

Abstract
Activation of tumor suppressors for the treatment of human cancer has been a long sought, yet elusive, strategy. PTEN is a critical tumor suppressive phosphatase that is active in its dimer configuration at the plasma membrane. Polyubiquitination by the ubiquitin E3 ligase WWP1 (WW domain–containing ubiquitin E3 ligase 1) suppressed the dimerization, membrane recruitment, and function of PTEN. Either genetic ablation or pharmacological inhibition of WWP1 triggered PTEN reactivation and unleashed tumor suppressive activity. WWP1 appears to be a direct MYC (MYC proto-oncogene) target gene and was critical for MYC-driven tumorigenesis. We identified indole-3-carbinol, a compound found in cruciferous vegetables, as a natural and potent WWP1 inhibitor. Thus, our findings unravel a potential therapeutic strategy for cancer prevention and treatment through PTEN reactivation.
Graphical Abstract
INTRODUCTION:
Inhibition of oncogenic proteins represents a mainstay approach for cancer therapeutic development. By contrast, pharmacological modulation of tumor suppressor activity for the treatment of cancer has remained elusive. PTEN is a potent tumor suppressor gene, antagonizing the proto-oncogenic phosphoinositide 3-kinase (PI3K)–AKT signaling pathway and governing fundamental cellular processes. Cancer cells cannot afford to lose complete PTEN activity prematurely, because this would trigger cellular senescence, making PTEN an “obligate haploinsufficient” tumor suppressor gene. For this reason, PTEN is frequently dysregulated through monoallelic loss, aberrant subcellular localization, and/or posttranslational modification in human cancers as well as in cancer susceptibility syndromes such as PTEN hamartoma tumor syndrome (PHTS). Because PTEN overexpression in mice results in a tumor-suppressive metabolic state and life-span extension, the identification of molecular mechanisms to activate and reactivate PTEN function would offer important therapeutic opportunities for human health.
RATIONALE:
Although PTEN dimer formation and recruitment at the plasma membrane are indispensable for its function and activation, the mechanisms regulating these processes remain unknown. We thus sought to identify upstream regulators of PTEN dimerization and membrane localization, inhibition of which may restore PTEN activity and provide therapeutic opportunities against cancer.
RESULTS:
Through immunoprecipitation followed by mass spectrometry analysis, we identified the HECT-type E3 ubiquitin ligase WWP1 as a physical PTEN interactor. We found that WWP1 specifically triggers nondegradative K27-linked polyubiquitination of PTEN to suppress its dimerization, membrane recruitment, and tumor-suppressive functions both in vitro and in vivo.
WWP1 is genetically amplified and frequently overexpressed in multiple cancers, including those of prostate, breast, and liver, which may lead to pleiotropic inactivation of PTEN. We found that WWP1 may be transcriptionally activated by the MYC proto-oncogene and that genetic depletion of Wwp1 in both Myc-driven mouse models of prostate cancer in vivo and cancer cells in vitro reactivates PTEN function, leading to inhibition of the PI3K-AKT pathway and MYC-driven tumorigenesis. Depletion of Wwp1 significantly reduced PI3KAKT activity in mouse fibroblasts harboring monoallelic PTEN or PTEN mutations, as observed in PHTS patients. These findings demonstrate that WWP1 acts downstream of MYC and that perturbation of WWP1 is sufficient to restore PTEN tumor-suppressive activity.
We next identified indole-3-carbinol (I3C), a derivative of cruciferous vegetables, as a natural and potent WWP1 inhibitor through structure simulation and biochemical analyses. Pharmacological inactivation of WWP1 by I3C in either Myc-driven or Pten heterozygous mice reactivates PTEN, leading to potent suppression of tumorigenesis driven by the PI3K-AKT pathway. Therefore, genetic or pharmacological targeting of the WWP1-PTEN axis holds promise for patients affected by a number of cancers and other disorders associated with germline mutations of the PTEN gene.
CONCLUSION:
We have identified the MYC-WWP1 axis as a fundamental and evolutionary conserved regulatory pathway for PTEN and PI3K signaling. This pathway emerges not only as a rheostat for growth control in physiological conditions but also as a critical vulnerability hijacked for neoplastic transformation, which may be reversed by WWP1 pharmacological inactivation. These findings pave the way toward a long-sought tumor suppressor “reactivation” approach to cancer treatment. Because an increased expression level of MYC-WWP1 or PTEN impairment is widely pervasive in various human cancers, targeting this pathway toward PTEN reactivation may represent an “Achilles heel” of broad application. ▪
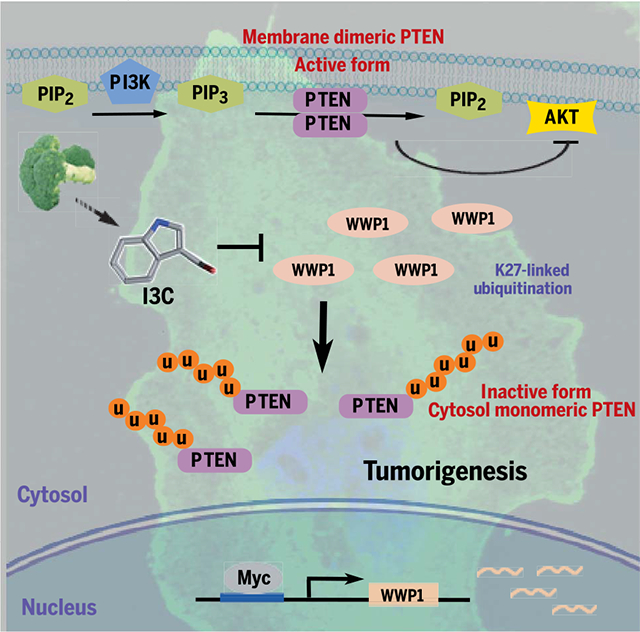
Model for WWP1-mediated PTEN K27-linked polyubiquitination in tumor development and progression. Deregulated MYC overexpression or MYC amplification promotes WWP1 expression and, in turn, triggers PTEN K27-linked polyubiquitination. Aberrant K27-linked polyubiquitination suppresses PTEN dimerization, plasma membrane recruitment, and tumor suppressive function, leading to tumor initiation and progression. Pharmacological inactivation of WWP1 by I3C, a derivative of cruciferous vegetables, reactivates PTEN, leading to suppression of tumorigenesis. PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; u, ubiquitin.
Cancer is a consequence of multiple genetic and epigenetic alterations that are either inherited or somatically acquired. Gain-of-function of proto-oncogenes or loss-of-function of tumor suppressor genes, or both, resulting from aberrant genetic alterations are dominant driving forces underlying tumorigenesis (1). Cancer therapy and drug discovery efforts have predominantly focused on targeting oncogenic events, whereas the activation of tumor suppressors has remained less explored as a mode of cancer treatment.
PTEN is one of the most frequently mutated, deleted, down-regulated, or silenced tumor suppressor genes in human cancer (2, 3). Partial loss of PTEN is observed at high frequency in cancers of various histological origins, reflecting the fact that PTEN is haploinsufficient in its tumor suppressive function (4), although its complete loss triggers cellular senescence, a potent fail-safe response (5). Germ-line mutations in PTEN can cause inherited syndromes characterized by developmental defects and cancer susceptibility, which are collectively referred to as PTEN hamartomas tumor syndrome (PHTS) (3, 6). That PTEN function is not often completely lost in cancer or PHTS offers an opportunity to explore its functional reactivation as a mode of cancer therapy, and indeed, enhanced function of PTEN is beneficial in multiple ways: Mice with increased abundance of PTEN are tumor resistant, metabolically fit, lean, and even long-lived (7). Thus, pharmacological activation of PTEN could potentially be beneficial for the treatment of human genetic disorders as well as cancer.
PTEN encodes a dual specificity phosphatase whose major substrate is phosphatidylinositol 3,4,5-trisphosphate (PIP3). PTEN dephosphorylates the D3-phosphate of the second messenger PIP3, which suppresses the activation of the proto-oncogenic phosphoinositide 3-kinase (PI3K)–AKT protein kinase signaling pathway, and thus controls cell proliferation, growth, and metabolism (8–11). PTEN is strictly regulated, and its deregulation through aberrant subcellular localization or posttranslational modifications, or both, occurs frequently in tumorigenesis (12). Mechanistically, monoubiquitination regulates the import of PTEN into the nucleus, where it exerts PIP3-independent functions (13, 14). But PTEN also exists as a dimer, and dimer formation and recruitment at the plasma membrane are crucial for its function and activation (15). However, the mechanisms that regulate PTEN dimerization and favor membrane recruitment remain largely unknown.
WWP1 E3 ligase mediates PTEN K27-linked polyubiquitination
We therefore sought to elucidate the underlying mechanisms and explore their roles in the activation of PTEN (15). To identify possible regulators, we immunoprecipitated exogenous PTEN from DU145 human prostate cancer cells and identified interacting proteins by mass spectrometry (MS) (Fig. 1A and table S1). We identified two members of the HECT-type family of ubiquitin E3 ligases: neural precursor cell–expressed, developmentally down-regulated 4 ubiquitin E3 ligase (NEDD4–1), which is a known PTEN interactor (13), and WW domain–containing ubiquitin E3 ligase 1 (WWP1). WWP1 exhibited the strongest binding capability among the NEDD4 family ubiquitin E3 ligases, suggesting a potential role for WWP1 in PTEN regulation (fig. S1A). We verified binding between endogenous WWP1 and PTEN in DU145 cells in in vivo reciprocal immunoprecipitation assays (Fig. 1B).
Fig. 1. Identification and characterization of the WWP1 E3 ligase for PTEN K27-linked polyubiquitination.
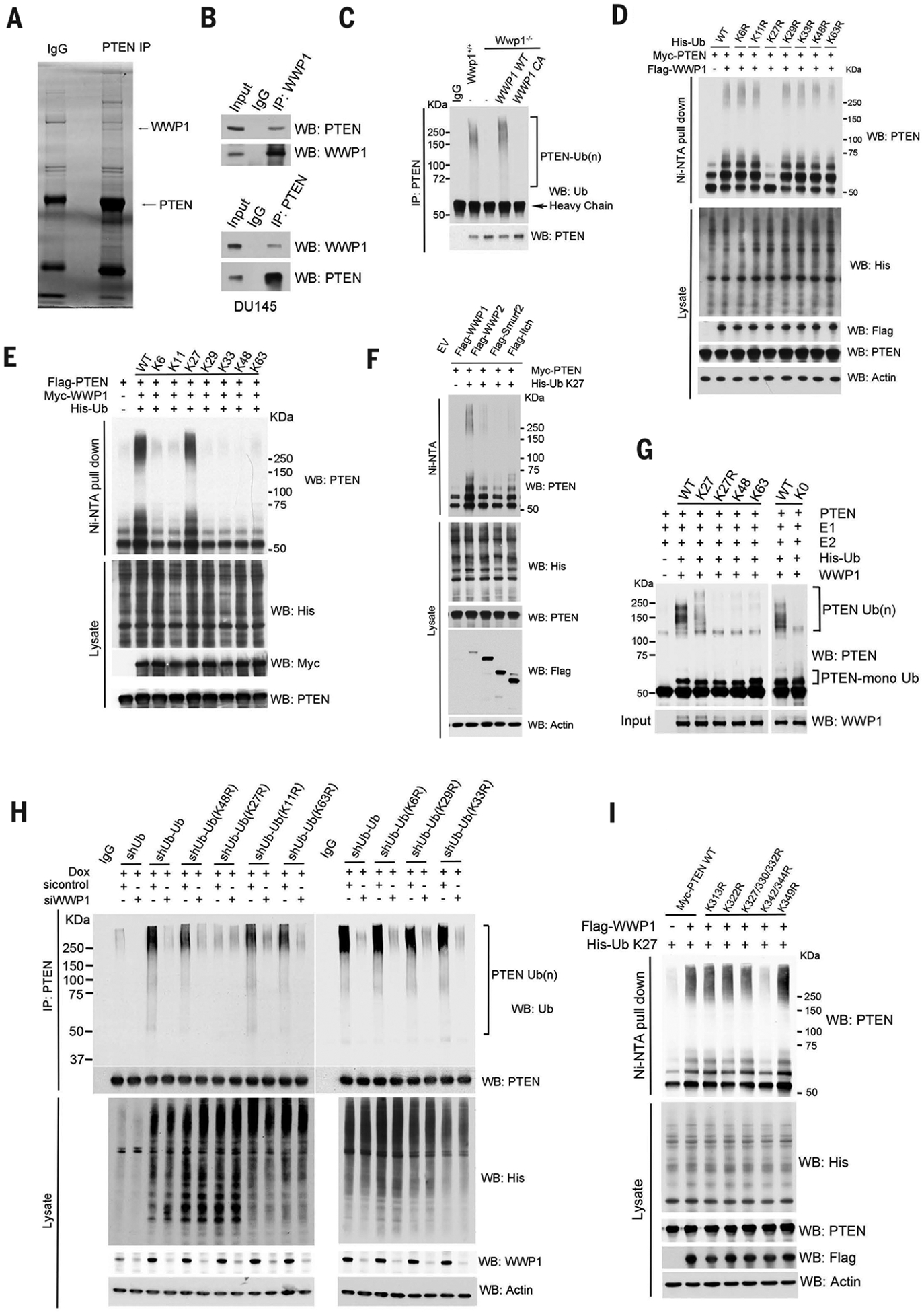
(A) Lysates from DU145 cells transfected with hemagglutinin (HA)–PTEN were immunoprecipitated (IP) with an anti-PTEN antibody followed by mass spectrometric peptide sequencing. WWP1 and PTEN were identified. IgG, immunoglobulin G. (B) WWP1 interacts with PTEN endogenously. DU145 cells were immunoprecipitated with either anti-WWP1 or anti-PTEN antibody and then analyzed by Western blot (WB). Input is 5% of the total lysates used in IP. (C) Analysis of endogenous PTEN ubiquitination (Ub) in Wwp1−/− MEFs with stable reconstitution of either WWP1 WT or its catalytically inactive mutant (C890A, abbreviated CA). (D and E) Effects of the indicated ubiquitin KR (Lys to Arg) (D) or K-only (E) ubiquitin mutants on WWP1-mediated PTEN polyubiquitination. HEK293T cells were transfected with the indicated constructs, and PTEN ubiquitination was analyzed. The ubiquitinated proteins were pulled down under denaturing conditions by nickel–nitrilotriacetic acid (Ni-NTA) agarose and analyzed by Western blot. (F) Analysis of PTEN K27-linked polyubiquitination in PC3 cells expressing the indicated NEDD4 family ubiquitin ligases as in (D). EV, cells transfected with empty vector plasmids. (G) In vitro ubiquitination of PTEN by WWP1 E3 ligase. Flag-PTEN purified from HEK293 cells was subject to in vitro ubiquitination reaction in the presence of E1, E2, E3, and ubiquitin or various ubiquitin mutants and then examined by Western blot with anti-PTEN antibody. The input control of WWP1 was determined by Western blotting and is shown at the bottom. (H) Analysis of endogenous PTEN ubiquitination by small interfering RNA (siRNA)–mediated WWP1 knock down in U2OS ubiquitin replacement cells treated with 1 μg/ml doxycycline for 48 hours. shUb, shRNAs targeting ubiquitin. (I) Effects of the indicated ubiquitin KR mutants on WWP1-mediated PTEN polyubiquitination. PC3 cells were transfected with indicated constructs, and PTEN ubiquitination was analyzed as in (D). In (D), (F), (H), and (I), actin is used as a loading control.
WWP1 contains a C2 domain, four WW domains, and a HECT domain; it is frequently overexpressed and genetically amplified in various cancers, including cancers of the prostate and breast, and hepatocellular carcinoma (16–19). We assessed the functional relevance of physical interaction between PTEN and WWP1 in triggering PTEN ubiquitination. Overexpression of wild-type (WT) WWP1 in human embryonic kidney–293T (HEK293T) cells increased PTEN polyubiquitination, whereas the expression of a catalytically inactive mutant [C890A (Cys890→Ala)] abrogated this effect, indicating the indispensable role of the catalytic activity of WWP1 in triggering PTEN polyubiquitination (fig. S1B). Conversely, depletion of WWP1 by short hairpin RNAs (shRNAs) or CRISPR likewise diminished PTEN polyubiquitination (Fig. 1C and figs. S1C and S2G). In Wwp1−/− mouse embryonic fibroblasts (MEFs), expression of WT WWP1 triggered PTEN polyubiquitination, but expression of the catalytically inactive mutant (C890A) failed to do so (Fig. 1C). These studies support a crucial role of catalytically active WWP1 in promoting PTEN polyubiquitination.
Despite the impact of WWP1 on PTEN polyubiquitination, the protein turnover rate of PTEN was not affected when WWP1 was ectopically expressed (fig. S1D). We therefore sought to determine the type of ubiquitin chain that was generated on PTEN by WWP1. We mutated each of the lysine residues on ubiquitin to test their individual effects on PTEN polyubiquitination in WWP1-overexpressing cells. Only K27R (Lys27→Arg) ubiquitin inhibited WWP1-mediated PTEN polyubiquitination (Fig. 1D). We used a K27-only ubiquitin, which retained only the K27 lysine residue (with the other six lysine residues mutated to arginine), to corroborate that WWP1 catalyzed PTEN K27-linked polyubiquitination of WWP1. Although K6-, K11-, K29-, K33-, K48-, and K63-ubiquitin did not trigger WWP1-mediated PTEN polyubiquitination, the K27-ubiquitin promoted chain formation on PTEN similar to that observed with WT ubiquitin (Fig. 1E). Moreover, in vivo ubiquitination assays revealed that overexpression of WWP1, but not other NEDD4 family E3 ligases (WWP2, Smurf2, and Itch), stimulated K27-linked PTEN polyubiquitination (Fig. 1F). Overexpression of WWP2 (20), but not that of WWP1 or other members of the NEDD4 family of E3 ligases, promoted K48-linked PTEN polyubiquitination (fig. S1E).
In in vitro ubiquitination assays, in the presence of purified PTEN, WWP1, and WT ubiquitin, efficient polyubiquitination of PTEN was detected. The K27-only ubiquitin mutant also promoted chain formation on PTEN. By contrast, K27R-, K48-, K63-, and K0-ubiquitins were not effective substrates for WWP1-mediated PTEN polyubiquitination (Fig. 1G). The ubiquitin E3 ligase NEDD4–1 interacts with PTEN and triggers its monoubiquitination (table S1) (13), so we included it as a positive control in our in vitro ubiquitination assays. NEDD4–1 consistently increased PTEN monoubiquitination but had a negligible effect on K27-linked polyubiquitination, whereas WWP1 efficiently triggered PTEN polyubiquitination (fig. S1F). In vitro pull-down and in vivo immunoprecipitation assays showed that WWP1 interacted with NEDD4–1 both in vitro and in vivo (fig. S1, G and H), and combined expression of NEDD4–1 and WWP1 synergistically increased PTEN polyubiquitination (fig. S1F). We concluded that WWP1 is a key E3 ligase triggering PTEN K27-linked polyubiquitination.
To further demonstrate the generation of a K27 ubiquitin chain on PTEN, we next performed an MS analysis of ubiquitinated PTEN peptides isolated from control or WWP1-overexpressing cells by PTEN immunoprecipitation, digested of protein with trypsin, and enriched with an antibody to K-ε-GG (lysine-ε−glycine-glycine). This analysis revealed that abundance of ubiquitin peptide with a di-Gly modification on the K27 residue was increased upon overexpression of WWP1 E3 ligase, whereas ubiquitin modification on other Lys residues, such as K11, K48, and K63, showed smaller increases (fig. S1, I and J). To validate the regulation of PTEN polyubiquitination by WWP1 endogenously, we used an ubiquitin replacement system that allowed simultaneous depletion of endogenous ubiquitin and expression of exogenous ubiquitin, or its variants in amounts comparable to those of endogenous ubiquitin, by administration of doxycycline (fig. S1K) (21). Replacement of endogenous ubiquitin with a K27R mutant decreased PTEN polyubiquitination, whereas replacement with other ubiquitin mutants showed little or no effect. Moreover, WWP1 depletion reduced PTEN polyubiquitination, although this effect was abrogated when endogenous ubiquitin was replaced by the K27R mutant (Fig. 1H). Taken together, these data indicate the essential roles of WWP1 in triggering PTEN K27-linked polyubiquitination.
We used a mutagenesis approach to inactivate the major individual lysine residues or polylysine patches exposed at the PTEN protein surface (fig. S2A) (22). Mutation of K342 or K344 (but not other lysine residues) to arginine, either alone or in combination, decreased WWP1-mediated K27-linked polyubiquitination of PTEN (Fig. 1I and fig. S1L), whereas the protein turnover rates of the PTEN K342R and K344R mutants were similar to that of WT PTEN (fig. S1M). Thus, our data provide multiple lines of evidence corroborating the formation of a K27-linked polyubiquitin chain on PTEN K342 and K344 residues mediated by the WWP1 E3 ligase. This atypical polyubiquitination did not interfere with the stability of the PTEN protein, which is consistent with reports showing that K27-linked polyubiquitination does not signal protein degradation (23–25). The K342 and K344 lysine patch is exposed outside of PTEN (fig. S2, A and B) and is well conserved across species, indicating that the WWP1-mediated K27-linked polyubiquitination may be evolutionarily conserved and important in the regulation of PTEN function.
K27-linked PTEN polyubiquitination by WWP1 suppresses its dimerization, membrane recruitment, and function
Ubiquitination can impose spatial limits on protein-protein interaction (26), which prompted us to examine whether WWP1-mediated K27-linked PTEN ubiquitination regulates PTEN dimer formation. To this end, we monitored PTEN dimerization in vitro by purification of Flag-tagged unmodified or ubiquitinated PTEN on beads and then tested its interaction with glutathione S-transferase (GST)–PTEN fusion protein purified from bacteria. Overexpression of WT ubiquitin with the WWP1 E3 ligase resulted in K27-linked polyubiquitination of PTEN, which disrupted the interaction of Flag-PTEN with GST-PTEN. By contrast, overexpression of the K27R mutant ubiquitin with WWP1 did not affect the interaction of Flag-PTEN with GST-PTEN; thus, K27-linked polyubiquitination of PTEN appears to inhibit its dimerization (Fig. 2A). Given that PTEN dimerization is associated with PTEN membrane recruitment and activation (15), we investigated the effects of WWP1-mediated K27-linked PTEN polyubiquitination on PTEN membrane recruitment and activity. Fractionation experiments showed that overexpression of WWP1 suppressed PTEN membrane recruitment (Fig. 2B), resulting in subsequent activation of the protein kinase AKT (Fig. 2C). By using the aforementioned ubiquitin replacement system, we found that replacement of endogenous ubiquitin with a K27R mutant (but not WT ubiquitin) not only inhibited PTEN polyubiquitination (Fig. 1H) but also induced PTEN localization to the plasma membrane, which in turn suppressed activation of AKT (fig. S2, C and D).
Fig. 2. K27-linked PTEN polyubiquitination suppresses PTEN dimerization, membrane recruitment, and function.
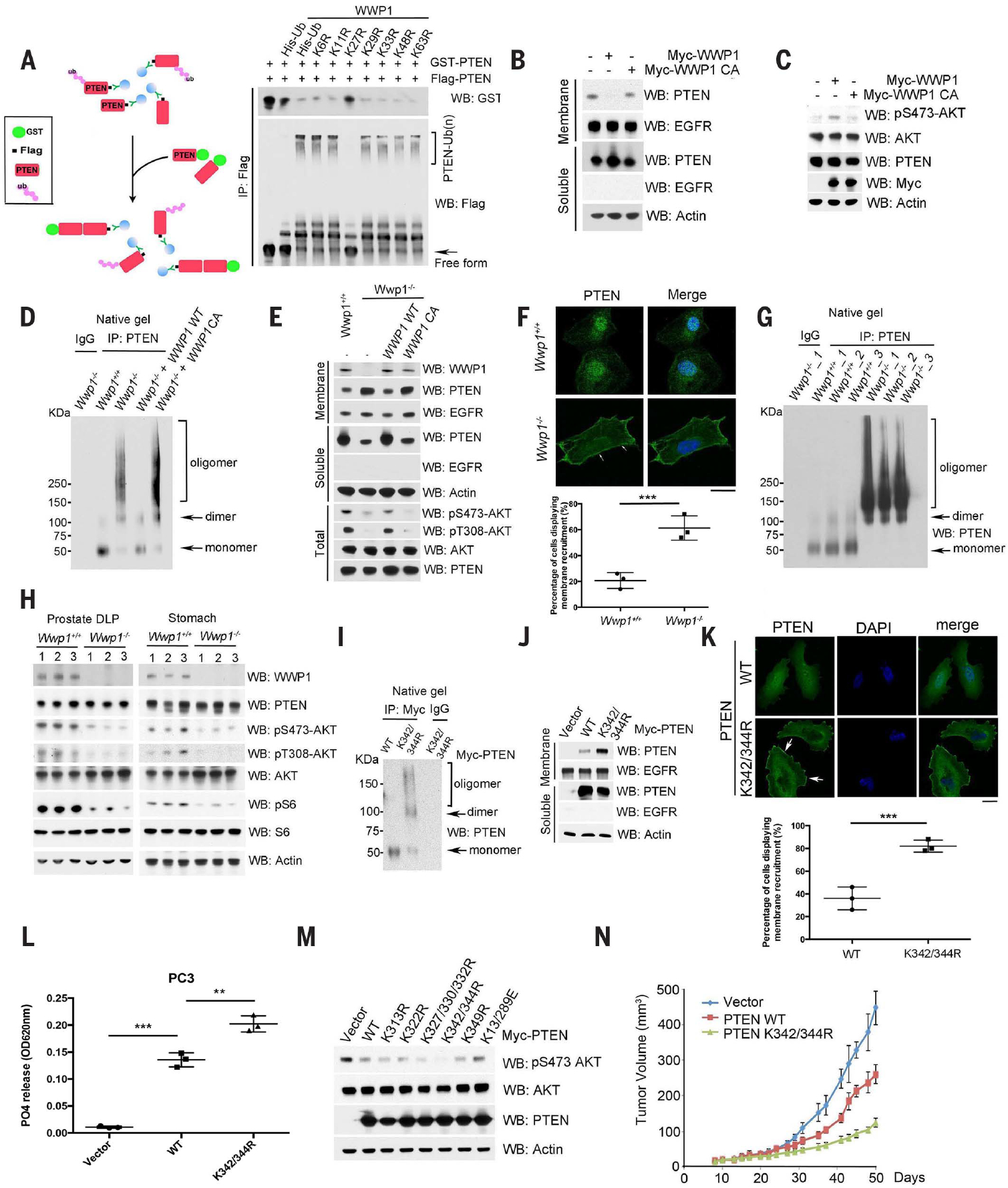
(A) Schematic description of the in vitro binding analysis of Flag-tagged unmodified or ubiquitinated PTEN with GST-PTEN from bacteria (left). In vitro pull-down assay with indicated Flag-tagged unmodified or ubiquitinated PTEN and GST-PTEN (right). Flagged untagged PTEN or ubiquitinated PTEN was purified from HEK293 cells transfected with Flag-PTEN or WWP1, along with individual His-ubiquitin variants, using M2 beads, whereas GST-PTEN was purified from bacteria. (B) Membrane and soluble fractions isolated from DU145 cells transfected with indicated constructs were analyzed by Western blot. Epidermal growth factor receptor (EGFR) serves as a membrane marker and actin as the internal control. (C) Analysis of AKT activation in DU145 cells. Total lysates were resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and then probed with the indicated antibodies. (D) Evaluation of PTEN dimerization in Wwp1−/− MEFs with stable reconstitution of either WWP1 WT or its catalytic dead mutant (C890A) by native gel electrophoresis. Total lysates from cells transduced with indicated constructs were immunoprecipitated with a rabbit anti-PTEN antibody, and then the immunocomplexes were natively eluted from the beads. The eluted samples were immediately run on the native gel. (E) Membrane and soluble fractions isolated from Wwp1+/+ and Wwp1−/− MEFs reconstituted with the indicated constructs were analyzed by Western blot. EGFR served as a membrane marker, and actin as the internal control. pS473-AKT and pT308-AKT are the markers of AKT activation. (F) Subcelluar localization of PTEN in Wwp1+/+ or Wwp1−/− MEFs. Confocal images of Wwp1+/+ or Wwp1−/− MEFs stained with 4′,6-diamidino-2-phenylindole (DAPI, blue) and PTEN (green) (top). The white arrows indicate PTEN plasma membrane localization. Scale bar, 20 μm. The percentage of cells displaying PTEN plasma membrane localization was quantified (bottom). Data are shown as mean ± SD for triplicate experiments, with 50 cells per group per experiment (***P < 0.0005, Student’s t test). (G) Evaluation of PTEN dimerization potential in DLP tissues derived from Wwp1 WT or Wwp1 knockout mice (n = 3). (H) Analysis of PI3K-AKT-mTOR signaling cascade in DLP tissues derived from Wwp1 WT or Wwp1 knockout mice (n = 3). Actin was used as a loading control. pS6 and S6 are markers to indicate the activation of the mTOR pathway. (I) Effects of the PTEN K342/K344R mutant on PTEN dimerization in PC3 cells. PC3 cells transfected with indicated constructs were serum starved for 6 hours and then treated with 100 ng/ml insulin for 10 min. Total lysates from cells were immunoprecipitated with a rabbit anti-Myc antibody, and then the immunocomplexes were natively eluted from the beads. The eluted samples were immediately run on a native gel, and Western blot using PTEN antibody showed the monomer and dimer of PTEN, as indicated by arrows. (J) Membrane localization of PTEN K342/K344R mutant in PC3 cells as in (I). Membrane and soluble fractions isolated from PC3 cells with indicated constructs were analyzed by Western blot. EGFR serves as the marker for the membrane fraction and actin as the internal control for the soluble fraction. Vector indicates cells transfected with empty vector plasmids. (K) Subcellular localization of the PTEN K342/K344R mutant in PC3 cells. Confocal images of PC3 cells stably expressing indicated PTEN WT or the K342/K344R mutant stained with DAPI and PTEN (green) (top). White arrows indicate PTEN plasma membrane localization. Scale bar, 20 μm. The percentage of cells displaying PTEN plasma membrane localization was quantified (bottom). Data are shown as mean ± SD for triplicate experiments, with 50 cells per group per experiment (***P < 0.0005, Student’s t test). (L) Effects of indicated PTEN KR mutants on PTEN lipid phosphatase activities in PC3 cells with indicated constructs. Data are shown as mean ± SD (***P < 0.0005, **P < 0.005, triplicate experiments, Student’s t test). OD620nm, absorbance at 620 nm. (M) Effects of indicated PTEN KR mutants on AKT activation in PC3 cells with indicated constructs. Total lysates were harvested and then probed with indicated antibodies. Actin was used as a loading control. (N) Effects of the PTEN K342/K344R mutant on tumor growth of PC3 cells as used in (K) in a xenograft mouse model. Error bars represent SEM (n = 5 mice per group).
To further corroborate the role of WWP1 in PTEN dimerization, membrane recruitment, and function, we generated Wwp1−/− MEFs by a CRISPRCas9 gene-editing approach. Genetic ablation of WWP1 abrogated PTEN polyubiquitination (Fig. 1C) and led to an increase in PTEN dimerization and membrane recruitment (as evaluated by native gel, membrane fractionation, and confocal analyses), which in turn increased PTEN lipid phosphatase activity and suppressed activation of AKT (Fig. 2, D to F, and fig. S2, E and F). To confirm that these effects were mediated by WWP1 catalytic activity, we added back either WT or a catalytically inactive mutant (C890A) WWP1 into the Wwp1−/− MEFs. WT WWP1 reverted all these phenotypes, but C890A-WWP1 failed to do so, indicating that WWP1 catalytic activity is required for the mediation of these phenotypes (Fig. 2, D and E). Genetic deletion of Wwp1 in mice resulted in increased dimerization of PTEN and subsequent suppression of the PI3K-AKT–mTOR (mammalian target of rapamycin) signaling pathway (Fig. 2, G and H). Similarly, depletion of WWP1 in multiple types of cancer cells with heterozygous or WT PTEN status decreased PTEN polyubiquitination (fig. S2G) and resulted in an increase in PTEN dimerization and membrane recruitment and subsequent suppression of AKT activation (fig. S2, H and I), indicating that this pathway is active in these cancer cells. Thus, several lines of evidence support a role for WWP1-mediated PTEN polyubiquitination in the regulation of PTEN ubiquitination, dimerization, membrane recruitment, and function.
In line with the critical role of the K342 and K344 residues on PTEN in WWP1-mediated K27-linked polyubiquitination (Fig. 1I), we also found that the PTEN K342/K344R ubiquitination-defective mutant, which has both the K342R and K344R mutations, was predominantly dimeric (Fig. 2I), which further corroborated the suppressive impact of K27-linked polyubiquitination on PTEN dimerization. Accordingly, the PTEN K342/K344R mutant accumulated in the plasma membrane, as detected by membrane fractionation and confocal analyses (Fig. 2, J and K). Of the PTEN mutants and WT controls analyzed, the PTEN K342/K344R mutant proved most active toward its phosphoinositide substrate PIP3 and exhibited the strongest inhibition of AKT activation (Fig. 2, L and M).
To determine the influence of PTEN K27-linked polyubiquitination on cell proliferation, tumorigenic potential, and tumor growth in vivo, we compared the tumor suppressive function of WT with that of a ubiquitination-defective PTEN K342/K344R mutant. The PTEN K342/K344R mutant, which resisted polyubiquitination by WWP1, inhibited cell proliferation and anchorage-independent growth more effectively than did WT PTEN (fig. S3, A and B). In a xenograft tumor model, tumors carrying the PTEN K342/344R mutant grew more slowly than those with WT PTEN (Fig. 2N and fig. S3C). Consistently, immunohistochemistry (IHC) analyses of tumors derived from PTEN WT or the K342/K344R mutant revealed that PTEN K342/K344R displayed greater localization to the plasma membrane, whereas PTEN WT was dispersed within the cells (fig. S3D). Together, these data indicate that both in vitro and in vivo, WWP1-mediated K27-linked PTEN polyubiquitination on K342 and K344 residues inhibits the tumor suppressor function of PTEN by dissociating it from the plasma membrane compartment.
MYC activates WWP1 gene expression toward PTEN suppression both in vitro and in vivo
Because activation of the PI3K-AKT signaling pathway and amplification of proto-oncogenic MYC frequently occur in cancers and correlate with a high pathological grade and poor prognosis (27–30), we tested whether MYC inhibited PTEN function and thereby promoted activation of PI3K-AKT pathways. Because MYC functions as a transcription factor, we tested whether WWP1 is a MYC target gene. Through analyses of Transcription factor Affinity Prediction (TRAP) software (http://trap.molgen.mpg.de/cgi-bin/home.cgi) and chromatin immunoprecipitation (ChIP)-sequencing databases (https://encodeproject.org), we identified potential MYC-responsive elements in the promoter of the WWP1 gene (Fig. 3A). ChIP assays confirmed that endogenous MYC was bound to the predicted regions on the promoter of WWP1, with the transcription factor JunB and ribosomal protein L30 (RPL30) serving as positive and negative controls, respectively (Fig. 3A). In line with this observation, CRISPR-mediated deletion of the MYC-responsive element within the WWP1 promoter decreased WWP1 expression and AKT activation, which in turn suppressed anchorage-independent growth (fig. S4, A to C). Conversely, overexpression of MYC increased abundance of WWP1 mRNA and protein in a dose-dependent manner, indicating that WWP1 may be a MYC target gene (Fig. 3, B and C). We assessed whether MYC affects PTEN K27-linked polyubiquitination as a result of MYC-mediated accumulation of WWP1 and performed tandem di-Gly MS analyses of endogenous ubiquitinated PTEN peptides obtained from MYC-stable cells with or without depletion of WWP1. Control cells overexpressing MYC exhibited an increase in ubiquitin peptides with a di-Gly modification on the K27 residue, whereas they showed little effect on other ubiquitin chain types, such as K11, K48, and K63. Conversely, cells overexpressing MYC, but lacking WWP1, showed a specific decrease in K27 ubiquitin peptides, but not other ubiquitin chain types (Fig. 3, D and E). We obtained similar results in cells by in vivo ubiquitination analyses (Fig. 3F); together, these data support the function of WWP1 in promoting PTEN K27-linked polyubiquitination when MYC is deregulated. As a consequence of its up-regulation of WWP1, overexpression of MYC disrupted PTEN dimerization and membrane accumulation in a dose-dependent manner, as assessed by immunoprecipitation and membrane fractionation analyses (fig. S4, D and E). Activation of AKT was increased upon MYC over-expression, in the absence of any change in abundance of PTEN (Fig. 3C). Conversely, depletion of MYC or WWP1 led to the suppression of PI3K-AKT signaling, again with negligible changes observed in PTEN, further corroborating that MYC is an upstream regulator of the WWP1-PTEN pathway (Fig. 3, G and H).
Fig. 3. MYC transactivates WWP1 gene expression toward PTEN suppression.
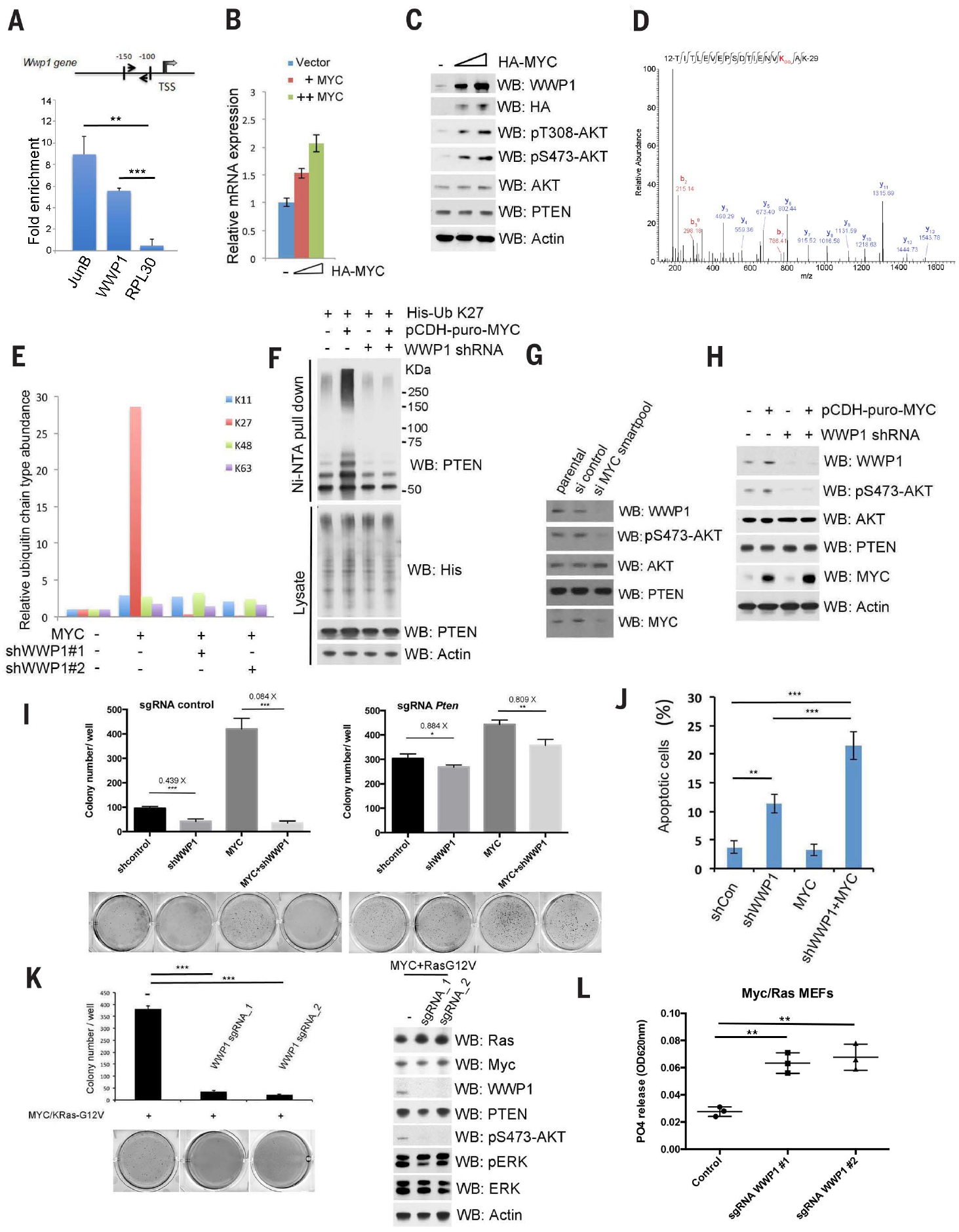
(A) Schematic description of the MYC responsive element on the WWP1 promoter (top). Chromatin level of MYC at the promoter of human WWP1 was measured in DU145 cells. Fold enrichment of MYC was determined by quantitative chromatin immunoprecipitation (qChIP) assays. JunB and RPL30 served as positive and negative controls, respectively. TSS, transcription start site. Data are shown as mean ± SD (***P < 0.0005, **P < 0.005, triplicate experiments, Student’s t test). (B) Reverse transcription–quantitative polymerase chain reaction (RT-qPCR) analysis of WWP1 in DU145 cells expressing the indicated constructs. The mRNA level of WWP1 was determined by RT-qPCR and is presented as a fold increase, as compared with the vector control (−). The HA-MYC levels are indicated by the triangle, from left to right. (C) Analysis of WWP1 and PTEN expression and AKT activation in DU145 cells expressing different amounts of HA-MYC. Total lysates were resolved by SDS-PAGE and then probed with indicated antibodies. The “−” indicates the vector control. (D) Tandem mass spectrum of a peptide derived from endogenous ubiquitinated PTEN in DU145 cells stably expressing the indicated constructs showed ubiquitin conjugation at the K27 residue of ubiquitin. (E) Ratio of indicated ubiquitin linkages detected by MS analysis of endogenous ubiquitinated PTEN purified from MYC overexpression or MYC overexpression–shWWP1 cells to that from control cells (without MYC overexpression). The abundance of each ubiquitin linkage was calculated as described in the materials and methods. (F) Analysis of PTEN K27-linked polyubiquitination in DU145 cells stably expressing MYC and/or WWP1 shRNAs. pCDH-puro-MYC, lentiviral expression of MYC. (G) Analysis of WWP1 and PTEN expression and AKT activation in DU145 cells expressing indicated siRNA SMARTpool. Total lysates were resolved by SDS-PAGE and then probed with indicated antibodies. (H) Analysis of WWP1 and PTEN expression and AKT activation in DU145 cells stably expressing MYC and/or WWP1 shRNAs. (I) Effects of WWP1 with or without PTEN on MYC-induced colony-forming activity in soft agar. The colony numbers are quantified and presented as mean ± SD (***P < 0.0005, **P < 0.005, *P < 0.05, triplicate experiments, Student’s t test). sgRNA, single-guide RNA. (J) Apoptosis assay of DU145 cells stably expressing MYC and/or WWP1 shRNAs. The percentage of apoptotic cells were stained with annexin V–PTEN and 7ADD and then quantified by fluorescence-activated cell sorting. Data are shown as mean ± SD (***P < 0.0005, **P < 0.005, triplicate experiments, Student’s t test). shCon, control. (K) Effects of WWP1 on Ras and MYC-induced colony-forming activity in soft agar or signaling pathway in cells. The colony numbers are quantified and presented as mean ± SD (***P < 0.0005, **P < 0.005, triplicate experiments, Student’s t test) (left). Total lysates were resolved by SDS-PAGE and then probed with indicated antibodies (right). ERK, extracellular signal–regulated kinase; pERK, phosphorylated ERK. (L) Effects of WWP1 on PTEN lipid phosphatase activities in MEFs with indicated constructs. Data are shown as mean ± SD (**P < 0.005, triplicate experiments, Student’s t test). In (C), (F), (H), and (K), actin was used as a loading control.
We investigated the biological impact of this WWP1-PTEN axis on MYC-induced tumorigenesis. Depletion of WWP1 led to decreased formation of soft-agar colonies, an effect which was more prominent in MYC-overexpressing cells (Fig. 3I, left) but significantly impaired when PTEN was depleted by CRISPR (Fig. 3I, right, and fig. S4F), suggesting the relevance of PTEN in MYC-WWP1–mediated tumorigenesis. This was accompanied by increased apoptosis (Fig. 3J and fig. S4G). Similarly, transformation induced by MYC and Ras and AKT activation were also significantly impaired with the increase in PTEN lipid phosphatase activity when endogenous WWPI was depleted by CRISPR (Fig. 3, K and L). On the other hand, MYC and WWP1 overexpression showed a cooperative effect in promoting anchorage-independent growth (fig. S4H). Collectively, multiple lines of evidence support an oncogenic role for WWP1.
Aberrancies of the MYC-WWP1-PTEN signaling axis in human cancer
To assess the pathophysiological relevance of the MYC-WWP1-PTEN interaction in human cancer, we examined the abundance of WWP and PTEN polyubiquitination in various human prostate cancer (PCa) cell lines. Expression of WWP1 was positively correlated with endogenous PTEN polyubiquitination and inversely correlated with PTEN accumulation in the membrane fraction but had no correlation with PTEN accumulation in either the total or the soluble fraction (fig. S4, I and J).
We next performed bioinformatics analyses to determine the correlations between MYC, WWP1, and PTEN activity in human prostate specimens with phospho-AKT, phospho-S6K, and phospho-S6 as a readouts. MYC transcript abundance correlated with abundance of WWP1 (fig. S5A). Amounts of WWP1 transcript correlated with amounts of phospho-AKT, phospho-S6K, and phospho-S6, indicating an impact on activity of the PI3K-AKT-mTOR pathway in WWP1-high patients (fig. S5, B to D). MYC levels also correlated with phospho-AKT (fig. S5E). Furthermore, abundance of MYC and WWP1 significantly correlated with phosphorylation of AKT, S6K, and S6 (fig. S5, F to H), indicating effects of the MYC-WWP1 axis in activation of the PI3K-AKT-mTOR pathway.
WWP1 is localized on chromosome 8q21, a region not only frequently amplified in human cancers of various histological origins but also near the MYC amplicon (16–19). Our analysis of human PCa specimens revealed amplification of both WWP1 with MYC (fig. S5I) and showed that amplification of both WWP1 and MYC (which could further potentiate the activation of this proto-oncogenic pathway toward PTEN inactivation) occurred in PTEN-intact PCa specimens (fig. S5J). In support of this notion, gene set enrichment analyses (GSEA) from The Cancer Genome Atlas (TCGA) PCa dataset exhibited a positive correlation of MYC or WWP1 levels with Kyoto Encyclopedia of Genes and Genomes (KEGG) PI3K-AKT transcriptional profiles (fig. S5K). Collectively, these findings support the relevance of aberrant MYC-WWP1-PTEN interaction in human PCa.
Inactivation of Wwp1 in the mouse suppresses MYC-driven prostate tumorigenesis
To assess in vivo whether loss of WWP1 inhibits MYC-driven prostate tumors, we used Hi-Myc mice, which express Hi-Myc in a prostate epithelium–specific manner that results in complete penetrance of high-grade prostatic intraepithelial neoplasia (PIN) by 3 months of age, with progression to invasive adenocarcinoma within 5 to 12 months of age (31). We crossed these mice with Wwp1 homozygous mice to obtain cohorts of Hi-Myc; Wwp1−/− mutants or Hi-Myc;Wwp1+/+ controls. Consistent with the results obtained in our cell lines, homozygous Wwp1 deletion significantly decreased Hi-Myc–driven tumorigenesis in terms of both tumor size and weight (Fig. 4, A and B). Histological analyses corroborated that prostates from Wwp1 homozygous mice had substantially decreased invasive carcinoma at 5 months of age as compared with those from Wwp1 WT counterparts (Fig. 4C). Homozygous Wwp1 deletion also inhibited the PI3K-AKT-mTOR signaling pathway and abundance of Ki67, a cellular marker for proliferation, without affecting abundance of PTEN in prostate-derived lysates (Fig. 4, C and D), and this inhibition was accompanied by concomitant induction of apoptosis in the dorsolateral prostate (DLP) of mice, as revealed by cleaved caspase-3 staining (Fig. 4D and fig. S6A). Immunofluorescence analyses of PTEN protein in DLP tissue revealed that Wwp1 homozygous mice showed accumulation of PTEN at the plasma membrane in prostate epithelial cells compared with Wwp1 WT counterparts (Fig. 4E). Global transcriptional analysis by RNA-sequencing (RNA-seq) analyses revealed that PI3K-AKT transcriptional profiles from both KEGG and Reactome databases were decreased upon Wwp1 genetic knockout, as compared with those of the WT counterparts (Fig. 4F and fig. S6B). Thus, genetic ablation of Wwp1 triggered PTEN reactivation and subsequent suppression of the PI3K-AKT pathway, leading to inhibition of Myc-driven tumorigenesis in vivo.
Fig. 4. Characterization of WWP1 effects on Hi-Myc driven prostate tumors in mice.
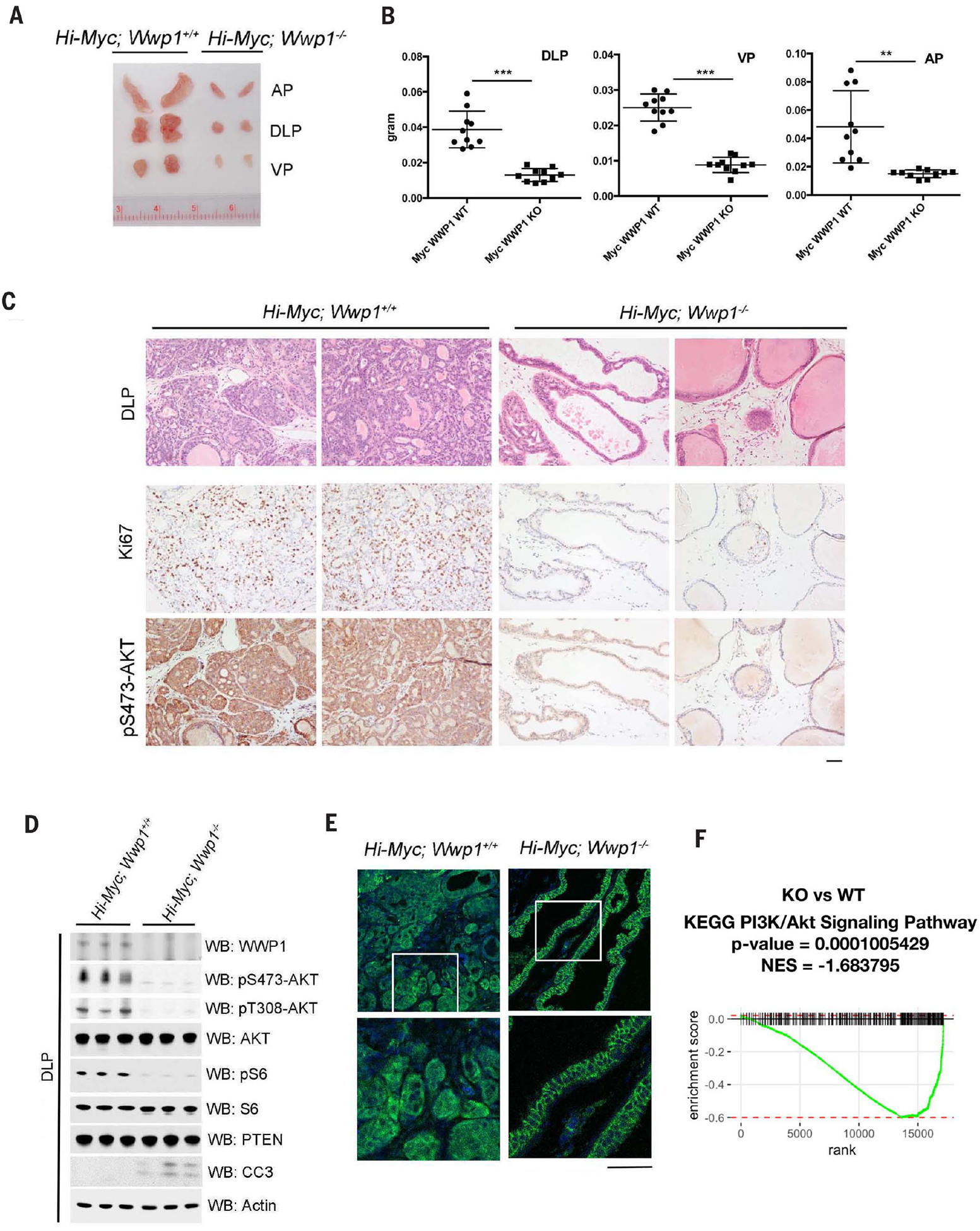
(A) Gross anatomy of representative urogenital tracts from Hi-Myc; Wwp1+/+ or Hi-Myc; Wwp1−/− mice. The mice were analyzed at 5 months of age. (B) Analysis of the weight of the prostate lobes derived from Hi-Myc; Wwp1+/+ or Hi-Myc; Wwp1−/− mice. The mice were analyzed at 5 months of age (n = 5 mice per genotype). Data are shown as mean ± SD (***P < 0.0005, **P < 0.005, Student’s t test). KO, knockout. (C) Hematoxylin and eosin (H&E) and IHC staining of DLPs from Hi-Myc; Wwp1+/+ or Hi-Myc; Wwp1−/− mice with indicated antibodies. The mice were analyzed at 5 months of age. Scale bar, 50 μm. (D) Western blot analysis of DLP lysates from Hi-Myc; Wwp1+/+ or Hi-Myc; Wwp1−/− mice. The mice were analyzed at 5 months of age (n = 3 mice per genotype). Actin was used as a loading control. CC3, cleaved caspase-3. (E) Confocal analysis of DLPs from indicated mice stained with PTEN (green) and DAPI (blue). The white-outlined areas in the top row are enlarged 2.6-fold (bottom row) to show the subcellular localization of PTEN. Representative pictures are shown (n = 2 mice per genotype). Scale bars, 50 μm. (F) GSEA of RNA-seq data from the DLPs of Wwp1 knockout versus Wwp1 WT mice using the PI3K-Akt signaling pathway gene set annotated in the KEGG. Mice from all four groups contain the same Hi-Myc genetic background unless otherwise noted. NES, normalized enrichment score.
Identification of indole-3-carbinol as a WWP1 inhibitor
Indole-3-carbinol (I3C) is a natural compound with negligible toxicity produced by the breakdown of glucosinolate glucobrassicin, which can be found in cruciferous vegetables such as broccoli, cauliflower, cabbage, collard greens, brussel sprouts, and kale (32–36). I3C appears to fit into the HECT domain of NEDD4–1 by in silico molecular modeling and thereby interacts with NEDD4–1 with a calculated equilibrium dissociation constant (Kd) of about 88.1 μM to effectively inhibit NEDD4–1 activity (37).
Because the HECT catalytic domain of WWP1 is structurally similar to that of NEDD4–1, we tested whether I3C might also inhibit WWP1. The NEDD4–1 HECT domain allows for the binding of the covalent inhibitor (covalent inhibitor 3) to its N-terminal lobe (38). When we modeled the binding of I3C to WWP1 by superimposing I3C and WWP1 onto a crystal structure of the covalent inhibitor 3 and NEDD4–1, we identified a binding pocket formed by WWP1 residues F577 (Phe577), L630 (Leu630), Y628 (Tyr628), C629 (Cys629), N650 (Asn650), and Y656 (Tyr656). The indole core of I3C could fit into the center of the N-terminal domain pocket of WWP1 and thus interact extensively with the surrounding hydrophobic residues, including F577 and Y656 (Fig. 5A and fig. S7A), which indicates a possible inhibitory function of I3C toward the E3 ligase activity of WWP1. A microscale thermophoresis (MST) binding analysis supported this possibility. The MST binding assay revealed that I3C binds to the WWP1 HECT domain with a dissociation constant of 450 nM, which is stronger than its binding affinity for NEDD4–1 (Fig. 5B). We also developed a F577/Y656A WWP1 mutant to disrupt the binding of I3C with WWP1, which was functionally equivalent to WT WWP1 in triggering PTEN polyubiquitination (fig. S7B); MST analysis revealed that I3C binds this WWP1 mutant at a dissociation constant of around 7719 nM, an order of magnitude weaker than its binding to WT WWP1 (Fig. 5B).
Fig. 5. Therapeutic targeting of WWP1 E3 ligase in vitro and in vivo.
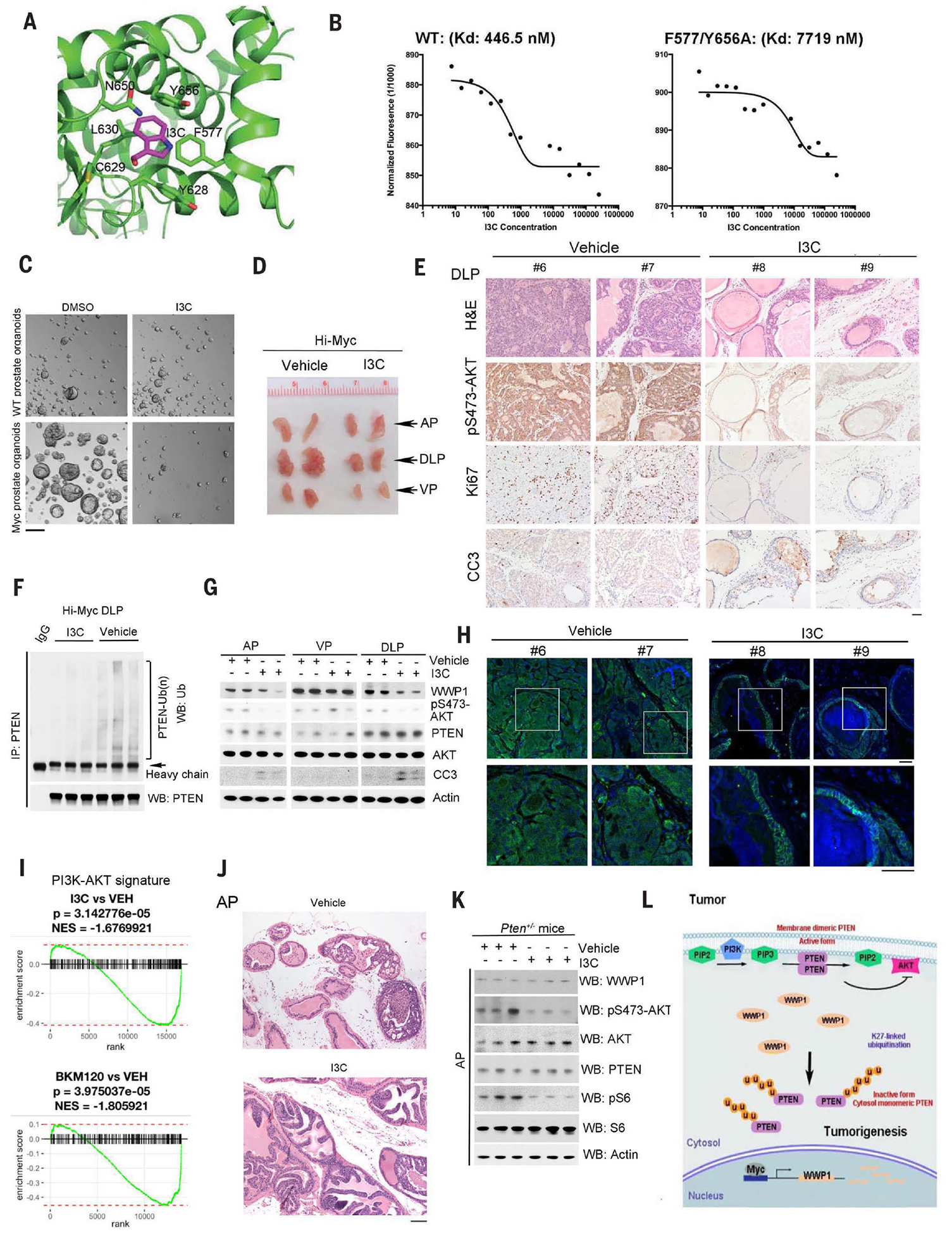
(A) In silico modeling of predicted interactions with the HECT domain of WWP1. (B) MST analysis determined the Kd of I3C toward His-WWP1 HECT WT or the His-WWP1 HECT F577/Y656A mutant. Concentration is reported in nanomolar. (C) Analysis of the effects of I3C on prostate organoid–forming ability from WT or Hi-Myc mice treated with or without 10 or 20 μM I3C for 3 days. Scale bar, 100 μm. DMSO, dimethyl sulfoxide. (D) Gross anatomy of representative urogenital tracts from Hi-Myc mice treated with vehicle or I3C. The mice, at 5 months of age, were treated intraperitoneally with I3C (20 mg/kg), three times a week for 1 month starting on day 0. Representative pictures are shown in the pane (n = 9 mice per group). (E) H&E and IHC staining of DLPs from Hi-Myc mice treated with vehicle or I3C with indicated antibodies. Scale bar, 50 μm. (F) In vivo ubiquitination analysis of endogenous PTEN ubiquitination levels of DLPs from Hi-Myc mice treated with vehicle or I3C. (G) Western blot analysis of AP, DLP, and VP lysates from Hi-Myc mice, as shown in (D), with the indicated antibodies. Actin was used as a loading control. (H) Confocal analysis of DLPs from indicated mice stained with PTEN (green) and DAPI (blue). The white-outlined areas in the top row are enlarged 2.6-fold (bottom row) to show the subcellular localization of PTEN. Representative pictures are shown (n = 2 mice per genotype). Scale bars, 50 μm. (I) GSEA of RNA-seq data from the DLPs of (i) I3C-treated mice versus vehicle (VEH)–treated mice and (ii) BKM120-treated mice versus vehicle-treated mice using the PI3K-Akt signaling pathway gene set annotated in the KEGG. Mice from all four groups contain the same Hi-Myc genetic background unless otherwise noted. (J) H&E staining of APs from Pten+/− mice treated with vehicle or I3C. At 7.5 months of age, the mice were treated intraperitoneally with I3C (20 mg/kg), three times a week for 1 month starting on day 0. Representative pictures are shown (n = 3 mice per group). Scale bar, 100 μm. (K) Western blot analysis of AP lysates from Pten+/− mice, as shown in (J), with the indicated antibodies. Actin was used as a loading control. (L) Model for WWP1-mediated PTEN K27-linked polyubiquitination in cell growth, tumor development, and progression. Deregulated MYC overexpression or MYC amplification promotes WWP1 expression and, in turn, triggers PTEN K27-linked polyubiquitination. Aberrant K27-linked polyubiquitination suppresses PTEN dimerization, plasma membrane recruitment, and tumor-suppressive function, leading to the tumor initiation and progression. PIP2, phosphatidylinositol 4,5-bisphosphate; u, ubiquitin.
We assessed the differential effects of I3C toward WWP1 or NEDD4–1 in Wwp1−/− or Nedd4–1−/− MEFs and found that cells lacking Wwp1 were more resistant to I3C treatment than cells lacking Nedd4–1 or WT cells (fig. S7C). In line with these results, reconstitution of WT WWP1 in Wwp1-null MEFs sensitized cells to I3C treatment, whereas the WWP1 F577/Y656A mutant failed to do so (fig. S7D). Collectively, these findings indicate that WT WWP1 may be a direct target of I3C, with binding mediated through its F577 and Y656 residues.
Therapeutic targeting of K27-linked PTEN polyubiquitination potently suppresses tumorigenesis
We examined whether inhibition of WWP1 by I3C would affect PTEN K27-linked polyubiquitination. Ubiquitination assays revealed that I3C inhibited WWP1-mediated PTEN K27-linked polyubiquitination both in vitro and in vivo (fig. S7, E and F), but not WWP2-mediated K48-linked polyubiquitination (fig. S7G). Based on the inhibitory effects of K27-linked polyubiquitination on PTEN plasma membrane recruitment, we tested whether inhibition of WWP1 as a result of I3C treatment would affect PTEN subcellular localization in cells. In line with the data above, inhibition of WWP1 via I3C induced PTEN plasma membrane accumulation, as evidenced by confocal immunofluorescence analysis (fig. S8A).
The observation that depletion of WWP1 suppresses MYC-induced tumorigenesis prompted us to examine whether MYC overexpressing cells could develop “oncogenic addiction” toward the WWP1-PTEN axis to support cell growth, which would be revealed by enhanced sensitivity to WWP1 inhibition. To this end, we examined the effect of I3C on both prostate organoids and spheres derived from WT and Hi-Myc mice at 3 months of age (31). Serial passaging of these samples revealed that Hi-Myc prostate epithelial cells displayed enhanced stem and progenitor self-renewal capacity, growth, and size as compared with cells from WT mice (Fig. 5C and fig. S8, B to D). I3C treatment suppressed both the number and size of prostate organoids and sphere-forming ability in Hi-Myc prostates, but not in WT counterparts, largely owing to increased apoptosis (Fig. 5C and fig. S8, D and E). Moreover, Pten deletion by CRIPSR in Hi-Myc organoids conferred partial resistance to I3C, again corroborating that I3C exerts its function at least in part in a PTEN-dependent manner (fig. S8F). In line with our previous data, prostate spheres from Hi-Myc mice displayed higher abundance of WWP1 and AKT activation, without affecting abundance of PTEN. Similarly, treatment with I3C abolished MYC-driven AKT activation and led to apoptosis in Hi-Myc prostate spheres, but not in WT prostate spheres (fig. S8E). Our data from these cellular models demonstrate that Hi-MYC expression induces oncogenic addiction to the WWP1-PTEN pathway, which sensitizes cells to I3C treatment.
To examine the therapeutic relevance of the MYC-WWP1-PTEN interaction in vivo, we performed preclinical studies with I3C in Hi-Myc mice (31). Hi-Myc mice develop complete-penetrance PIN lesions that evolve into invasive adenocarcinoma by 5 to 12 months of age (31). We treated cohorts of Hi-Myc mice at 5 months of age with either vehicle or I3C [20 mg/kg (mg per kg body weight); three times per week] for one month. Control Hi-Myc mouse prostates displayed heterogeneous disease progression, and DLPs displayed extensive invasive carcinoma, whereas the ventral prostate (VP) and anterior prostate (AP) developed PIN lesions to a similar extent (fig. S9A). However, I3C-treated Hi-Myc mouse prostates had decreased tumor weight and size (Fig. 5D and fig. S9B). Histological, IHC, in vivo ubiquitination, and Western blotting analyses confirmed that I3C treatment not only inhibited PTEN polyubiquitination but also inhibited AKT activity and abundance of Ki67 without affecting abundance of PTEN; this inhibition was accompanied by increased apoptosis in the AP and DLP, but not VP, of Hi-Myc mice (Fig. 5, E to G). Immunofluorescence analyses of PTEN revealed that in prostate epithelial cells, the administration of I3C induced greater PTEN accumulation at the plasma membrane (Fig. 5H). We also performed pharmacokinetic analysis with I3C. Male C57BL/6 mice were treated with I3C after intraperitoneal administration at 20 mg/kg every day or every other day. Plasma concentrations of I3C reached a steady state and maintained a steady plasma concentration over almost 2 weeks (fig. S9C). The steady-state concentration was around 387 ± 277 nM, which is in line with the Kd value between I3C and WWP1 observed in vitro. All treated mice were healthy without apparent clinical symptoms. Collectively, these results demonstrate that I3C targeting of WWP1 E3 ligase triggers reactivation of the PTEN tumor suppressive function, promoting its plasma membrane recruitment, and suppression of MYC-driven tumorigenesis in vivo.
We ascertained the global transcriptional output upon Wwp1 inactivation by I3C to determine which pathways were affected. We performed whole-transcriptome analysis using murine prostate tissue samples from vehicle or I3C-treated Hi-Myc mice. Consistent with our Western blot analyses, a GSEA using KEGG gene set annotations revealed that the PI3K pathway signatures were decreased in cells treated with I3C (Fig. 5I), like they were after genetic knock out of Wwp1 (Fig. 4F). To determine the selectivity of the observed effects on the PI3K-AKT pathway, we also performed whole-transcriptome analysis of mouse prostates from Hi-Myc mice treated with three PI3K inhibitors: BKM120, BYL719, and GDC-0032 (39). GSEA analyses showed similar down-regulation of PI3K-AKT transcriptional profiles in prostate treated with either I3C or PI3K inhibitors (Fig. 5I and fig. S9D). Moreover, we observed a clustering of I3C- and PI3K inhibitor–perturbed samples by principal components analyses, corresponding to 35% of the variance across the samples (fig. S9E). Taken together, the down-regulation of PI3K signaling pathway transcriptional outputs due to WWP1 inhibition combined with the clustering of PI3K-AKT signature profiles after perturbation by I3C and PI3K inhibitors indicates that genetic knock out of Wwp1 and pharmacological perturbation by I3C result in a similar transcriptional effect through the inhibition of the PI3K-AKT signaling pathway.
We identified the pathway that controls PTEN dimer formation and membrane recruitment. This MYC-WWP1–mediated regulatory pathway does not trigger PTEN proteasomal degradation. However, K27-linked ubiquitination by WWP1 suppresses PTEN activity by preventing its dimerization. Indeed, this ubiquitin chain appears to control of protein dimerization, which is consistent with the notion that K27-linked polyubiquitination is an inefficient proteolytic signal (23–25) (Fig. 5L). The TRAF6 ubiquitin ligase targets AKT for K63-linked polyubiquitination to potentiate its membrane recruitment and activation (40). Thus, nonproteolytic ubiquitination of the key components of the PI3K-AKT signaling pathway could represent a general mechanism for regulating their subcellular localization and membrane recruitment.
We have identified WWP1 as a target of MYC and show that WWP1 is coamplified with MYC in the human cancer specimens from TCGA PCa dataset (Fig. 3 and fig. S5I). Genetic ablation of WWP1, either in cells by CRISPR or in mice by gene targeting, resulted in PTEN dimerization and membrane recruitment accompanied by suppression of AKT signaling (Fig. 2, D to H), whereas genetic or pharmacological inactivation of WWP1 in vivo in Hi-Myc mice corroborates a critical role of WWP1 in tumorigenesis through PTEN antagonism (Figs. 4 and 5). Because MYC is a critical transcriptional factor involved in multiple biological processes—including replication, cell division, protein synthesis, and metabolism (41)—targeting downstream targets essential for its oncogenic activity may represent an effective way to treat MYC-driven tumors while minimizing potential side effects.
The genetic inactivation of WWP1 is compatible with life (42), yet its function is essential for PI3K-AKT-mTOR signaling activation and tumorigenesis. As compared with WT mice, Wwp1 knockout mice display some of the hallmark features of mice whose PTEN function has been increased by the germline addition of extra copies of the Pten gene through bacterial artificial chromosome technology (7). Such mice and Wwp1 knockout mice are both viable, developmentally normal, and fertile, although smaller in body size and weight. Both types of mice display tumor resistance, suppressed activation of PI3K signaling, and suppressed expression of key metabolic enzymes, such as PFKFB3 and PKM2 (fig. S10, A and B). That genetic Pten superactivation and Wwp1 ablation are compatible with adult life also points to pharmacological inhibition of WWP1 as a possibly well-tolerated means of triggering PTEN reactivation or superactivation for cancer prevention and therapy.
Conceptually, our findings provide an example of a “tumor suppressor reactivation” approach to cancer treatment. Aberrant MYC expression resulted in oncogenic addiction to the WWP1-PTEN axis to the extent that upon genetic or pharmacological WWP1 inactivation, the survival and growth of cancer cells were suppressed (Figs. 3 to 5). PTEN is also frequently down-regulated or monoallelically lost in human cancers and cancer susceptibility syndromes, such as PHTS, irrespective of MYC. Intriguingly, depletion of WWP1 reduced PI3K and AKT activity in MEFs with monoallelic Pten or mutated Pten as observed in PHTS patients (fig. S10C), suggesting that monoallelic PTEN reactivation may be sufficient to suppress PTEN heterozygous or mutation-driven tumorigenesis. In support of this notion, pharmacological inactivation of WWP1 by I3C substantially inhibited heterozygous Pten loss-mediated PI3K-AKT-mTOR activation and PIN phenotypes in vivo (Fig. 5, J and K, and fig. S10D). Therefore, inhibition of the WWP1-PTEN pathway holds promise for patients coping not only with tumors driven by MYC overexpression or aberrant PTEN function but also with disorders caused by germline mutations of the PTEN gene (3).
Materials and Methods
Human cell lines
Human cell lines used in this study were purchased from the American Type Culture Collection (ATCC; www.atcc.org).
Murine models
The Beth Israel Deaconess Medical Center IACUC Committee on Animal Research approved all animal experiments. The transgenic mice used in this study (Hi-Myc-mice; FVB background), in which the prostate specific expression of human c-Myc is driven by the rat probasin promoter with two androgen response elements, were obtained from the Mouse Repository of National Cancer Institute. Mice were randomly chosen and used to examine the tumor at the indicated age. Wwp1–/– mice and its paired Wwp1+/+ mice (on a C57/BL6 background) were obtained from L. Matesic (University of South Carolina, Columbia, SC, USA) (42). The Hi-Myc; Wwp1–/– and its counterpart WT mice were generated by the interbreeding Hi-Myc; Wwp1+/– mice, which were born by crossing Hi-Myc mice with Wwp1−/−mice. Pten+/− mice were on a C57/BL6 background. The prostate glands were scored as follows: normal tissues (glands with a monolayer of epithelial cells without cellular neoplastic features) and neoplastic tissues (glands with a monolayer of epithelial cells with evident cellular neoplastic features and glands with a multiple layer of epithelial cells) (43). The extent of normal prostate epithelium in each prostate section was quantitatively expressed as the percentage of normal glands over total glands. The pathologist determined the histological grade blindly.
Xenotransplantation
For assaying tumor growth in the xenograft model, 7-week-old NCr nude mice housed in specific pathogen-free environments were injected s.c. with 2.5 × 106 PC3 derivatives (n = 5 for each group) mixed with RPMI medium and Matrigel (vol/vol, 1:1). The care and treatment of animals were approved by the Beth Israel Deaconess Medical Center IACUC Committee on Animal Research.
Plasmids, reagents, and antibodies
Human PTEN cDNA was cloned into the pLVX-Puro vector to generate a PTEN lentivirus expression plasmid (Clontech). LentiCRISPR v2 plasmid was a gift from Feng Zhang (Addgene #52961) (44). pCDH-puro-cMyc (46970) was purchased from Addgene. All mutant constructs of PTEN and WWP1 were generated using a QuickChange Lightning Site-Direct Mutagenesis (Agilent Technologies). All mutations were confirmed by sequencing. His-Ubiquitin, His-Ubiquitin KR mutants, His-Ubiquitin K-only mutants, Myc-PTEN and GST-PTEN constructs have been previously described (15, 45). pLenti-HA-WWP1 WT-Puro, pLenti-HA-WWP1 C890A-Puro, pLenti-HA-WWP1 F577A/Y566A-Puro, pcDNA3-HA-MYC, pcDNA3-Myc-PTEN, MYC-WWP1 WT, MYC-WWP1 C890A, and Flag-NEDD4 family ligases constructs, such as Flag-WWP1, Flag-WWP2, Flag-Smurf2, and Flag-Itch were gifts from W. Wei’s lab. The two individual siRNA duplexes targeted to WWP1 and control nontarget siRNA were purchased from Sigma Aldrich, whereas siRNA SMARTpool targeted to MYC was purchased from Dharmacon. Lentivirus based constructs expression shRNAs targeting human WWP1 (TRCN0000003395; TRCN0000003396) were obtained from GE Dharmacon. Lipofectamine 2000, RPMI, DMEM, Opti-MEM reduced serum media and fetal bovine serum (FBS) were purchased from Invitrogen. Anti-Flag-M2 affinity gel, insulin, and puromycin were purchased from Sigma Aldrich. Insulin was used at 100 or 200 ng/ml. Polybrene was purchased from Santa Cruz Biotechnology, Inc. Indole-3-carbinol (I3C) was purchased from Sigma Aldrich. For western blotting: Anti-Myc-Tag (2276), anti-PTEN (9559), Anti-MYC for western blot and ChIP assay (13987), anti-EGFR (4267), anti-Ubiquitin (3936), anti-Cleaved Capase3 (9661), anti-Phospho-AKT (pSer473, 9271; pThr308, 9275), anti-AKT (pan AKT, 4685), anti-PKM2 (4053), anti-PFKFB3 (13123), anti-pS6 (2211), anti-S6 (2217), anti-Smurf2 (2174) antibodies were all purchased from Cell Signaling Technology. Mouse Anti-PTEN antibody (6H2.1) was purchased from Cascade Bioscience; Anti-GFP (A-11120) was purchased from Invitrogen; Anti-WWP1 (human) (H00011059-M01) was purchased from Novus Biologicals; Anti WWP1 (ab43791) was purchased from Abcam; Anti-Actin (A3853) and Anti-Flag-M2) were purchased from Sigma Aldrich; Anti-HA-Tag (16B12) was purchased from Covance; Anti-WWP2 (A302–935A) was purchased from Bethyl. For immunohistochemistry in the tissue microarray (TMA) analysis: anti-PTEN (6H2.1) was purchased from Cascade Bioscience.
Cell Culture, cell transfection, and establishment of stable cell lines
All cell lines were obtained from ATCC and checked for mycoplasma by MycoAlert Mycoplasma Detection Kit (Lonza). Wwp1–/– MEFs were generated by using CRISPR-Cas9 gene-editing approach. U2OS-derived ubiquitin replacement cell lines described previously(21), were grown in DMEM containing 10% tetracycline-free FCS (GIBCO). HCT116, HepG2, MDA-MB-231, 293, 293T, and primary MEF cells were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin, and streptomycin (Invitrogen). PC3, LNCaP, C4–2, 22rv1, and VCaP cells were cultured in RPMI medium containing 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin, and streptomycin (Invitrogen). RWPE-1 and PWR-1E cells were cultured in a K-SFM medium supplemented with recombinant human Epidermal Growth Factor (rhEGF) and Bovine Pituitary Extract (BPE). Transfections were performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instruction. In brief, 5 × 105cells were transfected with 5 μg of DNA plasmids or 20 nM of siRNA in a six-well dish. Cells were recovered in the completed media for 12 hours and then harvested at the indicated times. Stable cell lines were generated by lentivirus transduction.
Lentivirus production and infection
To generate recombinant lentivirus, 293T cells were cotransfected with VSVG, PMDL, REV, and indicated lentivirus based constructs. The virus-containing supernatant was harvested. For infection, the viral stock was supplemented with 10 μg/ml of polybrenem and the infected cells were selected by 2 μg/ml of puromycin.
Cell proliferation assay
At 8 hours posttransfection, cells were trypsinized, resuspended, and seeded in three separate 12-well plates at a final density of 20,000/well. Starting the following day (d0), one plate per day was washed once with PBS, fixed in 10% formalin solution for 10 minutes at room temperature, and kept in PBS at 4°C. On the last day, all of the wells were stained with crystal violet. After lysis with 10% acetic acid, optical density was read at 595 nm.
In vivo ubiquitination assay
To analyze in vivo ubiquitination of PTEN, cells were transfected with various constructs, together with His-Ubiquitin and Myc-PTEN. Cells were lysed by buffer A [6 M guanidine-HCl, Na2HPO4/NaH2PO4 (pH 8.0), and 10 mM imidazole], and lysates were incubated with Ni-NTA agarose for 1.5 hours at 4°C. The beads were washed once with buffer A, twice with buffer A/TI [1 vol buffer A: 3 vol buffer TI (25 mM Tris-HCl, pH 6.8, and 20 mM imidazole)], and three times with buffer TI, and then analyzed by western blot. In all experiments, an equal amount of His-Ubiquitin expression was verified by western blot analysis.
In vitro ubiquitination assay
For in vitro ubiquitination, 400 ng of purified Flag-PTEN from 293 cells was incubated with 40 ng E1 (UBE1) (E-305 Boston Biochem), 500 ng E2 (UbcH5a) (E2–616 Boston Biochem), 10 μg His-Ub variants (Boston Biochem), 8 mM ATP, 1× ligase reaction buffer [Fisher Scientific (Thermo Fisher Scientific) # B71], and 1× Energy regeneration system (Boston Biochem #B-10), 400 ng NEDD4 (Sigma Aldrich # SRP0226), and 400 ng WWP1 (Sigma Aldrich # SRP0229) at 37°C for 1 hour in 25 μl of reaction mixture.
Annexin-V–amino actinomycin double staining
For detection of apoptosis, cells, infected with indicated viruses, were co-stained with Annexin-V-phycoerythrin (PE) and amino actinomycin D (7-AAD; Annexin V-PE apoptosis detection kit I, BD Biosciences) according to the manufacturer’s instructions. Stained cells were sorted with fluorescence-activated cell sorting (FACS).
Mass spectrometry
DU145 cells transfected with HA-PTEN were immunoprecipitated with anti-PTEN antibody and the PTEN-associated proteins were resolved by SDS-PAGE on 4–12% gradient gel (Invitrogen) for coomassie blue staining. Specific bands were cut out from the gel and subjected to mass-spectrometric peptide sequencing.
Western blotting and immunoprecipitation
For western blotting, cells were lysed in RIPA buffer (Boston BioProducts) supplemented with protease (Roche) and phosphatase (Roche) inhibitor. Proteins were separated on NuPAGE 4–12% Bis-Tris gradient gels (Invitrogen), transferred to polyvinylidine difluoride membranes (Immobilon P, Millipore) and the blots were probed with the indicated antibodies. For immunoprecipitation, U2OS, DU145, PC3, 293T, and MEF cells were transfected with the indicated expression vectors by using LIPOFECTAMIN 2000 (Life Technologies). Twenty-four hours after transfection, cells were lysed in RIPA buffer with protease (Roche) and phosphatase (Roche) inhibitor. Of total lysates, 500 mg were precleared for 30 minutes at 4°C and then immunoprecipitated with anti-Myc (Cell Signaling Technology 9B11, 1:500), or anti-PTEN (Cell Signaling Technology 9559, 1:500) antibody overnight at 4°C. The Protein-A or Protein-G sepharose beads (GE Healthcare) were then added and incubated for another 2 hours. The immunoprecipitates were washed with RIPA buffer three times. In denaturing conditions, standard Laemmli-Buffer with 5% final concentration of β-mercaptoethanol was added to the samples, which were then boiled and separated on NuPAGE 4–12% Bis-Tris gradient gels (Invitrogen).
Native gel analysis
To determine if PTEN dimerization occurred, cells were lysed in lysis buffer containing 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP40, 1 mM EDTA, 1 mM protease (Roche), and phosphatase inhibitor (Roche) for further immunoprecipitation. For the native elution, prechilled 0.1 M glycine pH 2.5 was used to elute immunocomplexes for 10 minutes at 4°C, further neutralized with 1 M Tris-HCl pH 8.0. Tris-Glycine-Native Sample buffer (Boston BioProducts, BP-120) was added, and samples were immediately run on NativePAGE Gel (Cat. No. BN1004BOX, Thermo Fisher Scientific).
In vitro binding assay
Recombinant GST-PTEN (15) was expressed in BL21 (DE3) bacteria under Ampicillin selection. For protein expression, after 5 hours of IPTG induction, bacteria were lysed, sonicated, cleared by centrifuge, and subjected to GST-pull down. Complexes were eluted with 5 volumes of 10 mM reduced GSH (G4251, Sigma) in 50 mM Tris-HCl pH 8.0. Eluted fractions were transferred into dialysis cassettes (66380, Thermo Scientific) and left overnight at 4°C on gentle rotation. The day after, samples were recovered and concentrated by using centrifuge filters, Amicon Ultra (UFC201024PL, EMD Millipore), and according to the manufacturer’s procedures. For GST-pull down, Ubiquitinated Flag-PTEN was isolated from 293 cells transfected with indicated plasmids by using M2 beads. Of recombinant GST-PTEN, 250 ng was incubated with the unmodified Flag-PTEN or ubiquitinated Flag-PTEN in RIPA buffer (Boston BioProducts) for 2 hours at 4°C. The beads were washed and bound proteins were analyzed by Western blotting.
NanoLC-MS/MS analysis of ubiquitinated PTEN
Ubiquitinated PTEN was isolated by anti-Flag M2 beads from cells transfected with Flag-PTEN, His-ubiquitin, together with or without WWP1 E3 ligase. The bound proteins were eluted with denaturing buffer containing 8 M urea, 20 mM HEPES, 1 mg/ml aprotinin, 1 mg/ml leupeptin, 1 mM N-ethylmaleimide (Sigma), and 1 mM PMSF. Eluted proteins were digested with trypsin for overnight, treated with trifluoroacetic acid (TFA), and clarified by centrifugation. The supernatant was desalted on a Sep-Pak C18 column (Waters), and lyophilized peptides were dissolved in IP buffer containing 50 mM MOPS, pH 7.2, 10 mM sodium phosphate, and 50 mM NaCl. Ubiquitinated peptides were enriched by incubation overnight with protein A agarose conjugated with an anti-K-ε-GG antibody (Cell Signaling Technology, Inc.), which specifically recognizes the di-glycyl remnant produced on ubiquitinated lysine residues after trypsin digestion. Beads were washed with IP buffer followed by water, and bound peptides were eluted with 0.15% TFA, desalted by Stage tip chromatography, and lyophilized for MS analysis.
NanoLC–nanoESI-MS/MS analysis was performed on a nanoAcquity system (Waters) connected to the Orbitrap Elite hybrid mass spectrometer (Thermo Electron) equipped with a PicoView nanospray interface (New Objective). Peptide mixtures were loaded onto a 75-μm ID, 25-cm length C18 BEH column (Waters) packed with 1.7-mm particles with a pore of 130 Å and were separated at 35°C using a segmented gradient from 5 to 35% acetonitrile in 0.1% formic acid at a flow rate of 300 nl/min for 90 min. The mass spectrometer was operated in the data-dependent mode. Briefly, surveying full scan, MS spectra were acquired in the orbitrap (m/z 350 to 1600) with the resolution set to 120 K at m/z 400 and automatic gain control (AGC) target at 106. The 15 most intense ions were sequentially isolated for HCD MS/MS fragmentation and detection in the orbitrap with previously selected ions dynamically excluded for 60 s. For MS/MS, we used a resolution of 15,000, an isolation window of 2 m/z, and a target value of 50,000 ions, with maximum accumulation times of 200 ms. Fragmentation was performed with normalized collision energy of 30% and an activation time of 0.1 ms. Ions with singly and unrecognized charge state were also excluded. All data generated were searched against the customized Swiss-Prot Homo sapiens database and His-tagged ubiquitin protein sequences (20,169 entries total) database using the Mascot search engine (v.2.5.1; Matrix Science, Boston, MA, USA) through Proteome Discoverer (v 2.1.0.81; Thermo Scientific). Search criteria used were trypsin digestion, variable modifications set as carbamidomethyl (C), oxidation (M), GlyGly (K), and LeuArgGlyGly (K), allowing up to two missed cleavages, mass accuracy of 10 ppm for the parent ion, and 0.02 Da for the fragment ions. Two target values for a decoy database search were applied: strict FDR of 0.01 and a relaxed FDR of 0.05. Ubiquitination sites and peptide sequence assignments contained in MASCOT search results were validated by manual confirmation from raw MS/MS data. For label-free quantification, precursor ions areas were extracted using Precursor Ions Area Detector node in Proteome Discoverer 2.1.0.81 with a 2 ppm mass precision (the experimental m/z and retention times were recorded for precursor area quantification).
NanoLC–nanoESI-MS/MS analysis of ubiquitinated PTEN endogenously
Endogenous ubiquitinated PTEN was isolated by anti-PTEN beads (Santu Cruz, SC7974-AC) from DU145 cells stably expressing MYC with or without WWP1 shRNAs. The bound proteins were eluted with denaturing buffer containing 8 M urea, 20 mM HEPES, 1 mg/ml aprotinin, 1 mg/ml leupeptin, 1 mM N-ethylmaleimide (Sigma), and 1 mM PMSF. Eluted proteins were digested with trypsin for overnight, treated with trifluoroacetic acid (TFA), and clarified by centrifugation. The supernatant was desalted on a Sep-Pak C18 column (Waters), and lyophilized peptides were dissolved in IP buffer containing 50 mM MOPS, pH 7.2, 10 mM sodium phosphate, and 50 mM NaCl. Ubiquitinated peptides were enriched by incubation overnight with protein A agarose conjugated with an anti-K-ε-GG antibody (Cell Signaling Technology, Inc.), which specifically recognizes the di-glycyl remnant produced on ubiquitinated lysine residues after trypsin digestion. Beads were washed with IP buffer followed by water, and bound peptides were eluted with 0.15% TFA, desalted by Stage tip chromatography, and lyophilized for MS analysis.
NanoLC–nanoESI-MS/MS analysis was performed on a EASY-nLC™ 1200 system connected to an Thermo Orbitrap Fusion Luoms mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a Nanospray Flex ion source (Thermo Fisher Scientific, Bremen, Germany). Peptide mixtures were loaded onto a 75-mm ID, 25-cm length PepMap C18 column (Thermo Fisher Scientific) packed with 2-μm particles with a pore with of 100 Å and were separated using a segmented gradient in 60 min from 5 to 45% solvent B (80% acetonitrile with 0.1% formic acid) at a flow rate of 300 nl/min. Solvent A was 0.1% formic acid in water. The mass spectrometer was operated in the data-dependant mode. Briefly, survey scans of peptide precursors from 350 to 1600 m/z were performed at 60 K resolution with a 2 × 105 ion count target. Tandem MS was performed by isolation window at 1.6 Da with the quadrupole, HCD fragmentation with normalized collision energy of 30. The MS2 ion count target was set to 5 × 104, and the max injection time was 100 ms. Only those precursors with charge state 2 to 7 were sampled for MS2. The instrument was run in top speed mode with 3-s cycles; the dynamic exclusion duration was set to 15 s with a 10 ppm tolerance around the selected precursor and its isotopes. Monoisotopic precursor selection was turned on.
PD 2.2 label-free data analysis
All data generated were searched against the Swiss- Prot Homo sapiens database using the Mascot search engine (v.2.6.1; Matrix Science, Boston, MA, USA) through Proteome Discoverer (v 2.2.0.388; Thermo Scientific, Waltham, MA, USA). Search criteria used were trypsin digestion, variable modifications set as carbamidomethyl (C), oxidation (M), acetylation (protein N-terminal) and ubiquitinated lysine (K) allowing up to two missed cleavages, mass accuracy of 10 ppm for the parent ion, and 0.02 Da for the fragment ions. The false discovery rate (FDR) was set to 1% for peptide identifications. Ubiquitination sites and peptide sequence assignments contained in MASCOT search results were validated by manual confirmation from raw MS/MS data. For label-free quantification, precursor ions areas were extracted using Minora Feature Detector node in Proteome Discoverer 2.2 with a 10 ppm mass precision (align the LC/MS peaks mapping to the isotope pattern and retention time). The ratios for each peptide were normalized by the total identified peptides, and the ratios were used for protein-level quantification.
ChIP assay
DU145 cells were fixed by addition of 37% formaldehyde to a final concentration of 1% formaldehyde and incubated at room temperature for 10 min. Crosslinking was stopped by the addition of glycine to a final concentration of 0.125 M. Cells were then scraped, and samples were prepared using SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling, #9003) according to the manufacturer’s protocol. The chromatin fractions were incubated in each case with 10 mg of antibodies to one of the following: MYC (Cell Signaling, #13987), human RPL30 or normal rabbit IgG (both provided by Cell Signaling kit, Cell Signaling) at 4°C overnight with Magnetic Protein G Beads. After extensive washing and final elution, the product was treated at 65°C overnight to reverse the crosslinking. Input DNA and immunoprecipitated DNA were purified using a kit column and analyzed by qPCR using SYBR Green Supermix (Bio-Rad) with the following sets of primers (both proximal and distal promoter regions): human WWP1 promoter (forward, 5′-GTCCGGAGTTGGAGGCTTT-3′; reverse, 5′-GACCCCACACCTCCCTTC-3′), human JunB (forward, 5′-AAGCCCACAGAGAGAGGTGGAAG-3′; reverse, 5′-CCAGAAGGTGGTGCCTTTTTATTG-3′). All results were normalized to the respective input values.
Generating the PTEN knockout DU145 cell line using the CRISPR-Cas9 technology
The PTEN knockout DU145 cell line was generated following the protocol described previously1. The sgRNAs (hPTEN-exon5-sgRNA-F: CACCgGCATATTTATTACATCG; hPTEN-exon5-sgRNA-R: AAAcCGATGTAATAAATATGCc) targeting the genomic sequence of PTEN were designed using the CRISPR design tool (http://crispr.mit.edu) and cloned into Lenti-CRISPRv2. The control Lenti-CRISPRv2-sgRNAs (CGCTTCCGCGGCCCGTTCAA) was a kind gift from Dr. William Kaelin at Dana Farber Cancer Institute. For virus packaging, these constructs were transfected into HEK 293T cells with packing vectors (pCMV-D8.9 and VSVG plasmids) using lipofectamine 2000 reagent. Virus-containing media were collected and filtered before being used for infection of DU145 cells. For viral infection, DU145 cells with 50% confluence were infected with virus containing sgPTEN with 4 μg/ml polybrene (Sigma-Aldrich). After 48 hours, the cells were passaged and selected using 1 μg/ml puromycin (Sigma-Aldrich) for 72 hours to eliminate the uninfected cells before harvesting for Western blot validation.
CRISPR-Cas9 gene-editing approach
Construction of lenti–CRISPR-Cas9 vectors targeting mouse WWP1 was performed following the protocol associated with the backbone vector, LentiCRISPR v2 (Addgene, #52961). The software (http://crispr.mit.edu/) predicted the following sequences with priority given to sequences that matched the early coding exons of targeted genes. The nonbold part of the sequence is gene specific. Wwp1 sgRNA_1 (fwd: 5′-CACCGATCAGCTGCTCGTCCCATTT-3′; rv: AAACAAATGGGACGAGCAGCTGATC), Wwp1 sgRNA_2 (fwd: CACCGTAATACTCGAACTACTACAT; rv: AAACATGTAGTAGTTCGAGTATTAC). Two mouse Pten sgRNAs were from T. Jacks’s group (46). mPten sgRNA_1: AAACAAAAGGAGATATCAAG, mPten sgRNA_2: GCTAACGATCTCTTTGATGA. hPTEN sgRNA: GCATATTTATTACATCG.
CRISPR-Cas9–mediated deletion of MYC responsive element within the WWP1 promoter
sgRNAs that target WWP1 promoter region were individually inserted into lentiCRISPRv2 hygro and lentiCRISPRv2 puro, respectively. Transfection plasmids in DU145 cells were carried out with Lipofectamine 2000 Reagent (Thermo) according to the manufacturer’s instructions. Genomic DNAs of selected positive cells were extracted for the genotyping validation with appropriate set of primer listed below. (The nonbold part of the sequence is WWP1 promoter specific.)
sgRNA_1: GAGGCTGCTCTAAGCTCGCGC
sgRNA_3: GGCTGGGGGTGGCGCGTGGAC
sgRNA_4: GCTGGGGGTGGCGCGTGGACG
Genotyping primers:
Primer F1: ATCTGCTTTATCACCGGGCT
Primer R1: TCGGATGCTGCCTTAGGAAA
Primer R2: CAGCAGCCACTCGGAACC
Cellular fractionation
Membrane versus cytosolic fractionation of 293T, MEF, or PC3 cells transfected with the indicated constructs was performed using the Proteo-Extract Native Membrane Protein Extraction Kit (Calbiochem) and according to the manufacturer’s procedures.
Immunofluorescence analysis
PC3 and DU145 cells stably expressing the indicated constructs were plated on coverslip. The following day, cells were washed with ice-cold PBS, fixed in 4% paraformaldehyde, permeabilized with 0.02% Triton X-100, and then blocked with PBS supplemented with 20% goat serum. Cells were incubated with anti-PTEN antibody (6H2.1, 1:100) from Cascade Bioscience, diluted in PBS containing 10% goat serum overnight and then with Alexa488-conjugated secondary antibody together with 1 mg/ml of DAPI for 1 hour. Images were acquired with a LSM510META Confocal Laser System at the BIDMC microscopy core facility.
Expression and purification of wild-type and mutant WWP1 in bacteria
WWP1 was cloned into pET28a with an N-terminal His6 tag. The recombinant plasmids were transformed into E. coli BL21(DE3) RipL for expression. The cells were cultured to OD 0.6 at 37°C, and were induced by 0.5 mM IPTG at 18°C for 16 hours before harvest. Cell pellets were re-suspended in 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 15 mM imidazole and were disrupted by sonication on ice. The cell lysate was centrifuged at 17,000 rpm for 1 hour to remove cell debris. The supernatant was incubated with nickel chelating beads for 30 min in cold room. The nickel chelating beads were washed by 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 30 mM Imidazole for 10-bed volume. WWP1 protein was eluted from nickel chelating beads by 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 300 mM Imidazole, 10% glycerol.
Protein labeling and microscale thermophoresis (MST) analysis
The binding affinity of WWP1 and I3C was measured at 25°C in a binding buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol by MicroScale Thermophoresis using MONOLITH NT.115. WWP1 was applied to buffer exchange column and was eluted by PBS. 200 μl WWP1 at a concentration of 10 μM was incubated with threefold excess fluorescent dye at room temperature in the dark for 30 min. After incubation, the labeling reaction was adjusted to 500 μl and loaded onto a column to exchange into the binding buffer of 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol. The labeled WWP1 was mixed 1:1 with I3C in a two-fold dilution series from 7.63 nM to 250,000 nM for the measurement. The data were plotted by the GraphPad Software.
Structure modeling of the WWP1 in complex with I3C
The structure modeling was done using Coot (47). First, the structure of WWP1 HECT domain (PDB ID: 5HPS) (48) was superimposed with NEDD4–1 HECT domain (PDB ID: 5C91) (38) in Coot. Second, the I3C was superimposed onto a covalent inhibitor bound in a pocket at the NEDD4–1 N-terminal lobe. Third, the WWP1 HECT domain and the I3C molecule were saved as a new file for structural analysis and presented using PyMOL (www.pymol.org).
Inhibition of cell proliferation
Cells were seeded with a density of 3000 cells per well in 96-well flat-bottomed plates and incubated for 24 hours in 10% FBS–supplemented RPMI culture medium. Cells were then treated with various doses of I3C. Control cells received DMSO at a concentration equal to that in drug-treated cells. After 5 days, the number of cells was stained by medium containing 0.5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide for 2 hours incubation at 37°C, followed by removing the media before adding 200 ml DMSO. Absorbance was determined at 570 nm.
Prostate sphere assay
The culture and passage of prostate spheres were carried out as described (49). Dissociated prostate epithelial cells were prepared from mice at 12 weeks of age. To initiate sphere formation, unsorted mouse prostate cells were prepared in PrEGM medium (Lonza) at a density of 5 × 104 cells per ml. Of the cell suspension, 40 ml was mixed with 60 ml cold Matrigel (BD Bioscience), and pipetted around the rim of a well of a 12-well plate and allowed to solidify at 37°C for 30 min. One ml warm PrEGM was then added to each well. The spheres were cultured and monitored for 10 days with 50% medium change every 2 days. To passage spheres, Matrigel was digested by 1 mg/ml dispase solution (StemCell Technologie) for 30 min at 37°C. Digested cultures were collected, pelleted, resuspended, and subjected to sequential digestion by 2 mg/ml type I collagenase (Sigma) for 1 hour and 0.05% Trypsin-EDTA (Invitrogen) for 5 min at 37°C, and then passed through a 27-gauge syringe 5 to 10 times, and filtered through a 40-μm cell strainer. Cells were counted by hemocytometer and replated at the density of 5 × 104 cells per 12-well plate.
Mouse prostate organoid isolation and culture
Organoids were isolated and cultured as described previously (50). Briefly, The mouse prostates were isolated from 3-month-old WT or Hi-Myc mice. The whole prostates were minced and digested by 10 mg/ml collagenase at 37°C for 2 hours. Single-cell suspension was plated in 30-μl Matrigel drops in six-well plates covered with complete mouse organoid medium (1xB27, 1.25 mM N-acetyl-l-cysteine, 50 ng/ml EGF, 200 nM A83–01, 100 ng/ml Noggin, 500 ng/ml R-spondin 1, 1 nM dihydrotestosterone, 10 μM Y-27632 di-hydrochloride in adDMEM/F12 containing penicillin and streptomycin, 10 mM HEPES, and 2 mM GlutaMAX). Organoids were passaged once a week by triturating in 1 ml of TrypLE and replated in 30-μl Matrigel drops on six-well plates. For the I3C treatment, three Matrigel drops were plated in 12-well plates at day 0. Each drop contained 2 × 104 cells in 20 ul of Matrigel. The medium was replaced every 2 days in the presence and in the absence of various concentration of I3C. The number of organoids at each drop were counted at day 10.
Lentiviral infections were performed as described previously (51) using CRISPR lentiviral vectors targeting Pten or control Scramble. Briefly, lentiviral particles were prepared by transfection of 293T cells using Lipofectamin 2000, and 24 hours posttransfection, the medium was changed to a complete mouse organoid medium. The viral supernatant was collected 24 hours later and used to infect the organoid cells. Organoids were triturated in 1 ml of TrypLE, and 100,000 single cells were infected with complete organoid medium containing virus particles during centrifugation for 1 hour at 600g at 32°C in a 24-well low-adhesion culture plate. Cells were placed at 37°C, 5% CO2 for 3 hours to recover and subsequently centrifuged and plated in 20-μl Matrigel drops on six-well plates covered with complete mouse organoid medium. After 24 hours, medium was changed to 1 mg/ml puromycin, and cells were incubated for 2 days to ensure only infected cells remained.
Immunohistochemistry (IHC) assay
Individual tumors derived from NCr nude mice or GEMMs were dissected and fixed in 4% paraformaldehyde for IHC analysis. For staining, the tissues were embedded in paraffin according to standard procedures. Five-mm sections were cut and processed for H&E staining or were stained for PTEN (Cascade BioScience 6H2.1,1:250), pAKT (CST#4060, 1:100), Cleaved Caspase3 (CST#9661, 1:300), and Ki67 (Thermo, RM-9106, 1:200). The stained slides were visualized by a bright-field microscope.
PtdIns(3,4,5)P3 phosphatase assay
Primary MEFs and PC3 cell lysates were IP and subjected to native elution. For phosphatase assays, a solution with 25 mM Tris-HCl (pH 7.5), 140 mM NaCl, 1 mM DTT, and 100 mM diC8-PtdIns(3,4,5)P3 (Echelon) was prepared, and the assay ran at 37°C for 45 min. Free-phosphate release was measured with Green Reagent (Biomol) and according to the manufacturer’s instructions.
Intraperitoneal administration
The mice were treated intraperitoneally with I3C dissolved in 5% DMSO (20 mg/kg), three times a week for 14 days starting on day 0. Intraperitoneal administration allowed I3C to achieve maximal systemic exposure.
Gavage administration
BKM120, BYL719, and GDC-0032 were all purchased from Selleckchem and given via oral gavage in 100 μl. The mice were given 0.5% carboxymethyl cellulose (CMC) as vehicle control or PI3K inhibitors (BKM120: 30 mg/kg, BYL719: 30 mg/kg, and GDC-0032: 20 mg/kg) by gavage for 15 days (5 out of 7 days). The mice were euthanized after the treatment to check the transcriptional profiles of DLPs by RNA-seq analyses.
Pharmacokinetics analysis
Male C57BL/6 mice were used after the protocol approval by the Beth Israel Deaconess Medical Center IACUC Committee on Animal Research. Animals were housed during the acclimatization period (3 days) in a temperature of 22° to 25°C, humidity of 40 to 70% RH, and a 12-hour light–12-hour dark cycle. On the dosing day, Indole-3-Carbinol (I3C) was diluted to an appropriate dose concentration in DMSO (5%) + PBS (95%) and dosed 20 mg/kg (every 24 hours and every 48 hours) intraperitoneally using a 27-gauge needle. Blood samples were collected through a capillary, guided in retro-orbital plexus after gaseous anaesthesia at 0.083, 0.25, 1, 4, 12, and 24 hours and followed by every 24-hour dosing. The same is followed for 48-hour dosing regimen. The plasma fraction was separated by centrifugation (2500g, 15 min) and stored at −80°C until analysis. Plasma concentrations of I3C were determined using HPLC method. Nine standards of I3C calibration curve were generated in male C57BL/6 mice plasma. Telmisartan was used as internal standard for all the LC-MS/MS analysis.
The soft-agar colony formation assay
In six-well plates, 2 ml of bottom layer medium (0.6% agarose and 10% FBS) was added per well and cooled down for 0.5 hour at room temperature. Then 2 ml of top layer medium (0.3% agarose and 20% FBS) mixed with cells was added to each well and cooled down for 10 min at 4°C. The 1.5 × 105 cells were cultured at 37°C for 21 days. Then 1 ml of 0.005% Crystal Violet was added to each well for 1 hour at room temperature. Colonies were counted with ImageJ software.
Next-generation sequencing
NGS service (RNA) was performed by the Molecular Biology Core Facilities at the Dana Farber Cancer Institute.
Transcriptome profiling using RNA quantification sequencing
RNAs were extracted from GEMMs prostate DLPs (5 months of age) by using the the pureLink RNA Kit (Invitrogen). Ribosomal RNA removal was carried out using RiboMinus Human/Mouse Transcriptome Isolation Kit (Invitrogen) according to the manufacturer’s protocol. RNAs derived from GEMMs DLPs were subjected to next-generation sequencing (NGS) to generate deep coverage RNA-seq data. For each group, sequencing was performed on three biological samples. RNA-seq libraries were prepared using Illumina TruSeq RNA Sample Prep Kit V2 and subjected to deep sequencing with Illumina HiSeq 4000 at the Molecular Biology Core Facilities at the Dana Farber Cancer Institute. To achieve comprehensive coverage for each sample, we generated ~32 million to 39 million single-end reads.
Gene expression profiling
The Cancer Genome Atlas Prostate Adenocarcinoma (TCGA-PRAD) data were downloaded from the cBioPortal website (www.cbioportal.org/public-portal) (52, 53). After samples of PTEN homologous loss were excluded, samples with WWP1 or MYC expression below Z score 0 were counted as low, above Z score 0 counted as high. For the analyses, GraphPad Prism 6 software was used. The analysis of correlation of MYC and WWP1 mRNA expression was done by Pearson correlation coefficients. Two-tailed unpaired Welch’s t tests were conducted to calculate the significance of phospho-Akt (S473), pS6K, and pS6 protein level. Analysis of coamplification of MYC and WWP1 was carried out performing a Fisher’s exact test.
Statistical analysis
For analysis of average data, datasets were compared using unpaired two-tailed Student’s t tests. For the correlation of TMA staining with clinical parameters, datasets were compared using Pearson chi-square correlation. P values of <0.05 were considered to be statistically significant. *P < 0.05; **P < 0.005; ***P < 0.0005. All statistical tests were executed using GraphPad Prism software.
All primers used in this study
Primer sequences used in constructs and mutagenesis
PTEN K313R F: CGTGCAGATAATGACAGGGAATATCTAGTACTT
PTEN K313R R: AAGTACTAGATATTCCCTGTCATTATCTGCACG
PTEN K322R F: GTACTTACTTTAACAAGGAATGATCTTGACAAA
PTEN K322R R: TTTGTCAAGATCATTCCTTGTTAAAGTAAGTAC
PTEN K327/330/332R F: AAAAATGATCTTGACAGGGCAAATAGGGACAGGGCCAACCGATACTTT
PTEN K327/330/332R R: AAAGTATCGGTTGGCCCTGTCCCTATTTGCCCTGTCAAGATCATTTTT
PTEN K342/344R F: TACTTTTCTCCAAATTTTAGGGTGA -GGCTGTACTTCACAAAA
PTEN K342/344R R: TTTTGTGAAGTACAGCCTCACCCT -AAAATTTGGAGAAAAGTA
PTEN K349R F: AAGCTGTACTTCACAAGGACAGTAGAGGAGCCG
PTEN K349R R: CGGCTCCTCTACTGTCCTTGTGAAGTACAGCTT
WWP1 F577A F: GTGTCCCGGCAGACATTGGCAGAAGATTCCTTCCAACAG
WWP1 F577A R: CTGTTGGAAGGAATCTTCTGCCAATGTCTGCCGGGACAC
WWP1 Y656A F: AATCCAGACCATCTTTCAGCATTCTGTTTCATTGGTCGT
WWP Y656A R: ACGACCAATGAAACAGAATGCTGAAAGATGGTCTGGATT
Primer sequences used in ChIP assay
Wwp1 promoter F: GTCCGGAGTTGGAGGCTTT
Wwp1 promoter R: GACCCCACACCTCCCTTC
JunB promoter F: AAGCCCACAGAGAGAGGTGGAAG
JunB promoter R: CCAGAAGGTGGTGCCTTTTTATTG
Primer sequences used in CRISPR
Wwp1 sgRNA_1: ATCAGCTGCTCGTCCCATTT
Wwp1 sgRNA_2: TAATACTCGAACTACTACAT
mPten sgRNA_1: AAACAAAAGGAGATATCAAG
mPten sgRNA_2: GCTAACGATCTCTTTGATGA
hPTEN sgRNA: GCATATTTATTACATCG
WWP1 promoter sgRNA_1: AGGCTGCTCTAAGC-TCGCGC
WWP1 promoter sgRNA_3: GCTGGGGGTGGCGCGTGGAC
WWP1 promoter sgRNA_4: CTGGGGGTGGCGCGTGGACG
Primer sequences used in qRT-PCR
Human WWP1 F: TGCTTCACCAAGGTCTGATACT
Human WWP1 R: GCTGTTCCGAACCAGTTCTTTT
Supplementary Material
ACKNOWLEDGMENTS
We thank Z. J. Chen for sharing U2OS-derived ubiquitin replacement cell lines with our team (21). We are also grateful to all of the members of the Pandolfi lab for their support and constructive criticisms, as well as T. Garvey, L. Southwood, E. Stack, and K. Rodzenko for editing the manuscript. We thank A. V. Menon and K. Berry for mice genotyping and maintenance. LTQ-Orbitrap experiments and additional technical assistance were performed by the Academia Sinica Common Mass Spectrometry Facilities located at the Institute of Biological Chemistry. We thank the BIDMC confocal imaging and Mass Core Facility.
Funding: This work was supported by NIH grants R01 CA82328 and R35 CA197529 to P.P.P. Y.-R.L. was supported in part by the Postdoctoral Research Abroad Program Fellowship, Taiwan National Science Council (NSC), and DOD Prostate Cancer Research Program (PCRP) Postdoctoral Training Award (W81XWH-16-1-0249). A.P. is supported by a National Breast Cancer Foundation Career Development Fellowship.
Footnotes
Competing interests: P.P.P., W.W., and S.J. are cofounders of Rekindle Pharmaceuticals. The company is developing new therapies for cancer. P.P.P., W.W., and Y.-R.L. are inventors on patent application (#WO2018044875A1) held and submitted by Beth Israel Deaconess Medical Center that covers the concept presented in the study, “Compositions and methods for treating cancer.” All other authors declare no competing interests.
Data and materials availability: All data are available in the manuscript or the supplementary materials. Fastq files were generated from RNA-seq of mouse DLPs or retrieved from GEO (GSE126789). Wwp1−/− mice are available to not-for-profit research entities from L.M. under a material agreement with the National Cancer Institute–NIH and with the University of South Carolina.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Weinberg RA, The molecular basis of oncogenes and tumor suppressor genes. Ann. N. Y. Acad. Sci 758, 331–338 (1995). doi: 10.1111/j.1749-6632.1995.tb24838.x [DOI] [PubMed] [Google Scholar]
- 2.Li J et al. , PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 (1997). doi: 10.1126/science.275.5308.1943 [DOI] [PubMed] [Google Scholar]
- 3.Bonneau D, Longy M, Mutations of the human PTEN gene. Hum. Mutat 16, 109–122 (2000). doi: [DOI] [PubMed] [Google Scholar]
- 4.Berger AH, Knudson AG, Pandolfi PP, A continuum model for tumour suppression. Nature 476, 163–169 (2011). doi: 10.1038/nature10275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Z et al. , Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 (2005). doi: 10.1038/nature03918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollander MC, Blumenthal GM, Dennis PA, PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 11, 289–301 (2011). doi: 10.1038/nrc3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Cao I et al. , Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 149, 49–62 (2012). doi: 10.1016/j.cell.2012.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maehama T, Dixon JE, The tumor suppressor PTEN /MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem 273, 13375–13378 (1998). doi: 10.1074/jbc.273.22.13375 [DOI] [PubMed] [Google Scholar]
- 9.Stambolic V et al. , Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 (1998). doi: 10.1016/S0092-8674(00)81780-8 [DOI] [PubMed] [Google Scholar]
- 10.Sun H et al. , PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. U.S.A 96, 6199–6204 (1999). doi: 10.1073/pnas.96.11.6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). doi: 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee Y-R, Chen M, Pandolfi PP, The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol 19, 547–562 (2018). doi: 10.1038/s41580-018-0015-0 [DOI] [PubMed] [Google Scholar]
- 13.Trotman LC et al. , Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 (2007). doi: 10.1016/j.cell.2006.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song MS et al. , Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199 (2011). doi: 10.1016/j.cell.2010.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papa A et al. , Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157, 595–610 (2014). doi: 10.1016/j.cell.2014.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Matesic LE, The Nedd4-like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev. 26, 587–604 (2007). doi: 10.1007/s10555-007-9091-x [DOI] [PubMed] [Google Scholar]
- 17.Chen C et al. , Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene 26, 2386–2394 (2007). doi: 10.1038/sj.onc.1210021 [DOI] [PubMed] [Google Scholar]
- 18.Zhang XF et al. , Overexpression of WWP1 promotes tumorigenesis and predicts unfavorable prognosis in patients with hepatocellular carcinoma. Oncotarget 6, 40920–40933 (2015). doi: 10.18632/oncotarget.5712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C, Zhou Z, Ross JS, Zhou W, Dong JT, The amplified WWP1 gene is a potential molecular target in breast cancer. Int. J. Cancer 121, 80–87 (2007). doi: 10.1002/ijc.22653 [DOI] [PubMed] [Google Scholar]
- 20.Maddika S et al. , WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol 13, 728–733 (2011). doi: 10.1038/ncb2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu M, Skaug B, Zeng W, Chen ZJ, A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFα and IL-1β. Mol. Cell 36, 302–314 (2009). doi: 10.1016/j.molcel.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JO et al. , Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334 (1999). doi: 10.1016/S0092-8674(00)81663-3 [DOI] [PubMed] [Google Scholar]
- 23.Geisler S et al. , PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol 12, 119–131 (2010). doi: 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- 24.Peng DJ et al. , Noncanonical K27-linked polyubiquitination of TIEG1 regulates Foxp3 expression and tumor growth. J. Immunol 186, 5638–5647 (2011). doi: 10.4049/jimmunol.1003801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao Z et al. , Ubiquitin ligase TRIM62 regulates CARD9-mediated anti-fungal immunity and intestinal inflammation. Immunity 43, 715–726 (2015). doi: 10.1016/j.immuni.2015.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H et al. , K33-linked polyubiquitination of T cell receptor-z regulates proteolysis-independent T cell signaling. Immunity 33, 60–70 (2010). doi: 10.1016/j.immuni.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian J, Jenkins RB, Bostwick DG, Detection of chromosomal anomalies and c-myc gene amplification in the cribriform pattern of prostatic intraepithelial neoplasia and carcinoma by fluorescence in situ hybridization. Mod. Pathol 10, 1113–1119 (1997). [PubMed] [Google Scholar]
- 28.Sato K et al. , Clinical significance of alterations of chromosome 8 in high-grade, advanced, nonmetastatic prostate carcinoma. J. Natl. Cancer Inst 91, 1574–1580 (1999). doi: 10.1093/jnci/91.18.1574 [DOI] [PubMed] [Google Scholar]
- 29.Koh CM et al. , MYC and prostate cancer. Genes Cancer 1, 617–628 (2010). doi: 10.1177/1947601910379132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clegg NJ et al. , MYC cooperates with AKT in prostate tumorigenesis and alters sensitivity to mTOR inhibitors. PLOS ONE 6, e17449 (2011). doi: 10.1371/journal.pone.0017449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellwood-Yen K et al. , Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4, 223–238 (2003). doi: 10.1016/S1535-6108(03)00197-1 [DOI] [PubMed] [Google Scholar]
- 32.Sarkar FH, Li Y, Harnessing the fruits of nature for the development of multi-targeted cancer therapeutics. Cancer Treat. Rev 35, 597–607 (2009). doi: 10.1016/j.ctrv.2009.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmad A, Sakr WA, Wahidur Rahman KM, Novel targets for detection of cancer and their modulation by chemopreventive natural compounds. Front. Biosci. (Elite Ed.) E4, 410–425 (2012). doi: 10.2741/e388 [DOI] [PubMed] [Google Scholar]
- 34.Aggarwal BB, Ichikawa H, Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle 4, 1201–1215 (2005). doi: 10.4161/cc.4.9.1993 [DOI] [PubMed] [Google Scholar]
- 35.Ahmad A, Sakr WA, Wahidur Rahman KM, Anticancer properties of indole compounds: Mechanism of apoptosis induction and role in chemotherapy. Curr. Drug Targets 11, 652–666 (2010). doi: 10.2174/138945010791170923 [DOI] [PubMed] [Google Scholar]
- 36.Firestone GL, Sundar SN, Minireview: Modulation of hormone receptor signaling by dietary anticancer indoles. Mol. Endocrinol 23, 1940–1947 (2009). doi: 10.1210/me.2009-0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aronchik I, Kundu A, Quirit JG, Firestone GL, The antiproliferative response of indole-3-carbinol in human melanoma cells is triggered by an interaction with NEDD4–1 and disruption of wild-type PTEN degradation. Mol. Cancer Res 12, 1621–1634 (2014). doi: 10.1158/1541-7786.MCR-14-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kathman SG et al. , A small molecule that switches a ubiquitin ligase from a processive to a distributive enzymatic mechanism. J. Am. Chem. Soc 137, 12442–12445 (2015). doi: 10.1021/jacs.5b06839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hopkins BD et al. , Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). doi: 10.1038/s41586-018-0343-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang WL et al. , The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 (2009). doi: 10.1126/science.1175065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dang CV, MYC on the path to cancer. Cell 149, 22–35 (2012). doi: 10.1016/j.cell.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shu L, Zhang H, Boyce BF, Xing L, Ubiquitin E3 ligase Wwp1 negatively regulates osteoblast function by inhibiting osteoblast differentiation and migration. J. Bone Miner. Res 28, 1925–1935 (2013). doi: 10.1002/jbmr.1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lunardi A et al. , A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat. Genet 45, 747–755 (2013). doi: 10.1038/ng.2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). doi: 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan WC et al. , K33-linked polyubiquitination of Coronin 7 by Cul3-KLHL20 ubiquitin E3 ligase regulates protein trafficking. Mol. Cell 54, 586–600 (2014). doi: 10.1016/j.molcel.2014.03.035 [DOI] [PubMed] [Google Scholar]
- 46.Xue W et al. , CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384 (2014). doi: 10.1038/nature13589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K, Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). doi: 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 48.Zhang W et al. , System-wide modulation of HECT E3 ligases with selective ubiquitin variant probes. Mol. Cell 62, 121–136 (2016). doi: 10.1016/j.molcel.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lukacs RU, Goldstein AS, Lawson DA, Cheng D, Witte ON, Isolation, cultivation and characterization of adult murine prostate stem cells. Nat. Protoc 5, 702–713 (2010). doi: 10.1038/nprot.2010.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drost J et al. , Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc 11, 347–358 (2016). doi: 10.1038/nprot.2016.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karthaus WR et al. , Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159, 163–175 (2014). doi: 10.1016/j.cell.2014.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao J et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013). doi: 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cerami E et al. , The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). doi: 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.