

Gene expression and chromatin conformation reliably distinguish cell types, enabling equation-free control of human cells.
Abstract
The relationship between microscopic observations and macroscopic behavior is a fundamental open question in biophysical systems. Here, we develop a unified approach that—in contrast with existing methods—predicts cell type from macromolecular data even when accounting for the scale of human tissue diversity and limitations in the available data. We achieve these benefits by applying a k-nearest-neighbors algorithm after projecting our data onto the eigenvectors of the correlation matrix inferred from many observations of gene expression or chromatin conformation. Our approach identifies variations in epigenotype that affect cell type, thereby supporting the cell-type attractor hypothesis and representing the first step toward model-independent control strategies in biological systems.
INTRODUCTION
Genetically identical human cells are classified by their distinct behaviors into cell types, implying that nongenetic factors—including chromatin organization—contribute to their distinctive gene expression patterns. Being stably heritable through cell division, both chromatin organization and the unique pattern of gene expression are therefore epigenetic (1). Observing these epigenetic degrees of freedom, or epigenotype, of a wide variety of cells has become increasingly widespread thanks to technological advances in gene expression microarrays (2) and, more recently, genome-wide chromatin conformation capture (Hi-C, which measures the genome-wide frequency of physical contact between pairs of loci) (3) and RNA sequencing (RNA-seq) (4). Collections of data from these experiments are available in public databases, of which two especially large ones are the Gene Expression Omnibus (GEO) (5) and the Sequence Read Archive (SRA) (6). Yet, existing approaches remain too limited in scope to distinguish a large number of cell types on the basis of epigenotype, hampering the discovery of underlying physical principles that would facilitate manipulating cell behavior, cell reprogramming, and developing regenerative therapies. On the other hand, statistical physics (7) and nonlinear dynamics (8), combined with machine learning (9–11), offer a promising framework to determine cell type solely from macromolecular data.
Inferring cell type from epigenotype is a challenging problem, largely because the complexity and scale of the intracellular networks considered here (consisting of 104 to 106 genes or gene products) preclude the use of many existing approaches. These approaches range from direct simulation (12, 13) for networks smaller than 10 genes, to Boolean models (14, 15) for networks up to 100 genes, to nonlinear embedding methods (16, 17) for networks up to 1000 genes, to inverse Ising models (18, 19) for networks larger than 1000 genes. Inverse Ising models and other recent network identification approaches (20, 19) must contend with effective interactions between genes, such as those induced by the competition for cellular resources (21) and are sensitive to missing links in the reconstructed network when making predictions about cell behavior. On the other hand, approaches to predict the growth rate of microorganisms from gene expression (10, 11), and cell-fate decisions in mice from epigenetic markers (9), suggest that prediction of cell behavior from whole-cell measurements should be possible.
Here, motivated by these latter approaches, we present a data-driven approach that benefits from machine-learning techniques to infer cell type based on genome-wide observations of gene expression or chromatin conformation without the benefit of a network model. The gene-gene or contact-contact correlation matrices provide the structure of the data, obviating the need for an explicit network model. We show that our approach preserves cell type homogeneity (less variability exists within than between cell types), local consistency (states nearby measurements of a cell type likely belong to it), and data efficiency (state-space regions belonging to cell types may be estimated with few measurements). Applying this approach to both gene expression and Hi-C datasets, we distinguish cell types better than existing methods, even when considering a large set of cell types representative of the variety of human normal and cancer tissues.
Assigning gene expression or chromatin conformation states to a phenotype may be regarded as a coding problem in information theory, in which we want to choose the set of L binary features that most reliably classify the cell types when measurements may be incorrect with probability t. Here, we use “feature” to refer to either a single gene or eigengene, where an eigengene is a projection along a single eigenvector of correlations between genes. For Hi-C, features are either contacts between pairs of loci or eigenloci, which are defined analogously to eigengenes. Nonredundant codes transmit the most information per feature transmitted, but are also the most error prone, as the probability of correct transmission scales with (1 − t)L. In the approach described here, we quantify the cell type homogeneity, local consistency, and data efficiency criteria and use them to identify sets of features that reliably encode cell types. The flexibility of our approach is apparent from the application to two different methods for characterizing cell state across a diverse collection of cell types, and its reliability is demonstrated by comparing against other approaches.
RESULTS
Dataset description
We obtain human gene expression data from GEO (5), all publicly available Hi-C data from SRA (6), and RNA-seq data from the Genome-Tissue Expression database, referring to these datasets as GeneExp, Hi-C, and GTEx, respectively. Each dataset Xuvi consists of u ∈ 1, . . , N features, v ∈ 1, . . , M experiments, and i ∈ 1, . . K cell types where “experiment” is used to refer to a single measurement of all features. Here, (N, M, K) take the values of (17,525, 8842, 102) for GeneExp, (103,827, 453, 11) for Hi-C, and (20,689, 9850, 26) for GTEx (details available in Materials and Methods). We develop our model on the GeneExp and Hi-C datasets and apply it to the GTEx dataset. Furthermore, the columns are ordered by cell type such that if , then i = j, where j is an index over the K cell types and Mj is the number of associated experiments in the dataset.
We investigate the prevalence of correlations in macromolecular data by generating randomly resampled and correlated resampled data for each dataset and combine the resampled data with the actual data, as described in Materials and Methods. In fig. S1, dark blues in the diagonal going from the lower left to the upper right represent correct predictions of the state. Note that correlated data often are confused for the real data (at a rate of >70%, much higher than the corresponding rate for uncorrelated data), indicated by the blue in the top left and middle left squares, respectively. We further investigate the possibility of using correlations to the distinguish cell types in synthetically generated data when they are defined by differences along correlation eigenvectors (fig. S2, A and B), differences along genes (fig. S2, C and D), or a combination of both (fig. S2, E and F). Concluding that using correlations could be advantageous in distinguishing cell types, we choose to construct a redundant encoding based on correlations between genes and loci to define cellular state, as indicated in Fig. 1A.
Fig. 1. Schematic illustration of our approach to distinguish cell types.
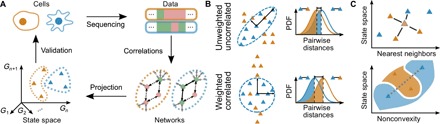
(A) The epigenomic measurements of two different cells in blue and orange (top left) yield different epigenotypes (top right) from which an condition-specific effective network (bottom right) is determined from correlations in the data, where solid or dashed lines indicate relationships that are enforced or not enforced but possible, respectively, under the specified conditions. Projection to the state space of correlation eigenvectors approximates the attractors. (B) The probability distribution functions of distances between pairs of measurements of the same and different types are compared at selected percentiles (shaded regions) to determine whether pairs of the same type are more similar than pairs of different types. (C) The performance is evaluated by using KNN to predict unseen data (top) and by measuring the frequency with which chords cross cell type boundaries (gray dashed line, bottom panel).
Description of the approach
We summarize our approach to translate biomolecular data into cell type in Fig. 1. Our goal is to select a subset of features that reliably encode cell type. To this end, we formalize the cell type homogeneity, local consistency, and data efficiency criteria.
Cell type homogeneity asserts that there should be less variation within types than between them. Thus, we compare the distribution of distances between measurement pairs within a given “test” cell type (labeled with c) with the corresponding distributions between the test cell type and all other “query” cell types. In the test cell type, certain pathways will be active, leading to stronger correlations between the constituent genes. We propose that after projecting to the correlation eigenvectors, the data should be rescaled in terms of the variance along each eigenvector, as we describe next.
Formally, let , where μu and σu are the mean and standard deviation (SD) of each row. Using boldface to indicate the suppression of the matrix indices, we decompose the correlations using
| (1) |
where U (V) are the left (right) eigenvectors of the feature correlations and where X is substituted for in the case of Hi-C. Let c be a set of column indices corresponding to a particular cell type (or possibly all cell types), and let be the SD of row u calculated using only these indices. Then, we assign weights using
| (2) |
Noting that U = [G1, . . , GM], where Gℓ are the axes in Fig. 1A, we obtain the eigenspace representation of the data
| (3) |
where λ(c) is a diagonal matrix whose entries are given by Eq. 2. In the weighted versions of our approach, c is the test cell type in one-versus-all classification or any cell type in {1, . . , K} for all-versus-all classification, while in the unweighted version, λ(c) = I for all c, and in the weighted case λ(c).
To construct the pairwise distance distributions, let J1 = 1 and for i > 1, where Mk is the number of experiments associated with the kth cell type. Then, the cumulative distribution function of pairwise distances is
| (4) |
with the added condition that w ≠ v if i = j, where is the indicator function, which is 1 if the argument is true and 0 otherwise. Let be the inverse of , where the argument p ∈ [0,1] is the percentile of the distribution. Then, cell type homogeneity is quantified as
| (5) |
which is illustrated in Fig. 1B. In our feature selection procedure, Eq. 5 represents “soft” constraint by using it as a regularization term in dimension reduction to reflect the possibility that closely (i.e., functionally) related cell types have overlapping distance distributions.
Local consistency, the idea that the most similar macromolecular profiles should be accurate predictors of cell identity, undergirds our approach to construct a mapping between genome-wide measurements and cell type. We proceed by dividing our dataset Xuvi into a training set Puvi and test set Qlmj. For ease of notation, we represent the training set data matrix Puvi as ordered pairs of experiments and cell type labels (xv, i) = Puvi with the boldface type to indicate that index over the feature labels u is suppressed and i ∈ {1, . . , K}. Let Dm be the set of column indices of the training data matrix corresponding to the k-nearest neighbors (KNN) of the test experiment and label (zm, j) = Qlmj.
Taking k = 9 (k = 7 for Hi-C) since we restrict our datasets to have at least 10 measurements per cell type, the KNN estimate for the cell type probabilities of zm is
| (6) |
as illustrated in Fig. 1C (top panel). The resulting cell type prediction is then .
For data efficiency, we define a chord between any two measurements of cell type i such that , where s ∈ [0,1] and ℓ ∈ {S}. We sample a total of P = 10,000 points along each of Q realizations of Cvv′(s) as follows: let κ = 2P//(Q2 − Q), where // denotes integer division. Then, we randomly select (κ + 1)(Q2 − Q)/2 − P chords, which are sampled at , and the remaining chords are sampled at . Applying KNN, we obtain , where a is an index over the P chords. The third criterion is
| (7) |
as illustrated in Fig. 1C (bottom panel).
The data efficiency criterion (Eq. 7) estimates the probability that the convex hull defined by the measurements of a cell type also belong to it. Equations 6 and 7 both reflect the fact that cell identity tends to be robust to small perturbations. The data decomposition and rescaling, in concert with the KNN method and the three criteria, constitute our approach, which is implemented in the source code available at https://github.com/twytock/Distinguishing_Cell_Types.
Comparison with other methods
We compare KNN with two other machine-learning techniques, support vector classifiers (SVC) and random forests (RF), to verify that it performs best. In Table 1, KNN, SVC, and RF are compared with two other existing methods based on Hopfield Neural Networks (HNN) (18) and spectral clustering [Partition Decoupling Method (PDM)] (22). In this comparison, we perform all-versus-all classification of cell types. In KNN, we calculate λ(c) in Eq. 3 using all the data as we are performing an all-to-all comparison. The remaining methods use . Let v ∈ {Ek}, k ∈ 1, . . , A be the set of columns belonging to the kth GEO Series Accession (GSE), SRA Sequencing Read Project (SRP), or GTEx subject ID. To test the accuracy of each method, we take {1, . . , M}\{Ek} and {Ek} as training and test sets and compare the predicted and actual cell types for each {Ek}. We use the shorthand “leave one GSE out” (LOGO) to refer to this validation strategy, as it reflects the situation of the method being applied to a new experiment about which it has no information, as described in the Materials and Methods. We note that PDM is an unsupervised method; therefore, we need to interpret the clusters generated as described in the Supplementary Materials.
Table 1. Comparison between our machine-learning techniques and existing methods applied to both datasets measured by the percentage correct classifications under LOGO cross-validation.
| Method | GeneExp (%) | Hi-C (%) |
| KNN | 68.4 | 63.4 |
| SVC | 57.8 | 43.7 |
| RF | 39.7 | 40.0 |
| HNN | 5.6 | 11.5 |
| PDM | 18.8 | 59.3 |
In addition to the methods in Table 1, we also compared our approach with that of principal components analysis (PCA), which can be used to reduce the data dimensionality while maintaining most of the variance. As in our approach, the principal components are calculated using Eq. 1, where X is the covariance matrix, Σ are the associated eigenvalues, and the rows of U are the eigenvectors. In PCA, dimension reduction proceeds by finding s such that . Here, the elements of Σ and associated rows of U are ordered by decreasing magnitude, and t is a threshold representing the fraction of the total variance accounted for by the first s rows. In contrast, our forward feature selection procedure selects sets of features based on an objective function (Materials and Methods).
Figure S3 shows the improvement of our feature selection method on PCA. We show that the feature sets S4 for GeneExp in fig. S3A and S3 for Hi-C in fig. S3C perform significantly better than their PCA counterparts when 5, 10, 15, and 20% of the data are held out. We also show that feature selection converges to the accuracy it achieves for large feature sets for small numbers of features in both datasets (fig. S3, B and D). PCA achieves a higher average value for the Hi-C dataset for feature sets with more than 15 features, highlighting that the forward feature selection procedure can get caught in a local maximum. Although fewer cell types are represented in the Hi-C dataset, we can still verify that our forward selection algorithm outperforms PCA for small numbers of features in fig. S3 (C and D). Because feature selection differs from PCA for both datasets, we suggest that the difference from PCA is a generic feature of our overall approach in the context of cell types.
We also compared the KNN method applied to the GTEx dataset with two methods designed for single-cell RNA-seq: SC3 (23) and MetaNeighbor (24). The former is an unsupervised method that achieves 63.0% accuracy compared with 92.5% for the KNN method. The latter is supervised, but its fit criterion is the area under the receiver operator characteristic (AUROC), which is 0.966 compared with 0.986 for the KNN method (Fig. 2A). In addition, the KNN method outperforms PCA; in particular, a classifier using KNN-selected eigengenes trained on 10% of the data outperforms a classifier using PCA-selected eigengenes trained on 95% of the data (Fig. 2B). The classification rates as a function of predicted and actual cell types are presented in Fig. 2C for the GTEx dataset.
Fig. 2. Assessment of our method applied to the GTEx dataset and comparison with alternatives.
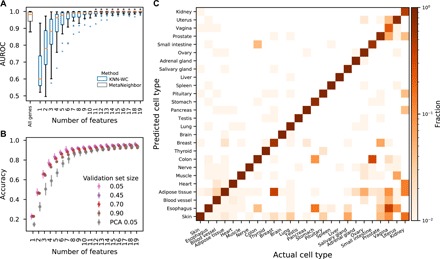
(A) AUROC for each cell type presented as a box plot for each number of features. Asterisks indicate significant improvement (P < 0.05, Kolmogorov-Smirnov test) relative to the MetaNeighbor performance. (B) Accuracy of LOGO validation as a function of the number of features and the size of the test set expressed as a fraction of all experiments. Optimization-selected features perform better than PCA-selected ones, especially for models with few features. (C) LOGO validation accuracy using nine features, where the cell types are listed in order of the number of experiments.
Comparison between versions of the method
Because KNN offers superior performance on all datasets, we confirm that both the eigenvector representation (Eq. 1) and the rescaling transformation (Eq. 2) are necessary. To achieve this, we benchmark the previously described weighted correlated (WC) version against an unweighted uncorrelated version (UU) and a weighted uncorrelated version (WU). The WC version of the KNN technique uses both Eq. 1 and Eq. 2 against that of a WU version, which uses Eq. 2 only, and an UU version, which uses neither. Note that using an unweighted correlated benchmark is equivalent to UU because they are related by a unitary transformation. Table S1 demonstrates the superiority of WC to both alternatives in distinguishing cell types using both datasets for all three criteria as detailed below.
The results reported in the Eq. 5 row of table S1 are broken down by cell type in Fig. 3 and fig. S4A. Over 90% of squares in the plot are gray, indicating that most cell types satisfy the cell type homogeneity criterion. In the GeneExp dataset, breast cancers, colon cancers, monocytes, lymphocytes, and leukemias are almost devoid of overlaps with other cell types for all three versions of the method. The homogeneity as characterized by the WC version, in particular, has few overlaps between cancer cell type groups and any other. Meanwhile, the epithelial cell tissues tend to overlap substantially, reflecting their functional similarity as shown in the second row from the bottom and second column from the left in the checkerboard. In addition, neural precursor cells (86th row from the bottom) are difficult to distinguish from others, particularly for the uncorrelated versions. This lack of distinguishability is consistent with the neural precursors’ known reprogramming capacity as manipulation of a single transcription factor is sufficient to induce a pluripotent state (25). These findings suggest that our approach is preserving aspects of the gene expression space relevant to cell function.
Fig. 3. Distinguishing cell types by the cell type homogeneity criterion for the GeneExp dataset.
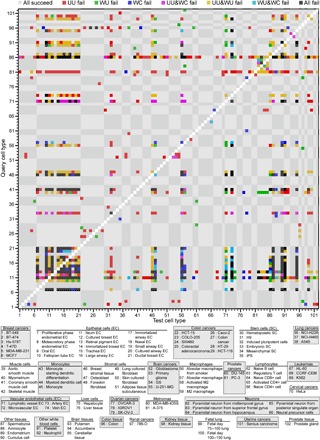
Equation 5 quantifies cell type homogeneity according to the UU, WU, and WC versions of measuring distance. The gray and white checkered background corresponds to the cell type groupings enumerated in table S2, and tick labels indicate the cell type associated with each row and column based on the key below the figure. The color coding defined in the legend above the figure marks the cases in which one or more of the versions failed for each query (row) and test (column) cell type. Gray indicates that the identification was successful for all three versions (91.4% of all cases). Self-comparisons (white diagonal) were not evaluated.
In the Hi-C dataset, the WC version has substantially fewer overlaps in fig. S4A. Of the 110 comparisons, there are only 7 overlaps for the WC version. The manner in which cell type heterogeneity manifests itself reveals biological similarities. The UU version shows cell type heterogeneity for types with few observations (top rows) when compared with types with many (left columns). On the other hand, the WU version fails to distinguish highly variable cell types with a substantial number of measurements, such as K562. While the leukemia cell line K562 shows substantial overlap with the other cell lines for all versions, in the WC version, two of those overlaps are with GM12878 and monocyte-derived macrophages. The latter are a B cell line and a macrophage line, respectively. Because the overlaps are all between cell lines derived from white blood cells, the overlap may be due to functionally relevant similarities in chromatin structure.
In Fig. 4, we demonstrate the superior predictive ability of the WC version by showing that it maintains its predictive power as the size of the test set grows for both datasets. In this case, the test set is constructed 25 times by randomly selecting a set of indices {g} ∈ 1, . . , A such that ∑k ∈ {g} ∣{E}k∣ ≥ fM, where f is the test fraction, with the constraint that all K cell types are in {1, .., M}\∪k ∈ {g}{E}k. In Fig. 4A, the WC version performs significantly better than the UU or WU versions for test fractions of 0.15 and 0.20. Similar results are obtained for the Hi-C dataset in Fig. 4C. The WC version is significantly more accurate than UU for f = 0.05, more accurate than WU for 0.15, and more accurate than both for f = 0.10 and f = 0.20.
Fig. 4. Comparison of the UU (blue), WC (orange), and WU (green) versions of the KNN technique applied to the GeneExp and Hi-C datasets.
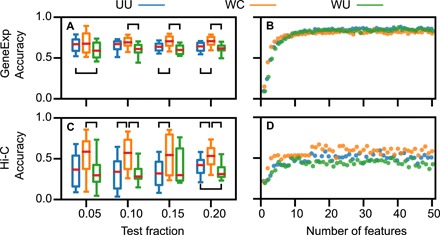
(A) Boxplots summarizing the distribution of classification accuracy over n = 25 test sets plotted as a function of the set size indicated as a fraction of all experiments for the GeneExp dataset. Red lines, boxes, and whiskers denote the median, interquartile range, and 5th to 95th percentile range, respectively. (B) Mean accuracy plotted as a function of the number of features for the GeneExp dataset. (C and D) Same as (A and B), respectively, but for the Hi-C dataset. Brackets indicate statistically significant differences between version accuracies as reported in table S1.
The WC version also achieves higher accuracy when optimizing for a small number of features, as shown in Fig. 4 (B and D). Letting αℓ and βℓ be the mean and SD of the accuracy for a KNN model with ℓ features and fixed size of the test set, we impose a cutoff for model complexity at
| (8) |
resulting in ℓ = 4 for GeneExp, ℓ = 3 for Hi-C, and ℓ = 9 for GTEx. We note that this criterion corresponds to an increase in accuracy being statistically significant at the 95% confidence level. For larger values of ℓ, the accuracy becomes highly dependent on the construction of the training and test sets, suggesting that the performance of the method is comparable for ℓ > 10.
Figure 5 breaks down the KNN results for both datasets when the LOGO validation is used on models of ℓ features. The cell type groups are ordered left to right and bottom to top by the number of experiments in the dataset in all panels. The presence of darker squares along the diagonal in the lower left shows that more data make cell type easier to classify. In Fig. 5A, experiments are assigned to the correct cell type group with 76.9% accuracy for the WC version, as indicated by the presence of orange color along the diagonal of the right panel. Monocytes (column 7), lymphocytes (column 12), leukemias (column 13), liver tissue (column 16), kidney tissue (column 25), and renal cancer (column 25) are classified without errors using the WC version, reflecting their uniqueness compared with the other cell type groups. The color in the second row from the bottom in all three panels shows that a variety of cell type groups are often misclassified as epithelial cells, reflecting this cell type group’s heterogeneity. In addition, the lack of data for the last three groups in the top right accounts for the method’s inability to classify them correctly. Under the WC version, the brain tissue samples (column 22) tend to be classified as neurons, brain cancers, or epithelial cells, while the remaining missing square along the diagonal corresponds to other tissue sample (column 20), which is a miscellaneous group of cell types.
Fig. 5. Comparison of LOGO validation for the three versions of the KNN technique applied to the GeneExp and Hi-C datasets.
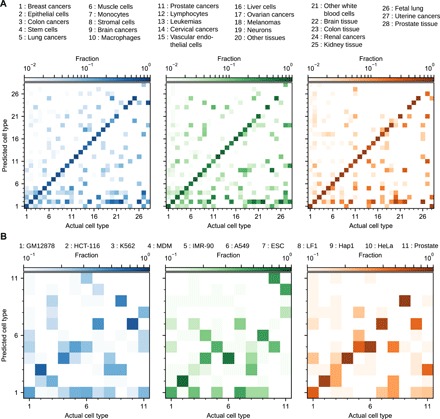
(A) Validation for the GeneExp dataset using 4 features. The colors indicate the version of the method used to classify the cell types (blue for UU, green for WU, and orange for WC), while the opacity indicates fraction of the total number of experiments belonging to the x axis cell type that are predicted to belong to the y axis cell type. (B) Same as (A), but for the Hi-C dataset using 3 features.
For the Hi-C data in Fig. 5B, the classifier maintains accuracy after reducing the entire ensemble of chromatin contacts to three dimensions. In the WC version (orange color), 7 of 11 cell types have their largest fraction in a column along the diagonal, while misclassifications occur between a lung cancer (A549) and two lung cell lines (LF1 and IMR-90, sixth and eighth columns from the left, fifth row from the bottom). The misclassification of HeLa cells as embryonic stem cells is interesting, possibly hinting at the common replicative potential of both cell lines. Prostate tissue, on the other hand, has the smallest number of samples in the dataset, making it difficult to classify.
Results for the WC version of the KNN method applied to the GTEx dataset are presented in Fig. 2C. In this case, nine features are used to classify the cell types reflecting the increased sensitivity of RNA-seq as a method compared with gene expression microarrays. Classification errors are primarily associated with functionally similar tissues (small intestine, stomach, colon, and esophagus) and tissues for which the number of experiments is small.
We break down the results of table S1 and Fig. 5 by cell type in fig. S5. In contrast with pairwise-distance distinguishability, cell types fail to be locally indistinguishable in a less organized fashion, reflecting individual measurement variability. Nevertheless, most misclassifications happen within cell type groups (diagonal of the checkerboard), particularly for the WC version of the method, suggesting that when this version misclassifies cell types, it often classifies them as a functionally related type. This point is further evidence that the WC version is preserving aspects of the gene expression related to cell function.
The number of experiments of each cell type, reported in table S2, affects how accurately the cell type can be predicted. Figure S5 reveals that the most prevalent cell type of each group tends to provoke the majority of the misclassifcations (i.e., the top row of each checkerboard row and the upper diagonal of the diagonal blocks of the checkerboard). This follows from the fact that an outlier point in feature space is more likely to be near the most common cell type than any other. Together, these results support our ansatz that Gℓ and λ(c) constitute a metric that improves the ability to classify cell types, because changes in gene expression and chromatin conformation must work in concert to effect changes in cell behavior.
Table S1 presents the overall fraction chords connecting points of the same cell type that exhibit nonconvexity, with 77.5% of the chords being convex in the GeneExp dataset and 89.5% being convex in the Hi-C dataset for the WC version of our approach. Specifically, figs. S4B and S6 break down the fraction of nonconvex chords by cell type. The WC version exhibits greater convexity relative to the UU and WU versions, and with it, more certainty that the interior of the convex hull is part of the cell type.
In fig. S6, we see most of the nonconvexity occurring in cell types is structured by cell type group, because the block of pairs with more nonconvex chords than threshold align with the checkerboard boundaries. We observe that lung cancers, muscle cells, stromal cells, brain cancers, lymphocytes, melanomas, neurons, fetal lung cells, and uterine cancers all have overlapping chords for the three versions of our approach.
Comparing the smaller Hi-C dataset in fig. S4B with the larger GeneExp dataset, we see that the advantage of the WC version becomes more pronounced. Only the lung fibroblast cell line LF1, the prostate tissue, and the K562 cell line overlap more than two other cell types for the WC version. On the other hand, the WU and UU versions show substantial overlaps with the other cell types. Notably, the IMR-90 cell line does not appear to overlap with the LF1 cell line, despite both of these being developed from lung fibroblasts. Since IMR-90 was isolated four decades ago and LF1 was more recently, this lack of similarity in chromatin structure may be a side effect of culturing a cell long term.
Since the overall counts of cell type pairs are not immediately apparent by eye in the preceding figures, they are enumerated in fig. S7. For all three criteria and for both datasets, the WC version has the smallest number of errors. Because there are fewer cell types in the Hi-C dataset compared with the GeneExp dataset, fig. S7D has smaller numbers than fig. S7A.
DISCUSSION
The prediction accuracy of >60% achieved here is greater than what is expected from RNA-protein correlations, given that fluctuations in mRNA only account for <45% of the variance of the protein abundance (26). Moreover, given the number of cell types and variables in the GeneExp, Hi-C, and GTEx datasets, it is unclear a priori that machine-learning approaches would work. Typically, such approaches are developed to classify a small number of distinct items with the number of measurements for each item much larger than the number of variables per measurement. For Hi-C data in particular, the prediction accuracy is surprisingly high since chromatin structure is another step away from protein expression. Previous analyses of Hi-C data tend to focus on short-range contacts (i.e., contacts between loci that are <500 kb apart), like CTCF-mediated topologically associating domains (TADs), which are often conserved across cell types and species (27); they are, thus, less useful for characterizing cell type compared with alternatives like histone methylation data from chromatin immunoprecipitation sequencing (28, 29). Nevertheless, our analysis is able to predict cell type from Hi-C data by taking into account the physical interactions between distant portions of the genome. This is shown in fig. S8, where only contacts between loci respectively greater than (fig. S8A) or less than (fig. S8B) a certain distance are included. Understanding the role that chemical interactions play in shaping the long-range physical structure of DNA is an intriguing application case for our method. Thus, the efficacy of our approach is greater than anticipated from biological and computational considerations.
The success of the approach in reducing dimension to three to nine features while maintaining predictive accuracy has several biological implications. First, this success stands in contrast to the previous lack of success in establishing biomarkers that reliably identify cancer subtypes (30). Second, the selected features differ from those selected by PCA in that they contain subdominant eigenvalues of the correlation matrix, reflecting the multiscale nature of cell type (Fig. 2B and fig. S3) arising from the hierarchical character of differentiation. This is particularly pronounced in Figs. 2 and 3 and figs. S5 and S6, where misclassifications tend to cluster between similar cell types, particularly for the WC approach. These trends suggest that closely related cell type are being distinguished by subtle changes (features associated with smaller eigenvalues) and distant cell types are being distinguished by broader changes (features associated with larger eigenvalues). Third, the inclusion of smaller eigenvalues, which contribute to noise sensitivity in so-called “sloppy” models (31), reinforces the necessity of using larger datasets and highlights the shortcomings of PCA in terms of distinguishing cell type. Fourth, successful dimensional reduction suggests that the selected features constrain variability in the unobserved cellular degrees of freedom (11, 32), which is consistent with previous equation-free nonlinear embedding methods that distinguish network behaviors (16, 17). The compression of genome-wide data to three to nine features is also consistent with the observed small scaling exponent between the number of cell types and the number of genes (33, 34) and supports the hypothesis that cell types are attractors of the underlying intracellular network dynamics (35). The empirically determined convex hull approximates the basin of attraction of the cell type attractor.
The successful application of our method to (protein-coding) RNA-seq data across a diverse set of tissue types reflects both its accuracy and flexibility. The increase in predictive power from 76.9% in the GeneExp dataset to 92.5% in the GTEx dataset suggests that most misclassifications are attributable to the less sensitive nature of microarray experiments compared with RNA-seq. Second, the favorable comparisons between our method and others applied to the GTEx dataset strengthen the conclusion that our method can predict cell type better than existing ones. Furthermore, application of our method to three datasets suggests that it could also be applied to noncoding RNAs to understand their functional role of in shaping cell types (36). It is possible to use the annotations to attribute functional information by masking the information for specific sets of genes and observing the change in predictive accuracy (Supplementary Materials). The success of our method also demonstrates its expected ability to interpret phenotype in forthcoming experiments in the context of large databases of existing cell type patterns.
Here, we showed that the correlation decomposition in Eq. 1 and rescaling factors in Eq. 2 increase the fraction of points in the convex hull identified with the cell type in figs. S4B and S6. These transformations are motivated by the usage of a nonlinear transformation to improve the convexity of predictions of network properties from data (37). In other words, these transformations enhance the resolution of the basin. Thus, our approach offers a solution to the challenging problem of estimating basins of attraction for high-dimensional systems from data and provides evidence for the notion that cell types are identifiable from genome-wide expression or chromatin conformation despite the high dimension of these measurements.
Two additional opportunities derive from our approach. First is the development of a semisupervised version that could identify previously unrecognized cell behaviors, using ideas from (22, 23). Second is the application of manifold discovery techniques like t-distributed stochastic neighbor embedding (38) to further refine the selected features and enhance data visualization.
Our approach advances the field of network medicine, which seeks to integrate large bioinformatic datasets to direct research into disease treatment (39). The global scope of our approach, in tandem with the resulting evidence for the cell type attractor hypothesis, is the first step in developing model-independent control strategies in cellular networks. Such strategies consist of identifying cell type attractors, curating the macromolecular responses of the cell to perturbations and finding combinations of these responses that together steer the cell from one attractor to another. Thus, in addition to distinguishing cell types based solely on genome-wide measurements, our approach could orient the development of rational strategies for cell reprogramming, the identification of therapeutic interventions, and other applications involving a combinatorially large number of options.
MATERIALS AND METHODS
Data preprocessing
All of the data used in this study are publicly available on the GEO and SRA databases maintained by the NIH (for a list of accession numbers, see source code). For the gene expression data, we chose to look for experiments conducted on the Affymetrix HG-U133+2 platform (GPL570 GEO accession), because its use was widespread and the probes could be mapped to the hg19 build of the human genome. We applied the following five different filters for experiments on this platform:
(I) We searched for experiments in which genes were perturbed to gain insight into how the cellular network processes information and, thus, to infer how the genes are correlated with one another under different conditions.
(II) We chose to gather gene expression assays using the NCI-60 cell lines as a proxy of human cancers because these cell types are commonly available and used to screen drugs and other compounds for their effectiveness in treating cancer.
(III) We obtained gene expression data from the Cancer Cell Line Encyclopedia to sample a wider variety of cancer cell lines.
(IV) We also included “normal” cell types and intermediate cell states obtained by searching for reprogramming experiments.
(V) We retrieved data from a study that attempted to identify transcription factors that control cell identity to broaden the spectrum of cell types included.
Data from source (I) were used only for the purpose of constructing the correlation matrix, while only unperturbed cells in sources (II) to (V) were used to train and validate the model. Together, the combined dataset, collectively referred to as “GeneExp,” comprises 102 distinct cell types with >10 observations. We downloaded the raw data from GEO and preprocessed it with a custom CDF file based on the hg19 build of the human genome to select the probes that correspond to genes (40). After preprocessing the gene expression using robust median averaging (41), we “batch corrected” the data, which attempts to filter out systematic experimental effects (42). The accession numbers are listed in table S2.
The chromatin conformation data, referred to as Hi-C, were also obtained from GEO/SRA by searching for “Hi-C” or “HiC” while filtering the organism to Homo sapiens (access date: 25 September 2018). The files were iteratively corrected, as described previously (3), using the tools available at https://github.com/mirnylab/. Chromosomal contacts were binned at 100-kb resolution so that experiments with lower-resolution sequencing coverage could be included.
The RNA-seq data, referred to as GTEx, were obtained from the GTEx Portal website: https://www.gtexportal.org/home/. We used the version 8 gene count data and associated annotations. For RNA-seq data derived from lysate or cell pellet, the data were normalized for the total number of reads in each experiment, filtered to include only genes from high-quality experiments (SMATSSCR<2) that were expressed in >1% of all experiments at >10 times the minimum expression level. The remaining data were log-transformed and batch corrected on the basis the SMGEBTCH identifier. We also filtered out any cell types with fewer than eight experiments. The preprocessing according to the described criteria resulted in 9850 samples with 20,689 gene identifiers representing 26 cell types (SMTS identifier) from 980 subjects.
The data processing results in gene expression levels, which have SDs approximately independent of their mean, making decomposition of advantageous. However, the distribution of Hi-C counts has a long tail because nearby loci come into contact exponentially more frequently than distant loci, so decomposing X rather than in Eq. 1 is more appropriate as the standard deviation of interlocus contacts are dependent on the mean.
Testing correlation predominance
To test whether correlations imparted noticeable structure in each dataset, a classifier was trained on a dataset consisting of the actual data, uncorrelated random data, and correlated random data. First, uncorrelated and correlated data were generated by randomly permuting actual measurements of each feature before and after projecting onto the eigenvectors, respectively, resulting in a dataset consisting of M instances of each experimental observations, uncorrelated simulated profiles, and correlated simulated profiles. From this set of 3M instances, a training set of size 2M was drawn, comprising one-third of each real data, uncorrelated data, and correlated data, with each instance labeled on the basis of how it was generated. A KNN classifier trained on these data predicted the generation method for each profile in the test data. For the GeneExp dataset, classification was performed using correlation eigenvectors with an associated eigenvalue λ > 1, a total of 1063, to reduce the impact of noise.
We also explored our method’s performance on synthetic data as a function of the signal-to-noise ratio (SNR). Simulated data for 100 “genes” from 2 “cell types” were generated using a fixed correlation structure derived from randomly resampling the correlations from the GeneExp dataset. Cell types were distinguished by introducing differences into randomly selected genes/eigengenes at SNRs ranging from 0.05 to 20. All models were trained on a small set of fixed size and evaluated on a validation set of 10,000 randomly generated profiles. Both training and test sets had equal numbers of cell types. This analysis was performed for three cases:
(I) Eigengene-defined cell types. The data for one to four (randomly selected) eigengenes were perturbed by adding the SNR/2 to one cell type and subtracting it from the other.
(II) Gene-defined cell types. The data for one to four genes were perturbed by adding the SNR∕2 to one cell type and subtracting it from the other.
(III) Correlation-defined cell types with confounding genes. The data for one eigengene are perturbed at an SNR of 5, and the data for one gene is perturbed by cell type in the training set only, at the prescribed SNR.
The size of the training set per cell type was two, three, or five in cases (I) and (II) and three, four, or five in case (III), which are the smallest numbers required to distinguish cell types in each case. In cases (I) and (II), the SNR is simply the nominal SNR used to generate the data. In case (III), the , where μi is the mean difference between the cell types along eigengene i before the gene perturbation is added, and is this quantity after it is added, as reported in fig. S2 (E and F). This accounts for the instances in which an eigengene with a small associated eigenvalue is dominated by the gene perturbation.
KNN cross-validation
In our cross-validation analysis, we apply Eq. 6 to each experiment of the test set and compare the resulting predictions with the known measurements wjm. We use a one-versus-all classification scheme in which the test cell type had one label and the remaining cell types had another because is the same regardless of the number of remaining cell types.
We adopted different standards for Eq. 6 and strategies for choosing the test set and training set depending on whether we were performing cross-validation or testing our approach’s performance on unseen data. In the case of cross-validation, we adopt the one-versus-all standard, in which measurements of a cell type were assigned to the test class and all remaining measurements were assigned to the query class.
1) Cross-validation proceeded by dividing the dataset into three pieces, called “folds,” constrained to be equal both in size and in the ratio of test to query cell types. We cross-validate three times, once with each fold as the test set. We then calculated to evaluate the overall accuracy across all single cell type frames of reference. This cross-validation scheme is used in the feature selection method.
2) For the purposes of figs. S4A, S5, and S7 (A and D), we use the LOGO strategy under the standard of all-versus-all classification. In this standard, each cell type is assigned to its own class. In the figures, we color the block white if , with threshold R = 0.05 for GeneExp and R = 0.1 for Hi-C. Note that some GSE/SRPs contain all of the observations of a given cell type. In such cases, the KNN method automatically fails for that cell type under that particular test set. Therefore, our success could be even higher if we had restricted to cases where all cell types are available in the training set.
3) The largest GSE/SRP/Subject ID was <5% of the data, so we extended the LOGO procedure to construct larger test sets as reported in Fig. 4 (A and C) and fig. S3 (A and C). For these figures, we randomly selected GSEs/SRPs until the test set was at least as large as the desired fraction f ∈ {0.05,0.10,0.15,0.20} (or up to 0.90 in the case of the GTEx dataset) with the restriction that all cell types must be represented in the training data.
Feature selection
Our framework is a hybrid of metric learning and supervised learning techniques. Thus, our objective function consists of a loss term based on the accuracy of the classifier described in Eq. 6 and a regularization term based on Eq. 5. To make explicit the impact of the set of features S, we rewrite Eq. 6 as
| (9) |
and note that S is already explicit in Eq. 5. Letting , our objective function is
| (10) |
where γ = 0.5 is a scalar regularization parameter that controls the strength of Eq. 5, giving it approximately half the importance of Eq. 6. Values of γ ≫ 1 will select features solely based on satisfaction of Eq. 5, while γ ≪ 1 will ignore this requirement in favor of Eq. 6.
With the objective function defined, we describe the forward feature selection algorithm. Recall that N is the number of features of the dataset. We first define {U1} = {{i}, i ∈ {1, . . , N}}. Our scheme for dimension reduction proceeds by finding S1 = arg min{S∈U1}F(S), then constructing {U2} = {{S1, i} i ∈ {1, . . , N}\S1}. Continuing iteratively, sets of features of arbitrary length Sℓ may be constructed. We continue until ℓ = 50, which is long after the addition of features has stopped improving the classification accuracy in the LOGO tests (Fig. 4).
Supplementary Material
Acknowledgments
Funding: This work was supported by CR-PSOC grant no. 1U54CA193419. T.P.W. also acknowledges support from NSF-GRFP fund no. DGE-0824162 and NIH/NIGMS grant no. 5T32GM008382-23. Author contributions: T.P.W. and A.E.M. designed the research and wrote and edited the manuscript. T.P.W. wrote the software, curated the dataset, and analyzed the results. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to the paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaax7798/DC1
Supplementary Information
Fig. S1. Confusion matrices for discerning actual and simulated data.
Fig. S2. Method testing results as a function of the SNR under three scenarios (rows) for two criteria (columns).
Fig. S3. Comparison of forward selection with PCA.
Fig. S4. Distinguishing cell types for the Hi-C dataset.
Fig. S5. KNN classification accuracy by cell type for the GeneExp dataset under LOGO cross-validation.
Fig. S6. Fraction of nonconvex chords for each cell type.
Fig. S7. Compilation of the number of squares of each color found in the preceding figures.
Fig. S8. Accuracy as a function of genomic distance between loci and number of features for the Hi-C dataset.
Table S1. Version comparison results and KS test P values.
Table S2. Cell type counts, tick labels for Figs. 2C, 3, and 5 and figs. S5 and S6, and database accession numbers for the GeneExp and Hi-C datasets.
Reference (43)
REFERENCES AND NOTES
- 1.Cortini R., Barbi M., Caré B. R., Lavelle C., Lesne A., Mozziconacci J., Victor J.-M., The physics of epigenetics. Rev. Mod. Phys. 88, 025002 (2016). [Google Scholar]
- 2.Shi L., Reid L. H., Jones W. D., Shippy R., Warrington J. A., Baker S. C., Collins P. J., de Longueville F., Kawasaki E. S., Lee K. Y., Luo Y., Sun Y. A., Willey J. C., Setterquist R. A., Fischer G. M., Tong W., Dragan Y. P., Dix D. J., Frueh F. W., Goodsaid F. M., Herman D., Jensen R. V., Johnson C. D., Lobenhofer E. K., Puri R. K., Schrf U., Thierry-Mieg J., Wang C., Wilson M., Wolber P. K., Zhang L., Amur S., Bao W., Barbacioru C. C., Lucas A. B., Bertholet V., Boysen C., Bromley B., Brown D., Brunner A., Canales R., Cao X. M., Cebula T. A., Chen J. J., Cheng J., Chu T. M., Chudin E., Corson J., Corton J. C., Croner L. J., Davies C., Davison T. S., Delenstarr G., Deng X., Dorris D., Eklund A. C., Fan X. H., Fang H., Fulmer-Smentek S., Fuscoe J. C., Gallagher K., Ge W., Guo L., Guo X., Hager J., Haje P. K., Han J., Han T., Harbottle H. C., Harris S. C., Hatchwell E., Hauser C. A., Hester S., Hong H., Hurban P., Jackson S. A., Ji H., Knight C. R., Kuo W. P., LeClerc J. E., Levy S., Li Q. Z., Liu C., Liu Y., Lombardi M. J., Ma Y., Magnuson S. R., Maqsodi B., McDaniel T., Mei N., Myklebost O., Ning B., Novoradovskaya N., Orr M. S., Osborn T. W., Papallo A., Patterson T. A., Perkins R. G., Peters E. H., Peterson R., Philips K. L., Pine P. S., Pusztai L., Qian F., Ren H., Rosen M., Rosenzweig B. A., Samaha R. R., Schena M., Schroth G. P., Shchegrova S., Smith D. D., Staedtler F., Su Z., Sun H., Szallasi Z., Tezak Z., Thierry-Mieg D., Thompson K. L., Tikhonova I., Turpaz Y., Vallanat B., Van C., Walker S. J., Wang S. J., Wang Y., Wolfinger R., Wong A., Wu J., Xiao C., Xie Q., Xu J., Yang W., Zhang L., Zhong S., Zong Y., Slikker W. Jr., The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–1161 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imakaev M., Fudenberg G., McCord R. P., Naumova N., Goloborodko A., Lajoie B. R., Dekker J., Mirny L. A., Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozsolak F., Milos P. M., RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett T., Troup D. B., Wilhite S. E., Ledoux P., Rudnev D., Evangelista C., Kim I. F., Soboleva A., Tomashevsky M., Marshall K. A., Phillippy K. H., Sherman P. M., Muertter R. N., Edgar R., NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res. 37, D885–D890 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leinonen R., Sugawara H., Shumway M.; International Nucleotide Sequence Database Collaboration , The sequence read archive. Nucleic Acids Res. 39, D19–D21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hopfield J. J., Neural networks and physical systems with emergent collective computational abilities. Proc. Natl. Acad. Sci. U.S.A. 79, 2554–2558 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Packard N. H., Crutchfield J. P., Farmer J. D., Shaw R. S., Geometry from a Time Series. Phys. Rev. Lett. 45, 712–716 (1980). [Google Scholar]
- 9.Lu R., Markowetz F., Unwin R. D., Leek J. T., Airoldi E. M., MacArthur B. D., Lachmann A., Rozov R., Ma’ayan A., Boyer L. A., Troyanskaya O. G., Whetton A. D., Lemischka I. R., Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature 462, 358–362 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Airoldi E. M., Miller D., Athanasiadou R., Brandt N., Abdul-Rahman F., Neymotin B., Hashimoto T., Bahmani T., Gresham D., Steady-state and dynamic gene expression programs in Saccharomyces cerevisiae in response to variation in environmental nitrogen. Mol. Biol. Cell 27, 1383–1396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wytock T. P., Motter A. E., Predicting growth rate from gene expression. Proc. Natl. Acad. Sci. U.S.A. 116, 367–372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Assaf M., Roberts E., Luthey-Schulten Z., Goldenfeld N., Extrinsic noise driven phenotype switching in a self-regulating gene. Phys. Rev. Lett. 111, 058102 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Lu M., Onuchic J., Ben-Jacob E., Construction of an effective landscape for multistate genetic switches. Phys. Rev. Lett. 113, 078102 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Wang R.-S., Saadatpour A., Albert R., Boolean modeling in systems biology: An overview of methodology and applications. Phys. Biol. 9, 055001 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Saadatpour A., Albert R., A comparative study of qualitative and quantitative dynamic models of biological regulatory networks. EPJ Nonlinear Biomed. Phys. 4, 5 (2016). [Google Scholar]
- 16.Donner R. V., Zou Y., Donges J. F., Marwan N., Kurths J., Ambiguities in recurrence-based complex network representations of time series. Phys. Rev. E 81, 015101 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Crutchfield J. P., Between order and chaos. Nat. Phys. 8, 17–24 (2011). [Google Scholar]
- 18.Lang A. H., Li H., Collins J. J., Mehta P., Epigenetic landscapes explain partially reprogrammed cells and identify key reprogramming genes. PLOS Comput. Biol. 10, e1003734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dettmer S. L., Nguyen H. C., Berg J., Network inference in the nonequilibrium steady state. Phys. Rev. E 94, 052116 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Han X., Shen Z., Wang W.-X., Di Z., Robust reconstruction of complex networks from sparse data. Phys. Rev. Lett. 114, 028701 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Rondelez Y., Competition for catalytic resources alters biological network dynamics. Phys. Rev. Lett. 108, 018102 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Braun R., Leibon G., Pauls S., Rockmore D., Partition decoupling for multi-gene analysis of gene expression profiling data. BMC Bioinformatics 12, 497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiselev V. Y., Kirschner K., Schaub M. T., Andrews T., Yiu A., Chandra T., Natarajan K. N., Reik W., Barahona M., Green A. R., Hemberg M., SC3: Consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483–486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crow M., Paul A., Ballouz S., Huang Z. J., Gillis J., Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat. Commun. 9, 884 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J. B., Greber B., Araúzo-Bravo M. J., Meyer J., Park K. I., Zaehres H., Schöler H. R., Direct reprogramming of human neural stem cells by OCT4. Nature 461, 649–653 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M., Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Tang Z., Luo O. J., Li X., Zheng M., Zhu J. J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B., Michalski P., Piecuch E., Wang P., Wang D., Tian S. Z., Penrad-Mobayed M., Sachs L. M., Ruan X., Wei C.-L., Liu E. T., Wilczynski G. M., Plewczynski D., Li G., Ruan Y., CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ernst J., Kheradpour P., Mikkelsen T. S., Shoresh N., Ward L. D., Epstein C. B., Zhang X., Wang L., Issner R., Coyne M., Ku M., Durham T., Kellis M., Bernstein B. E., Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marco E., Meuleman W., Huang J., Glass K., Pinello L., Wang J., Kellis M., Yuan G.-C., Multi-scale chromatin state annotation using a hierarchical hidden Markov model. Nat. Commun. 8, 15011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haury A.-C., Gestraud P., Vert J.-P., The influence of feature selection methods on accuracy, stability and interpretability of molecular signatures. PLOS ONE 6, e28210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Transtrum M. K., Machta B. B., Sethna J. P., Geometry of nonlinear least squares with applications to sloppy models and optimization. Phys. Rev. E 83, 036701 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Yang Y., Wang J., Motter A. E., Network observability transitions. Phys. Rev. Lett. 109, 258701 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Bell G., Mooers A. O., Size and complexity among multicellular organisms. Biol. J. Linn. Soc. 60, 345–363 (1997). [Google Scholar]
- 34.Bonner J. T., Perspective: The size-complexity rule. Evolution 58, 1883–1890 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Huang S., Eichler G., Bar-Yam Y., Ingber D. E., Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys. Rev. Lett. 94, 128701 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Sarropoulos I., Marin R., Cardoso-Moreira M., Kaessmann H., Developmental dynamics of lncRNAs across mammalian organs and species. Nature 571, 510–514 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horvát S., Czabarka É., Toroczkai Z., Reducing degeneracy in maximum entropy models of networks. Phys. Rev. Lett. 114, 158701 (2015). [DOI] [PubMed] [Google Scholar]
- 38.van der Maaten L., Hinton G., Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008). [Google Scholar]
- 39.Sonawane A. R., Weiss S. T., Glass K., Sharma A., Network medicine in the age of biomedical big data. Front. Genet. 10, 294 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai M., Wang P., Boyd A. D., Kostov G., Athey B., Jones E. G., Bunney W. E., Myers R. M., Speed T. P., Akil H., Watson S. J., Meng F., Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 33, e175 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P., Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Johnson W. E., Li C., Rabinovic A., Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Fugmann S. D., Form follows function — The three-dimensional structure of antigen receptor gene loci. Curr. Opin. Immunol. 27, 33–37 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/12/eaax7798/DC1
Supplementary Information
Fig. S1. Confusion matrices for discerning actual and simulated data.
Fig. S2. Method testing results as a function of the SNR under three scenarios (rows) for two criteria (columns).
Fig. S3. Comparison of forward selection with PCA.
Fig. S4. Distinguishing cell types for the Hi-C dataset.
Fig. S5. KNN classification accuracy by cell type for the GeneExp dataset under LOGO cross-validation.
Fig. S6. Fraction of nonconvex chords for each cell type.
Fig. S7. Compilation of the number of squares of each color found in the preceding figures.
Fig. S8. Accuracy as a function of genomic distance between loci and number of features for the Hi-C dataset.
Table S1. Version comparison results and KS test P values.
Table S2. Cell type counts, tick labels for Figs. 2C, 3, and 5 and figs. S5 and S6, and database accession numbers for the GeneExp and Hi-C datasets.
Reference (43)