

Abstract
The pH dependence of the trypsin-catalyzed hydrolysis of N-α-benzyloxycarbonyl-l-lysine p-nitroanilide has been studied at 25 °C. kcat/KM was maximal at alkaline pH values but decreased with decreasing pH. kcat/KM was dependent on free enzyme pKa values of 6.75 ± 0.09 and 4.10 ± 0.13, which were assigned to the ionization of the active site histidine-57 and aspartate-189, respectively. Protonation of either group abolished catalytic activity. kcat is shown to equal the acylation rate constant k2 over the pH range studied. k2 decreased on the protonation of two groups with pKa values of 4.81 ± 0.15 and 4.23 ± 0.19. We assign the pKa of 4.23 to the ionization of the aspartate-189 residue and the pKa of 4.81 to the oxyanion of the tetrahedral intermediate formed during acylation. We conclude that during acylation, breakdown of the catalytic tetrahedral intermediate is rate-limiting and that there is a strong interaction between the imidazolium ion of histidine-57 and the oxyanion of the catalytic tetrahedral intermediate, which perturbs their pKa values. From the pH dependence of k3, we conclude that deacylation depends on a pKa of 6.41 ± 0.22 and that the ionization of the carboxylate group of aspartate-189 does not have a significant effect on the rate of deacylation (k3). A catalytic mechanism is proposed to explain the pH dependence of catalysis.
1. Introduction
Trypsin and trypsin-like serine proteases specifically catalyze the hydrolysis of peptide bonds involving the carbonyl carbon of the α-carboxylate group of the positively charged amino acid residues lysine or arginine. Trypsin is a serine protease involved in protein digestion. Due to its high specificity for positively charged amino acid residues trypsin is widely used for peptide sequencing in proteomics.1,2 Trypsin-like serine proteases are involved in a range of biological processes and diseases, e.g., the trypsin like enzyme urokinase plasminogen activator plays an important role in both fibrinolysis3 and cancer progression.4,5 In this study, we utilize pH studies to investigate the catalytic mechanism of trypsin.
Catalysis by the serine proteases can be described by the minimal three-step kinetic mechanism (eq 1) below
| 1 |
where ES is the Michaelis complex and ES′ is the acyl intermediate. Ks is the disassociation constant of the Michaelis complex (Ks = k–1/k+1 = [E][S]/[ES]). k2 and k3 are the first-order rate constants for acylation and deacylation, respectively. Catalysis obeys the Michaelis–Menten equation (d[P]/dt = kcat[E0][S0]/([S0] + KM)). The Michaelis parameters are complex assemblies of rate constants with kcat= k2k3/(k2 + k3) and KM = k3[(k–1 + k2)/k+1]/(k2 + k3), and if k–1 ≫ k2, then KM = k3Ks/(k2 + k3) and kcat/KM = k2/Ks. Therefore, the mechanistic significance of the pH dependence of these Michaelis parameters is often not clear. However, pioneering studies with chymotrypsin at pH 76 have shown that with ester substrates with good leaving groups (P1) deacylation (k3) is rate limiting and so
while with amide and anilide substrates with poor leaving groups (P1), acylation (k2) can be rate limiting with
Therefore, with highly reactive ester substrates, it should be possible to determine k3 from kcat values, while with less reactive amide or anilide substrates, it should be possible to determine k2 and Ks values from kcat and KM, respectively.
At pH 2.66, the reaction of trypsin with the reactive ester substrate Z-lys-pnp was slow enough for both k2 and k3 to be measured and it was shown that acylation was much more rapid than deacylation (k2/k3 = 27.6).7 However, it was found that at higher pH values, the ratio k2/k3 decreased and k2 was not much greater than k3.8,9 Therefore, it cannot be assumed that k2 is always very much greater than k3 with reactive ester substrates and so pH studies are essential if we are to fully understand the kinetics of catalysis at different pH values. The less reactive para-nitroanilide substrates are thought to be better models than the more reactive para-nitrophenol ester substrates for natural peptide substrates. However, a detailed kinetic analysis of the trypsin-catalyzed hydrolysis of the equivalent less reactive para-nitroanilide substrate Z-Lys-pna has not been carried out.
Therefore, in the present study a detailed study of the trypsin-catalyzed hydrolysis of the anilide substrate Z-Lys-pna has been undertaken. The effect of pH on the ratio k3/k2 and on values of KM and Ks has been quantified. Also, the effect of pH on kcat/KM and on the rates of acylation (k2) and deacylation (k3) has been determined. From these studies, the pKa values affecting catalysis in the free enzyme as well as during acylation and deacylation have been determined. In any pH study, it is difficult to dismiss the possibility that ionizations outside the active site can also affect catalytic activity. However, in this study, there is no evidence of this and all observed ionizations are assigned to active site groups.
The mechanistic significance of these results are discussed and a catalytic mechanism is proposed, which explains the pH dependence of catalysis. The mechanistic proposals and their background are briefly summarized in the following paragraph.
Some earlier studies10−14 have suggested that the pKa of the catalytic histidine is decreased during acylation (pKa < 7), while more recent studies15−23 have suggested that the histidine pKa must be raised (pKa > 11) so that it can be an effective general base catalyst during acylation and that the pKa of the oxyanion of the catalytic tetrahedral intermediate (THI) is lowered so that trypsin can be an effective enzyme at physiological pH values. In the present work, pKa values of 4.8 and 4.2 were detected from the pH studies of the acylation step of catalysis (k2 in eq 1). The pKa of 4.8 is assigned to the oxyanion of the catalytic tetrahedral intermediate formed during acylation and not to the pKa of the catalytic histidine that is assigned a pKa > 11 in ES and the tetrahedral intermediate. Aspartate-189 that binds the ε-amino group of the substrate in the S1 specificity pocket is assigned a pKa of 4.1–4.2 and is not significantly perturbed on substrate binding or on tetrahedral intermediate formation during acylation.
2. Results
2.1. Determination of the Catalytic Parameters kcat, kcat/KM, and kcat/KM at Different pH Values
The Michaelis parameters kcat,KM, and kcat/KM for the trypsin-catalyzed hydrolysis of Z-Lys-pna were determined by computer-fitting the initial rate data obtained at a given pH to the hyperbolic form of the Michaelis–Menten equation (d[P]/dt = Vm·[S0]/([S0] + KM)) using the method of Wilkinson.24
Examples of the experimental data and the fitted lines are given in Figure 1. The trypsin concentrations were increased at lower pH values to compensate for the decreases in kcat and kcat/KM. KM values increased as the pH decreased, and so higher substrate concentrations were used for determining KM values at lower pHs (Figure 1).
Figure 1.

Determination of the catalytic parameters for the trypsin-catalyzed hydrolysis of Z-Lys-pna at different pH values. Initial rates (d[P]/dt) were fitted to eq 1 (d[P]/dt = Vm·[S0]/([S0] + KM)). The pH, trypsin concentration, and the fitted values of Vm, Vm/KM, and KM were: (A) pH 3.13, 83.5 μM, 0.0768 ± 0.0207 μM s–1, 2.22 ± 0.9 μs–1, 34.6 ± 10.5 mM; (B) pH 3.82, 25.6 μM, 0.388 ± 0.094 μM s–1, 15.8 ± 7.0 μs–1, 24.5 ± 9.2 mM; (C) pH 4.40, 4.68 μM, 0.444 ± 0.037 μM s–1, 32.3 ± 5.7 μs–1, 13.8 ± 2.1 mM; (D) pH 5.98, 0.39 μM, 0.344 ± 0.029 μM s–1, 52.8 ± 8.9 μs–1, 6.51 ± 0.95 mM; (E) pH 6.94, 0.427 μM, 0.202 ± 0.005 μM s–1, 395 ± 32 μs–1, 0.512 ± 0.039 mM; (F) pH 9.05, 0.379 μM, 0.182 ± 0.005 μM s–1, 463 ± 34 μs–1, 0.394 ± 0.027 mM.
2.2. Effect of pH on kcat/KM for the Trypsin-Catalyzed Hydrolysis of Z-Lys-pna
kcat/KM values were determined by dividing Vm/KM values by the enzyme concentration in the reaction mixture. kcat/KM values were dependent on the sequential ionization of two groups (kcat/KM = (kcat/KM)max/(1 + [H+]/Kb + [H+]2/Ka·Kb)): one with a pKa 6.75 ± 0.09 and the other pKb = 4.10 ± 0.13 (Figure 2). The pH dependence of kcat/KM reflects ionizations in either the free enzyme or the free substrate. Previous studies with other substrates have detected similar free enzyme pKa values of ∼7 and ∼4.9,25−30
Figure 2.

Effect of pH on log kcat/KM for the trypsin-catalyzed hydrolysis of Z-Lys-pna at 25 °C. The solid line was calculated using the equation kcat/KM = (kcat/KM)max/(1 + [H+]/Kb + [H+]2/Ka·Kb), and the fitted values of (kcat/KM)max = 1391 ± 72 M–1·s–1, pKa = 4.10 ± 0.13 and pKb = 6.75 ± 0.09.
2.3. Calculation of k3 for the Trypsin-Catalyzed Hydrolysis of Z-Lys-pnp and Z-Lys-pna
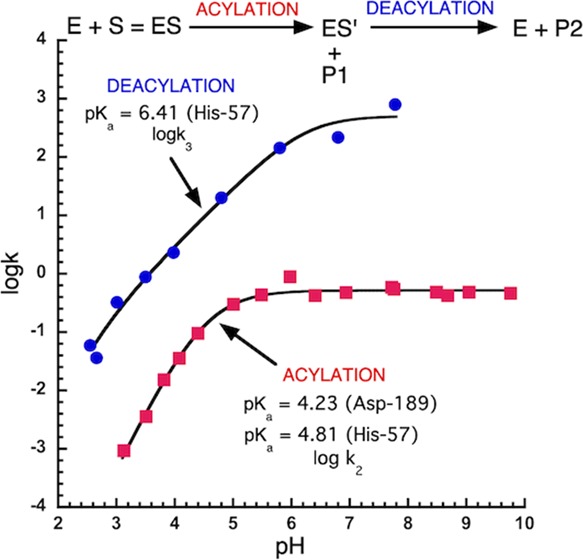
The trypsin-catalyzed hydrolysis of both Z-Lys-pna and Z-Lys-pnp proceeds via a common acyl intermediate (Z-Lys-trypsin, ES′ in eq 1 and structure d1 in Scheme 1). Therefore, both will have the same deacylation rate constant (k3). Using pre-steady-state kinetics, the k2 values for the trypsin-catalyzed hydrolysis of Z-Lys-pnp have been determined.25 Steady-state kinetics have been used to determine kcat values for Z-Lys-pnp.25kcat = k2k3/(k2 + k3), and this equation can be rearranged to give the equation, k3 = k2kcat/(k2 – kcat), which has been used to calculate the k3 values (line 1 in Figure 3A) for Z-Lys-pna and Z-Lys-pnp from the experimentally determined values of k2 and kcat for Z-Lys-pnp.25 The experimental data was fitted to the equation, k3 = k3max/([1 + [H+]/Kb]), assuming k3 was dependent on a single ionizing group (AH = A– + H+) and the fitted values were k3 = 561 ± 104 s–1 and pKa = 6.41 ± 0.22 (Figure 3A, line 1).
Scheme 1. Catalytic Mechanism of Trypsin.

E and S are the free enzyme and substrate. ES is the enzyme substrate complex or the Michaelis complex. THI is the catalytic tetrahedral intermediate, and ES′ is the acyl enzyme intermediate. P1 (R′NH2) and P2 (RCOOH) are the first and second products, respectively, of the trypsin-catalyzed hydrolysis of the peptide RCONHR′.
Figure 3.

pH dependence of the catalytic parameters kcat (k2), k3 and the ratio k3/k2 for the trypsin-catalyzed hydrolysis of Z-Lys-pna at 25 °C. (A) (1) The filled circles are k3 values calculated using Z-Lys-pnp data20 (see text for further details). The solid line was calculated using the equation k3 = k3max/([1 + [H+]/Kb]), and the fitted values of k3max = 561 ± 104 s–1, pKb = 6.41 ± 0.22. (2) The filled squares are kcat values (kcat = k2) for Z-Lys-pna, and the solid line was calculated using the equation kcat = (kcat)max/([1 + [H+]/Kb + [H+]2/Ka·Kb]) and the fitted values of (kcat)max = 0.517 ± 0.014. s–1, pKa = 4.23 ± 0.19 and pKb = 4.81 ± 0.15. (B) The ratio k3/k2 was calculated using the fitted values for k2 and k3 determined in (A). For k3, k3max = 561 s–1, pKa = 6.41. For k2, k2max = 0.517 s–1, pKa = 4.23, pKb = 4.81.
2.4. Determination of kcat and k2 for the Trypsin-Catalyzed Hydrolysis of Z-Lys-pna
In the present work, kcat values have been determined for Z-Lys-pna from pH 3.1–9.8 (Figure 3A, line 2). kcat = k2k3/(k2 + k3), which can be rearranged to give k2 = k3kcat/(k3 – kcat). As k3 is the same for both Z-Lys-pnp and Z-Lys-pna, we can use the fitted parameters obtained from the pH dependence of k3 for Z-Lys-pnp to calculate the value of k2 for Z-Lys-pna for each value of kcat determined for Z-Lys-pna from pH 3.1–9.8. Therefore, as k3 and kcat are known for Z-Lys-pna, k2 values could be calculated for Z-Lys-pna. However, this calculation was not necessary to calculate k2 because k3 was 2–3 orders of magnitude greater than k2 (Figure 3B) and so within the experimental error (<2%) kcat = k2. Therefore, kcat values for the trypsin-catalyzed hydrolysis of Z-Lys-pna can be assumed to equal k2 (Figure 3A, line 2). Experimental kcat data (kcat = k2) was fitted to the equation kcat = (kcat)max/(1 + [H+]/Kb + [H+]2/Ka·Kb) assuming kcat was dependent on the sequential ionization of two ionizing groups (line 2 in Figure 3A). We conclude that k2 has a maximal value of 0.517 ± 0.014 s–1 and is dependent on the sequential ionization of two ionizing groups (line 2 in Figure 3A) with pKa values of 4.81 ± 0.15 and the 4.23 ± 0.19 (Table 1).
Table 1. Catalytic Parameters for the Trypsin-Catalyzed Hydrolysis of Z-Lys-pna.
| parameter | kmax | pKa | pKb | Ksa | Ksb | Ksc |
|---|---|---|---|---|---|---|
| kcat/KM (M–1 s–1)a | 1390 ± 72 | 4.10 ± 0.13 | 6.75 ± 0.09 | |||
| k2 (s–1)a | 0.517 ± 0.014 | 4.23 ± 0.19 | 4.81 ± 0.15 | |||
| k3 (s–1)a | 561 ± 104 | 6.41 ± 0.22 | ||||
| Ks (mM)b | 4.23 | 4.82 | 24.0 | 32.2 | 0.373 | |
| 1/Ks (mM–1)b | 4.10 | 6.75 | 24.0 | 32.4 | 0.372 |
Errors are standard errors of the fitted parameters.
The Ks values used to determine pKa, pKb, Ksa, Ksb, and Ksc were calculated from the fitted values of kcat and kcat/KM (see text for details).
2.5. Effect of pH on the Ratio k3/k2 for the Trypsin-Catalyzed Hydrolysis of Z-Lys-pna
The fitted values of k2 and k3 (lines 1 and 2 in Figure 3A) were used to calculate the ratio of k3/k2 from pH 2.6 to 9.8 (Figure 3B). The ratio k3/k2 was dependent on pH having a minimal value of ∼55 at pH 4.5 but reaching values of 1200 and 1080 at pH 2.6 and 9.8, respectively (Figure 3B). Therefore, for the trypsin-catalyzed hydrolysis of Z-Lys-pna, k3 ≫ k2 from pH 2.6 to 9.8 and so k2 is rate limiting for Z-Lys-pna and the expression for KM (KM = Ks·k3/(k2 + k3)) simplifies to KM = Ks from pH 2.6 to 9.8. Therefore, for Z-lys-pna, KM values are equal to Ks values.
2.6. Determination of Ks and its pH Dependence
Analyzing the KM versus pH data for dependence on one pKa (equation Ks(obs) = Ksa/(1 + Ka/[H+]) + Ksb/(1 + [H+]/Ka)) gives a minimum Ks value of 0.333 ± 0.025 mM and a maximum Ks value of 23.5 ± 1.9 mM, with a pKa of 5.12 ± 0.17 (Figure 4A). For optimal accuracy when determing KM using the Michaelis–Menten equation, substrate concentrations should ideally be in the range KM/5 to 5 × KM to optimize accuracy. However, the maximum concentration of Z-Lys-pna was ∼20 mM and so at low pHs, substrate concentrations were similar to the KM values and therefore accurate KM values could not be determined (Figure 4A). In contrast, the experimental data for kcat/KM (Figure 2) and kcat (k2 in Figure 3) were both in good agreement with the fitted lines. This is because both kcat and kcat/KM decrease rapidly and do not level off like KM values to a fixed value. Consequently, kcat and kcat/KM values cover a larger range of values than KM values. The log plots of kcat/KM and kcat will reflect two pKa values, pKE1 and pKE2 for kcat/KM and pKES1 and pKES2 for kcat. In contrast, a plot of KM versus pH should give pKa values of 4.2 and 4.8 (Table 1). However, the experimental KM data (Figure 4A) is not good enough to resolve these two pKa values and determine the pH-independent values of Ks.
Figure 4.

Effect of pH on Ks. (A) Ks values are the experimentally determined KM values determined by fitting initial rate values to the Michaelis–Menten equation, as described in the Experimental Section. The solid line was calculated using the equation Ks(obs) = Ksa/(1 + Ka/[H+]) + Ksb/(1 + [H+]/Ka) and the fitted values of Ksa = 23.5 ± 1.9 mM, Ksb = 0.333 ± 0.025 mM, and pKa = 5.12 ± 0.17. (B) Ks values (solid circles) were calculated at 0.1 pH intervals by dividing the fitted values for k2 (Figure 3A) by the fitted values for kcat/KM (Figure 2), kcat/KM = k2/Ks. The solid line was calculated using the equation Ks(obs) = (Ksa·[H+]2 + Ksb·Ka·[H+] + Ksc·Ka·Kb)/([H+]2 + Ka[H+] + Ka·Kb) and the fitted values Ksa = 24.0 mM, Ksb = 32.2 mM, Ksc = 0.373 mM, pKa = 4.23, pKb = 4.82. (C) 1/Ks values (solid circles) were calculated at 0.1 pH intervals using the values of Ks in (B). The solid line was calculated using the equation (1/Ks(obs)) = {(1/Ksa)·[H+]2 + (1/Ksb)·Ka·[H+] + (1/Ksc)·Ka·Kb}/([H+]2 + Ka·[H+] + Ka·Kb) and the fitted values Ksa = 24.0 mM, Ksb = 32.4 mM, Ksc = 0.372 mM, pKa = 6.75, pKb = 4.10.
Therefore, to obtain KM (KM = Ks) values consistent with the fitted values of kcat (solid line 2 in Figure 2A) and kcat/KM (solid line in Figure 1), KM values were calculated using these fitted values and the equation KM = kcat × KM/kcat. The values of Ks (Ks = KM) were calculated at 0.1 pH intervals from pHs 2.6–9.8 (solid circles in Figure 4B).
To determine the pH-independent Ks values (Ksa, Ksb, and Ksc in Scheme 1), Ks values (Ks = KM) were fitted (Figure 4B) to the equation for a doubly ionizing system (Ks(obs) = (Ksa·[H+]2 + Ksb·Ka·[H+] + Ksc·Ka·Kb)/([H+]2 + Ka·[H+] + Ka·Kb)) and this gave three pH-independent Ks values of 24 mM (Ksa), 32.2 mM (Ksb), and 0.373 mM (Ksc) (Scheme 1 and Table 1). The fitted values of pKa and pKb determined from the pH dependence of Ks had values of 4.23 and 4.82, in good agreement with the pKa values obtained from the pH dependence of k2 (Table 1). This is expected as the pH dependence of Ks (or KM) like kcat reflects ionizations within the ES complex or THI intermediate (Scheme 1). The pH dependence of 1/Ks like that of kcat/KM reflects ionizations in the free enzyme. Fitting the 1/Ks values (Figure 4C) to a doubly ionizing system gave pKa values of 6.75 and 4.1, which, as expected, were the same as observed for the pH dependence of kcat/KM (Table 1), which also reflects ionizations in the free enzyme. The agreement of the pKa values determined from the pH dependence of Ks and 1/Ks with the pKa values obtained from the pH dependence of kcat and kcat/KM, respectively, confirms that this approach should allow the determination of estimates of the pH-independent Ks values Ksa, Ksb, and Ksc that are consistent with the experimentally determined values of kcat and kcat/KM used to calculate the Ks values used. This is supported by the fact that the pH-independent Ks values Ksa, Ksb, and Ksc obtained from the pH dependence of Ks and 1/Ks were essentially the same (Table 1).
3. Discussion
The pH dependence of kcat/KM showed that the active group was A2– in eq 2 and that it was inctivated by protonation to form AH– and AH2 (eq 2)
| 2 |
The free enzyme pKa values 6.75 ± 0.09 and pKb = 4.10 ± 0.13 obtained from the pH dependence of kcat/KM and 1/Ks (Table 1) have been attributed to the trypsin residues histidine-57 and aspartate-189, respectively.9,25−30 In both cases, it is the ionized form of these groups that are catalytically active (structure a1 in Scheme 1). Histidine-57 is part of the catalytic triad and its ionized form acts as a general base catalyst during catalysis (b1 in Scheme 1). Once it is protonated (AH–, eq 2), it can no longer act as a general base catalyst for tetrahedral intermediate formation (b1 in Scheme 1) and so catalysis is inhibited. However, if the histidine pKa is raised in the ES complex to a value >11 (structures b1 and b2 in Scheme 1) so that it can be an effective general base catalyst for tetrahedral intermediate formation (structures b1 to c1 in Scheme 1), then this pKa will not be observed in our pH studies from pH 3–10. Protonation of histidine-57 in the free enzyme (structures a1 to a2 in Scheme 1) is also known to decrease substrate and inhibitor binding and so this should also contribute to substrate catalysis being inhibited with a pKa of 6.75.22,31,32
The negatively charged side chain carboxylate group of the aspartate-189 residue is located at the bottom of the S1 specificity site where it can form an ion pair with the positively charged side chains of lysine or arginine substrates. pKa values of 7.1 and 4.55 have been obtained from the pH dependence of k2/Ks for the p-nitrophenol substrate Z-Lys-pnp with trypsin.25 With the neutral substrate Z-Ala-pnp, no substrate interaction with aspartate-189 is expected and so the fact that only one pKa value of 6.9 was obtained from the pH dependence of k2/Ks with this neutral substrate25 confirms the assignment of the pKa value of ∼4 to aspartate-189 in the free trypsin. Likewise, the fact that pH studies of k2 showed that the pKa of ∼4 was detected with the lysine substrate Z-Lys-pnp but not with the neutral substrate Z-Ala-pnp also confirms the assignment of pKa of ∼4 to aspartate-189 in the trypsin ES complex with Z-Lys-pnp.25 Therefore, the pKa values of 4.1 and 4.23 obtained from the pH dependence of kcat/KM and k2 in the present study with Z-lys pna are assigned in the same way to apartate-189 in free trypsin and to the trypsin ES complex or the trypsin tetrahedral intermediate (THI in Scheme 1), respectively (Table 1).
The protonation of a group with a pKa of 4.1 in the free enzyme and 4.23 in ES and or the trypsin tetrahedral intermediate (THI in Scheme 1) decreases the Ks from 32 to 24 mM (Table 1). Therefore, if this pKa of 4.10–4.2 is due to ion pair formation between the substrate’s lysine side chain and aspartate-189, then ion pair formation does not increase substrate binding but instead leads to a small decrease in binding by increasing Ks from 24 to 32 mM. Small positively charged guanidines or alkylamines inhibit the hydrolysis of positively charged substrates by trypsin and they enhance the hydrolysis of neutral substrates by increasing kcat while KM undergoes minimal perturbation.25,33,34 This led to the suggestion that positively charged ligands and substrates activate trypsin by binding in the S1 specificity site.33 Structural studies have confirmed that positively charged ligands binding to aspartate-189 in the S1 specificity pocket induce conformational changes that activate trypsin.35−38 Replacing aspartate-189 with serine resulted in a a small 2–6-fold change in KM but a much larger, ∼100 000, decrease in kcat/KM, confirming that the primary role of lysine and arginine substrates binding to aspartate-189 is to activate trypsin.39 Therefore, these results show that the binding energy between the carboxylate group of aspartate-189 and the positively charged side chains of lysine or arginine substrates is mainly used to activate trypsin and has a minimal effect on Ks. This explains why protonation of aspartate-189 (pKa 4.1–4.2) has such a small effect on Ks (42.4–24.0 mM, Table 1) and yet such a large effect on k2, resulting in the stoichiometric inhibition of trypsin (line 2 in Figure 3A).
With the positively charged Z-Lys-pna substrate, an additional pKa of 4.81 was obtained from the pH dependence of the acylation rate constant k2 (eq 1). Protonation of this group led to an ∼86-fold increase in Ks from 0.37 to 32.4 mM (Table 1). A similar ∼86-fold increase in Ks was observed when histidine-57 was protonated in the free enzyme with a pKa of 6.75 (Table 1), which would appear to suggest that the pKa of histidine-57 changes from 6.75 in the free enzyme to 4.82 in the tetrahedral intermediate adduct (pKTHI1 in Scheme 1) and that the ∼86-fold decrease in KM is due to binding energy being used to lower the pKa of histidine-57 from 6.75 to 4.82. It also shows that the protonation state of histidine-57 has a major role in substrate binding. An ∼3-fold increase in KM below pH 7 was also observed with N-benzyloxycarbonyl-l-arginine-p-toluidide and trypsin,40 but in this case, the decrease in histidine pKa was much smaller (pKa 6.38). Larger 13-fold increases in KM values have also been observed when histidine-57 in chymotrypsin is protonated with hydrazide substrates.41 It has been suggested that these decreases in binding were due to the positively charged histidine-57 interacting with the leaving group amine.13 As expected with trypsin and neutral p-nitrophenol substrates that do not have a leaving group amine, there is no increase in Ks when histidine-57 is protonated.25 However, with chymotrypsin, decreases in inhibitor binding at low pH have also been observed with 2-p-toluidinylnaphthaline-6-sulfonate,32 proflavin,31 and peptide-derived glyoxal inhibitors.22 This suggests that a neutral imidazole group of the histine-57 residue is required for optimal binding of these substrates and inhibitors.
There is a considerable body of evidence that shows that the serine proteases stabilize zwitterionic tetrahedral adducts that mimic the the catalytic zwitterionic tetrahedral intermediate. These zwitterionic tetrahedral adducts refer to the negatively charged oxyanion of the tetrahedral intermediate and the positively charged imidazolium ion of histidine-57 and ignore all other charged groups on the enzyme. In these zwitterionic tetrahedral adducts, the pKa of the majority of histidine-57 (analogous to pKTHI2 in Scheme 1) is raised16,18,20−22,42 and the pKa (analogous to pKTHI1 in Scheme 1) of the majority of the oxyanion is lowered.16,18,19,22,43,44 Specific peptide-derived glyoxal inhibitors are tightly bound as neutral zwitterionic tetrahedral complexes (structure c1 in Scheme 1), which are thought to mimic the catalytic tetrahedral intermediate.22,43,45 In these zwitterionic glyoxal complexes, 1H NMR and 13C NMR have been used to show that the pKa of histidine-57 is >11 and the oxyanion pKa is ∼4.22,43 The increase in Ki values at low pH was dependent on a pKa of ∼4, and this pKa was assigned to protonation of the oxyanion in the glyoxal-chymotrypsin tetrahedral adduct.22 Therefore, if the breakdown of the zwitterionic tetrahedral intermediate in the acylation step of the trypsin-catalyzed hydrolysis of Z-Lys-pna (species c1 in Scheme 1) is rate-limiting, then the pKa of 4.81 (Table 1) in trypsin catalysis should be assigned to protonation of the oxyanion in the catalytic zwitterionic tetrahedral intermediate (pKTHI1 in Scheme 1). It also shows that tight binding of inhibitors and substrates is possible when histidine-57 is protonated provided the oxyanion is also present to neutralize the charge on the imidazolium ion of histidine-57 (ImH+ O–). If the oxyanion is protonated (ImH+ OH), the charge on the protonated imidazolium ion will no longer be neutralized by the oxyanion and so the positively charged imidazolium ion of histidine-57 will inhibit binding. Therefore, Ki values will increase as the oxyanion is protonated, as observed with peptide glyoxal inhibitors that bind to chymotrypsin as zwitterionic tetrahedral adducts mimicking the catalytic tetrahedral intermediate.22 This also explains why Ks values with trypsin increase by essentially the same amount when histidine-57 is protonated (1/Ks in Table 1) in the free enzyme (a1 to a2 in Scheme 1) and also in the acylation complex (Ks in Table 1) when the oxyanion of the zwitterionic tetrahedral intermediate is protonated (structures c1 to c2 in Scheme 1).
It is generally accepted that the nucleophilicity of the hydroxyl group of serine-195 is enhanced by the imidazole group of histidine-57 acting as a general base catalyst (structure b1 in Scheme 1). It has been argued that as the pKa of the serine hydroxyl group is ∼15,15,16,22,23,42,46 then for general base catalysis by histidine to be effective (structure b1 in Scheme 1), its pKa should raised to a similar value of ∼15 on forming the enzyme substrate complex (ES in Scheme 1)15,16,22,23 and not lowered to a value <7.
Earlier studies on the pH dependence of chymotrypsin catalysis appeared to contradict these results as it appeared that the pKa of histidine-57 had been decreased to a value <7 within the ES complex.10−14 The reassignment of this pKa to the oxyanion in the present work resolves this contradiction. As protonation of the oxyanion inhibits catalysis, it is essential that the oxyanion pKa is reduced to ensure the enzyme is catalytically active at physiological pH values. This therefore explains why the serine proteases have evolved to lower the oxyanion pKa so effectively.16,18,19,22,43
When an interaction occurs between two ionizing groups, four species would be formed with four microscopic pKa values (c1, c2, c3, c4 in Scheme 1). So, for example, if in Scheme 1 the concentrations of all species (c1, c2, c3, c4 in Scheme 1) are equal, then 50% of histidine-57 (pKTHI4 in Scheme 1) and 50% of the oxyanion (pKTHI1 in Scheme 1) will both have pKa values of 4.8. Likewise, 50% of histidine-57 (pKTHI2 in Scheme 1) and 50% of the oxyanion (pKTHI3 in Scheme 1) will have pKa values of >11. So this interaction could explain how histidine-57 could have microscopic pKa values of both ∼4.8 (pKTHI4) and >11 (pKTHI2) in the THI (Scheme 1). Likewise, the oxyanion could have microscopic pKa values of ∼4.8 (pKTHI1) and >11 (pKTHI3) in the THI (Scheme 1). However, the serine proteases preferentially stabilize zwitterionic terahedral intermediates (structure c1 in Scheme 1) and so it is expected that at least 99% of the oxyanion and the imidazolium ion of histidine-57 will have pKa values of ∼4.8 and >11, respectively.
The deacylation rate constant (k3, in eq 1) was dependent on a singly ionizing group (AH = A–) with a pKa = 6.41 ± 0.22, which we assign to histidine-57. Therefore, we can conclude that the ionization of aspartate-189 does not appear to have a significant effect on the pH dependence of deacylation.
3.1. Mechanism for the pH Dependence of the Trypsin-Catalyzed Hydrolysis of Z-Lys-pna
The proposed mechanism is summarized in Scheme 1. In the free enzyme (a1, a2, and a3 in Scheme 1), the imidazole group of histidine-57 has a pKa of 6.75 and the carboxylate group of aspartate-189 (Scheme 1) that binds the side chains of the lysine or arginine residues of substrates or inhibitors has a pKa of 4.1. Binding of the substrate to form the enzyme–substrate complex (ES in Scheme 1) causes a strong interaction between the carboxylate group of aspartate-102 of the catalytic triad and the imidazolium group of histidine-57,16,18,22,47 which perturbs the pKa values of these groups to values <3 and >11, respectively, so they are no longer detected in pH studies. This increase in the pKa (pKa > 11) of the imidazolium group of the histidine-57 enables it to act as an effective general base catalyst (structure b1 in Scheme 1) for deprotonation of the hydroxyl group of the catalytic serine group (pKa ∼ 15), promoting its reaction with the substrate to form a zwitterionic tetrahedral intermediate (structure c1 in Scheme 1). The positively charged imidazolium group of histidine-57 helps lower the pKa of the oxyanion stabilizing the zwitterionic tetrahedral intermediate. A small reorientation the imidazole ring48 will then allow histidine-57 to act as general base catalyst for breakdown of the tetrahedral intermediate (structure c1 in Scheme 1) to form the acyl intermediate (structure d1 in Scheme 1). The rate-limiting step in acylation (k2) is this breakdown of this zwitterionic tetrahedral intermediate (structures c1 to d1 in Scheme 1). Protonation of the oxyanion with a pKa value of 4.8 (pKTHI1 in Scheme 1) will decrease the concentration of the catalytic zwitterionic intermediate (c1 in Scheme 1), inhibiting catalysis, and so k2 decreases with a pKa of 4.8. Protonation of aspartate-189 (pKTHI2 in Scheme 1) with a pKa of 4.2 disrupts its ion pair interaction with the ε-amino group of the substrate deactivating trypsin and promoting substrate disassociation from trypsin. This inhibits catalysis, and so k2 also decreases with a pKa of 4.2.
Deacylation (k3 step in Scheme 1) like acylation is also expected to proceed via a tetrahedral intermediate(not shown in Scheme 1). The imidazole group of histidine-57 has the potential to act as a general base catalyst for formation of the zwitterionic tetrahedral intermediate and as a general acid catalyst for its breakdown. As the rate of deacylation (k3) increases with increasing pH with a pKa of 6.4 (Figure 3A), this suggests the imidazole group is acting as a general base catalyst for tetrahedral intermediate formation. However, as water has a pKa value of 15.74, an imidazole group with a pKa of 6.4 would not be expected to be an effective general base catalyst. Aldehyde inhibitors of chymotrypsin form zwitterionic tetrahedral adducts with chymotrypsin where the oxyanion has a pKa ∼ 7 and histidine-57 has a pKa ∼ 8.49 It has been argued that the histidine pKa of ∼8 can be explained by the fact that these inhibitor adducts of chymotrypsin-like acyl intermediates do not have a large leaving group that can fix the catalytic histidine in position and raise its pKa > 11.49 If breakdown of this zwitterionic tetrahedral intermediate is rate limiting for deacylation, then another possibility is that the pKa of 6.4 reflects formation of the oxyanion of the zwitterionic tetrahedral intermediate as proposed for the acylation reaction. However, as the carboxylate group of the product of deacylation (P2) is a good leaving group, formation and not breakdown of the zwitterionic tetrahedral intermediate is expected to be rate limiting in the deacylation reaction. Also, as there is a very good leaving group in the deacylation step, a concerted reaction becomes a possibility.17
4. Experimental Section
4.1. Materials
Trypsin (type III, 2× crystalized, salt free from bovine pancreas) and all other reagents were obtained from Sigma-Aldrich Chemical Co., Gillingham, Dorset, U.K. Trypsin was 72% fully active by active site titration with p-nitrophenyl p-guanidobenzoate,50 as described by Malthouse et al.19 Z-Lys-pna was synthesized as described by Mackenzie et al.51
4.2. Kinetic Studies
The trypsin -catalyzed hydrolysis of Z-Lys-pna was studied at 25 °C in 3 mL volumes and 0.1 M ionic strength buffers.52 The buffers used were pH 3.13–4.38 (sodium formate), pH 4.40–5.49 (sodium acetate), 5.98–7.72 (potassium phosphate), 7.76–8.68 (Tris–HCl), pH 9.05 (sodium borate), and pH 9.76 (sodium carbonate). pH measurements were made either with a Radiometer combination electrode (GK 2401C) or by using a Beckman combination electrode model number 39522. Stock solutions of Z-Lys-pna were made up in 1 mM HCl (maximum solubility ∼18 mM) and quantified using E314 = 13 900 M–1·cm–1, as described by Mackenzie et al.51 Primary stock solutions of ∼1 mM fully active trypsin were prepared in 1 mM HCl, and this solution was diluted in 1 M HCl to prepare appropriate concentrations for the pH studies. A typical assay contained 1 mL of buffer (I = 0.3 M), 1.9–x mL of 1 mM HCl, and x mL of substrate in 1 mM HCl. Different substrate concentrations were obtained by adding different amounts of the stock substrate (x mL). Catalysis was initiated by adding 0.1 mL trypsin in 1 mM HCl. The concentration of trypsin was kept constant when determining the catalytic parameters at a given pH. However, fully active enzyme concentrations were increased (0.35–83.6 μM) as the pH was decreased to help compensate for the decrease in kcat and kcat/KM as the pH decreased. The fully active trypsin concentrations used in the assays were 11–84 μM (pH 3.1–5.0), 3.7–4.4μM (pH 4.4–5.5), and 0.35–0.39μM (pH 6.0–9.8). Initial rates of hydrolysis of Z-Lys-pna were followed by determining the amount of p-nitroaniline (E410 = 8800 M–1·cm–1) released over a 5–15 min period. It was ensured that there was always at least a 15-fold excess of the substrate over enzyme when initial rates were determined. kcat/KM data at pHs 3.56 and 4.38 were determined from initial rates obtained when [S0] ≪ KM. The effect of pH on the catalytic parameters was determined by fitting experimental data to the appropriate function as described by Cleland.53
Acknowledgments
The author is indebted to Dr. N.E. Mackenzie who synthesized and kindly provided the Z-Lys-pna used in these studies. This work was supported by University College Dublin and the Science and Engineering Research Council (U.K.).
Glossary
Abbreviations
- Z
benzyloxycarbonyl
- pna
p-nitroanilide
- pnp
p-nitrophenyl ester
Accession Codes
The UniProtKB accession ID for bovine trypsin is P00760.
The author declares no competing financial interest.
References
- Dapic I.; Baljeu-Neuman L.; Uwugiaren N.; Kers J.; Goodlett D. R.; Corthals G. L. Proteome analysis of tissues by mass spectrometry. Mass Spectrom. Rev. 2019, 38, 403–441. 10.1002/mas.21598. [DOI] [PubMed] [Google Scholar]
- Yang H.; Li Y. C.; Zhao M. Z.; Wu F. L.; Wang X.; Xiao W. D.; Wang Y. H.; Zhang J. L.; Wang F. Q.; Xu F.; Zeng W. F.; Overall C. M.; He S. M.; Chi H.; Xu P. Precision De Novo Peptide Sequencing Using Mirror Proteases of Ac-LysargiNase and Trypsin for Large-scale Proteomics. Mol. Cell. Proteomics 2019, 18, 773–785. 10.1074/mcp.TIR118.000918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S.; Acharya S. S. Inherited disorders of the fibrinolytic pathway. Transfus. Apheresis Sci. 2019, 58, 572–577. 10.1016/j.transci.2019.08.007. [DOI] [PubMed] [Google Scholar]
- Pusina S. Correlation of Serum Levels of Urokinase Activation Plasminogen (uPA) and Its Inhibitor (PAI-1) with Hormonal and HER-2 Status in the Early Invasive Breast Cancer. Med. Arch. 2018, 72, 335–340. 10.5455/medarh.2018.72.335-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. C.; Lin C. W.; Yang W. E.; Fan W. L.; Yang S. F. The urokinase-type plasminogen activator (uPA) system as a biomarker and therapeutic target in human malignancies. Expert Opin. Ther. Targets 2016, 20, 551–566. 10.1517/14728222.2016.1113260. [DOI] [PubMed] [Google Scholar]
- Zerner B.; Bond P. M.; Bender M. L. Kinetic Evidence for the Formation of Acyl-Enzyme Intermediates in the alpha-Chymotrypsin-Catalysed Hydrolysis of Specific Substrates. J. Am. Chem. Soc. 1964, 86, 3674–3679. 10.1021/ja01072a016. [DOI] [Google Scholar]
- Bender M. L.; Kezdy F. J.; Feder J. Kinetics of Trypsin-Catalyzed Hydrolysis of P-Nitrophenyl Alpha-N-Benzyloxycarbonyl-L-Lysinate Hydrochloride. J. Am. Chem. Soc. 1965, 87, 4953–4954. 10.1021/ja00949a048. [DOI] [PubMed] [Google Scholar]
- Ascenzi P.; Menegatti E.; Bortolotti F.; Guarneri M.; Antonini E. Steady-state and pre-steady-state kinetics of the trypsin-catalysed hydrolysis of alpha-CBZ-L-lysine-p-nitrophenyl ester. Biochim. Biophys. Acta 1981, 658, 158–164. 10.1016/0005-2744(81)90259-X. [DOI] [PubMed] [Google Scholar]
- Malthouse J. P. G.; Scott A. I. Cryoenzymology of trypsin. A detailed kinetic study of the trypsin-catalysed hydrolysis of N-alpha-benzyloxycarbonyl-L-lysine p- nitrophenyl ester at low temperatures. Biochem. J. 1983, 215, 555–563. 10.1042/bj2150555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker L.; Wang J. H. On the mechanism of action at the acylation step of the alpha-chymotrypsin-catalyzed hydrolysis of anilides. J. Biol. Chem. 1968, 243, 3729–3734. [PubMed] [Google Scholar]
- Inagami T.; Patchornik A.; York S. S. Participation of an acidic group in the chymotrypsin catalysis. J. Biochem. 1969, 65, 809–819. 10.1093/oxfordjournals.jbchem.a129081. [DOI] [PubMed] [Google Scholar]
- Caplow M. Chymotrypsin catalysis. Evidence for a new intermediate. J. Am. Chem. Soc. 1969, 91, 3639–3645. 10.1021/ja01041a037. [DOI] [PubMed] [Google Scholar]
- Fersht A. R.; Requena Y. Mechanism of the alpha-chymotrypsin-catalyzed hydrolysis of amides. pH dependence of kc and Km. Kinetic detection of an intermediate. J. Am. Chem. Soc. 1971, 93, 7079–7087. 10.1021/ja00754a066. [DOI] [PubMed] [Google Scholar]
- Zeeberg B.; Caswell M.; Caplow M. Concerning a reported change in rate-determining step in chymotrypsin catalysis. J. Am. Chem. Soc. 1973, 95, 2734–2735. 10.1021/ja00789a081. [DOI] [PubMed] [Google Scholar]
- Blow D. M. Structure and Mechanism of Chymotrypsin. Acc. Chem. Res. 1976, 9, 145–152. 10.1021/ar50100a004. [DOI] [Google Scholar]
- Finucane M. D.; Malthouse J. P. G. A study of the stabilization of tetrahedral adducts by trypsin and delta-chymotrypsin. Biochem. J. 1992, 286, 889–900. 10.1042/bj2860889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4523. 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- Liang T. C.; Abeles R. H. Complex of a-Chymotrypsin and N-Acetyl-L-leucyl-L-phenylalanyl Trifluoromethyl Ketone: Structural Studies with NMR Spectroscopy. Biochemistry 1987, 26, 7603–7608. 10.1021/bi00398a011. [DOI] [PubMed] [Google Scholar]
- Malthouse J. P. G.; Primrose W. U.; Mackenzie N. E.; Scott A. I. 13C NMR Study of the Ionizations within a Trypsin-Chloromethyl Ketone Inhibitor Complex. Biochemistry 1985, 24, 3478–3487. 10.1021/bi00335a014. [DOI] [PubMed] [Google Scholar]
- O’Connell T. P.; Malthouse J. P. G. Determination of the ionization state of the active-site histidine in a subtilisin-(chloromethane inhibitor) derivative by 13C-NMR. Biochem. J. 1996, 317, 35–40. 10.1042/bj3170035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primrose W. U.; Scott A. I.; Mackenzie N. E.; Malthouse J. P. G. A 13C-NMR investigation of ionizations within a trypsin-inhibitor complex. Evidence that the pKa of histidine-57 is raised by interaction with the hemiketal oxyanion. Biochem. J. 1985, 231, 677–682. 10.1042/bj2310677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spink E.; Cosgrove S.; Rogers L.; Hewage C.; Malthouse J. P. G. 13C and 1H NMR studies of ionizations and hydrogen bonding in chymotrypsin-glyoxal inhibitor complexes. J. Biol. Chem. 2007, 282, 7852–7861. 10.1074/jbc.M611394200. [DOI] [PubMed] [Google Scholar]
- Uritsky N.; Shokhen M.; Albeck A. Stepwise Versus Concerted Mechanisms in General-Base Catalysis by Serine Proteases. Angew. Chem., Int. Ed. 2016, 55, 1680–1684. 10.1002/anie.201507772. [DOI] [PubMed] [Google Scholar]
- Wilkinson G. N. Statistical estimations in enzyme kinetics. Biochem. J. 1961, 80, 324–332. 10.1042/bj0800324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini E.; Ascenzi P. The mechanism of trypsin catalysis at low pH. Proposal for a structural model. J. Biol. Chem. 1981, 256, 12449–12455. [PubMed] [Google Scholar]
- Kasserra H. P.; Laidler K. J. Mechanisms of Action of Trypsin and Chymotrypsin. Can. J. Chem. 1969, 47, 4031–4039. 10.1139/v69-669. [DOI] [Google Scholar]
- Kasserra H. P.; Laidler K. J. pH Effects in Trypsin Catalysis. Can. J. Chem. 1969, 47, 4021–4029. 10.1139/v69-668. [DOI] [Google Scholar]
- Spomer W. E.; Wootton J. F. The hydrolysis of alpha-N-benzoyl-L-argininamide catalyzed by trypsin and acetyltrypsin. Dependence on pH. Biochim. Biophys. Acta 1971, 235, 164–171. 10.1016/0005-2744(71)90044-1. [DOI] [PubMed] [Google Scholar]
- Stewart J. A.; Dobson J. E. Trypsin-catalyzed hydrolysis of N-benzoly-L-arginine ethyl ester at low pH. Biochemistry 1965, 4, 1086–1091. 10.1021/bi00882a016. [DOI] [PubMed] [Google Scholar]
- Stewart J. A.; Dobson J. E.; Lee H. S. Evidence for a Functional Carboxyl Group in Trypsin and Chymotrypsin. J. Am. Chem. Soc. 1963, 85, 1537–1538. 10.1021/ja00893a034. [DOI] [Google Scholar]
- Bruylants G.; Wintjens R.; Looze Y.; Redfield C.; Baetik K. Protonation linked equilibria and apparent affinity constants: the thermodynamic proile of the α-chymotrypsin-proflavin interaction. Eur. Biophys. J. 2007, 37, 11–18. 10.1007/s00249-007-0148-0. [DOI] [PubMed] [Google Scholar]
- McClure W. O.; Edelman G. M. Fluorescent probes for conformational states of proteins. II. The binding of 2-p-toluidinylnaphthalene-6-sulfonate to alpha-chymotrypsin. Biochemistry 1967, 6, 559–566. 10.1021/bi00854a025. [DOI] [PubMed] [Google Scholar]
- Inagami T.; Murachi T. The Mechanism of the Specificity of Trypsin Catalysis. 3. Activation of the Catalytic Site of Trypsin by Alkylammonium Ions in the Hydrolysis of Acetylglycine Ethyl Ester. J. Biol. Chem. 1964, 239, 1395–1401. [PubMed] [Google Scholar]
- Inagami T.; York S. S. Effect of alkylguanidines and alkylamines on trypsin catalysis. Biochemistry 1968, 7, 4045–4052. 10.1021/bi00851a036. [DOI] [PubMed] [Google Scholar]
- Bode W.; Schwager P. The refined crystal structure of bovine beta-trypsin at 1.8 A resolution. II. Crystallographic refinement, calcium binding site, benzamidine binding site and active site at pH 7.0. J. Mol. Biol. 1975, 98, 693–717. 10.1016/S0022-2836(75)80005-2. [DOI] [PubMed] [Google Scholar]
- Chambers J. L.; Stroud R. M. The Accuracy of Refined Protein Structures: Comparison of Two Independently Refined Models of Bovine Trypsin. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1979, 35, 1861–1874. 10.1107/S0567740879007925. [DOI] [Google Scholar]
- Krieger M.; Kay L. M.; Stroud R. M. Structure and specific binding of trypsin: comparison of inhibited derivatives and a model for substrate binding. J. Mol. Biol. 1974, 83, 219–230. 10.1016/0022-2836(74)90388-X. [DOI] [PubMed] [Google Scholar]
- Szabo E.; Bocskei Z.; Naray-Szabo G.; Graf L. The three-dimensional structure of Asp189Ser trypsin provides evidence for an inherent structural plasticity of the protease. Eur. J. Biochem. 1999, 263, 20–26. 10.1046/j.1432-1327.1999.00452.x. [DOI] [PubMed] [Google Scholar]
- Graf L.; Jancso A.; Szilagyi L.; Hegyi G.; Pinter K.; Naray-Szabo G.; Hepp J.; Medzihradszky K.; Rutter W. J. Electrostatic complementarity within the substrate-binding pocket of trypsin. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 4961–4965. 10.1073/pnas.85.14.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagami T. A kinetic study of the trypsin catalyzed hydrolysis of a specific anilide substrate. J. Biochem. 1969, 66, 277–279. 10.1093/oxfordjournals.jbchem.a129144. [DOI] [PubMed] [Google Scholar]
- Lucas E. C.; Caplow M.; Bush K. J. Chymotrypsin catalysis. Evidence for a new intermediate. J. Am. Chem. Soc. 1973, 95, 2670–2673. 10.1021/ja00789a043. [DOI] [PubMed] [Google Scholar]
- Shokhen M.; Khazanov N.; Albeck A. The cooperative effect between active site ionized groups and water desolvation controls the alteration of acid/base catalysis in serine proteases. ChemBioChem 2007, 8, 1416–1421. 10.1002/cbic.200700241. [DOI] [PubMed] [Google Scholar]
- Djurdjevic-Pahl A.; Hewage C.; Malthouse J. P. G. A 13C-NMR study of the inhibition of delta-chymotrypsin by a tripeptide-glyoxal inhibitor. Biochem. J. 2002, 362, 339–347. 10.1042/bj3620339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D. B.; O’Connell T. P.; Mahon M. M.; Koenig A.; Milne J. J.; Fitzpatrick T. P.; Malthouse J. P. G. 13C-NMR Study of How the Oxyanion pKa Values of Subtilisin and Chymotrypsin Tetrahedral Adducts Are Affected by Different Amino Acid Residues Binding in Enzymes Subsites S1–S4. Biochemistry 1999, 38, 6187–6194. 10.1021/bi990126c. [DOI] [PubMed] [Google Scholar]
- Spink E.; Hewage C.; Malthouse J. P. G. Determination of the structure of tetrahedral transition state analogues bound at the active site of chymotrypsin using 18O and 2H isotope shifts in the 13C NMR spectra of glyoxal inhibitors. Biochemistry 2007, 46, 12868–12874. 10.1021/bi701212u. [DOI] [PubMed] [Google Scholar]
- Bruice T. C.; Fife T. H.; Bruno J. J.; Brandon N. E. Hydroxyl group catalysis. II. The reactivity of the hydroxyl group of serine. The nucleophilicity of alcohols and the ease of hydrolysis of their acetyl esters as related to their pKa. Biochemistry 1962, 1, 7–12. 10.1021/bi00907a002. [DOI] [PubMed] [Google Scholar]
- Lin J.; Cassidy C. S.; Frey P. A. Correlations of the basicity of His 57 with transition state analogue binding, substrate reactivity, and the strength of the low-barrier hydrogen bond in chymotrypsin. Biochemistry 1998, 37, 11940–11948. 10.1021/bi980278s. [DOI] [PubMed] [Google Scholar]
- Zhou Y. Z.; Zhang Y. K. Serine protease acylation proceeds with a subtle re-orientation of the histidine ring at the tetrahedral intermediate. Chem. Commun. 2011, 47, 1577–1579. 10.1039/C0CC04112B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J. A.; Doherty W.; Evans P.; Malthouse J. P. Hemiacetal stabilization in a chymotrypsin inhibitor complex and the reactivity of the hydroxyl group of the catalytic serine residue of chymotrypsin. Biochim. Biophys. Acta 2014, 1844, 1119–1127. 10.1016/j.bbapap.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase T. Jr.; Shaw E. p-Nitrophenyl-p’-guanidinobenzoate HCl: a new active site titrant for trypsin. Biochem. Biophys. Res. Commun. 1967, 29, 508–514. 10.1016/0006-291X(67)90513-X. [DOI] [PubMed] [Google Scholar]
- Mackenzie N. E.; Malthouse J. P. G.; Scott A. I. Chemical synthesis and papain-catalysed hydrolysis of N-alpha- benzyloxycarbonyl-L-lysine p-nitroanilide. Biochem. J. 1985, 226, 601–606. 10.1042/bj2260601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C. N. H.Biochemists’ Handbook; Van Nostrand: Princeton, NJ, 1961. [Google Scholar]
- Cleland W. W. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979, 63, 103–138. [DOI] [PubMed] [Google Scholar]