

Abstract
The pH‐responsive nature of two self‐assembled NDI‐peptide amphiphile conjugates is reported. The diethoxy substituted NDI showed a pH‐dependent assembly behaviour, as expected. In contrast, the isopropylamino‐ and ethoxy‐substituted NDI based supramolecular polymer was stable at acidic and basic aqueous conditions. This finding highlights how subtle changes in the molecular design of π‐stacked chromophore‐peptide conjugates have a drastic impact on their equilibrium structure and ultimately functional properties.
Keywords: self-assembly, peptides, supramolecular polymers, fluorescence, chromophores
Small changes ‐ big impact! Peptide tethered π‐conjugate chromophores combine the stimuli‐responsive nature of self‐assembled oligopeptide designs with the tunable optical properties of organic semiconductors. In this contribution, we discuss how subtle changes in the molecular design of core substituted naphthalene diimide‐peptide conjugates have a drastic impact on the pH‐responsive nature of the supramolecular polymerization process in water.
Self‐assembly of peptide‐chromophore conjugate amphiphiles has been investigated for the design of functional supramolecular materials in order to combine the stimuli‐responsive nature of self‐assembled oligopeptide domains with the tunable optical properties of organic chromophores. Highly directional hydrogen bonding interactions between peptide segments as the structure‐directing motif provide access to ordered supramolecular polymers.1 Furthermore, the side‐chains of the amino acid sequences are able to embed tunable stimuli‐responsive features into the peptide‐chromophoric assemblies. For example, peptide‐naphthalene diimide based self‐assembled nanotubes were shown to act as receptors for C60 and C70 fullerenes.2 Parquette and coworkers synthesized n‐type chromophore‐peptide conjugates for electron transfer and optoelectronic devices.3 Ulijn and coworkers used enzyme‐responsive peptide sequences to have structural and spatiotemporal control over chromophore nanostructures using bio‐catalytic reactions.4 A controlled pH decrease coupled to pH‐responsive peptide‐chromophore molecules was designed by Adams and Smith to achieve programmable self‐sorted photoconductive gels.5 The Stupp and Meijer labs showed that tethered chromophores can be used to probe the exchange kinetics of peptide amphiphiles.6 In the present work we synthesize core substituted naphthalene diimide (cNDI)‐peptide‐conjugate amphiphiles, using the organic chromophore as a functional probe, but also as a rigid π‐stacking segment. The latter therefore allows us to modulate the self‐assembly and stimuli‐responsive nature of the peptide sequences in order to control and direct the 1D supramolecular polymerization of the peptide‐NDI amphiphiles in water.
Earlier from our group, we have reported the self‐assembly of charged dendritic peptide amphiphiles containing alternating phenylalanine (hydrophobic)‐lysine (hydrophilic) sequences into 1D supramolecular polymers, on screening the charges via varying the pH or by the use of charge complementary comonomers.7 However, the stimuli‐responsive self‐assembly of these peptide amphiphiles is highly dependent on the delicate balance between the attractive interactions of the hydrophobic segments and the charge repulsion between the side chains.8 We envisage that appending charged, pH‐responsive peptide domains with fluorescent π‐conjugated chromophores, would allow the spectroscopic and microscopic probing of the self‐assembly process and would also provide additional structural insights into the role of additional π‐π stacking and hydrophobic interactions in modulating the pH‐responsive nature of these assemblies. With this objective, we have tethered ß‐sheet forming peptide segments with alternating phenylalanine (hydrophobic) and lysine (hydrophilic) amino acid sequences, to diethoxy substituted cNDI (NDI‐diOEt‐cat 11) and ethoxy and isopropylamine substituted cNDI (NDI‐OEtiPA‐cat 13) through the imide position of the NDIs (Figure 1a). The peptide sequence 7 was synthesized by standard Fmoc‐protocol on solid phase and was coupled with solubilizing dendritic tetraethylene glycol chains 6. After Fmoc deprotection the free amine containing molecule 9 was reacted with either of the substituted cNDI based dicarboxylic acids 2 and 5. After cleavage of the Boc group the amphiphiles NDI‐diOEt‐cat 11 and NDI‐OEtiPA‐cat 13 were obtained. The final molecules and all intermediate compounds were characterized via NMR and mass spectrometry (see supporting information for details). We have recently reported fluorescent supramolecular polymers of core‐substituted NDI chromophores with J‐type molecular organization in organic solvents, and were expecting to use the luminescent chromophores as an efficient probe to study supramolecular assembly characteristics in water when conjugated with ß‐sheet forming peptides.9
Figure 1.
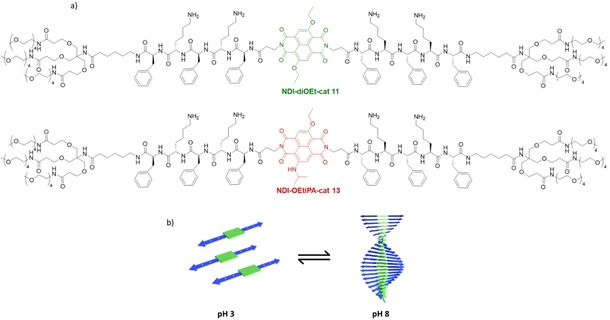
a) Molecular structures of NDI‐diOEt‐cat and NDI‐OEtiPA‐cat. b) Schematic illustration of pH‐responsive self‐assembly of cNDI‐peptide‐amphiphiles.
To investigate the pH‐responsive self‐assembling behavior of NDI‐diOEt‐cat and NDI‐OEtiPA‐cat in water using spectroscopic techniques, we first dissolved the charged amphiphiles in acidic pH (pH 3.0 and 3.98 respectively) and performed a pH titration by gradually adding base (Tris‐HCl buffer). The self‐assembly of both peptide‐cNDI amphiphilic conjugates could be spectroscopically probed using specific changes in cNDI spectral features which are characteristic for intermolecular interactions.10
The absorption spectrum of a 5×10−5 M solution of NDI‐diOEt‐cat in pH 3 citric acid buffer displayed sharp π‐π* transitions with vibronic features (λmax=350 nm and 368 nm) and a n‐π* transition with a maximum at λ=480 nm (Figure 2a). The higher absorbance of the λ=368 nm vibronic peak in comparison to the one at 350 nm along with the sharp spectral features are indicative of the molecularly dissolved nature of NDI‐diOEt‐cat in acidic solution due to charge repulsion between protonated lysine moieties. The monomeric nature of NDI‐diOEt‐cat in acidic pH is further supported by the absence of induced circular dichroism (ICD) in the spectral range of cNDI electronic transitions (Figure 2b). Increasing the pH of the solution of NDI‐diOEt‐cat, by the addition of Tris‐HCl buffer showed distinct spectroscopic changes in the cNDI absorption range characteristic of cNDI self‐assembly. The self‐assembled NDI‐diOEt‐cat at basic pH 8.5 displayed a reversed ratio of the intensities of the π‐π* vibronic transitions at λ=350 nm and 368 nm along with a decrease and broadening of the absorbance (Figure 2a). The self‐assembled NDI‐diOEt‐cat further displayed the appearance of an induced circular dichroism and negative band at λ=480 nm, as well as a bisignated CD signal at the π‐π* absorption maximum, indicating helically π‐π stacked cNDI chromophores (Figure 2b).11 Additionally, the presence of a negative CD band at λ=215 nm indicates the formation of ß‐sheets in the peptide dendron domains of the self‐assembling motifs (Figure SI1a). Furthermore, titration studies using a gradual increase in the pH value revealed that self‐assembly of NDI‐diOEt‐cat starts at pH 6 (Figure 2), which is much lower than the pH value expected purely from the pKa of isolated lysine side chains (pKa=10.6). The increased hydrophobicity of the amphiphiles, due to the cNDI core and hydrophobic aromatic side chains of the phenylalanine groups, shifts the apparent pKa value of the amine groups by about four orders of magnitude. These results are consistent with our earlier reports demonstrating shifts in the apparent pKa values of weakly acidic and basic dendritic peptide amphiphiles, depending on the thermodynamic driving force for the supramolecular polymerization process.7
Figure 2.
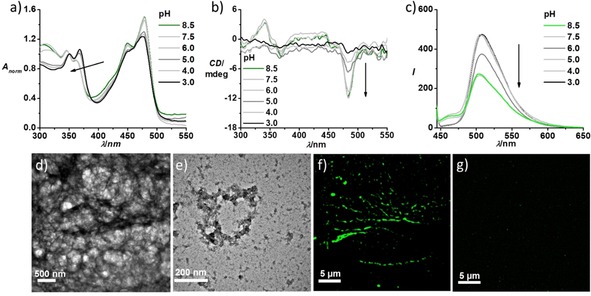
pH‐dependent a) absorption, b) CD and c) emission spectra (λex=430 nm) of NDI‐diOEt‐cat, which shows a change in the ratio between π‐π* vibronic bands, appearance of CD signal and quenching of emission (λex=430 nm) on increasing the pH of the solution. These spectral features refer to the self‐assembled structures at pH 8.5 and monomeric or molecular dissolved species at pH 3 due to charge repulsion between lysine moieties. TEM images of NDI‐diOEt‐cat at d) pH 8 (10 mM Tris HCl buffer) and at e) pH 3 (0.15 M citric acid buffer, 9 : 1 Ca/Na3C) show the presence of 1D supramolecular polymers and absence of elongated self‐assembled structures, respectively. TEM samples were prepared by placing a drop of the solution on carbon coated copper grids followed by drying at room temperature in a desiccator; samples were stained with uranyl acetate. SIM microscopy images (λex=488 nm) depict f) long fibre‐like green fluorescent supramolecular polymers at pH 8 and g) absence of any fluorescent self‐assembled structures at pH 3 ([NDI‐diOEt‐cat]=5×10−5 M, l=10 mm).
Next, we probed the changes in the luminescent properties upon varying the pH in order to get an insight into the emission of the NDI‐diOEt‐cat assembly. Excitation of the n‐π* band (λex=430 nm) of NDI‐diOEt‐cat monomers in acidic pH showed a broad emission with an emission maximum at λem=510 nm, characteristic of cNDI chromophores. After gradually increasing the pH of the solution we observed a quenching of the emission, which is indicative of π‐stacking between the cNDI chromophores during supramolecular polymerization. Normalized emission spectra of the monomeric (pH=3) and self‐assembled (pH=8.5) NDI‐diOEt‐cat did not show significant differences apart from a slight broadening of the emission in the self‐assembled state (Figure SI1b). However, the excitation spectrum collected at λ=510 nm for the self‐assembled state is broadened in comparison to monomer excitation spectrum, supporting the emissive nature of the self‐assembled chromophores (Figure SI1c). This was corroborated by the time resolved fluorescence life time decay profile of the self‐assembled chromophores collected at λ=510 nm, which showed a shorter life time of 4.74 ns compared to monomer life time of 13.5 ns (Figure SI1d). The fluorescent nature of these chromophore‐peptide stacks was utilized to visualize the assemblies using fluorescence microscopy (vide infra). Transmission electron microscopy of the negatively stained (uranyl acetate) NDI‐diOEt‐cat assemblies at pH 8 showed a dense network of micrometer long fibers (Figure 2d), which provides further evidence for the one‐dimensional supramolecular polymerization process.
The fluorescent NDI‐diOEt‐cat assemblies facilitated its visualization as green 1D structures in solution using structured illumination microscopy (SIM) (Figure 2f). To investigate the pH‐responsive disassembly and reversibility of the self‐assembly process of NDI‐diOEt‐cat, we added citric acid buffer to acidify the basic solution of self‐assembled NDI‐diOEt‐cat. Upon acidification, the spectra signatures resemble that of a monomeric state (Figure SI3) (the ratio between π‐π* vibronic transitions reverses, the induced CD signal disappears and the intensity of the photoluminescence increases). Morphological investigations of NDI‐diOEt‐cat (Figure 2e, g) at lower pH, did not show any ordered supramolecular structures. The morphological investigations agree with findings from spectroscopic signatures, suggesting hydrogen bonding, hydrophobic desolvation and π‐π stacking driven 1D self‐assembly at pH 8. Acidic pH leads to protonation of lysine moieties to trigger charge repulsion between self‐assembled monomers and the formation of a molecular dissolved or monomeric state.
In contrast to the pH‐responsive supramolecular polymerization of NDI‐diOEt‐cat, we were surprised to realize that NDI‐OEtiPA‐cat remains self‐assembled at both acidic and basic pH (Figure 3). Absorption spectra of a 2.5×10−5 M solution of NDI‐OEtiPA‐cat at pH 8.9 and pH 3.98 both gave rise to π‐π* vibronic transitions around λ=350 nm and 368 nm, characteristic of π‐ stacked cNDI chromophores (Figure 3a). Furthermore, the presence of an induced circular dichroism signal with a positive band at λ=578 nm (n‐π* transitions) and a bisignated CD signal at the π‐π* transition (Figure 3b) are characteristic of helically stacked chromophores.11 This confirms the self‐assembled nature of the NDI‐OEtiPA‐cat amphiphiles at both pH values. Further, negatively stained TEM images of NDI‐OEtiPA‐cat at both acidic pH=3 and basic pH=8 show the presence of high aspect ratio 1D supramolecular polymers (Figure 3d, e). SIM microscopy images of NDI‐OEtiPA‐cat reveal the presence of red‐emitting supramolecular polymers at pH 8 and pH 3 (Figure 3f, g).
Figure 3.
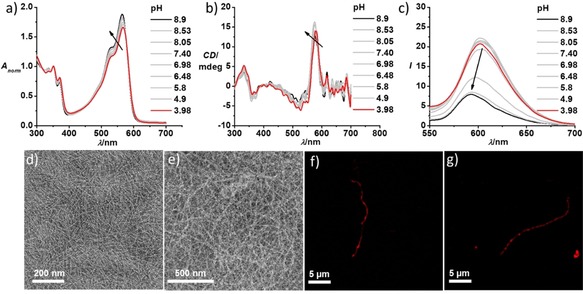
pH‐dependent a) normalized absorption spectra and b) CD spectra of NDI‐OEtiPA‐cat, showing a reversed ratio between π‐π* vibronic bands and the presence of an induced CD signal at both high and low pH values. This is indicative of a pH‐insensitive self‐assembly of NDI‐OEtiPA‐cat. c) Corresponding emission spectra (λex=530 nm) shows a red shift and increased emission upon decreasing the pH. TEM images of NDI‐OEtiPA‐cat show the presence of supramolecular polymers at both d) pH 8 (10 mM tris HCl buffer) and at e) pH 3 (0.15 M citric acid buffer, 9 : 1 Ca/Na3C). TEM samples were prepared by placing a drop of the solution on carbon coated copper grids followed by drying at room temperature in a desiccator; samples were stained with uranyl acetate. SIM microscopy images (λex=561 nm) also show fibre‐like red fluorescent supramolecular polymers at both f) pH 8 and at g) pH 3 ([NDI‐OEtiPA‐cat]=2.5×10−5 M, 0.15 M citric acid buffer, 9 : 1 Ca/Na3C for pH=3, 10 mM Tris‐HCl buffer for pH 9, l=10 mm).
However, careful observation of the absorption spectra reveals a slight shift of the n‐π* absorption maximum (λ=566 nm at pH 3.98 and λ=564 nm at pH 8.9) along with a decrease in the absorbance and corresponding change in the CD maximum (λ=580 nm at pH 3.98 and λ=575 nm at pH 8.9) on going from pH 3.98 to pH 8.9. These slight changes indicate that although the NDI‐OEtiPA‐cat molecules are self‐assembled at both pH values of interest, there are likely changes in the molecular organization within stacks due to the pH‐induced variation in electrostatic interactions between the charged peptide segments. This is also indicated by the changes in the photoluminescence, which showed a significant quenching of the emission upon increasing the pH from 3.98 to 8.9 (Figure 3c). We attribute the decrease in fluorescence to the stronger π‐π‐stacking between the cNDI cores, upon the neutralization of the charges in the peptide segments, thus minimizing the electrostatic repulsion between the molecules. This is further reflected in the time resolved fluorescence measurements (Figure SI4c) revealing an increased contribution of the shorter life time component of 0.1 ns at pH 8.6 (27 %) compared to the 0.12 ns contribution at pH 3.0 (15 %), which could be due to an enhanced exciton migration in the closely packed stacks at pH 8.6.12 These observations indicate that NDI‐OEtiPA‐cat stacks are not responsive to the changes in the pH, suggesting a very delicate balance between interactions in the hydrophobic domains and the charge repulsion which are required for a pH‐responsive assembly in this classes of cNDI‐peptide amphiphile conjugates. A change in the substitution of the NDI core from ethoxy to isopropylamino group altered the hydrophobic to hydrophilic balance in the molecular design and the electrostatic repulsion between the charged lysine moieties no longer overruled the strong hydrophobic desolvation, hydrogen bonding and π‐π stacking between chromophores. This is in agreement with the calculated logP (partition coefficient) values using online calculation service at www.molinspiration.com which gives a higher logP value for NDI‐OEtiPa‐cat core (2.46) than that of NDI‐diOEt‐cat core (1.69) (Figure SI6).13 This suggests an increased hydrophobicity for NDI‐OEtiPa‐cat than that of NDI‐diOEt‐cat.
In conclusion, we have investigated the stimuli‐responsive nature using two different self‐assembled π‐conjugated cNDIs, tethered with the same dendritic‐peptide amphiphiles in aqueous solution. The charged dendritic‐peptide segments were previously shown to exhibit pH‐responsive behavior during self‐assembly. However, the pH‐responsive behavior is dependent on the balance between attractive hydrophobic, π‐π stacking and hydrogen bonding interactions versus the repulsive interaction embedded in the side chains of lysine amino acids in the peptide segments. In this study, we have shown that incorporating the π‐conjugated segments of cNDI cores has a significant impact on the delicate balance between the attractive and repulsive interactions that modulate the pH‐responsive behavior of alternating oligopeptide sequences of phenylalanine and basic lysine. The diethoxy substituted cNDI derivatives show a pH‐responsive assembly behavior, as expected, but surprisingly the unsymmetrical isopropylamino‐ and ethoxy‐substituted cNDI based supramolecular polymer was stable towards protonation of the lysine side chains after lowering the pH. Thus, subtle changes after introducing a slightly larger isopropyl substituent and the more electron‐rich nature of the NDI, shifts the balance between attractive (π‐π stacking, hydrogen bonding, dispersive interactions) and repulsive Coulomb interactions. This renders the supramolecular polymerization process insensitive towards pH. Finally, the fluorescent nature of the π‐stacked cNDI chromophores helped the spectroscopic probing and microscopic visualization of the supramolecular polymerization process for both cNDI conjugates. The pH‐responsive nature of the peptide segments, along with the tunable optical properties of the core‐substituted NDI chromophores provide attractive molecular designs, which will be investigated in the future for spatio‐temporal resolution and multicomponent functional materials.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to thank JNCASR and the Department of Science and Technology, Government of India, for financial support. In addition, the funding from Sheikh Saqr Laboratory (SSL), JNCASR is also acknowledged. We also thank Prof. Sarit Agasti for the SIM facility. S.J.G. acknowledges the funding received from (DST‐DAAD, sanction order no‐INT/FRG/DAAD/P‐01/2018). P.B. acknowledges financial support from the DAAD (PPP‐DST project 57389690). A.S. and R.S. thank CSIR for a fellowship. This work was supported by the Max Planck Graduate Center with the Johannes Gutenberg‐University Mainz (MPGC). C.M. Berač is the recipient of a position through the DFG Excellence Initiative by the Graduate School Materials Science in Mainz (GSC 266).
A. Sarkar, J. C. Kölsch, C. M. Berač, A. Venugopal, R. Sasmal, R. Otter, P. Besenius, S. J. George, ChemistryOpen 2020, 9, 346.
Dedicated to Prof. Jean‐Marie Lehn on the occasion of his 80th birthday
Contributor Information
Prof. Dr. Pol Besenius, Email: besenius@uni-mainz.de.
Prof. Dr. Subi J. George, Email: george@jncasr.ac.in.
References
- 1.
- 1a. Aida T., Meijer E. W., Stupp S. I., Science, 2012, 335, 813–817; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Hendricks M. P., Sato K., Palmer L. C., Stupp S. I., Acc. Chem. Res. 2017, 50, 2440–2448; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Sato K., Hendricks M. P., Palmer L. C., Stupp S. I., Chem. Soc. Rev. 2018, 47, 7539–7551; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Ulijn R. V., Smith A. M., Chem. Soc. Rev. 2008, 37, 664–675; [DOI] [PubMed] [Google Scholar]
- 1e. Pashuck E. T., Cui H., Stupp S. I., J. Am. Chem. Soc. 2010, 132, 6041–6046; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Behanna H. A., Donners J. J. J. M., Gordon A. C., Stupp S. I., J. Am. Chem. Soc. 2005, 127, 1193–1200; [DOI] [PubMed] [Google Scholar]
- 1g. Cornelissen J. J. L. M., Donners J. J. J. M., de Gelder R., Graswinckel W. S., Metselaar G. A., Rowan A. E., Sommerdijk N. A. J. M., Nolte R. J. M., Science 2001, 293, 676–680; [DOI] [PubMed] [Google Scholar]
- 1h. Matmour R., De Cat I., George S. J., Adriaens W., Leclère P., Bomans P. H. H., Sommerdijk N. A. J. M., Gielen J. C., Christianen P. C. M., Heldens J. T., van Hest J. C. M., Löwik D. W. P. M., Feyter S. D., Meijer E. W., Schenning A. P. H. J., J. Am. Chem. Soc. 2008, 130, 14576–14583. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Pantos G. D., Wietor J. L., Sanders J. K. M., Angew. Chem. Int. Ed. 2007, 46, 2238–2240; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2288–2290; [Google Scholar]
- 2b. Wietor J.-L., Pantos G. D., Sanders J. K. M., Angew. Chem. Int. Ed. 2008, 47, 2689–2692; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2729–2732. [Google Scholar]
- 3.
- 3a. Shao H., Gao M., Kim S. H., Jaroniec C. P., Parquette J. R., Chem. Eur. J. 2011, 17, 12882–12885; [DOI] [PubMed] [Google Scholar]
- 3b. Shao H., Seifert J., Romano N. C., Gao M., Helmus J. J., Jaroniec C. P., Modarelli D. A., Parquette J. R., Angew. Chem. Int. Ed. 2010, 49, 7688–7691; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7854–7857; [Google Scholar]
- 3c. Shao H., Nguyen T., Romano N. C., Modarelli D. A., Parquette J. R., J. Am. Chem. Soc. 2009, 131, 16374–16376; [DOI] [PubMed] [Google Scholar]
- 3d. Shao H., Parquette J. R., Chem. Commun. 2010, 46, 4285–4287; [DOI] [PubMed] [Google Scholar]
- 3e. Tu S., Kim S. H., Joseph J., Modarelli D. A., Parquette J. R., J. Am. Chem. Soc. 2011, 133, 19125–19130. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Nalluri S. K. M., Berdugo C., Javid N., Frederix P. W. J. M., Ulijn R. V., Angew. Chem. Int. Ed. 2014, 53, 5882–5887; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5992–5997; [Google Scholar]
- 4b. Kumar M., Ing N. L., Narang V., Wijerathne N. K., Hochbaum A. I., V. Ulijn R., Nat. Chem. 2018, 10, 696–703. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Cross E. R., Sproules S., Schweins R., Draper E. R., Adams D. J., J. Am. Chem. Soc. 2018, 140, 8667–8670; [DOI] [PubMed] [Google Scholar]
- 5b. Ardoña H. A. M., Tovar J. D., Chem. Sci. 2015, 6, 1474; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Ardoña H. A. M., Draper E. R., Citossi F., Wallace M., Serpell L. C., Adams D. J., Tovar J. D., J. Am. Chem. Soc. 2017, 139, 8685–8692. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. da Silva R. M. P., v. d. Zwaag D., Albertazzi L., Lee S. S., Meijer E. W., Stupp S. I., Nat. Commun. 2016, 7, 11561; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Matmour R., De Cat I., George S. J., Adriaens W., Leclère P., Bomans P. H. H., Sommerdijk N. A. J. M., Gielen J. C., Christianen P. C. M., Heldens J. T., van Hest J. C. M., Löwik D. W. P. M., Feyter S. D., Meijer E. W., Schenning A. P. H. J., J. Am. Chem. Soc. 2008, 130, 14576–14583. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Frisch H., Unsleber J. P., Lüdeker D., Peterlechner M., Brunklaus G., Waller M., Besenius P., Angew. Chem. Int. Ed. 2013, 52, 10097–10101; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10282–10287; [Google Scholar]
- 7b. Frisch H., Nie Y., Raunser S., Besenius P., Chem. Eur. J. 2015, 21, 3304–3309; [DOI] [PubMed] [Google Scholar]
- 7c. Ahlers P., Frisch H., Besenius P., Polym. Chem. 2015, 6, 7245–7250; [Google Scholar]
- 7d. Spitzer D., Rodrigues L. L., Straßburger D., Mezger M., Besenius P., Angew. Chem. Int. Ed. 2017, 56, 15461–15465; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15664–15669; [Google Scholar]
- 7e. van Buel R., Spitzer D., Berač C. M., van der Schoot P., Besenius P., Jabbari-Farouji S., J. Chem. Phys. 2019, 151, 014902; [DOI] [PubMed] [Google Scholar]
- 7f. Berac C. M., Zengerling L., Straβburger D., Otter R., Urschbach M., Besenius P., Macromol. Rapid Commun. 2019, 1900476. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Krieg E., Bastings M. M. C., Besenius P., Rybtchinski B., Chem. Rev. 2016, 116, 2414–2477; [DOI] [PubMed] [Google Scholar]
- 8b. Chakraborty S., Berac C. M., Kemper B., Besenius P., Speck T., Macromolecules 2019, 52, 7661–7667. [Google Scholar]
- 9. Sarkar A., Dhiman S., Chalishazar A., George S. J., Angew. Chem. Int. Ed. 2017, 56, 13767–13771; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13955–13959. [Google Scholar]
- 10.
- 10a. Sakai N., Mareda J., Vauthey E., Matile S., Chem. Commun. 2010, 46, 4225–4237; [DOI] [PubMed] [Google Scholar]
- 10b. Würthner F., Ahmed S., Thalacker C., Debaerdemaeker T., Chem. Eur. J. 2002, 8, 4742–4750. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. George S. J., Tomović Ž, Schenning A. P. H. J., Meijer E. W., Chem. Commun. 2011, 47, 3451–3453; [DOI] [PubMed] [Google Scholar]; Tomović, Schenning A. P. H. J., Meijer E. W., Chem. Commun. 2011, 47, 3451–3453; [DOI] [PubMed] [Google Scholar]
- 11b. Cat I. D., Guo Z., George S. J., Meijer E. W., Schenning A. P. H. J., Feyter S. D., J. Am. Chem. Soc. 2012, 134, 3171–3177. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Schmid S. A., Abbel R., Schenning A. P. H. J., Meijer E. W., Herz L. M., Philos. Trans. R. Soc. London 2012, 370, 3787–3801. [DOI] [PubMed] [Google Scholar]
- 13. Garrey C. J., Schreiber S. L., Nat. Rev. Drug Discovery 2018, 17, 333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary