. doi: 10.1093/nar/gkg560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu CY, Greninger AL, Chen EC, Haggerty TD, Parsonnet J, Delwart E, Derisi JL, Ganem D. Cultivation and serological characterization of a human Theiler’s-like cardiovirus associated with diarrheal disease. J Virol. 2010;84:4407–4414. doi: 10.1128/JVI.02536-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chizhikov V, Rasooly A, Chumakov K, Levy DD. Microarray analysis of microbial virulence factors. Appl Environ Microbiol. 2001;67:3258–3263. doi: 10.1128/AEM.67.7.3258-3263.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chizhikov V, Wagner M, Ivshina A, Hoshino Y, Kapikian AZ, Chumakov K. Detection and genotyping of human group rotaviruses by oligonucleotide microarray hybridization. J Clin Microbiol. 2002;40:2398–2407. doi: 10.1128/JCM.40.7.2398-2407.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chuaqui RF, Bonner RF, Best CJM, Gillespie JW, Flaig MJ, Hewitt SM, Phillips JL, Krizman DB, Tangrea MA, Ahram M, et al. Post-analysis follow-up and validation of microarray experiments. Nat Genet. 2001;32:509–514. doi: 10.1038/ng1034. [DOI] [PubMed] [Google Scholar]
- 26.Churchill G. Fundamentals of experimental design for cDNA microarrays. Nat Genetics Suppl. 2002;32:490–495. doi: 10.1038/ng1031. [DOI] [PubMed] [Google Scholar]
- 27.Coiras MT, Aguilar JC, Garcia ML, Casas I, Perez-Brena P. Simultaneous detection of fourteen respiratory viruses in clinical specimens by two multiplex reverse transcription nested-PCR assays. J Med Virol. 2004;72:484–495. doi: 10.1002/jmv.20008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coiras MT, Lopez-Huertas MR, Lopez-Campos G, Aguilar JC, Perez-Brena P. Oligonucleotide array for simultaneous detection of respiratory viruses using a reverse-line blot hybridization assay. J Med Virol. 2005;76:256–264. doi: 10.1002/jmv.20350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coiras MT, Lopez-Huertas G, Lopez-Campos JC, Aguilar P, Perez B. Oligonucleotide array for simultaneous detection of respiratory viruses using a reverse-line blot hybridization assay. J Med Virol. 2005;76:256–264. doi: 10.1002/jmv.20350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colantuoni C, Henry G, Zeger S, Pevsner J. Local means normalization of microarray element signal intensities across an array surface: quality control and correction of spatially systematic artifacts. Biotechniques. 2002;32:1316–1320. doi: 10.2144/02326mt02. [DOI] [PubMed] [Google Scholar]
- 31.Conejero-Goldberg C, Wang E, Yi C, Goldberg TE, Jones-Brando L, Marincola FM, Webster MJ, Torrey EF. Infectious pathogen detection arrays: viral detection in cell lines and postmortem brain tissue. Biotechniques. 2005;39:741–751. doi: 10.2144/000112016. [DOI] [PubMed] [Google Scholar]
- 32.Corless CE, Guiver M, Borrow R, Edwards-Jones V, Fox AJ, Kaczmarski EB. Simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in suspected cases of meningitis and septicemia using real-time PCR. J Clin Microbiol. 2001;39:1553–1558. doi: 10.1128/JCM.39.4.1553-1558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dacheux L, Berthet N, Dissard G, Holmes EC, Delmas O, et al. Application of broad-spectrum resequencing microarray for genotyping rhabdoviruses. J Virol. 2010;84:9557–9574. doi: 10.1128/JVI.00771-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davignon L, Walter EA, Mueller KM, Barrozo CP, Stenger DA, Lin B. Use of resequencing oligonucleotide microarrays for identification of Streptococcus pyogenes and associated antibiotic resistance determinants. J Clin Microbiol. 2005;43:5690–5695. doi: 10.1128/JCM.43.11.5690-5695.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong F, Allawi HT, Anderson T, Neri BP, Lyamichev VI. Secondary structure prediction and structure-specific sequence analysis of single-stranded DNA. Nucleic Acids Res. 2001;29:3248–3257. doi: 10.1093/nar/29.15.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dudoit S, Yang YH, Callow MJ, Speed TP. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Science. 2000;283:83–87. [Google Scholar]
- 37.Dunbar SA. Applications of Luminex xMAP technology for rapid, high-throughput multiplexed nucleic acid detection. Clin Chim Acta. 2006;363:71–82. doi: 10.1016/j.cccn.2005.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eisen MB, Brown PO. DNA arrays for analysis of gene expression. Methods Enzymol. 1999;303:179–205. doi: 10.1016/S0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- 39.Epstein CB, Hale W, IV, Butow RA. Numerical methods for handling uncertainty in microarray data: an example analyzing perturbed mitochondrial function in yeast. Methods Cell Biol. 2001;65:439–452. doi: 10.1016/S0091-679X(01)65026-X. [DOI] [PubMed] [Google Scholar]
- 40.Feezor R, Baker HV, Mindrinos M, Hayden D, Tannahill CL, Brownstein BH, Fay A, MacMillan S, Laramie J, Xiao W, Moldawer LL, Cobb JP, Laudanski K, Miller-Graziano CL, Maier RV, Schoenfeld D, Davis RW, Tompkins RG. Whole blood and leukocyte RNA isolation for gene expression analyses. Physiol Genomics. 2004;19:247–254. doi: 10.1152/physiolgenomics.00020.2004. [DOI] [PubMed] [Google Scholar]
- 41.Fejzo M, Slamon DJ. Frozen tumor tissue microarray technology for analysis of tumor RNA, DNA, and proteins. Am J Pathol. 2001;159:1645–1650. doi: 10.1016/S0002-9440(10)63011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Field LA, Jordan RM, Hadix JA, Dunn MA, Shriver CD, Ellsworth RE, Ellsworth DL. Functional identity of genes detectable in expression profiling assays following globin mRNA reduction of peripheral blood samples. Clin Biochem. 2007;40:499–502. doi: 10.1016/j.clinbiochem.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Gentit P. Detection of plum pox virus: biological methods. EPPO Bull. 2006;36:251–253. doi: 10.1111/j.1365-2338.2006.00982.x. [DOI] [Google Scholar]
- 44.Ghedin E, Pumfery A, de la Fuente C, Miller Yao K, Lacoste N, Quackenbush V, Jacobson JS, Kashanchi F. Use of a multi-virus array for the study of human pathogens: gene expression studies and ChIP-chip analysis. Retrovirology. 2004;1:10. doi: 10.1186/1742-4690-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glasbey CA, Ghazal P. Combinatorial image analysis of DNA microarray features. Bioinformatics. 2003;19:194–203. doi: 10.1093/bioinformatics/19.2.194. [DOI] [PubMed] [Google Scholar]
- 46.Goodman N. Biological data becomes computer literate: new advances in bioinformatics. Curr Opin Biotechnol. 2002;13:68–71. doi: 10.1016/S0958-1669(02)00287-2. [DOI] [PubMed] [Google Scholar]
- 47.Gorelenkov V, Antipov A, Lejnine S, Daraselia N, Yuryev A set of novel tools for PCR primer design. Biotechniques. 2001;31:1326–1330. doi: 10.2144/01316bc04. [DOI] [PubMed] [Google Scholar]
- 48.Grace MB, McLeland CB, Gagliardi SJ, Smith JM, Jackson WE, III, Blakely WF. Development and assessment of a quantitative reverse transcription-PCR assay for simultaneous measurement of four amplicons. Clin Chem. 2003;49:1467–1475. doi: 10.1373/49.9.1467. [DOI] [PubMed] [Google Scholar]
- 49.Greenberg SA. DNA microarray gene expression analysis technology and its application to neurological disorders. Neurology. 2001;57:755–761. doi: 10.1212/WNL.57.5.755. [DOI] [PubMed] [Google Scholar]
- 50.Haan CA, Stadler K, Godeke GJ, Bosch BJ, Rottier PJ. Cleavage inhibition of the murine coronavirus spike protein by a furinlike enzyme affects cell–cell but not virus-cell fusion. J Virol. 2004;78(11):6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardegger D, Nadal D, Bossart W, Altwegg M, Dutly F. Rapid detection of Mycoplasma pneumoniae in clinical samples by real-time PCR. J Microbiol Methods. 2000;41:45–51. doi: 10.1016/S0167-7012(00)00135-4. [DOI] [PubMed] [Google Scholar]
- 52.Hegde P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, Hughes JE, Snesrud E, Lee N, Quackenbush J. A concise guide to cDNA microarray analysis. Biotechniques. 2000;29:548–562. doi: 10.2144/00293bi01. [DOI] [PubMed] [Google Scholar]
- 53.Ji H, Masse N, Tyler S, Liang B, Li Y, Merks H, Graham M, Sandstrom P, Brooks J. HIV drug resistance surveillance using pooled pyrosequencing. PLoS ONE. 2010;5:e9263. doi: 10.1371/journal.pone.0009263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kerr K, Churchill G. Statistical design and the analysis of gene expression microarray data. Genet Res. 2001;77:123–128. doi: 10.1017/s0016672301005055. [DOI] [PubMed] [Google Scholar]
- 55.Kim H, Zhao B, Snesrud EC, Haas BJ, Town CD, Quackenbush J. Use of RNA and genomic DNA references for inferred comparisons in DNA microarray analyses. Biotechniques. 2002;33:924–930. doi: 10.2144/02334mt06. [DOI] [PubMed] [Google Scholar]
- 56.Lai MM, Brayton PR, Armen RC, Patton CD, Pugh C, Stohlman SA. Mouse hepatitis virus A59: mRNA structure and genetic localization of the sequence divergence from hepatotropic strain MHV-3. J Virol. 1981;39:823–834. doi: 10.1128/jvi.39.3.823-834.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Landraud L, Brisse S (2010) Enterobacteriaceae. In: Cohen J, Powderly WG (eds) Infectious diseases, 3rd edn. Mosby Elsevier, London, pp 1690–1703
- 58.Leong WF, Tan H, Ooi C, Koh EE, Chow DR. Microarray and real-time RT-PCR analyses of differential human gene expression patterns induced by severe acute respiratory syndrome (SARS) coronavirus infection of Vero cells. Microbes Infect. 2005;7:248–259. doi: 10.1016/j.micinf.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leski TA, Lin B, Malanoski AP, Wang Z, Long NC. Testing and validation of high density resequencing microarray for broad range biothreat agents detection. PLoS ONE. 2009;4:e6569. doi: 10.1371/journal.pone.0006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J, Chen S, Evans DH. Typing and subtyping influenza virus using DNA microarrays and multiplex reverse transcriptase PCR. J Clin Microbiol. 2001;39:696–704. doi: 10.1128/JCM.39.2.696-704.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, He Z, Zhou J. Selection of optimal oligonucleotide probes for microarrays using multiple criteria, global alignment and parameter estimation. Nucleic Acids Res. 2005;33:6114–6123. doi: 10.1093/nar/gki914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin B, Blaney KM, Malanoski AP, Ligler AG, Schnur JM, Metzgar D, Russell KL, Stenger DA. Rapid testing for over 20 respiratory pathogens simultaneously in clinical samples using resequencing arrays. Washington, DC: Naval Research Laboratory; 2006. [Google Scholar]
- 63.Lin BZ, Wang GJ, Vora JA, Thornton JM, Schnur DC, Thach KM, Blaney AG, Ligler AP, Malanoski, Santiago J, Walter EA, Agan BK, Metzgar D, Seto D, Daum LT, Kruzelock R, Rowley RK, Hanson EH, Tibbetts C, Stenger DA. Broad-spectrum respiratory tract pathogen identification using resequencing DNA microarrays. Genome Res. 2006;16:527–535. doi: 10.1101/gr.4337206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lipshutz RJ, Fodor SP, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genet. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- 65.Lönnstedt I, Speed TP. Replicated microarray data. Stat Sinica. 2002;12:31–46. [Google Scholar]
- 66.Madhi SA, Klugman KP. A role for Streptococcus pneumonia in virus-associated pneumonia. Nat Med. 2004;10:811–813. doi: 10.1038/nm1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malanoski AP, Lin B, Wang Z, Schnur JM, Stenger DA. Automated identification of multiple micro-organisms from resequencing DNA microarrays. Nucleic Acids Res. 2006;34:5300–5311. doi: 10.1093/nar/gkl565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martinez MJ, Aragon AD, Rodriguez AL, Weber JA, Timlin MB, Sinclair DM, Haaland DM, Werner-Washburne M (2003) Identification and removal of contaminating fluorescence from commercial and in-house printed DNA microarrays. Nucleic Acids Res. 31:e18 [DOI] [PMC free article] [PubMed]
- 69.McDonough EA, Barrozo CP, Russell KL, Metzgar D. A multiplex PCR for detection of Mycoplasma pneumoniae, Chlamydophila pneumoniae, Legionella pneumophila, and Bordetella pertussis in clinical specimens. Mol Cell Probes. 2005;19:314–322. doi: 10.1016/j.mcp.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 70.Mehlmann M, Townsend MB, Stears RL, Kuchta RD, Rowlen KL. Optimization of fragmentation conditions for microarray analysis of viral RNA. Anal Biochem. 2005;347:316–323. doi: 10.1016/j.ab.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 71.Mehlmann M, Dawson ED, Townsend MB, Smagala JA, Moore CM, Smith CB, Cox NJ, Kuchta RD, Rowlen KL. Robust sequence selection method used to develop the FluChip diagnostic microarray for influenza virus. J Clin Microbiol. 2006;44:2857–2862. doi: 10.1128/JCM.00135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 73.Metzgar D, Myers CA, Russell KL, Faix D, Blair PJ, et al. Single assay for simultaneous detection and differential identification of human and avian influenza virus types, subtypes, and emergent variants. PLoS ONE. 2010;5:e8995. doi: 10.1371/journal.pone.0008995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MGED—Microarray gene expression data society home. http://www.mged.org. Accessed 6 Jan 2012
- 75.Mills J, Roth KA, Cagan RL, Gordon JI. DNA microarrays and beyond: completing the journey from tissue to cell. Nat Cell Biol. 2001;3(E):175–178. doi: 10.1038/35087108. [DOI] [PubMed] [Google Scholar]
- 76.Molling P, Jacobsson S, Backman A, Olcen P. Direct and rapid identification and genogrouping of meningococci and porA amplification by LightCycler PCR. J Clin Microbiol. 2002;40:4531–4535. doi: 10.1128/JCM.40.12.4531-4535.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Molling P, Jacobsson S, Backman A, Olcen P. Direct and rapid identification and genogrouping of meningococci and porA amplification by LightCycler PCR. J Clin Microbiol. 2002;40:4531–4535. doi: 10.1128/JCM.40.12.4531-4535.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muller RA, Ditzen K, Hille M, Stichling R, Ehricht T, Illmer G, Ehninger J. Detection of herpesvirus and adenovirus coinfections with diagnostic DNA-microarrays. J Virol Methods. 2008;155:161–166. doi: 10.1016/j.jviromet.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 79.Murphy D. Gene expression studies using microarrays: principles, problems, and prospects. Adv Physiol Educ. 2002;26:256–270. doi: 10.1152/advan.00043.2002. [DOI] [PubMed] [Google Scholar]
- 80.Natarajan K, Shepard LA, Chodosh J. The use of DNA array technology in studies of ocular viral pathogenesis. DNA Cell Biol. 2002;21:483–490. doi: 10.1089/10445490260099782. [DOI] [PubMed] [Google Scholar]
- 81.Pellois X, Zhou O, Srivannavit T, Zhou E, Gulari X. Gao, individually addressable parallel peptide synthesis on microchips. Nat Biotechnol. 2002;20:922–926. doi: 10.1038/nbt723. [DOI] [PubMed] [Google Scholar]
- 82.Puppe W, Weigl JA, Aron G, Grondahl B, Schmitt HJ, Niesters HG, Groen J. Evaluation of a multiplex reverse transcriptase PCR ELISA for the detection of nine respiratory tract pathogens. J Clin Virol. 2004;30:165–174. doi: 10.1016/j.jcv.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 83.Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002;32:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- 84.Raj A, Oudenaarden A. Single-molecule approaches to stochastic gene expression. Ann Rev Biophys. 2009;38:255–270. doi: 10.1146/annurev.biophys.37.032807.125928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raychaudhuri S, Stuart JM, Altman RB (2000) Principal components analysis to summarize microarray experiments: application to sporulation time series. Pac Symp Biocomput 455–466 [DOI] [PMC free article] [PubMed]
- 86.Relman DA. The search for unrecognized pathogens. Science. 1999;284:1308–1310. doi: 10.1126/science.284.5418.1308. [DOI] [PubMed] [Google Scholar]
- 87.Rimour S, Hill D, Militon C, Peyret P. Go arrays: highly dynamicand efficient microarray probe design. Bioinformatics. 2005;21:1094–1103. doi: 10.1093/bioinformatics/bti112. [DOI] [PubMed] [Google Scholar]
- 88.Robb JA, Bond CW. Pathogenic murine coronaviruses. I. Characterization of biological behavior in vitro and virus-specific intracellular RNA of strongly neurotropic JHMV and weakly neurotropic A59V viruses. Virology. 1979;94:352–370. doi: 10.1016/0042-6822(79)90467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roepman P, Wessels LF, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P, Tilanus MG, Koole R, Hordijk GJ, van der Vliet PC, Reinders MJ, Slootweg PJ, Holstege FC. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet. 2005;37(2):182–186. doi: 10.1038/ng1502. [DOI] [PubMed] [Google Scholar]
- 90.Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 91.Rubina AY, Pan’kov SV, Dementieva EI, Pen’kov DN, Butygin AV, Vasiliskov VA, Chudinov AV, Mikheikin AL, et al. Hydrogel drop microchips with immobilized DNA: properties and methods for large-scale production. Anal Biochem. 2004;325:92–106. doi: 10.1016/j.ab.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 92.Sanchez A, Kondev J. Transcriptional control of noise in gene expression. Proc Natl Acad Sci USA. 2008;105:5081–5086. doi: 10.1073/pnas.0707904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, Kohn KW, Reinhold WC, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- 94.Shinde D, Lai Y, Sun F, Arnheim N. Taq DNA polymerase slippage mutation rates measured by PCR and quasi-likelihood analysis: (CA/GT)n and (A/T)n microsatellites. Nucleic Acids Res. 2003;31:974–980. doi: 10.1093/nar/gkg178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sickinger E, Stieler M, Kaufman B, Kapprell H-P, West D, Sandridge A, Devare S, Schochetman G, Hunt JC, Daghfal D, the AxSYM Clinical Study Group Multicenter evaluation of a new, automated enzyme-linked immunoassay for detection of human immunodeficiency virus-specific antibodies and antigen. J Clin Microbiol. 2004;42:21–29. doi: 10.1128/JCM.42.1.21-29.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Small J, Call DR, Brockman FJ, Straub TM, Chandler DP. Direct detection of 16S rRNA in soil extracts by using oligonucleotide microarrays. Appl Environ Microbiol. 2001;67:4708–4716. doi: 10.1128/AEM.67.10.4708-4716.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Stafford GFP, Brun M. Three methods for optimization of cross laboratory and cross-platform microarray expression data. Nucleic Acids Res. 2007;35:e72. doi: 10.1093/nar/gkl1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sobek J, Bartscherer K, Jacob A, Hoheisel JD, Angenendt P. Microarray technology as a universal tool for high-throughput analysis of biological systems. Comb Chem High Throughput Screen. 2006;9:365–380. doi: 10.2174/138620706777452429. [DOI] [PubMed] [Google Scholar]
- 99.Stabenau A, McVicker G, Melsopp C, Proctor G, Clamp M, Birney E. The Ensembl core software libraries. Genome Res. 2004;14:929–933. doi: 10.1101/gr.1857204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stafford P, Brun M. Three methods for optimization of crosslaboratory and cross- platform microarray expression data. Nucleic Acids Res. 2007;35(10):e72–140. doi: 10.1093/nar/gkl1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sterrenburg E, Turk R, Boer JM, van GB, Ommen den JT (2002) A common reference for cDNA microarray hybridizations. Nucleic Acids Res 30:e116 [DOI] [PMC free article] [PubMed]
- 102.Striebel HM, Birch-Hirschfeld E, Egerer R, Földes-Papp Z. Virus diagnostics on microarrays. Curr Pharm Biotechnol. 2003;4:401–415. doi: 10.2174/1389201033377274. [DOI] [PubMed] [Google Scholar]
- 103.Tan SL, Ganji G, Paeper B, Proll S, Katze MG. Systems biology and the host response to viral infection. Nat Biotechnol. 2007;25:1383–1389. doi: 10.1038/nbt1207-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tang P, Chiu C. Metagenomics for the discovery of novel human viruses. Futur Microbiol. 2010;5:177–189. doi: 10.2217/fmb.09.120. [DOI] [PubMed] [Google Scholar]
- 105.Tang BS, Chan KH, Cheng VC, Woo PC, Lau SK, Lam CC, Chan TL, Wu AK, Hung IF, Leung SY, Yuen KY. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. J Virol. 2005;79(10):6180–6193. doi: 10.1128/JVI.79.10.6180-6193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tembe W, Zavaljevski N, Bode E, Chase C, Geyer J, Wasieloski L, Benson G, Reifman J. Oligonucleotide fingerprint identification for microarray-based pathogen diagnostic assays. Bioinformatics. 2007;23:5–13. doi: 10.1093/bioinformatics/btl549. [DOI] [PubMed] [Google Scholar]
- 107.Trost E, Hackle H, Maurer M, Trajanoski Z. Java editor for biological pathways. Bioinformatics. 2003;19:786–787. doi: 10.1093/bioinformatics/btg052. [DOI] [PubMed] [Google Scholar]
- 108.Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH. Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001;29:2549–2557. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Van de Peppel J, Kemmeren P, Van B H, Radonjic M, Van L, Holstege D (2003) Monitoring global messenger RNA changes in externally controlled microarray experiments. EMBO Rep 4(4):387–393 [DOI] [PMC free article] [PubMed]
- 111.Varga A, James D. Detection and differentiation of plum pox virus using real-time multiplex PCR with SYBR green and melting curve analysis: a rapid method for strain typing. J Virol Methods. 2005;123:213–220. doi: 10.1016/j.jviromet.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 112.Vet JAM, Majithia AR, Marras SAE, Tyagi S, Dube, Poiesz SB, Krame JR. Multiplex detection of four pathogenic retroviruses using molecular beacons. Proc Natl Acad Sci USA. 1999;96:6394–6399. doi: 10.1073/pnas.96.11.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, Naeem A, Zaidi S, Delwart E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol. 2009;83:4642–4651. doi: 10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vinje J, Koopmans MP. Simultaneous detection and genotyping of “Norwalk-like” viruses by oligonucleotide array in a reverse line blot hybridization format. J Clin Microbiol. 2000;38:2595–2601. doi: 10.1128/jcm.38.7.2595-2601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wagner E, Garcia Ramirez JJ, Stingley SW, Aguilar SA, Buehler L, Devi-Rao GB, Ghazal P. Practical approaches to long oligonucleotide-based DNA microarray: lessons from herpesvirus. Prog Nucleic Acid Res. 2002;71:445–491. doi: 10.1016/S0079-6603(02)71048-9. [DOI] [PubMed] [Google Scholar]
- 116.Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, De Risi L. Microarray-based detection and genotyping of viral pathogens. Proc Natl Acad Sci USA. 2002;99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang D, Urisman A, Liu YT, Springer M, Ksiazek TG, Erdman DD, Mardis ER, Hickenbotham M, Magrini V, Eldred J, et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003;1(2):E2. doi: 10.1371/journal.pbio.0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang HY, Malek RL, Kwitek AE, Greene AS, Luu TV, Behbahani B, Frank B, Quackenbush J, Lee NH. Assessing unmodified 70-mer oligonucleotide probe performance on glass-slide microarrays. Genome Biol. 2003;4:R5. doi: 10.1186/gb-2003-4-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Z, Daum LT, Vora GJ, Metzgar D, Walter EA, Canas LC, Malanoski AP, Lin B, Stenger DA. Identifying influenza viruses with resequencing microarrays. Emerg Infect Dis. 2006;12:638–646. doi: 10.3201/eid1204.051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Weil MR, Macatee T, Garner HR. Toward a universal standard: comparing two methods for standardizing spotted microarraydata. Biotechniques. 2002;32:1310–1314. doi: 10.2144/02326mt01. [DOI] [PubMed] [Google Scholar]
- 121.Whelen AC, Persing DH. The role of nucleic acid amplification and detection in the clinical microbiology laboratory. Annu Rev Microbiol. 1996;50:349–373. doi: 10.1146/annurev.micro.50.1.349. [DOI] [PubMed] [Google Scholar]
- 122.Wilson KH, Wilson WJ, Radosevich JL, DeSantis TZ, Viswanathan VS, Kuczmarski TA, Andersen GL. High-density microarray of small-subunit ribosomal DNA probes. Appl Environ Microbiol. 2002;68:2535–2541. doi: 10.1128/AEM.68.5.2535-2541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wittberger D, Berens C, Hammann C, Westhof E, Schroeder R. Evaluation of uranyl photocleavage as a probe to monitor ion binding and flexibility in RNAs. J Mol Biol. 2000;300:339–352. doi: 10.1006/jmbi.2000.3747. [DOI] [PubMed] [Google Scholar]
- 124.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J. Speed TP: normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30(4):e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang IV, Chen E, Hasseman JP, Liang W, Frank BC, Wang S, Sharov V, Saeed AI, et al. Within the fold: assessing differential expression measures and reproducibility in microarray assays. Genome Biol. 2002;3:62.1–62.13. doi: 10.1186/gb-2002-3-11-research0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yee HY, Speed TP. Design issues for cDNA microarray experiments. Nat Rev Genet. 2002;3:579–588. doi: 10.1038/nrg863. [DOI] [PubMed] [Google Scholar]
- 128.Zammatteo N, Hamels S, De Longueville F, Alexandre I, Gala JL, Brasseur F, Remacle J. New chips for molecular biology and diagnostics. Biotechnol Annu Rev. 2002;8:85–101. doi: 10.1016/S1387-2656(02)08005-5. [DOI] [PubMed] [Google Scholar]
- 129.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]