

SUMMARY
Non-clustered δ1- and δ2-protocadherins, close relatives of clustered protocadherins, function in cell adhesion and motility and play essential roles in neural patterning. To understand the molecular interactions underlying these functions, we used solution biophysics to characterize binding of δ1- and δ2-protocadherins, determined crystal structures of ectodomain complexes from each family, and assessed ectodomain assembly in reconstituted intermembrane junctions by cryoelectron tomography (cryo-ET). Homophilic trans (cell–cell) interactions were preferred for all δ-protocadherins, with additional weaker heterophilic interactions observed exclusively within each subfamily. As expected, δ1- and δ2-protocadherin trans dimers formed through antiparallel EC1–EC4 interfaces, like clustered protocadherins. However, no ectodomain-mediated cis (same-cell) interactions were detectable in solution; consistent with this, cryo-ET of reconstituted junctions revealed dense assemblies lacking the characteristic order observed for clustered protocadherins. Our results define non-clustered protocadherin binding properties and their structural basis, providing a foundation for interpreting their functional roles in neural patterning.
Graphical Abstract
In Brief
Non-clustered δ-protocadherins are adhesion molecules linked to a number of neurological disorders. Harrison et al. apply biophysical and structural methods across the family to show preferential self-binding through a canonical interface and disordered assemblies of adhesive dimers between membranes that diverge from ordered assemblies of close relatives clustered protocadherins.
INTRODUCTION
Members of the cadherin superfamily function in a wide variety of calcium-dependent cell adhesion and recognition processes and are characterized by consecutive extracellular cadherin (EC) repeats. Classical type I, type II, and desmosomal cadherins contain four to five EC repeats and form adhesive trans interactions between cells via N-terminal EC1 domains (Brasch et al., 2012). Outside of these well-characterized proteins, cadherin superfamily members containing more than five EC repeats have been loosely termed protocadherins (pcdhs). The largest groups within the protocadherin branch are the clustered protocadherins (pcdhα, pcdhβ, and pcdhγ), encoded by a contiguous gene cluster subject to alternative splicing (Mountoufaris et al., 2018; Rubinstein et al., 2017), and the non-clustered δ-protocadherins - subjects of the current study—whose genes are dispersed in the genome (Kim et al., 2007; Light and Jontes, 2017; Morishita and Yagi, 2007; Redies et al., 2005). Both families encode single-pass transmembrane proteins containing six or seven EC domains and are closely related in their extracellular regions (~30%–40% identity). Clustered protocadherins function primarily in neurite self-avoidance (Lefebvre et al., 2012; Mountoufaris et al., 2017) and branching (Molumby et al., 2016), distinct from the primarily adhesive roles of classical and desmosomal cadherins (Brasch et al., 2018; Harrison et al., 2016; Meng and Takeichi, 2009), while functional roles of non-clustered δ-protocadherins remain to be fully characterized.
The non-clustered δ-protocadherins comprise nine typical members in human and mouse, divided into δ1- and δ2-subfamilies with four and five members, respectively (Hulpiau and van Roy, 2009). δ1-protocadherins contain seven EC repeats and are characterized by conserved CM1, CM2, and CM3 sequence motifs of unknown structure and function in their cytoplasmic domains (Redies et al., 2005; Vanhalst et al., 2005). δ2-protocadherins have six EC repeats and contain only CM1 and CM2 (Hulpiau and van Roy, 2009). In addition to the nine canonical family members, the related proteins pcdh12 and pcdh20 represent atypical δ-protocadherins that diverge primarily in their intracellular regions (Hulpiau and van Roy, 2009). Each δ-protocadherin is expressed in a spatiotemporally regulated pattern in the nervous system and other tissues (Blevins et al., 2011; Cooper et al., 2016; Etzrodt et al., 2009; Gaitan and Bouchard, 2006; Kim et al., 2011; Lin et al., 2012). Overlapping expression in vivo suggests that functional specificity could be derived from expression of multiple δ-protocadherins in the same cell (Bisogni et al., 2018; Kim et al., 2007). In vitro studies have demonstrated that most δ-protocadherins mediate homophilic cell aggregation (Bisogni et al., 2018; Pederick et al., 2018), consistent with functions involving cell adhesion and recognition. In addition, a number of δ-protocadherins are associated with neurological diseases (Kahr et al., 2013; Kim et al., 2011).
Likely the best understood of the δ-protocadherins is the δ2- family member pcdh19. Mutations in the Pcdh19 gene underlie X-linked Pcdh19-girls clustering epilepsy (Pcdh19-GCE), which manifests in heterozygous females, but not in hemizygous males. Pederick et al. (2018) showed that mosaic expression of wild-type and inactive pcdh19 in heterozygous mutant female mice led to abnormal sorting in which neural progenitors expressing wild-type pcdh19 separated from those expressing the mutant, demonstrating that pcdh19 homophilic adhesion can effect cell localization. Functional roles dependent on homophilic recognition have also been suggested for pcdh17 in collective axon extension (Hayashi et al., 2014) and pcdh7 in axon guidance (Leung et al., 2013).
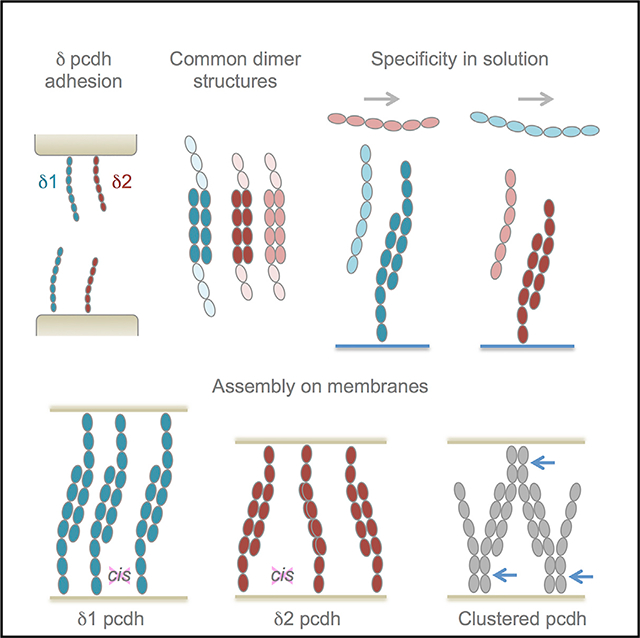
Crystal structures of pcdh19 from zebrafish (Cooper et al., 2016) and a crystal structure of human pcdh1 published while this paper was under review (Modak and Sotomayor, 2019) revealed the trans binding mechanism for δ1- and δ2-protocadherins to be consistent with that observed for the related α-, β-, and γ-clustered protocadherins (Goodman et al., 2016a, 2016b). In each trans-dimer structure, EC1–EC4 regions of partner molecules bind in an antiparallel orientation to form a dimer mediated by two EC1:EC4 and two EC2:EC3 interfaces related by 2-fold symmetry. Clustered protocadherins have been shown to additionally form asymmetrical EC5–EC6:EC6 cis dimers (Goodman et al., 2017) that are proposed to function in combination with trans interactions to allow combinatorially expressed isoforms to impart distinct cellular identities to individual neurons (Brasch et al., 2019; Rubinstein et al., 2015). It has not been clear whether δ-protocadherins participate in similar cis interactions.
Here we use solution biophysics to analyze homophilic and heterophilic binding of human δ1- and δ2-protocadherins, showing trans dimerization to be preferentially homophilic, with weaker heterophilic interactions within subfamilies; and present crystal structures of trans-binding fragments from four δ2- and one δ1-protocadherin to identify regions likely to govern trans-interaction specificity. We also show that, distinct from clustered protocadherins, δ-protocadherins form no detectable ectodomain–cis interactions in solution, lack conservation of cis-interface regions, and concentrate between membranes as trans dimers without higher-order zipper-like structures characteristic for clustered-protocadherin.
RESULTS
δ-Protocadherin trans Interactions Are Preferentially Homophilic
We characterized trans binding specificity across the human δ-protocadherin family using surface plasmon resonance (SPR). Ectodomain fragments composed of adhesive domains EC1–EC4 were prepared for δ1-pcdh-1, −7, −9, and −11, and for δ2-pcdh-10, −17, −18, and −19, comprising all typical members of each branch (Figure 1A) except for pcdh8, which was not analyzed in SPR due to poor solubility. We attempted to produce atypical members pcdh12 and −20, which are phylogenetically related to δ2- and δ1-protocadherins, respectively (Figure 1A). Pcdh20 could not be expressed, but pcdh12 was produced as a full ectodomain fragment (EC1–EC6). To test all potential interactions, separate surfaces were prepared from biotinylated forms of each protocadherin binding fragment and analytes were tested over each (Figure 1B).
Figure 1. SPR Analysis of trans Binding Interactions in the δ-protocadherin Family.
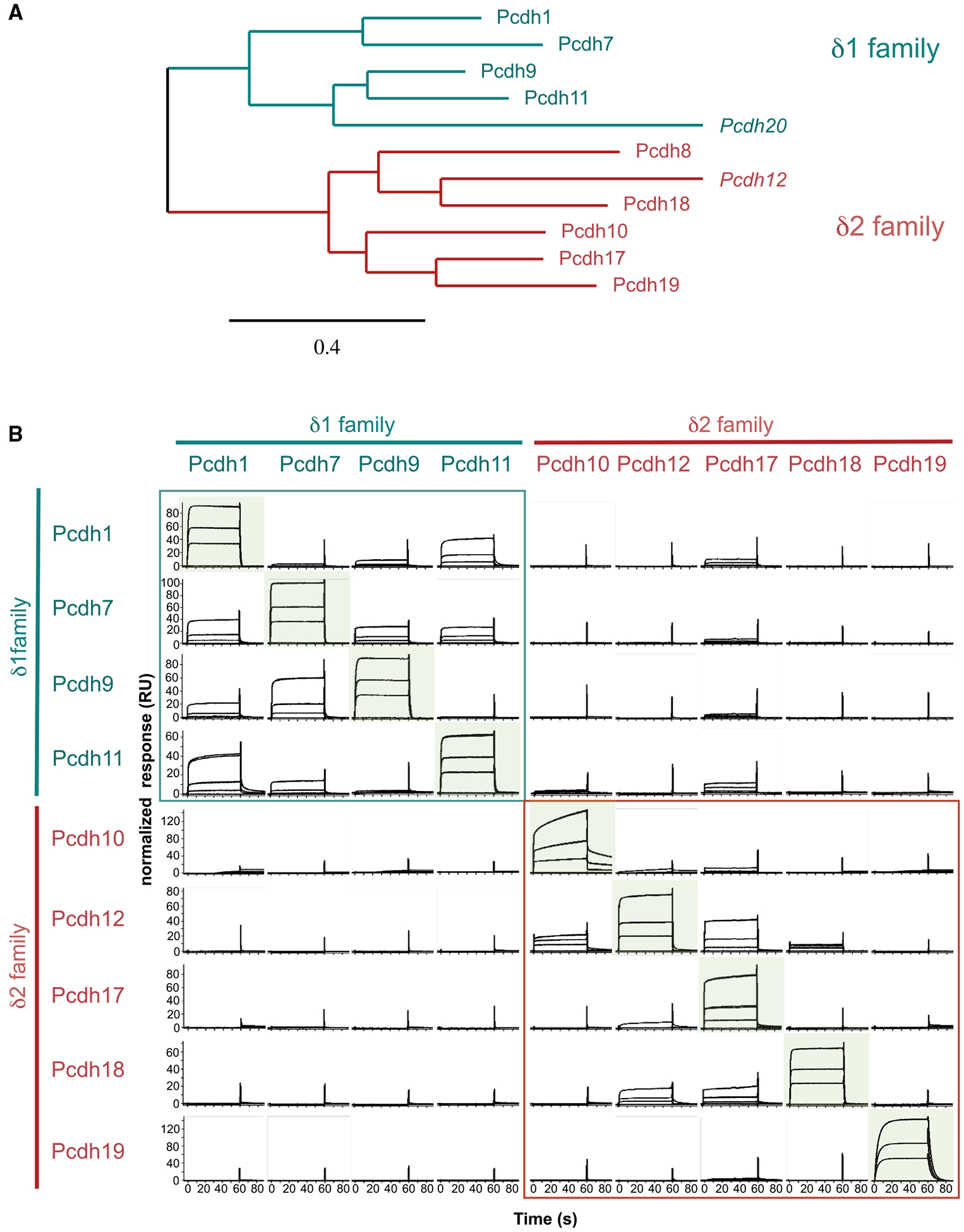
(A) Phylogenetic tree of human δ-protocadherins from aligned full-length amino acid sequences. Atypical members italicized. Scale indicates protein distance.
(B) SPR binding profiles of δ-protocadherin analytes (columns) over surfaces coated with the same set of proteins (rows).
Analyte concentrations of 27, 9, and 3 μM are plotted on each panel. Responses are normalized for molecular weight and scaled for each surface, permitting comparison across rows only. Homophilic combinations are highlighted in green; heterophilic interactions within δ1- and δ2-subfamilies are boxed in teal and red. See Figure S1.
Specific binding was detected over all protocadherin surfaces (rows) with each displaying a distinct binding profile (Figure 1B). Over a pcdh1 surface, the highest binding response was for homophilic interactions with soluble pcdh1 (Figure 1B, top row). A weaker heterophilic binding response was also detected to pcdh11, another member of the δ1-subfamily, while other analytes, including all δ2-protocadherins, showed no binding to pcdh1 above background (Figure 1B, top row). Heterophilic binding of pcdh1 with two other members of the δ1-subfamily, pcdh7 and-9, was additionally detectable at low levels when pcdh1 was present in the analyte phase (Figure 1B, left column).
Pcdh-7, −9, −11, −10, −12, −17, −18, and −19 displayed analogous binding profiles within which their respective homophilic interactions gave the highest binding responses (Figure 1B, diagonal). Weaker heterophilic binding was also observed for a subset of interactions (Figure 1B). This was restricted within subfamilies and no binding between δ1- and δ2-protocadherins was observed in any combination, suggesting them to be incompatible (Figure 1B). Within the δ1-family, heterophilic interactions were extensive and were observed for all subtype combinations except for that of pcdh9 with pcdh11 (Figure 1B). Heterophilic binding within the δ2-family was observed only for a limited subset and response levels were markedly low compared to the respective homophilic interactions (Figure 1B). Binding of pcdh12 to pcdh17 and −18 was detected in both orientations of analyte and surface, while other δ2-heterophilic combinations showed poor correspondence between the two orientations, likely reflecting weak binding close to the limits of detection (Figure 1B). Uniquely, pcdh19 displayed exclusively homophilic binding with no heterophilic responses detected above background (Figure 1B).
We also tested binding of pcdh1, −7, −10, and −18 EC1–EC4 fragments to surfaces of representative clustered protocadherins α7 EC1–EC5, β6 EC1–EC4, γA8 EC1–EC4, γB2 EC1–EC5, and γC4 EC1–EC4 (Figure S1). No binding of the δ-protocadherins over these surfaces was detected, while homophilic binding showed strong responses, suggesting that these families do not trans-interact.
δ-Protocadherins Have Differential Homophilic Affinities in the Low Micromolar Range
We determined homophilic binding affinities for all δ-protocadherins using sedimentation equilibrium analytical ultracentrifugation (AUC), since determination of KD values from SPR data is unreliable for proteins that self-associate (Rich and Myszka, 2007). Full ectodomain fragments encompassing domains EC1–EC7 (δ1) or EC1–EC6 (δ2) were prepared for AUC analysis. Most proteins yielded apparent molecular weights in AUC intermediate between monomer values determined from mass spectrometry and dimer values, suggesting monomer–dimer equilibria (Table 1). Complementary analyses using size exclusion chromatography with multi-angle light scattering (SEC-MALS) also showed single major elution peaks for each δ-protocadherin with apparent molecular weights between monomer and dimer, supporting this interpretation (Figure S2). No evidence of higher-order oligomers (>dimer) was observed using either technique.
Table 1.
Dissociation Constants (KD) for Homodimerization of δ-protocadherin Extracellular Regions and Fragments Determined by Analytical Ultracentrifugation
| Protein | Schematic | Oligomeric State | Monomer Mw (kDa)a | Apparent Mw (kDa)b | Kd (µM) |
|---|---|---|---|---|---|
| δ1-family | |||||
| Pcdh-1 | dimer | 95.7 | 177 ± 3.4 | 1.2 ± 0.55c | |
| Pcdh-7moused | dimer | 101.6 | 168 ± 4.9 | 8.2 ± 2.5 | |
| Pcdh-9 | dimer | 103.5 | 204 ± 3.7 | 2.03 ± 0.43 | |
| Pcdh-11e | dimer | 113.5 | 190 ± 1.4 | 7.8 ± 0.17 | |
| δ2-family | |||||
| Pcdh-8 | monomer | 78.7 | 83.1 ± 0.7 | N/A | |
| Pcdh-8mouse | monomer | 79.4 | 78.3 ± 0.2 | N/A | |
| Pcdh-8.1Xenopus | dimer | 76.7 | 134.7 ± 5.9 | 3.5 ± 2.4 | |
| Pcdh-8.2Xenapus | dimer | 76.6 | 138.2 ± 0.9 | 1.7 ± 0.10 | |
| Pcdh-10 | dimer | 77.2 | 134 ± 2.0 | 3.6 ± 0.61 | |
| Pcdh-12 | dimer | 78.8 | 133 ± 0.1 | 5.4 ± 0.20 | |
| Pcdh-17 | dimer | 87.3 | 168 ± 4.9 | 4.8 ± 0.54 | |
| Pcdh-18 | dimer | 82.0 | 148 ± 2.8 | 2.7 ± 0.67 | |
| Pcdh-19 | dimer | 78.0 | 139 ± 3.8 | 0.48 ± 0.10 | |
| Fragments | |||||
| Pcdh-1 EC1-EC4 | dimer | 50.5 | 97.7 ± 2.2 | 0.53 ± 0.46 | |
| Pcdh-7 EC1-EC4 | dimer | 56.5 | 104 ± 1.5 | 2.70 ± 1.0 | |
| Pcdh-7 EC1-EC4mouse | dimer | 56.4 | 101 ± 1.4 | 4.1 ± 1.3 | |
| Pcdh-9 EC1-EC4 | dimer | 56.1 | 104 ± 1.5 | 2.0 ± 0.52 | |
| Pcdh-10 EC1-EC4 | dimer | 50.2 | 96.4 ± 0.4 | 0.68 ± 0.13 | |
| Pcdh-1 EC1-EC3 | monomer | 35.9 | 36.8 ± 0.9 | N/A | |
| Pcdh-10 EC1-EC3 | weak dimer | 39.5 | 45.8 ± 1.4 | 280 ±110 | |
| Pcdh-1 EC5-EC7 | monomer | 47.1 | 45.8 ± 0.04 | N/A | |
| Pcdh-10 EC5-EC6 | monomer | 28.7 | 29.4 ± 0.2 | N/A | |
| Pcdh-8 EC5-EC6 | monomer | 26.9 | 27.8 ± 0.1 | N/A | |
See also Figure S2.
Average molecular weight determined from mass spectrometry peak maximum.
Determined from AUC data using an ideal monomer model.
Errors represent standard deviation from two or more experiments.
All constructs derive from human except where stated.
Pcdh11Y isoform is reported.
Fitting of the AUC data to monomer-dimer models yielded KD values for homodimerization spanning the low micromolar range (Table 1). KD values across both subfamilies differed over an ~20-fold range with no general trend separating δ1 from δ2. Within the δ1-family, pcdh1 and −9 formed tighter homodimers relative to pcdh7 and −11 by ~4-fold (Table 1). In the δ2-subfamily, KD values for most members were comparable within error except for pcdh18, which formed modestly higher affinity dimers and pcdh19, which formed the highest affinity dimers observed in these experiments (KD 0.48 μM, Table 1). While pcdh8 EC1–EC6 fragments from human and mouse both displayed impaired solubility, their soluble fractions behaved as apparent monomers in AUC and SEC-MALS analyses, suggesting the possibility that pcdh8 may not form dimers. However, we observed that full-length human pcdh8 could induce cell aggregation in qualitative assays using transfected K562 cells (Figure S2), as reported previously for the mouse ortholog (Bisogni et al., 2018). We also found that the Xenopus ortholog of pcdh8, pcdh8.1 (MGC84237), and its paralog pcdh8.2 (PAPC) formed dimers in AUC (Table 1). Thus, human and mouse pcdh8 trans dimerization appears sufficient to produce measurable cell surface adhesion, but may have weak affinity below the threshold of detection in solution. Weak adhesion has been described previously for arcadlin, the rat ortholog of pcdh8, and its homophilic binding has been suggested to enhance endocytosis of N-cadherin rather than mediate stable adhesion (Yasuda et al., 2007); similar roles for mouse and human pcdh8 remain to be tested.
δ1- and δ2-Protocadherins Adopt Canonical Antiparallel trans Dimers
The mechanism of δ2-protocadherin trans binding was revealed previously in a crystal structure of zebrafish pcdh19 EC1–EC4 (Cooper et al., 2016), which adopted an antiparallel trans dimer similar to clustered protocadherins (Goodman et al., 2016a, 2016b). Formation of a similar trans dimer by δ1-protocadherins was also reported in a recent crystal structure of human pcdh1 EC1–EC4 (Modak and Sotomayor, 2019), representing the same crystal form as that described here. To compare binding determinants across the δ-protocadherin family, we determined EC1–EC4 fragment crystal structures of human pcdh1, −10 (two crystal forms), −17, −18, and −19, with resolutions 2.3–3.7 Å (Table S1).
In each of the six δ-protocadherin structures (Figure 2), individual protomers adopted approximately linear arrangements of domains EC1–EC4 rigidified by coordination of three calcium ions in each interdomain linker, as is characteristic for the cadherin family. N-linked glycan moieties were observed in all structures and included conserved glycosylation of the B-strand of EC3 (e.g., Asn248 in pcdh1), suggesting possible biological significance. Additionally, O-linked glycans were observed on the EC2 G-strand in pcdh-10 and −17, corresponding to cadherin-specific O-mannosylation sites identified previously by mass spectrometry (Larsen et al., 2017). No glycans were significantly buried in protein–protein interfaces. Individual loops in pcdh10 (EC2 F-G loop) and pcdh17 (EC4 C-D loop) corresponding to glycine-rich insertions of 19 and 15 amino acids (Figure 2) not present in other δ-protocadherins appeared disordered, and showed no interpretable electron density. The function of these loops remains to be determined, but they are distal from trans interface regions (see below), and are unlikely to directly modify binding.
Figure 2. Structures of Adhesive EC1–EC4 Fragments of δ1- and δ2-Protocadherins.
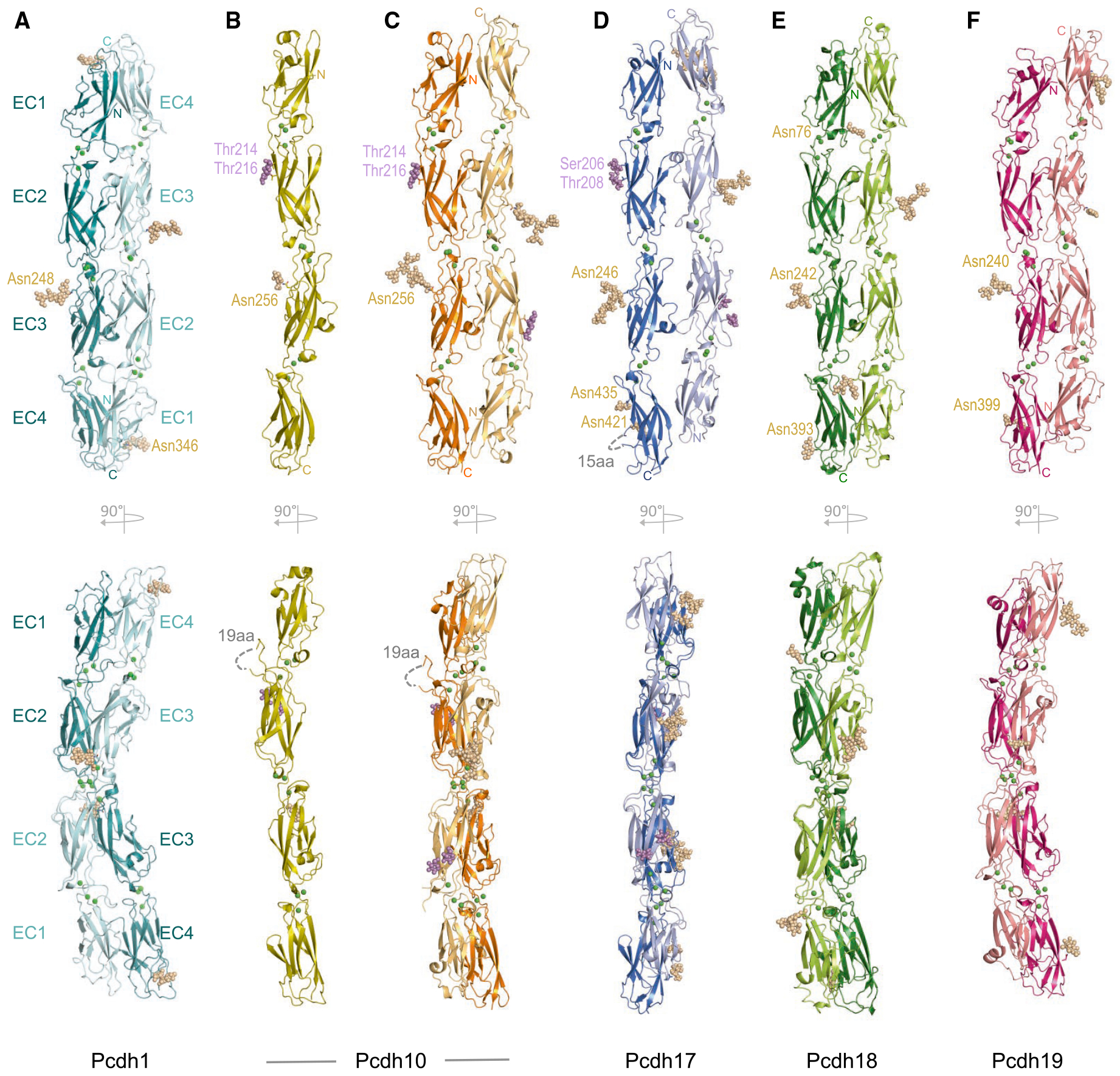
(A–F) Ribbon representations showing two orthogonal views (upper and lower panels) of human EC1–EC4 fragment structures of (A) pcdh1; (B) pcdh10 monomer; (C) pcdh10 dimer; (D) pcdh17; (E) pcdh18.; and (F) pcdh19.
Single trans dimers, formed between symmetry-related protomers (A) or in the crystallographic asymmetric unit (C–F) are shown. Interdomain calcium ions are shown as green spheres; N-linked and O-linked glycans as wheat and magenta spheres. See Figures S3 and S4 and Tables S1 and S2.
Structures belonging to both the δ1- (pcdh1) and δ2- (pcdh-10, −17, −18, and −19) subfamilies contained antiparallel 2-fold symmetric or pseudosymmetric dimers in their crystal lattices (Figures 2A, 2C, and 2F). In addition to the dimer, pcdh10 also crystallized in a monomeric crystal form (Figure 2B), likely due to low pH conditions in the crystallization experiment (see STAR Methods). Overall dimer arrangements were broadly similar for all structures, consistent with δ1- and δ2-family members forming trans dimers by the same general mechanism (Figure 2). Dimer topologies corresponded to those described for zebrafish pcdh19 (Cooper et al., 2016) and for clustered protocadherins (Goodman et al., 2016a, 2016b). Briefly, partner EC1–EC4 regions overlap fully in an antiparallel dimer stabilized by EC1:EC4 and EC2:EC3 interfaces on both sides of a 2-fold symmetry axis centered on the EC2–EC3 linker (Figure 2). EC1:EC4 interfaces form between the C-F-G face of EC1 and the A-B-E-D face of EC4 while EC2:EC3 interfaces form between the A-B-E-D face of EC2 and the C-F-G face of EC3 (Figure S3A). Symmetric EC3:EC3 contacts between paired FG loops form the center of the dimer where paired protomers cross (Figure S3A). In pcdh-1, −18, and −19, a disulfide-stabilized E-F loop in EC1 protrudes sufficiently to form additional minimal contacts with the E-F loop of the partner EC3 (Figure S3A). Buried surface areas (BSAs) for individual dimers (Table S2) ranged from 2,024 Å2 (pcdh17) to 4,176 Å2 (pcdh19). High BSA for pcdh19, in combination with a higher proportion of hydrophobic buried surface (70%, Table S2), likely underlies the strong homophilic affinity observed in AUC (see Table 1).
Differences in Overall Dimer Arrangements among δ-Protocadherins
Despite conservation of overall dimer topology, differences in precise dimer orientation and interface loop conformations were observed between δ-protocadherins. Dimers of pcdh-10, −18, and −19 superposed moderately well (Figures S3B and S3E, root mean square deviation (RMSD) 2.7–3.2 Å over 772–794 Cα) and were closest in conformation to the previously reported structure of zebrafish pcdh19 (Cooper et al., 2016; Figure S3E; 2.2 Å over 791 Cα between zebrafish and human pcdh19).
Pcdh17 adopted a markedly more parallel alignment of paired protomers (Figure S3B) with increased separation near the EC2–EC3 linker and a shifted EC1:EC4 interface (Figure S3). However, a packing interaction with a symmetry-related protomer observed in the pcdh17 crystal lattice would partially occlude a canonical EC1:EC4 interface, suggesting the unusual conformation of the overall dimer to likely be a result of crystal packing (Figure S4A).
δ1-family member pcdh1 also showed differences in dimer conformation compared to δ2-pcdh-10, −18, and −19, adopting a bent dimer conformation compared to the canonical arrangement (Figure S3B). This arises in part from a more acute interdomain angle between EC2 and EC3 (Figure S3C). Accompanying this deviation, domains EC2 and EC3 interacted less closely (Figures S3A and S3D) and contribute a reduced BSA of 652 Å2 per dimer (22% of total) compared to 1,137–2,136 Å2 (42%–66%) for pcdh-10, −18, and −19 (Table S2). In particular, while contacts near the base of EC2 were preserved, those near the top of the EC2:EC3 interface involving EC2 D-E and EC3 C-D loops were more distant or solvent-exposed in pcdh1 (Figures S3A and S3D). While additional structures of δ1-protocadherins will be required to determine if this dimer arrangement is general and exclude the possibility that it results from crystal packing in our pcdh1 crystals, the subtle conformational differences could account in part for binding incompatibility between δ1- and δ2-protocadherins.
Local conformational differences in loops located in the dimer interface were also observed between δ-protocadherin subtypes, suggesting small structural variations in interface regions could contribute to binding preferences (Figure S4C). Superposition of individual EC domains of pcdh-1, −10, −17, −18, and −19 (RMSD 0.5–2.5 Å over 58–105 aligned Cα) revealed differences in interfacial C-D, D-E, and F-G loops of EC1; the DE loop of EC2; the C-D loop of EC3; and the A-strand and D-E loop of EC4 (Figure S4C). Notably, pcdh1 contains an extended D-E loop in EC1 (Figure S4C) whose additional length is conserved in the δ1-branch and could, in principle, stabilize the more open dimer conformation observed for pcdh1. As expected, close correspondence in domain structure between monomer and dimer crystal forms of pcdh10 was observed (RMSD 0.179–0.282 Å over 79–98 residues per domain), however, local shifts of the EC2 D-E loop (~4.9 Å) and EC4 B-C loop (~1.2 Å) toward the partner protomer (Figure S4D) suggest the possibility of subtle conformational changes in these regions upon binding.
Conserved and Variable Molecular Interactions in δ-Protocadherin trans Dimers
We compared molecular interactions in dimer structures of pcdh1, −10, −18, and −19 to identify conserved and variable regions. Those of pcdh17 (Figure S4B) were not compared due to the uncertain physiological relevance of the divergent dimer orientation observed in the structure.
Conserved Interactions
In all structures, the EC1:EC4 interface was centered on a small conserved interaction primarily between the G-strand of EC1 and B-strand of EC4 (Figure 3A). In the pcdh1 dimer, Leu94 in the EC1 G-strand closely contacts Leu359 in the partner EC4 B-strand, while surrounding residues Pro92 (EC1), and Ile335, Ala356, Ala388, and Phe400 (EC4) contribute additional contacts to form a small hydrophobic “core” interface (Figure 3A). An analogous hydrophobic region in δ2-pcdh dimer structures (pcdh-10, −18, and −19) is centered on apposed residues Leu88/93/Ile91 and Leu362/354/349 corresponding to Leu94 and Leu359 in pcdh1 (Figures 3A and S5), and is surrounded by hydrophobic residues corresponding to Pro92 and Ala356 in pcdh-1 (e.g., Leu86 and Val359 in pcdh10) or specific to the δ2-branch (e.g., Val39, Phe81 in pcdh10). Central residues Leu94 and Leu359 (pcdh1 numbering) are highly conserved across the whole δ-protocadherin family and most surrounding hydrophobic residues are conserved in character (Figure S5), as described previously (Cooper et al., 2016; Nicoludis et al., 2015; Nicoludis et al., 2016). Adjacent to the hydrophobic region in EC1, an acidic residue (Glu80 in pcdh1) is buried in the interface in all structures (Figure 3A), and is also conserved across all subtypes (Figure S5). In pcdh1, Glu80 forms a potential salt bridge with Arg333 in EC4, which is also conserved in other members of the δ1-branch (Figures 3A and S5). Arg333 is not conserved in δ2 sequences, instead, potential salt bridges with Glu80 were observed with non-equivalent residue positions (Arg356 and Arg351 in pcdh18 and −19) or were replaced by potential H-bonding interactions (pcdh10, Figure 3A).
Figure 3. Conserved and Variable Molecular Interactions in the trans Dimer Interface.
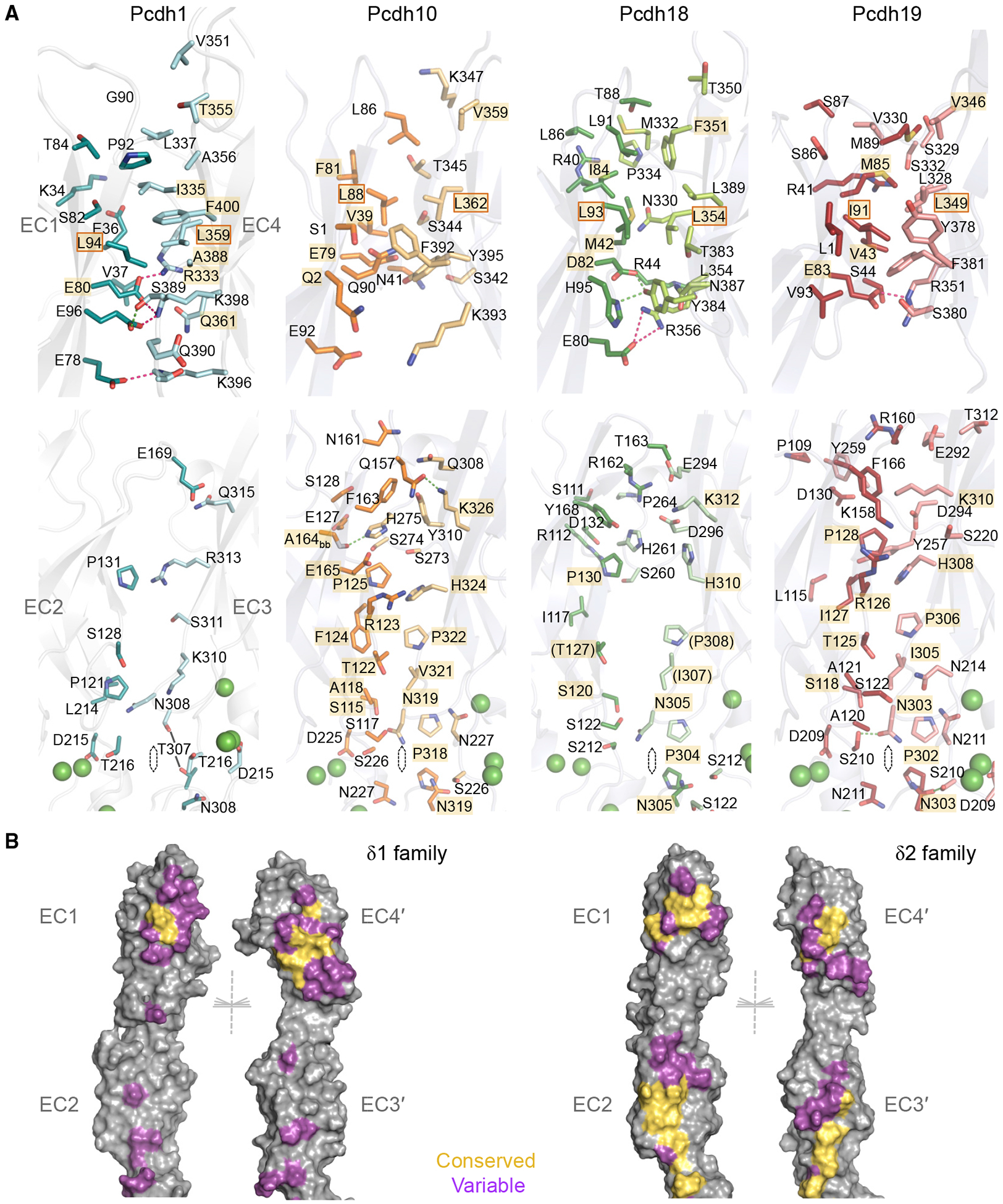
(A) Residue views of interface regions EC1:EC4 (top) and EC2:EC3 (bottom) in trans dimers of pcdh-1, −10, −18, and −19 (left to right). Side chains of interfacial residues (>5% buried) are shown as sticks. Interactions conserved between multiple structures are highlighted in gold. Conserved hydrophobic residues chosen for mutation are boxed. Green spheres: calcium ions.
(B) Molecular surfaces of representative δ1 (pcdh1, left) and δ2 (pcdh10, right) trans dimer structures opened to display interfacial residues color-coded according to their conservation within the respective subfamily (yellow: conserved in character; magenta: variable). Non-interface residues are shown in gray. Half of each 2-fold symmetric dimer is shown for clarity.
See Figures S3–S5.
No other regions of interface conservation across both δ1- and δ2-branches were observed, however, the base of the EC2:EC3 interface where EC2 A/B and EC3 G-strands interact contained an extensive conserved region specific to the δ2-branch (Figure 3A). In each dimer, a conserved proline in EC2 (Pro318 in pcdh10) is symmetrically apposed with the equivalent proline in the partner molecule with a distance of 3.5–4.2 Å (Figure 3A). Above this, residues Asn319, Val321, Pro322, His324, and Lys326 in the EC3 G-strand are apposed with Ser226 in the EC2–EC3 linker and Thr122, Arg123, Phe124, and Pro125 in EC2 (Figure 3A, pcdh10 numbering). While there are variations in the distances between these apposed residues in the respective dimers (Figure 3A), they are conserved in character among pcdh-10, −12, −17, −18, and −19 (Figure S5), indicating their likely importance. In δ1-protocadherins, most of these residue positions are variable and differ from those of δ2-sequences (Figure S5), suggesting that part of the incompatibility between the two subfamilies may derive from mismatches in these regions.
Variable Interactions
Outside the conserved regions described above, interfacial residues in the EC1:EC4 and EC2:EC3 interfaces are highly variable between δ-protocadherin dimer structures (Figure 3) and are thus likely to underlie homophilic binding specificities. Mapping of these onto molecular surfaces of representative δ1- and δ2-structures reveals variable regions distinct for each subfamily framing conserved sites (Figure 3B).
The periphery of the EC1:EC4 interface contains variable interface residues in both subfamilies (Figures 3 and S5), and may be particularly important for δ1 specificity since EC1:EC4 interactions predominate in the pcdh1 dimer structure (Table S2; Figure 3B). In pcdh1, apposed hydrophobic residues Pro92 (EC1) and Ala356 (EC4) extend the area of hydrophobic contact above the conserved region in the dimer structure (Figure 3A). Substitution of Ala356 for lysine in other δ1-family members pcdh9 and - 11 suggests alternative interactions in these dimers, likely with nearby subtype-specific acidic residues at position 90 (Figure S5, pcdh1 numbering). Similarly, two intermolecular salt bridges below the conserved EC1:EC4 region in pcdh1, Glu96–Lys398 and Glu78–Lys396 (Figure 3A), are potentially conserved in pcdh7, but not in other δ1-protocadherins where the Glu96 position is a lysine (Figure S5). This would likely electrostatically clash with Lys398 in putative heterodimers, but may be accommodated in homodimers where residue 398 is a glutamine (pcdh9, −11) or glutamate (pcdh20). In δ2-protocadherins, residues corresponding to the salt-bridge–forming pair Glu96–Arg398 in pcdh1 are also highly variable in character and engage in polar (pcdh10, −18) or hydrophobic (pcdh19) interactions, suggesting they could contribute to homophilic preferences and to incompatibility between δ1- and δ2-subfamilies (Figure 3A). Other δ2 EC1:EC4 variable interactions are also primarily located at the base of EC1, and involve residues Ser342, Ser344, and Thr345 (Figure 3A, pcdh10 numbering).
The EC2:EC3 interface in pcdh1 involves few close interactions, but apposed residues near the EC2–EC3 linker are nonetheless variable within the δ1-subfamily (Figures S3 and S5). Most strikingly, at the center of the pcdh1 dimer Thr307 interacts symmetrically with the equivalent residue of the partner molecule at a distance of 4.9 Å (Figure 3A). This residue is δ1-subtype–specific, with Gln, Ser, Leu, and Cys residues found in pcdh-7, −9, −11, and −20 (Figure S5). Differences in side-chain length and character would likely lead to steric and hydrophobic:hydrophilic mismatches that could destabilize putative heterodimers. Supporting this possibility, self-interacting central residues in clustered protocadherin trans dimers have been shown to strongly influence binding specificity in mutagenesis studies (Goodman et al., 2016a).
In δ2-protocadherins the more extensive EC2:EC3 interface is conserved near its base as described above, but subtype-specific contacts are observed at the top of the interface where EC2 D-E and EC3 C-D loops contact the partner domains (Figure 3). The EC2 D-E loop engages primarily in electrostatic interactions with the partner EC3 domain in pcdh18 and −19, including a potential salt bridge (Arg162–Glu294 in pcdh18; Arg160–Glu292 in pcdh19) that is also conserved by sequence in pcdh-8 and −17 (Figure S5). In pcdh10, however, hydrogen-bonding interactions predominate in this region and both charged residues are substituted with glutamine (Gln157 and Gln308, Figure 3A). Pcdh12 contains a single substitution in the arginine position to glycine, suggesting alternative dimer packing for this subtype (Figure S5). Notably, δ1-protocadherins contain an arginine at the glutamate position (Arg297 in pcdh1), and the possibility that resulting electrostatic clashes could prevent binding between subfamilies has been discussed previously (Cooper et al., 2016). In EC3, C-D loops of pcdh-10, −17, and −18 share similar sequences (Figure S5) and conformations (Figure S4C), but that of pcdh19 adopts a subtly different conformation in the interface (Figures 3A and S4C), accommodating two pcdh19-specific interfacial aromatic residues (Tyr257 and Tyr259) that extend to form close contacts with EC2 and contribute to the more hydrophobic character of the pcdh19 interface.
Mutations in a Conserved Region of the EC1:EC4 Interface Abolish trans Binding
We next tested the effects of targeted point mutations on binding of EC1–EC4 fragments in SPR (Figure 4). While both EC1:EC4 and EC2:EC3 interface regions contribute substantial BSA (Table S2), we targeted the hydrophobic region of the EC1:EC4 interaction due to its family-wide conservation. Substitution of highly conserved residues Leu94 (EC1 side) or Leu359 (EC4 side) with lysine or aspartic acid ablated homophilic binding of pcdh1 (Figure 4A, L94K and L359D mutants). Equivalent EC4 mutations in pcdh7 (L414D) and pcdh9 (L362D) also ablated homophilic binding, consistent with involvement of the hydrophobic region throughout the δ1 family (Figure 4A). Heterophilic binding interactions between pcdh-7 and −9 or between pcdh-1 and −11 were also abolished by these mutations (Figure 4B), indicating that these form through the same interface.
Figure 4. SPR Analysis of Targeted trans Interface Mutations.
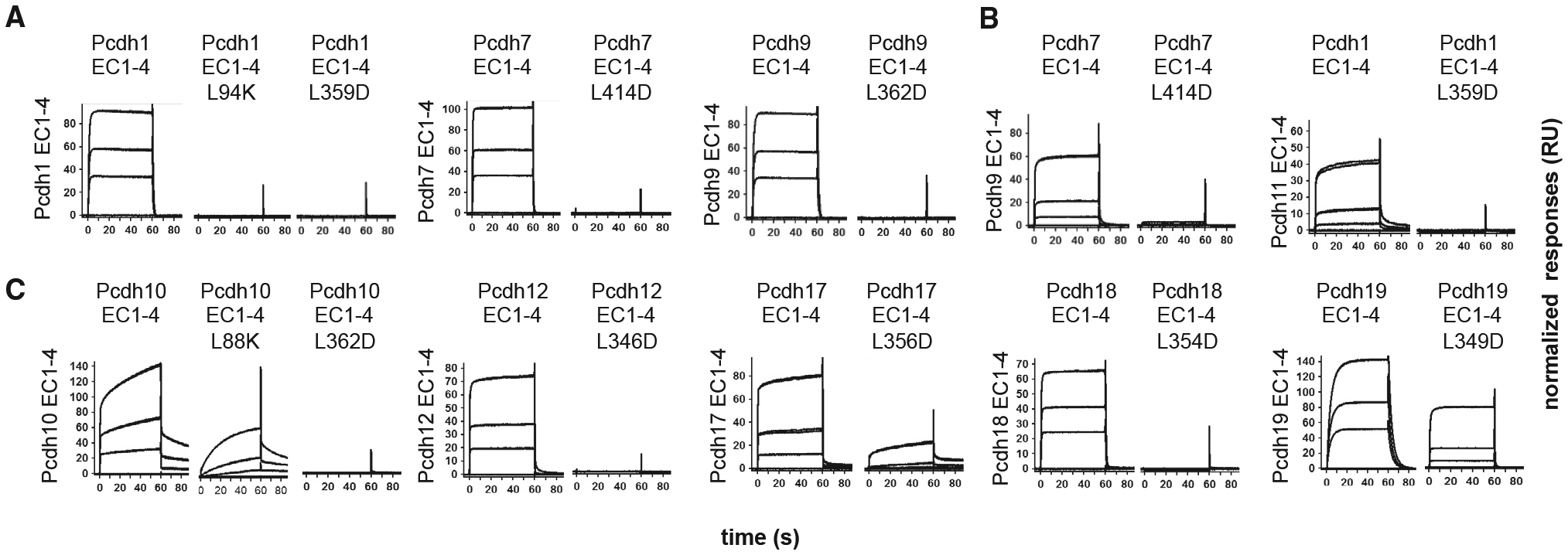
(A) Homophilic binding of wild-type and trans interface mutant δ1 EC1–EC4 fragments of pcdh-1, −7, and −9 over their respective wild-type surfaces.
(B) Effects of trans interface mutations in pcdh1 and −7 on heterophilic binding to pcdh-9 (left) and −11 (right).
(C) Homophilic binding of wild-type and trans interface mutants of δ2-pcdh-10, −12, −17, −18, and −19 over wild-type surfaces. Three analyte concentrations (27, 9, and 3 μM) are plotted and responses are scaled independently for each surface.
In δ2-protocadherins, equivalent mutations had comparable effects: homophilic binding was ablated in pcdh10 (L88K and L362D), −12 (L346D), and −18 (L354D) and diminished in pcdh17] (L356D) and −19 (L349D, Figure 4C). Inhibition of pcdh17 homophilic binding by mutation L356D suggests the interface likely adopts a more canonical conformation in solution than in the divergent crystal structure (Figure S4A), where Leu356 is solvent-exposed (Figure S4B). Nevertheless, the incomplete inhibition may suggest weaker contribution of EC1:EC4 to pcdh17 binding that could permit crystallization in the non-canonical orientation observed. Pcdh19 L349D mutant showed the mildest diminution of comparative binding responses in SPR, likely reflecting maintenance of the dimer by the more extensive contacts observed in the dimer overall (Figure 3A; Table S2).
Together, the mutational data confirm the structurally observed dimer to be generally conserved across the δ-protocadherin family and support previous suggestions that a hydrophobic “core” in the EC1:EC4 interface is a conserved characteristic of canonical protocadherin dimers (Cooper et al., 2016; Nicoludis et al., 2016; Rubinstein et al., 2015).
δ-Protocadherins Do Not Form Higher Order Oligomers in Solution
Our AUC and SEC-MALS analyses of complete ectodomains revealed no evidence for oligomers larger than dimers for any δ-protocadherin (Table 1; Figure S2). These observations for δ-protocadherins contrast with comparable AUC analyses of clustered β-, γB-, and C-type protocadherins for which tetramers were detected in solution (Goodman et al., 2016b; Rubinstein et al., 2015), formed by a combination of EC1–EC4-mediated trans interactions and EC5–EC6-mediated cis interactions (Goodman et al., 2016b). Thus, our data suggested that δ-protocadherins, despite their overall similarity to the clustered branch (Hulpiau and van Roy, 2009), might lack comparable ectodomain-mediated cis interactions strong enough to be detected in solution.
To test further if membrane proximal EC5–EC7 (δ1) or EC5–EC6 (δ2) domains contributed to associations measured in solution, we analyzed a range of truncated fragments in AUC. C-terminally truncated fragments of pcdh1, −7, −9, and −10 encompassing EC1–EC4 showed homodimerization affinities comparable to or stronger than those of the full-length ectodomains, indicating that EC5–EC6/7 domains do not enhance oligomerization of the longer fragments (Table 1). As expected, further truncation of pcdh1 and pcdh10 to EC1–EC3 to remove part of the trans binding EC1–EC4 region reduced homodimerization to very weak or undetectable levels (Table 1). Lastly, fragments containing only membrane-proximal regions, pcdh1 EC5–EC7, pcdh10 EC5–EC6, and pcdh8 EC5–EC6, were monomeric, consistent with an absence of strong EC5–EC6-mediated homophilic interactions (Table 1). Similar results were obtained for binding of the pcdh1 and pcdh10 fragments to respective full ectodomains in SPR (Figure S1C). Potential heterophilic interactions between membrane-proximal EC5–EC6 and EC5–EC7 fragments of pcdh-1, −8, and −10 fragments were also not detected when stoichiometric mixtures were assessed by SEC-MALS (Figure S2B).
cis-interaction Signatures of Clustered Protocadherins Are Absent from δ-Protocadherin Membrane-Proximal Region Structures
The lack of detectable cis interactions in solution did not exclude the possibility that weaker associations below the threshold of detection of AUC experiments (KD > ~1 mM) could form. To investigate this, we characterized prospective cis-interaction regions in three additional δ2 family structures: full ectodomains (EC1–EC6) of Xenopus pcdh8.1 and human pcdh10, and an EC5–EC6 fragment of human pcdh8 (Figure 5; Table S1).
Figure 5. Membrane-proximal Regions of δ2-Protocadherins Lack cis Interface Signatures.
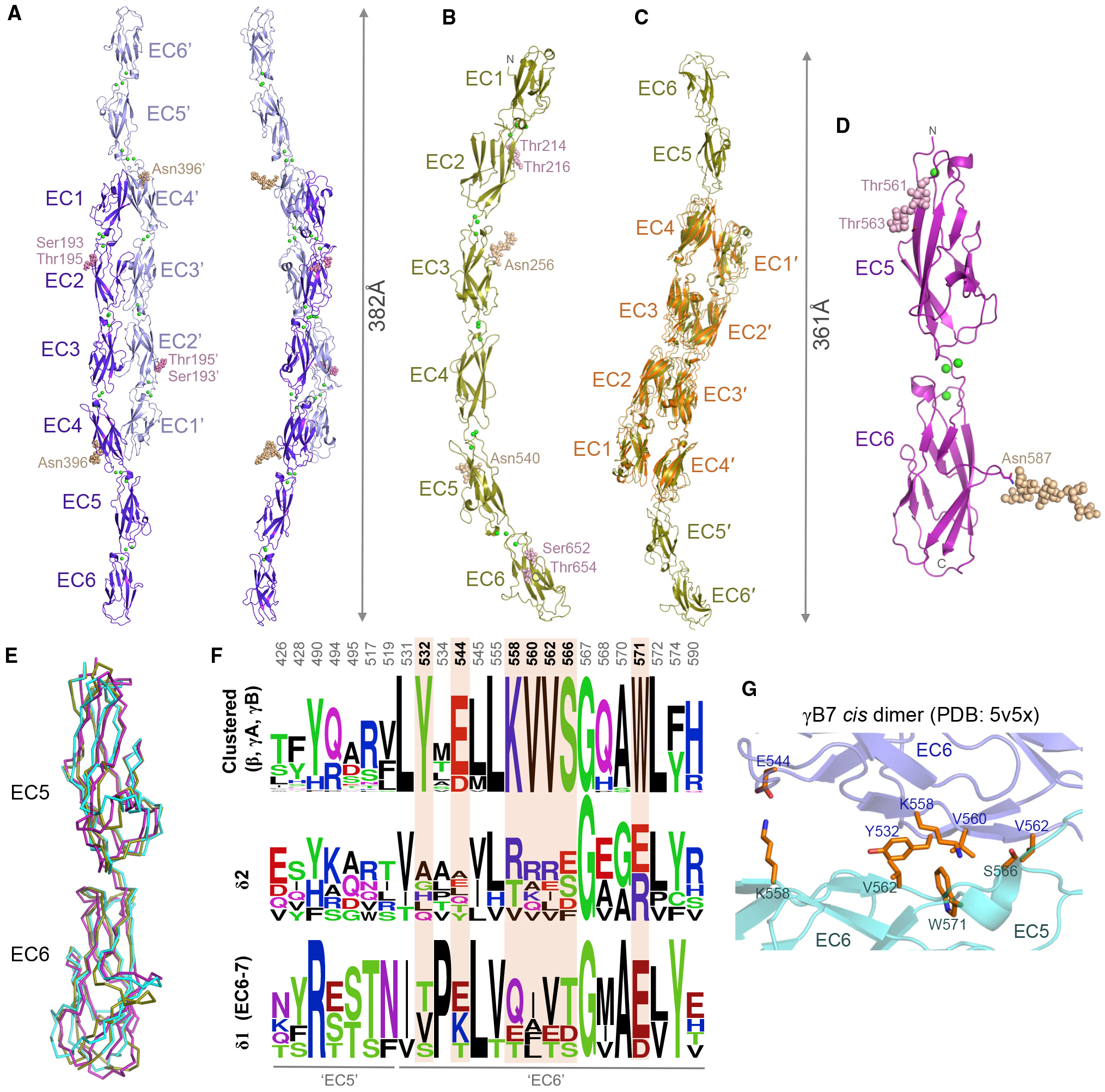
(A and B) Crystal structures of Xenopus pcdh 8.1 EC1–EC6 (A) and human pcdh10 EC1–EC6 (B), shown as ribbons. Green spheres: calcium ions; wheat and magenta spheres: N- and O-linked glycans.
(C) Superposition of two molecules of pcdh10 EC1–EC6 (gold) over the trans dimer structure of pcdh10 EC1–EC4 (orange).
(D) Crystal structure of human pcdh8 EC5–EC6 membrane-proximal fragment, shown as ribbon.
(E) Superposition of EC5–EC6 membrane-proximal regions of pcdh8 (magenta) and pcdh10 (gold) over EC5–EC6 from clustered pcdh γB7 (cyan, PDB: 5V5X; Goodman et al., 2017).
(F) Sequence logo plots of aligned mouse clustered protocadherins β, γA, and γB (top) or human δ-protocadherins (bottom). Only residue positions with side chains > 20% buried in the γB7 cis dimer (PDB: 5V5X) are shown. Positions conserved only in clustered protocadherinpcdhs are highlighted orange. Numbering refers to pcdh γB7.
(G) Close-up view of the cis interface of clustered pcdh γB7 (Goodman et al., 2017) showing differentially conserved interface residues from (E). Protomers colored slate and cyan.
Crystals of Xenopus pcdh8.1 and human pcdh10 ectodomains each showed highly anisotropic diffraction with resolution limits of 3.0/3.0/2.0 Å and 4.2/4.2/3.3 Å, respectively (Table S1). Both structures revealed an approximately linear overall arrangement of six EC domains including canonical calcium binding in all interdomain linkers (Figures 5A and 5B). Pcdh8.1 formed canonical trans dimers in the crystal lattice (Figure 5A), similar to those observed for human δ2-protocadherins, including close EC2:EC3 contacts and an EC1:EC4 interface centered on hydrophobic residues (Leu86 and Phe344) equivalent to Leu94 and Leu359 of pcdh1. Trans dimers were not observed in the pcdh10 EC1–EC6 crystal lattice (Figure 5B), likely due to acidic crystallization conditions (pH 5; see STAR Methods) similar to those of the monomeric EC1–EC4 fragment structure (Figure 2B). However, since our mutagenesis data showed that pcdh10 ectodomains likely form canonical trans dimers in solution (Figures 4C and S1C; Table 1), we used the EC1–EC4 trans dimer structure crystallized at neutral pH to model a full-length pcdh-10 trans dimer by superposition over domains EC1–EC4 (Figure 5C). The extended bow-like dimers for pcdh8.1 and pcdh10 show distances of 382 Å and 361 Å between paired C-termini, representing lower limits for predicted intermembrane spacing due to stalk regions of 24 (pcdh8.1) and 35 (pcdh10) amino acids preceding the predicted transmembrane regions that were not included in the crystallized constructs. Notably, neither pcdh8.1 nor pcdh10 EC1–EC6 crystal lattices revealed interfaces that could geometrically align protomers deriving from the same membrane surface in a cis orientation. A 2.9 Å resolution structure of human pcdh-8 EC5–EC6 also revealed canonical EC domain architecture and did not contain interfaces geometrically compatible with cis interactions (Figure 5D), in agreement with the monomeric nature of the EC5–EC6 fragment in AUC experiments (Table 1).
EC5–EC6 regions from human pcdh8 and pcdh10 structures superposed closely with the equivalent domains of clustered protocadherin γB7 EC3–EC6 (Goodman et al., 2017; RMSD 1.3–1.9 Å for 181–191 aligned Cα; Figure 5E), allowing surface residue positions involved in the γB7 cis interface to be compared. Sequence alignment of residue positions whose side chains are buried in the γB7 cis interface reveals seven positions with high conservation within β- and γ-clustered protocadherins but poor conservation in pcdh-8 and −10 and other δ-protocadherins (Figures 5F and S6). Mapping the differentially conserved positions on the structure of the γB7 cis interface reveals them to constitute a large proportion of the core of the interaction (Figure 5G). Furthermore, these include residues Tyr532 and Val560, shown by mutagenesis to be critical for cis interactions of clustered protocadherins (Goodman et al., 2017, 2016b; Figure 5G). Together, poor conservation of cis interaction surfaces and lack of detectable cis oligomerization in solution suggest that the δ-protocadherins do not assemble laterally through an interface comparable to that of clustered protocadherins.
Assembly of δ1- and δ2-Protocadherin Ectodomains in Reconstituted Junctions
We recently showed, using cryo-electron tomography (cryo-ET), that clustered protocadherin γB6 ectodomains form distinctive zipper-like assemblies of alternating cis and trans interactions at contact sites between membranes (Brasch et al., 2019). We would expect such assemblies to be absent from δ-protocadherin contacts based on the lack of cis interface conservation. We therefore visualized intermembrane adhesion by δ1- and δ2-protocadherins by attaching full ectodomains of pcdh1 (δ1-family) or pcdh10 (δ2-family) to liposome surfaces via C-terminal hexahistidine tags. Initial fluorescence microscopy experiments using rhodamine-labeled liposomes showed extensive aggregation mediated by pcdh1 and pcdh10, which was abolished for trans interface mutants pcdh1 L359D and pcdh10 L362D (Figure 6A). We next visualized vitrified liposome aggregates by cryo-ET (Table S3). Tomographic reconstructions of pcdh1-mediated aggregates (Figure 6B; Videos S1, S2, and S3) showed extensive junction-like structures between contacting liposomes, with flattening of apposed membranes and an intermembrane spacing of ~490 Å (Figure 6D). Dense accumulation of pcdh1 ectodomains protruding perpendicularly from both membranes was observed at these sites and a diffuse midline of increased density with a width of ~200 Å could be discerned (Figure 6B), corresponding closely to the predicted width of over-lapped EC1–EC4 regions in trans dimers, where density would be expected to be doubled. Individual pcdh1 ectodomains were resolved within the junctions (Figures 6B and 6D) and on free membranes (Figure 6B) with elongated overall conformations, as expected. Broadly similar junction formation was observed for pcdh10, including flattening of apposed membranes and dense accumulation of ectodomains (Figures 6C and 6D; Videos S4 and S5). Intermembrane spacing was ~377 Å, narrower than pcdh1 junctions where ectodomains contain one extra EC domain (Figure 6D). Midlines and individual ectodomains were less clearly resolved for pcdh10, and junctions appeared less uniform than those of pcdh1 (Figure 6D). Importantly, characteristic ellipsoidal views of zipper-like lattices observed previously for clustered protocadherin γB6 (Figure 6D; Brasch et al., 2019) were absent from tomographic reconstructions of both pcdh1 and pcdh10 (Figures 6B–6D). Moreover, while the δ-protocadherin junctions contained high densities of closely packed trans dimers they appeared to lack regular periodicity in the intermembrane densities that would indicate overall lattice-like order. This was particularly evident in “top views” of junctions between liposomes stacked along the z axis, in which the lateral position in the membrane of each protocadherin molecule can be seen: no apparent order was observed for pcdh1 and pcdh10 in contrast to the ordered grid-like pattern for clustered pcdh γB6 (Figure 6E; Brasch et al., 2019). These findings suggest that cis interactions seen for clustered protocadherins are not formed by δ-family protocadherins and, further, that their ectodomains may lack a propensity to organize into ordered structures between membranes.
Figure 6. Assembly of δ-protocadherin Ectodomains in Reconstituted Liposome Junctions.

(A) Fluorescence microscopy of liposome aggregation mediated by pcdh1 and pcdh10 ectodomains or trans dimer mutants pcdh1 L359D and pcdh10 L362D. Scale bar: 0.5 mm.
(B and C) Representative slices of reconstructed tomograms showing aggregated liposomes of pcdh1 (B) or pcdh10 (C). Protocadherin ectodomains enrich at liposome contact sites seen in “side views” (black arrowheads) where membranes appear parallel, and “top views” (arrows) where liposomes are stacked vertically. Unbound ectodomains protrude from non-junctional membranes (white arrowheads). See Videos S1, S2, S3, S4, and S5.
(D) Side views of pcdh1, pcdh10, and pcdh gB6 junctions showing ordered assembly only for pcdh gB6. Intensity plots below each image show intermembrane distances and shallow minima where trans dimers overlap (brackets). Lipid bilayers are indicated with dashed lines.
(E) Comparison of top views showing formation of a regular lattice by pcdh gB6 ectodomains only. Scale bars in (B)–(E): 400 Å. See Table S3.
DISCUSSION
Here we have biophysically and structurally characterized the ectodomain interactions of most typical members of the δ1- and δ2-protocadherin families. While δ-protocadherins are highly similar to the clustered protocadherins, they exhibit specific differences that likely relate to their distinct biological roles. The clustered, δ1-, and δ2-protocadherin families bind in trans through topologically similar antiparallel EC1–EC4 dimers, each composed of two large EC1:EC4 and EC2:EC3 and one small EC3:EC3 interface region. This binding mode has been well characterized for the clustered protocadherins (Goodman et al., 2016a, 2016b) and for δ2-family member Pcdh19 from zebrafish (Cooper et al., 2016), while structures of pcdh1 reported here and in Modak and Sotomayor (2019) extend the mechanism to the δ1-family. Despite topological similarity overall, we observed no cross-family interactions between δ1-, δ2-, and clustered protocadherins, likely reflecting differences in interface orientations and residue conservation. This “orthogonal” subfamily specificity resembles that observed previously between type I and type II classical cadherins (Katsamba et al., 2009) and may allow different subfamilies to establish overlapping but independent sets of adhesive cues. In δ1- and δ2-protocadherins, orthogonal specificity may derive from differences in preferred sub-interface orientations, though additional structures of δ1-family trans dimers will be necessary to establish that differences observed for pcdh1 are maintained for other subtypes. Additional incompatibility may derive from residue differences in the central EC3:EC3 interaction; the base of the EC2:EC3 interface that is conserved in δ2 only; and in the DE loop of EC2, where electrostatic clashes between some subtypes may arise, as noted previously (Cooper et al., 2016).
Within the δ1- and δ2-subfamilies, trans-binding specificities revealed in our SPR experiments are primarily homophilic, in agreement with cell aggregation studies (Bisogni et al., 2018; Pederick et al., 2018; Tai et al., 2010) and consistent with the homophilic cell sorting role uncovered for pcdh19 in vivo (Pederick et al., 2018). In addition, we detect substantial heterophilic binding among members of each subfamily. Overall, this specificity pattern is similar to those of the type I and type II classical cadherin families that function primarily in adhesion. Type I cadherins (including E-, N-, and P-cadherins) have highest trans-binding strength with self in most cases, but show significant cross-interactions with other family members (Katsamba et al., 2009; Vendome et al., 2014), while type II cadherins display extensive heterophilic interactions that frequently exceed homophilic binding (Brasch et al., 2018). Homophilic preference is more uniform for δ-protocadherins where homophilic SPR responses were strongest for all subtypes tested, potentially explaining the absence of detectable heterotypic aggregates between cells transfected with different single δ-protocadherins (Bisogni et al., 2018; Pederick et al., 2018). Nonetheless, heterophilic responses observed in the δ1-family approach those of homophilic responses, suggesting likely physiological relevance. The limited heterophilic responses observed between δ2-protocadherins produced far lower responses than homophilic binding, and thus their physiological significance is less clear. Heterophilic interactions, commonly found for protein families that function primarily in adhesion, can play a role at boundaries between different cell types sharing a single family member (Basu et al., 2018; Brasch et al., 2018; Generous et al., 2019; Labernadie et al., 2017; Togashi et al., 2011; Volk et al., 1987), or in refining cell-level interaction specificity when co-expressed (Carrillo et al., 2015; Cosmanescu et al., 2018; Patel et al., 2006; Xu et al., 2018). As described above, δ-protocadherins are often co-expressed in the same cell (Etzrodt et al., 2009), and effects of δ-protocadherin co-expression on cell-interaction specificity have been demonstrated in cell aggregation experiments (Bisogni et al., 2018; Pederick et al., 2018).
Outside of the trans-binding mechanism, we identified significant differences between δ- and clustered protocadherins. Clustered protocadherins are thought to emanate from the plasma membrane as cis dimers mediated by their membrane-proximal EC5 and EC6 domains (Goodman et al., 2017). Their ectodomains have been shown to form tetramers in solution (γB-, β-, and C-type; Goodman et al., 2016b; Rubinstein et al., 2015) and to assemble zipper-like structures of alternating cis and trans interactions in crystal structures and between membranes (pcdh γB4 and γB6; Brasch et al., 2019). By contrast, δ-protocadherin ectodomains form no detectable cis interactions in AUC, SPR, and SEC-MALS experiments (Table 1; Figures S1C and S2). We also observe no evidence for cis interactions in crystal structures of δ2-protocadherin full ectodomains or membrane-proximal regions (Figure 5) and canonical cis interface regions are poorly conserved (Figure 5), arguing against the possibility of weaker cis interactions not detectable in solution as observed for E-, N-, and C-cadherins (Harrison et al., 2011). More definitively, tomograms of reconstituted junctions formed by representative members of the δ1- and δ2-families display structures that lack evidence of ordered lattices or zippers and appear instead to be characteristic of adhesion proteins engaged only in trans interactions (Figure 6).
Lack of cis-dimerization is consistent with functional differences between clustered and δ-protocadherins. The clustered protocadherins, among other functions, encode diverse single-cell identities for some neuron types that underlie self-avoidance (Kostadinov and Sanes, 2015; Lefebvre et al., 2012; Molumby et al., 2017; Mountoufaris et al., 2017; Zipursky and Grueber, 2013). We previously proposed that, despite comprising only 58 isoforms, co-expressed clustered protocadherins can encode sufficient diversity by engaging in cis and trans interactions to polymerize a zipper-like structure between dendrites from the same neuron to signal repulsion, which would be terminated by isoform mismatches with non-self-dendrites (Brasch et al., 2019; Rubinstein et al., 2015). We suggest here that this mechanism, which critically depends on cis dimerization, represents a clustered protocadherin-specific adaptation to provide the diversity for self-avoidance that did not arise in δ-protocadherins, which are not thought to participate in this process.
Notably, there is previous evidence for some form of cis interaction between δ-protocadherins from experiments demonstrating co-immunoprecipitation of pcdh19 with pcdh10 and pcdh17 in co-transfected K562 cells (Pederick et al., 2018). Since these experiments involved full-length proteins, these interactions may represent associations between transmembrane or cytoplasmic domains, either direct or indirect, which our data for extracellular regions do not exclude.
Functional roles of δ-protocadherins are coming into view with increasing clarity. The remarkable cell patterns that arise in GCE through X-linked inheritance of inactive forms of the Pcdh19 gene leaves little doubt that it plays an important role in cell adhesion (Pederick et al., 2018). δ-protocadherins also display expression patterns that differentially track tissue structures (Etzrodt et al., 2009; Redies et al., 2005), and for which changes in expression often precede the emergence of anatomical features in development (Redies et al., 2005; Roy and Bandyopadhyay, 2014). Phenotypes potentially related to adhesion have been found for numerous other δ-protocadherins, as well (Bononi et al., 2008; Cooper et al., 2015; Hayashi et al., 2014; Hayashi and Takeichi, 2015; Hoshina et al., 2013; Uemura et al., 2007; Zhu et al., 2014). Overall, these observations are consistent with a common role in cell–cell adhesion and, indeed, we have shown that the trans-binding properties of δ-protocadherins are similar to those of other families of adhesion proteins. Further, our data suggest that non-clustered protocadherins diverge from their clustered relatives in lacking specialized higher-order extracellular assemblies mediated by their ectodomains. Mechanisms linking trans binding of δ-protocadherins to their cellular signaling functions remain to be elucidated.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lawrence Shapiro (lss8@columbia.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The Freestyle 293F cell line was obtained from Thermo Fisher Scientific and is derived from the parental Human Embryonic Kidney (HEK) 293 cell line, of which the sex is female. Freestyle 293F cells were cultured in suspension in Freestyle 293 Expression medium at 37°C and 10% CO2. Human K-562 bone marrow chronic myelogenous leukemia (CML) cells, of which the sex is female, were obtained from ATCC and cultured in DMEM/10% FBS at 37°C and 5% CO2.
METHOD DETAILS
Cloning and expression of recombinant proteins
Complementary DNA (cDNA) clones encoding full-length human δ protocadherins −17, −18 and −19 were a kind gift from Dr. Chan Aye Thu (Columbia University). Clones encoding human protocadherins −1, −8, 9, −10, −11Y, 12, and 20, mouse pcdh7, and Xenopus pcdh8.1 and pcdh8.2 were obtained from the DNASU plasmid repository or Transomic Inc. Mouse pcdh8 was obtained from a cDNA library derived from day 15 embryo (Clontech). All constructs hereafter refer to human orthologs except where specified and amino acid numbering refers to the mature protein following signal peptide cleavage predicted using the SignalP server (Almagro Armenteros et al., 2019). Regions encoding full ectodomains of pcdh1 (1-TRV-RGN−795), pcdh7 (1-KQL-RLS−849, mouse), pcdh9 (1-QEL-YLT−791), pcdh11Y (1-QEK-YVK−789), pcdh20 (1-SYS-MPT−829), pcdh8 (1-KTV-VTA−683, human and 1-KTV-VTA−683, mouse), Xenopus pcdh8.1 (1-KTV-LVT−641), Xenopus pcdh8.2 (1-EIA-LTD−646), pcdh10 (1-SQL-LVD−662), pcdh12 (1-LTV-FVT−668), pcdh17 (1-LKN-DMS−688), pcdh18 (1-KNL-IFE−651), and pcdh19 (1-LIN-NLS−657) or truncated ectodomain fragments pcdh1 EC1–3 (1-TRV-VKD−322), pcdh1 EC1–4 (1-TRV-VVD−441), pcdh1 EC5–7 (442-VND-RGN-795), mouse pcdh7 EC1–4 (1-KQL-VGD-497), pcdh9 EC1–4 (1-QEL-LED−438), pcdh11 Y EC1–4 (1-QEK-VKD−435), pcdh8 EC1–4 (1-KTV-VGD-460), pcdh8 EC5–6 (461-END-VTA−683), pcdh 10 EC1–3 (1-SQL-VLD−334), pcdh10 EC1–4 (1-SQL-VSD−438), pcdh10 EC5–6 (439-VND-LVD−662), pcdh17 EC1–4 (1-LKN-ILD−447), pcdh18 EC1–4 (1-KNL-IND−430), and pcdh19 EC1–4 (1-LIN-ITD−424) were amplified by PCR from the respective full-length templates. Ectodomain sequences of pcdhs −1, −7, −9, −11, −20, 17 and −19 and pcdh1 EC5–7 included up to 38 residues belonging to a ‘stalk’ region between the end of the last EC domain and the start of the start of the transmembrane region in the full-length protein whose inclusion was found to improve expression and solubility; these regions were not included for pcdh −8, −8.1, −8.2, − 10, −12, -and −18, however, an additional pcdh10 construct containing this region, pcdh 10 EC1–6ECD(1-SQL-DLT−698) was prepared for use in liposome experiments. Amplicons were inserted between Not1/BamH1 sites of the mammalian expression vector VRC8400 (Barouch et al., 2005) preceded by the signal sequence of human Binding immunoglobulin protein (BiP; MKLSLVAAMLLLL-SAARA) and followed by a hexahistidine tag and stop codon. Human pcdh7 EC1–4 (1-KQL-VGD-497) fragment, for which cDNA was not available, was prepared from the mouse construct by mutagenesis using the Quikchange method (Invitrogen). Trans interface mutant constructs were prepared in the same way. Fidelity of all expression constructs was checked by DNA sequencing and was matched to amino acid sequences of Uniprot entries Uniprot: Q08174 (pcdh1), A0A0A6YY83 (mouse pcdh7), O60245 (human pcdh7), Q9HC56 (pcdh9), Q9BZA8 (pcdh11), O95206 (human pcdh8), Q7TSK3 (mouse pcdh8), Q6GLU2 (Xenopus pcdh8.1), A0A1L8HHP1 (Xenopus pcdh8.2), Q9P2E7 (pcdh10), Q9NPG4 (pcdh12), O14917 (pcdh17), Q9HCL0 (pcdh18), and Q8TAB3 (pcdh19).
For protein expression, Freestyle HEK293 cells (Thermo Fisher Scientific) grown in suspension were transiently transfected using Polyethyleneimmine (Baldi et al., 2012) diluted in Opti-Mem reduced serum medium (Thermo Fisher Scientific). Transfected cells were maintained in Freestyle cell culture medium (Thermo Fisher Scientific) supplemented with 5mM CaCl2 16 hours after transfection to improve cadherin yield. After six days, secreted hexahistidine-tagged cadherins were collected from conditioned media by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography using a batch procedure (1h, 25°C) followed by further purification by size-exclusion chromatography using a Superdex 200 26/60 column (GE Healthcare) on an AKTA pure fast protein liquid chromatography system (GE Healthcare). Purified proteins were concentrated to between 2 and 22mg/mL in 150mM NaCl, 10mM Tris-Cl pH8.0, 3mM CaCl2 and flash frozen in liquid nitrogen for long-term storage.
Constructs encoding biotinylated EC1–4 (pcdhs −1, −7, −8, −9, −10, −11, −17, −18, −19) or EC1-EC6 (pcdh12) fragments were prepared by insertion of an Avi-tag (GLNDIFEAQKIEWHE)-encoding sequence between the hexa-histidine-tag and stop codon. These were co-transfected with a plasmid encoding the biotin-Ligase BirA from E. coli (Lys2-Lys321) that included a BiP signal sequence and a C-terminal endoplasmic reticulum-retention signal (DYKDEL) prepared previously (Harrison et al.,2016) according to the method of Barat and Wu (2007). Expression plasmid and BirA plasmid were mixed at a 9:1 ratio for transfection and 50 μM Biotin (Sigma) was added to the media after 16 hours. Purification was carried out exactly as for the non-biotinylated constructs and biotinylation was confirmed by western blot using NeutrAvidin-HRP (Pierce).
Trans binding fragments of clustered protocadherins α7 (EC1–5), β6 (EC1–4), γA8(EC1–4), γB2(EC1–5) and gC4(EC1–4) were prepared as described previously (Goodman et al., 2016a, 2016b; Rubinstein et al., 2015) using a similar method to that described for δ protocadherins. Biotinylated forms were prepared as described above.
SPR binding experiments
SPR binding experiments were performed using a Biacore T100 biosensor equipped with a Series S CM4 sensor chip, immobilized with NeutrAvidin over all four flow cells. NeutrAvidin immobilization was performed in HBS-P buffer (0.01 M HEPES, 0.15 M NaCl, 0.05% v/v Tween-20, pH 7.4) at 32°C, over all four surfaces using amine-coupling chemistry as described in Katsamba et al. (2009), resulting in approximately 10,000 RU of NeutrAvidin immobilized. Binding experiments were performed at 25°C in a running buffer containing 10 mM Tris-Cl pH 7.4, 150 mM NaCl, 3mM CaCl2, 0.5 mg/mL BSA and 0.005% (v/v) Tween-20.
Biotinylated pcdh1, −7, −9, −11, −10, −12, −17, −18, −19 proteins comprising domains EC1–6 (pcdh12) or EC1–4 (all others) were captured over individual NeutrAvidin-immobilized surfaces at 1100–1300 RU using a flow rate of 20 mL/min. A NeutrAvidin-immobilized flow cell was used as a reference in each experiment to subtract bulk refractive index changes. Pcdh analytes and their respective mutants, as shown in Figures 1 and 4, were prepared in running buffer and tested for binding at 3, 9 and 27 μM, in order of increasing concentration, with each concentration series tested in duplicate to confirm reproducibility. In each binding cycle, a protein sample was injected over the captured surfaces at 50 μL/min for 60 s, followed by 120 s of dissociation phase, a running buffer wash step and a buffer injection at 100 μL/min for 60 s. After three binding cycles, buffer was used as an analyte instead of a protein sample to double reference the binding responses by removing systematic noise and instrument drift. The data was processed using Scrubber 2.0 (BioLogic Software). Binding responses were normalized for molecular weight differences between the ectodomains of each protein.
EC1–4 fragments of pcdhs −1, −7, −10 and −18 were also tested for binding at 3, 9 and 27 μM over surfaces of biotinylated clustered pcdhs α7 (EC1–5), β6 (EC1–4), γA8(EC1–4), γB2(EC1–5) and γC4(EC1–4), captured at 1700–3000 RU. Experiments were performed at 25°C in a running buffer containing 10 mM Tris-Cl pH 8.0, 150 mM NaCl, 3mM CaCl2, 20 mM imidazole, 0.25 mg/mL BSA and 0.005% (v/v) Tween-20. Binding was tested using the same parameters as described above, except that a 40 s analyte contact time, followed by a 90 s dissociation phase was used. Clustered pcdhs α7 (EC1–5), β6 (EC1–4), γA8(EC1–4), γB2(EC1–5) and γC4(EC1–4) were tested for homophilic binding over their respective biotinylated surfaces at 0.1, 0.3, 0.89, 2.67, 8 and 24 μM.
Analytical ultracentrifugation
Equilibrium analytical ultracentrifugation experiments were performed using a Beckman XLA/I ultracentrifuge, with a Ti50An or Ti60An rotor. Prior to each experiment, all proteins were diluted with buffer (150 mM NaCl, 10 mM Tris-Cl pH 7.4, 3 mM CaCl2) and dialyzed for 16 hours at 4°C in the same buffer. 120 μL of proteins at three different concentrations (Abs 280nm at 10 mm of 0.65, 0.43 and 0.23, repectively), were loaded into six-channel equilibrium cells with parallel sides and sapphire windows. We performed all experiments at 25°C and collected UV data at 280 nm, using dialysis buffer as blank. Pcdh8 EC1–6 from mouse and human showed a tendency to non-specifically aggregate when samples thawed from −80° storage were analyzed so unfrozen protein collected immediately following purification was used. EC1–6 and EC1–7 full ectodomain fragments were spun for 20 hours at 11000 rpm, increasing to 14000 rpm, 17000 rpm, and 20000 rpm for 10 hours each. Four scans (one per hour) were collected at each speed for the three concentrations to yield 48 scans per sample. Two, three and four ectodomain fragments were analyzed using the same protocol, except that 15000 rpm, 19000 rpm, 23000 rpm and 27000 rpm were used. We calculated the buffer density and protein v-bars using the program SednTerp (Alliance Protein Laboratories), and analyzed the retrieved data using HeteroAnalysis 1.1.44 (http://biotech.uconn.edu/auf; Cole et al., 2008). We fitted data from all concentrations and speeds globally by nonlinear regression to either a monomer-dimer equilibrium model or an ideal monomer model. All experiments were performed at least in duplicate.
Size-exclusion chromatography with multi-angle light scattering (SEC-MALS)
SEC-MALS analyses were performed using a Superdex 200 Increase 3.2/300 size exclusion column on an AKTA FPLC system (GE healthcare) coupled to inline static light scattering (Dawn Heleos II, Wyatt Technology), differential refractive index (Optilab rEX, Wyatt Technology) and UV detection. Purified ectodomains were diluted to 24 μM in running buffer (150mM NaCl, 10mM Tris-Cl pH 7.4, 3mM CaCl2) and 50ul samples were run at a flow rate of 0.5ml/min at 25°C. Mixtures of membrane-proximal pcdh fragments were prepared in the same buffer at final concentrations of 100 μM for each protein and incubated for 20 minutes at 25°C prior to running under the same conditions. All proteins eluted as single peaks, shown in Figure S1. Data were analyzed using ASTRA software (Wyatt Technologies).
K562 cell aggregation assays
Full-length human Pcdhs were cloned into the pmax-mCherry expression vector encoding a C-terminal mCherry-tag, then transfected into K562 cells (ATCC CCL243) as previously described (Goodman et al., 2017; Thu et al., 2014). Briefly, K562 cells were cultured at 37°C with 5% CO2 in DMEM with GlutaMAX (GIBCO) supplemented with 10% FBS and 1% penicillin-streptomycin. After two days, cells were counted, centrifuged, and resuspended in SF Cell Line 4D-Nucleofector Solution SF with supplement according to manufacturer’s instructions (Lonza) at a density of ~1.5×104 cells/μL. For each experiment, 2 μg of the Pcdh expression construct were transfected into 20 μL of the K562 cell suspension by electroporation using an Amaxa 4D-Nucleofector (Lonza). Transfected cells were transferred to a 24-well plate in 500 μL of medium per well and incubated overnight at 37°C and 5% CO2. Cells then were mixed by gentle rocking for 4 hours and imaged with an Olympus IX73 fluorescent microscope to determine the extent of aggregation.
Protein crystallization and structure determination
Crystals were grown using a hanging drop method in which 0.6–1.2 μL of purified protein were mixed with 0.6–1.2 μL of crystallization solution and incubated at 23°C for 1–10 days. Crystallization conditions for human pcdh1 EC1–4 (13.3mg/ml) were 8.5% (w/v) PEG 4000, 20% (v/v) glycerol, 0.1M tris-bicine buffer pH8.5, 0.1M Morpheus monosaccharides mix (0.02 M D-glucose, 0.02 M D-mannose, 0.02 M D-galactose, 0.02 M L-fucose, 0.02 M D-xylose, 0.02 M N-acetyl-D-glucosamine; Molecular Dimensions) with no further cryoprotection; those for Xenopus pcdh8.1 EC1–6 were 10% (w/v) PEG 8000, 20% (v/v) ethylene glycol, 0.135M nitrate-phosphate-sulfate mix (0.045M sodium phosphate dibasic dihydrate, 0.045M Ammonium sulfate, 0.045 M sodium nitrate; Molecular Dimensions), 0.1M MES/imidazole buffer pH 6.5 with no further cryoprotection; those for human pcdh8 EC5–6 (13.5mg/ml) were 10% PEG 20 000 (w/v), 20% (v/v) PEG 550 monomethylether, 0.12M Morpheus ethylene glycols mix (0.03M diethyleneglycol, 0.03M triethyleneglycol, 0.03M tetraethyleneglycol, 0.03M pentaethyleneglycol; Molecular Dimensions), 0.1M MES/imidazole buffer pH 6.5 with no further cryoprotection; those for human pcdh10 EC1–4 (10.8mg/ml) monomer form were 14% PEG 400 (v/v), 0.1M sodium acetate pH 4.6, 0.1M CaCl2, with 30% ethylene glycol (v/v) added as cryoprotectant; those for pcdh10 EC1–4 (21.5mg/ml) dimer form were 12% (w/v) PEG 8000, 20% (v/v) ethylene glycol, 0.1M Tris-bicine buffer pH8.5, 0.1M Morpheus monosaccharides mix (0.02 M D-glucose, 0.02 M D-mannose, 0.02 M D-galactose, 0.02 M L-fucose, 0.02 M D-xylose, 0.02 M N-acetyl-D-glucosamine; Molecular Dimensions) with no further cryoprotection; those for human pcdh10 EC1–6 (15.1mg/ml) were 0.22M CaCl2, 0.1M Na Acetate pH 5.0 with 30% (v/v) glycerol as cryoprotectant and glutaraldehyde (0.5% v/v) added to the reservoir after crystal formation to aid crystal stability by cross-linking; conditions for human pcdh17 EC1–4 (8.8mg/ml) were 13%(w/v) PEG 4000, 0.3M NaCl, 0.1M MES pH 6.5, with 30% PEG 400 (v/v) added as cryoprotectant; those for human pcdh18 EC1–4 (12mg/ml) were 13% (w/v) PEG 20 000, 0.1M MES pH 6.5, with 30% (v/v) ethylene glycol added as cryoprotectant; and conditions for human pcdh19 EC1–4 (13.3mg/ml) were 7%(w/v) PEG 4000, 0.2M NaCl, 0.05M MES pH6.5, with 15% (v/v) butanediol (2R,3R) added as cryoprotectant.
Data were collected from single frozen crystals at 100K using a wavelength of 0.979Å at the Northeastern Collaborative Access Team beamlines 24-ID-E and –C at the Advanced Photon Source, Argonne National Laboratory, Argonne Illinois, USA. Data were processed and scaled using XDS (Kabsch, 2010) and merged using AIMLESS (Evans and Murshudov, 2013) as part of the ccp4-suite (Winn et al., 2011). Severe diffraction anisotropy was observed for pcdh10 EC1–6 and ellipsoidal truncation of the data was performed using the Staraniso server (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi) using the default I/sigI threshold of 1.2. Using this limit, the region of included reflections was approximated by an ellipsoid extending to 4.2, 4.2 and 2.7Å for pcdh10 EC1–6 and 3.0/3.0/2.0Å for pcdh8.1 along the principal axes a*, b*, and c*. While Staraniso indicated that data for pcdh10 EC1–6 extended to 2.7Å in the strongest direction, data were manually truncated at 3.3A (resolution limits 4.2/4.2/3.3 along a*, b*, c*) since inclusion of the additional ~2000 reflections was not found to substantially improve model residuals or geometry in paired refinement tests. For both structures, the ellipsoidally truncated data were used for refinement since the treatment strongly improved the interpretability of Fo-Fc maps. Unwanted ‘filling’ of the removed data by Fcalc in map calculation was circumvented by removal of the respective indices in the mtz file using SFTOOLS in the ccp4 suite (Winn et al., 2011), as recommended in the Staraniso documentation. Data quality statistics are listed in Table S1.
Structures were solved by molecular replacement using Phaser (McCoy et al., 2007) as part of the phenix suite (Adams et al., 2010). Domains EC1–2 and EC4 of pcdh α4 EC1–4 (PDB: 5dzw), and EC3 of pcdh β1 EC1–3 (PDB: 4zpl) were used as search models for pcdh1 EC1–4. Domains EC1–4 of pcdh α7 EC1–5 (5dzv) were used as a search model for pcdh10 monomer form and the refined pcdh10 structure was then subsequently used as a search model for the pcdh10 dimer form and for the EC1–4 structures of pcdhs − 17, −18, and −19. Pcdh8 EC5–6 was solved using the EC5 and EC6 domains from the structure of pcdh γB2 EC3–6 (PDB: 5szr); the refined structure of pcdh8 EC5–6 was then used in combination with the pcdh10 monomer structure to solve the full ectodomain structures of pcdh8.1 and pcdh10. Structures were refined by iterative rounds of model building in Coot (Emsley et al., 2010) or phenix autobuild (Terwilliger et al., 2008) and automated refinement using phenix.refine (Afonine et al., 2012). Local torsional restraints between non-crystallographic symmetry-related chains were used in the early stages of refinement for all structures containing multiple chains in the crystallographic asymmetric unit: pcdh10 EC1–4 dimer (five chains), pcdh17 EC1–4 (four chains), pcdh18 EC1–4 (two chains), and pcdh19 EC1–4 (three-chains). Additional restraints for pcdh10 EC1–4 dimer and pcdh10 EC1–6 structures were generated using the pcdh10 EC1–4 monomer structure refined to higher resolution as an external reference model during early stages (pcdh10 EC1–6) or throughout refinement (pcdh10 EC1–4). Secondary structure restraints were also applied in phenix.refine for all structures. For pcdh19 EC1–4, superposition of zebrafish pcdh19 EC1–4 (PDB: 5iu9) was used to guide manual adjustment of ambiguous regions in late stages of refinement. In the structure of Xenopus pcdh8.1 EC1–6, EC6 domains of both chains were poorly ordered so the EC6 domain from the structure of human pcdh8 EC5–6 was used to closely guide manual building. B factor models, chosen based on data resolution (available reflections per model atom) and by comparison of Rfree values for different protocols, comprised: individual isotropic B factors for pcdh1 EC1–4 (4.8 reflections per atom), pcdh10 EC1–4 monomer form (6.7 reflections per atom) and pcdh18 EC1–4 (5.4 reflections per atom); individual isotropic B factors with translation-libration-screw (TLS) refinement for pcdh10 EC1–6 (one TLS group, 3.8 reflections per atom) and Xenopus pcdh8.1 EC1–6 (nine TLS groups, 6 reflections per atom); and simpler TLS-only models for pcdh10 EC1–4 dimer form (8 TLS groups, 3 reflections per atom), pcdh17 EC1–4 (16 groups, 1.8 reflections per atom), pcdh19 EC1–4 (12 groups, 2.8 reflections per atom) and pcdh8 EC5–6 (2 groups, 2.9 reflections per atom). TLS groups comprised whole chains or one or more contiguous EC domains, with precise boundaries determined using the TLSMD server (Painter and Merritt, 2006). Refinement statistics are listed in Table S1. For pcdh10 EC1–6, the refinement statistics listed in Table S1 refer to agreement of the model with the ellipsoidally truncated data (20–4.4/4.4/3.3A); corresponding Rwork/Rfree values against spherically truncated data (20–3.3A) were 0.2853/0.3297.
Pymol 1.7.4.4 (DeLano, W.L. Pymol: An open-source molecular graphics tool. (2002)) was used for structure visualization, superposition of structures, and for preparation of images for publication. The PISA program (Krissinel and Henrick, 2007) was used to calculate interfacial buried surface areas.
Sequence analysis
Multiple sequence alignments were prepared using Multalin (Corpet, 1988) and annotated using ESPript (http://espript.ibcp.fr; Robert and Gouet, 2014). Phylograms were prepared using the phylogeny.fr server (Dereeper et al., 2008) based on the multiple sequence alignment of full-length sequences. Protein distances were estimated according to Whelan and Goldman (2001). Sequence Logo representations were generated using WebLogo 3.0 (Crooks et al., 2004) using a default y axis representing information content scaled to a maximum of 4 bits and without using correction for low sequence numbers. Sequences of mouse clustered protocadherins used for preparation of Logo plots corresponded to Uniprot entries: Uniprot: Q91XX8, Q91XX7, Q91XX6, Q91XX5, Q91XX4, Q91XX3, Q91XX2 (pcdh γB1–2, 4–8); Uniprot: Q91Y08, Q91Y00, Q91XZ7, Q91XZ6, Q91XZ5, Q91XZ4, Q91XZ3, Q91XZ2, E9Q5G2, Q91VE5, Q91UZ8, Q91Y07, Q91Y06, Q6PB90, Q91Y04, Q91Y03, Q91VD8, Q91Y02, Q91Y01, Q91XZ9, Q91V48, Q91XZ8 (pcdh β1–22); Uniprot: Q91XZ0, Q91XY6, Q91XY5, Q91XY4, Q91XY3, Q91XY2, Q91XY1, Q91XY0, Q91XX9, Q91XY9, Q91XY8, Q91XY7 (pcdh γA1–12).
Liposome aggregation assay and sample preparation for cryo-ET experiments
Liposomes were prepared using a hydration and extrusion method according to the manufacturers protocol (Avanti Lipids) with a final 8:2 molar ratio of 2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and the nickel salt of 1,2-dioleoyl-sn-glycero-3-([N(5-amino-1-carboxypentyl) iminodiaceticacid]-succinyl) (DOGS-NTA). For fluorescent microscopy experiments, 1% Rhodamine B 1,2-Dihexa-decanoyl-sn-Glycero-3-Phosphoethanolamine (rhodamine-DHPE) lipids (Thermo Fisher) were incorporated into the liposomes in addition to the standard composition of DOPC and DOGS-NTA. For all samples, after evaporation of chloroform under nitrogen, combined lipids were hydrated with assay buffer (100mM Potassium Chloride, 25mM HEPES pH 7.4, 3mM Calcium Chloride, 10% (v/v) glycerol) and large aggregates dispersed by five cycles of flash freezing in liquid nitrogen followed by rapid thaw at 37°C. Resuspended liposomes were extruded using membranes with a pore size of 100nm and stored at 4°C under a layer of nitrogen until use.
Aggregation assays were performed in a total volume of 200μL, with a final concentration of 500μM liposomes and 7μM C-terminal hexa-histidine tagged purified protocadherin ectodomains (pcdh1 EC1–7, pcdh1 EC1–7 L359D, pcdh10 EC1–6ECD, pcdh10 EC1–6 L362D, or mouse pcdh γB6 EC1–6). For ‘uncoated’ controls protein volume was replaced by sizing buffer. After addition of purified ectodomains to liposomes, aggregation assays were incubated for 7 hours. For fluorescent imaging, 5μL of each suspension were transferred to microscopy slides and imaged using a Nikon Eclipse 800 microscope using Qcapture at a magnification of 4x. For electron microscopy, the large aggregates were gently broken up by trituration with a 10 μL pipette tip prior grid preparation for electron microscopy experiments. Home-made, glow-discharged lacey carbon grids were prepared using 300 mesh copper grids (Electron Microscopy Sciences) and 0.025% (w/v) cellulose acetate (Sigma) dissolved in ethyl acetate (EMD Biosciences) and were incubated with 3μl of each suspension of aggregated liposomes for 10 s at 85% relative humidity, blotted for 2.5sec and flash frozen in liquid ethane using a semi-automated approach by utilizing a Gatan CP3 plunge freezer.
Tilt-series data collection and processing
All tilt-series were acquired using a Titan Krios (FEI/Thermo Fisher) at 300 keV outfitted with a direct electron detector Gatan K2 Summit direct electron detector, a post-column Gatan Bioquantum image filter (GIF; Gatan, Inc.) and a spherical aberration corrector (CEOS GmbH) calibrated to have negligible spherical aberration. In addition, most of the tilt-series were collected using a Volta phase plate (FEI/Thermo Fisher). Data was collected sequential with a tilt-range of either −60° to 60° with 2.2° increments or −55° to 55° in 2° increments for protocadherin1 and −10, respectively using the MSI-T2 application in Leginon (Suloway et al., 2005, 2009) with 100 ms frames for each tilt image at a nominal defocus range of 3.5 and 4 microns. Total cumulative dose per tilt-series collected was between 81 and 189 e−/Å2 with dose rates of approximately 8 e−/pixel/sec. Incident dose for the 0° tilt image was between 1.5 and 2.8 e−/Å2, and increased for higher tilt angles according to the cosine of the tilt angle. Tilt-series were collected at pixel sizes of 1.84Å and 2.8Å. Full-frame alignment was performed using MotionCor2 (Zheng et al., 2017) without dose weighting.
All tilt-series were aligned using Appion-Protomo (Lander et al., 2009; Noble and Stagg, 2015; Winkler and Taylor, 2006). After coarse alignment, tilt-series were manually aligned and then refined using a set of alignment thicknesses between 600 and 2,000Å. Refinement and manual alignment were iterated until refinements converged. The best aligned iteration was reconstructed for visual analysis using Tomo3D SIRT (Agulleiro and Fernandez, 2011, 2015) after moderate dose-compensation using the relation described in Grant and Grigorieff (2015). CTF correction was not performed. 3dmod (Kremer et al., 1996) was used to prepare the tomogram slices for Figure 6 and Videos S1, S2, S3, S4, and S5.
Intermembrane distance measurements
Intensity plot profiles were plotted using the analyze plot profile function in Fiji (Schindelin et al., 2012) for representative reconstituted junctions on tomogram slices. Measurements of 16 junctions each for pcdh 1 and pcdh 10 were taken from slices from the tomograms shown in Figure 6 and averaged. Reconstituted junctions were identified as described in the main text.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics of X-ray diffraction datasets and crystal structures are reported in Table S1 and were determined using AIMLESS (Evans and Murshudov, 2013) and Phenix (Adams et al., 2010).
DATA AND CODE AVAILABILITY
The accession numbers for the crystal structure coordinates and structure factors of human pcdh1 EC1–4, pcdh10 EC1–4 monomer, pcdh10 EC1–4 dimer, pcdh17 EC1–4, pcdh18 EC1–4, pcdh19 EC1–4, pcdh8 EC5–6, pcdh10 EC1–6, and Xenopus pcdh 8.1 EC1–6 reported in this paper are PDB: 6VFP, 6VFQ, 6VFW, 6VFT, 6VFR, 6VFU, 6VFV, 6VG4, and 6VG1, respectively. The accession codes for tomograms 1–3 of pcdh1-coated liposomes and tomograms 1–3 of pcdh10-coated liposomes reported in this paper are EMDB: 21188, 21189, 21190, 21191, 21192, 21193.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Non-clustered protocadherin proteins | This paper | N/A |
| Clustered protocadherin proteins | Rubinstein et al., 2015; Goodman et al., 2016a, 2016b; this paper | N/A |
| Polyethylenimine | Polysciences | Cat# 24765–2 |
| Freestyle 293 Expression Media | Thermo Fisher Scientific | Cat# 12338–018 |
| Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | Cat# 31985–070 |
| IMAC Sepharose 6 Fast Flow | GE Healthcare | Cat# 17092109 |
| Biotin | Sigma | Cat# B4501 |
| NeurAvidin-HRP | Thermo Fisher Scientific | Cat# 31030 |
| Tris Base | Fisher Scientific | Cat# BP152–5 |
| Sodium Chloride | Fisher Scientific | Cat# S271–10 |
| Calcium Chloride Dihydrate | JT Baker | Cat# 1336–01 |
| Imidazole | ACROS | Cat# 301870025 |
| NeutrAvidin protein | Thermo Fisher Scientific | Cat# 31000 |
| HEPES | Sigma | Cat# H3375 |
| Tween-20 | Sigma | Cat# P7949 |
| BSA | Sigma | Cat# A7906 |
| Series S CM4 chip | GE Healthcare | Cat# BR100539 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 16141079 |
| DMEM with GlutaMAX | Thermo Fisher Scientific | Cat# 10569010 |
| Penicillin Streptomycin | Thermo Fisher Scientific | Cat# 15070063 |
| Glycerol | ACROS | Cat# 332031000 |
| PEG 4,000 | ACROS | Cat# 434630010 |
| Bicine | Molecular Dimensions | Cat# MD2–003 |
| Morpheus monosaccharides mix | Molecular Dimensions | Cat# MD2-100-75 |
| PEG 8,000 | Sigma | Cat# 89510 |
| Ethylene Glycol | Fluka | Cat# 03760 |
| Morpheus NPS mix | Molecular Dimensions | Cat# MD2-100-72 |
| MES | Sigma | Cat# M3671 |
| PEG 20,000 | Sigma | Cat# 81300 |
| PEG 550 monomethyl ether | Sigma | Cat# 202487 |
| PEG 400 | Sigma | Cat# 91893 |
| Sodium Acetate | Sigma | Cat# S7545 |
| Bicine | Sigma | Cat# B3876 |
| Glutaraldehyde | Sigma | Cat# G5882 |
| (2R,3R)-(–)-2,3-Butanediol | Sigma | Cat# 237639 |
| DOPC lipids | Avanti Polar Lipids | Cat# 850375C |
| DOGS-NTA lipids | Avanti Polar Lipids | Cat# 790404C |
| Rhodamine-DHPE lipids | Thermo Fisher Scientific | Cat# L1392 |
| Potassium chloride | Sigma | Cat# P3911 |
| 300 mesh copper EM grids | Electron Microscopy Services | Cat# EMS300-Cu |
| Cellulose acetate | Sigma | Cat# 180955 |
| Ethyl acetate | EMD | Cat# EX0241–6 |
| Critical Commercial Assays | ||
| Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| Hispeed Plasmid Maxi Kit | QIAGEN | Cat# 12663 |
| SF Cell Line 4D-Nucleofector® X Kit S | Lonza | Cat# V4XC-2032 |
| Deposited Data | ||
| Crystal structure of human pcdh1 EC1–4 | This paper | PDB ID: 6VFP |
| Crystal structure of human pcdh10 EC1–4 monomer | This paper | PDB ID: 6VFQ |
| Crystal structure of human pcdh10 EC1–4 dimer | This paper | PDB ID: 6VFW |
| Crystal structure of human pcdh17 EC1–4 | This paper | PDB ID: 6VFT |
| Crystal structure of human pcdh18 EC1–4 | This paper | PDB ID: 6VFR |
| Crystal structure of human pcdh19 EC1–4 | This paper | PDB ID: 6VFU |
| Crystal structure of human pcdh8 EC5–6 | This paper | PDB ID: 6VFV |
| Crystal structure of Xenopus pcdh8.1 EC1–6 | This paper | PDB ID: 6VG1 |
| Crystal structure of human pcdh10 EC1–6 | This paper | PDB ID: 6VG4 |
| Human δ pcdh1 ectodomains on membranes, tomogram 1 | This paper | EMDB: EMD-21188 |
| Human δ pcdh1 ectodomains on membranes, tomogram 2 | This paper | EMDB: EMD-21189 |
| Human δ pcdh1 ectodomains on membranes, tomogram 3 | This paper | EMDB: EMD-21190 |
| Human δ pcdh10 ectodomains on membranes, tomogram 1 | This paper | EMDB: EMD-21191 |
| Human δ pcdh10 ectodomains on membranes, tomogram 2 | This paper | EMDB: EMD-21192 |
| Human δ pcdh10 ectodomains on membranes, tomogram 3 | This paper | EMDB: EMD-21193 |
| Experimental Models: Cell Lines | ||
| Human: FreeStyle 293-F cells | Thermo Fisher Scientific | Cat# R79007 |
| Human: K-562 bone marrow chronic myelogenous leukemia (CML) cells | ATCC | Cat# ATCC CCL-243 |
| Recombinant DNA | ||
| VRC-8400 vector | Vaccine Research Center (NIH), Gary Nabel | N/A |
| pmax-mCherry vector | Thu et al., 2014 | N/A |
| BirA plasmid | Harrison et al., 2016 | N/A |
| Human pcdh1 cDNA | DNASU | Cat# HsCD00399247 |
| Mouse pcdh7 cDNA | Transomic | Cat# BC131967 |
| Human pcdh9 cDNA | DNASU | Cat# HsCD00516491 |
| Human pcdh11Y cDNA | DNASU | Cat# HsCD00083030 |
| Human pcdh8 cDNA | DNASU | Cat# HsCD00045356 |
| Human pcdh10 cDNA | DNASU | Cat# HsCD00505992 |
| Human pcdh12 cDNA | DNASU | Cat# HsCD00641460 |
| Human pcdh17 cDNA | Dr. Chan Aye Thu | N/A |
| Human pcdh18 cDNA | Dr. Chan Aye Thu | N/A |
| Human pcdh19 cDNA | Dr. Chan Aye Thu | N/A |
| Human pcdh20 cDNA | DNASU | Cat# HsCD00399990 |
| Xenopus pcdh8.1 cDNA | Transomic | Cat# BC074360 |
| Xenopus pcdh8.2 (PAPC) cDNA | DNASU | Cat# XLCD00716163 |
| Software and Algorithms | ||
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de |
| CCP4 | Winn et al., 2011 | http://www.ccp4.ac.uk/ |
| AIMLESS | Evans and Murshudov, 2013 | http://www.ccp4.ac.uk |
| Phenix | Adams et al., 2010 | https://hkl-xray.com/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Staraniso Server | Global Phasing | https://staraniso.globalphasing.org/cgi-bin/staraniso.cgi |
| Pymol | Schrӧdinger | https://pymol.org/2/ |
| PDBePISA | Krissinel and Henrick, 2007 | http://www.ebi.ac.uk/pdbe/pisa/ |
| TLSMD server | Painter and Merritt, 2006 | http://skuld.bmsc.washington.edu/~tlsmd/ |
| Scrubber 2.0 | BioLogic Software | http://www.biologic.com.au |
| SednTerp | Dr. Thomas Laue | http://bitcwiki.sr.unh.edu/index.php/Main_Page |
| HeteroAnalysis | Cole et al., 2008 | https://core.uconn.edu/auf |
| Multalin | Corpet, 1988 | http://multalin.toulouse.inra.fr/multalin/ |
| ESPript | Robert and Gouet, 2014 | http://espript.ibcp.fr |
| WebLogo 3.0 | Crooks et al., 2004 | http://weblogo.threeplusone.com/ |
| Leginon | Suloway et al., 2005, 2009 | https://emg.nysbc.org/redmine/projects/leginon/wiki/Leginon_Homepage |
| MotionCor2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsfmotioncor2 |
| Appion-Protomo | Lander et al., 2009; Noble and Stagg, 2015; Winkler and Taylor, 2006 |
https://emg.nysbc.org/redmine/projects/appion/wiki/Appion_Home; http://www.electrontomography.org/ |
| Tomo3D SIRT | Agulleiro and Fernandez, 2015 | https://sites.google.com/site/3demimageprocessing/tomo3d |
| 3dmod | Kremer et al., 1996 | https://bio3d.colorado.edu/imod/ |
| Fiji | Schindelin et al., 2012 | https://imagej.net/Fiji |
Highlights.
δ-pcdh adhesive interactions are preferentially homophilic with distinct affinities
δ1- and δ2-pcdhs form canonical adhesive dimers with divergent specificity regions
Ectodomain-mediated cis interactions are not observed, in contrast to clustered pcdhs
Assemblies at contact sites differ markedly from those of clustered pcdhs
ACKNOWLEDGMENTS
We thank SEMC OPs for technical support at the microscopes. Clustered protocadherins were provided by Seetha Mannepalli, Fabiana Bahna, and Kerry Goodman. D. Neau, S. Banerjee, I. Kourinov, and S. Narayanasami helped with data collection at the APS NE-CAT 24-ID-C/-E beamlines, supported by the National Institutes of Health (NIH) (grant P41GM103403). We acknowledge support from the National Science Foundation (grant MCB-1914542 to B.H.) and NIH (grants R01MH114817 and R01GM118584 to L.S.; F32GM128303 to A.J.N.; and R01DK106548 to R.V.S.). cryo-ET was performed at the Simons Electron Microscopy Center (SEMS) and the National Resource for Automated Molecular Microscopy at the New York Structural Biology Center, supported by the Simons Foundation (grant SF349247), NYSTAR, and NIH (grant GM103310).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.003.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agulleiro JI, and Fernandez JJ (2011). Fast tomographic reconstruction on multicore computers. Bioinformatics 27, 582–583. [DOI] [PubMed] [Google Scholar]
- Agulleiro J-I, and Fernandez J-J (2015). Tomo3D 2.0—exploitation of advanced vector extensions (AVX) for 3D reconstruction. J. Struct. Biol 189, 147–152. [DOI] [PubMed] [Google Scholar]
- Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, and Nielsen H (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol 37, 420–423. [DOI] [PubMed] [Google Scholar]
- Baldi L, Hacker DL, Meerschman C, and Wurm FM (2012). Large-scale transfection of mammalian cells. Methods in Molecular Biology 801, 13–26. [DOI] [PubMed] [Google Scholar]
- Barat B, and Wu AM (2007). Metabolic biotinylation of recombinant antibody by biotin ligase retained in the endoplasmic reticulum. Biomol. Eng 24, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch DH, Yang ZY, Kong WP, Korioth-Schmitz B, Sumida SM, Truitt DM, Kishko MG, Arthur JC, Miura A, Mascola JR, et al. (2005). A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J. Virol 79, 8828–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Duan X, Taylor MR, Martin EA, Muralidhar S, Wang Y, Gangi-Wellman L, Das SC, Yamagata M, West PJ, et al. (2018). Heterophilic type II cadherins are required for high-magnitude synaptic potentiation in the hippocampus. Neuron 98, 658–668. [DOI] [PubMed] [Google Scholar]
- Bisogni AJ, Ghazanfar S, Williams EO, Marsh HM, Yang JY, and Lin DM (2018). Tuning of delta-protocadherin adhesion through combinatorial diversity. eLife 7, e41050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins CJ, Emond MR, Biswas S, and Jontes JD (2011). Differential expression, alternative splicing, and adhesive properties of the zebrafish δ1-protocadherins. Neuroscience 199, 523–534. [DOI] [PubMed] [Google Scholar]
- Bononi J, Cole A, Tewson P, Schumacher A, and Bradley R (2008). Chicken protocadherin-1 functions to localize neural crest cells to the dorsal root ganglia during PNS formation. Mech. Dev 125, 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch J, Goodman KM, Noble AJ, Rapp M, Mannepalli S, Bahna F, Dandey VP, Bepler T, Berger B, Maniatis T, et al. (2019). Visualization of clustered protocadherin neuronal self-recognition complexes. Nature 569, 280–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch J, Harrison OJ, Honig B, and Shapiro L (2012). Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol. 22, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasch J, Katsamba PS, Harrison OJ, Ahlsén G, Troyanovsky RB, Indra I, Kaczynska A, Kaeser B, Troyanovsky S, Honig B, and Shapiro L (2018). Homophilic and heterophilic interactions of type II cadherins identify specificity groups underlying cell-adhesive behavior. Cell Rep. 23, 1840–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo RA,Özkan E, Menon KP, Nagarkar-Jaiswal S, Lee PT, Jeon M, Birnbaum ME, Bellen HJ, Garcia KC, and Zinn K (2015). Control of synaptic connectivity by a network of Drosophila IgSF cell surface proteins. Cell 163, 1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL, Lary JW, P Moody T, and Laue TM (2008). Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol. 84, 143–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SR, Emond MR, Duy PQ, Liebau BG, Wolman MA, and Jontes JD (2015). Protocadherins control the modular assembly of neuronal columns in the zebrafish optic tectum. J. Cell Biol 211, 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SR, Jontes JD, and Sotomayor M (2016). Structural determinants of adhesion by protocadherin-19 and implications for its role in epilepsy. eLife 5, e18529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet F (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosmanescu F, Katsamba PS, Sergeeva AP, Ahlsen G, Patel SD, Brewer JJ, Tan L, Xu S, Xiao Q, Nagarkar-Jaiswal S, et al. (2018). Neuron-subtype–specific expression, interaction affinities, and specificity determinants of DIP/Dpr cell recognition proteins. Neuron 100, 1385–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzrodt J, Krishna-K K, and Redies C (2009). Expression of classic cadherins and delta-protocadherins in the developing ferret retina. BMC Neurosci. 10, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitan Y, and Bouchard M (2006). Expression of the delta-protocadherin gene Pcdh19 in the developing mouse embryo. Gene Expr. Patterns 6, 893–899. [DOI] [PubMed] [Google Scholar]
- Generous AR, Harrison OJ, Troyanovsky RB, Mateo M, Navaratnarajah CK, Donohue RC, Pfaller CK, Alekhina O, Sergeeva AP, Indra I, et al. (2019). Trans-endocytosis elicited by nectins transfers cytoplasmic cargo, including infectious material, between cells. J. Cell Sci 132, e235507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Rubinstein R, Dan H, Bahna F, Mannepalli S, Ahlsén G, Aye Thu C, Sampogna RV, Maniatis T, Honig B, and Shapiro L (2017). Protocadherin cis-dimer architecture and recognition unit diversity. Proc. Natl. Acad. Sci. USA 114, E9829–E9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Rubinstein R, Thu CA, Bahna F, Mannepalli S, Ahlsén G, Rittenhouse C, Maniatis T, Honig B, and Shapiro L (2016a). Structural basis of diverse homophilic recognition by clustered α- and β-protocadherins. Neuron 90, 709–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman KM, Rubinstein R, Thu CA, Mannepalli S, Bahna F, Ahlsén G, Rittenhouse C, Maniatis T, Honig B, and Shapiro L (2016b). γ-Protocadherin structural diversity and functional implications. eLife 5, e20930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, and Grigorieff N (2015). Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife 4, e06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, and Shapiro L (2016). Structural basis of adhesive binding by desmocollins and desmogleins. Proc. Natl. Acad. Sci. USA 113, 7160–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, et al. (2011). The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19, 244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Inoue Y, Kiyonari H, Abe T, Misaki K, Moriguchi H, Tanaka Y, and Takeichi M (2014). Protocadherin-17 mediates collective axon extension by recruiting actin regulator complexes to interaxonal contacts. Dev. Cell 30, 673–687. [DOI] [PubMed] [Google Scholar]
- Hayashi S, and Takeichi M (2015). Emerging roles of protocadherins: from self-avoidance to enhancement of motility. J. Cell Sci 128, 1455–1464. [DOI] [PubMed] [Google Scholar]
- Hoshina N, Tanimura A, Yamasaki M, Inoue T, Fukabori R, Kuroda T, Yokoyama K, Tezuka T, Sagara H, Hirano S, et al. (2013). Protocadherin 17 regulates presynaptic assembly in topographic corticobasal ganglia circuits. Neuron 78, 839–854. [DOI] [PubMed] [Google Scholar]
- Hulpiau P, and van Roy F (2009). Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol 41, 349–369. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahr I, Vandepoele K, and van Roy F (2013). delta-Protocadherins in health and disease. Prog. Mol. Biol. Transl. Sci 116, 169–192. [DOI] [PubMed] [Google Scholar]
- Katsamba P, Carroll K, Ahlsen G, Bahna F, Vendome J, Posy S, Rajebhosale M, Price S, Jessell TM, Ben-Shaul A, et al. (2009). Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc. Natl. Acad. Sci. USA 106, 11594–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Chung HS, Sun W, and Kim H (2007). Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 147, 996–1021. [DOI] [PubMed] [Google Scholar]
- Kim SY, Yasuda S, Tanaka H, Yamagata K, and Kim H (2011). Non-clustered protocadherin. Cell Adhes. Migr 5, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostadinov D, and Sanes JR (2015). Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit function. eLife 4, e08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Labernadie A, Kato T, Brugués A, Serra-Picamal X, Derzsi S, Arwert E, Weston A, González-Tarragó V, Elosegui-Artola A, Albertazzi L, et al. (2017). A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol 19, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau P-W, et al. (2009). Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol 166, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen ISB, Narimatsu Y, Joshi HJ, Siukstaite L, Harrison OJ, Brasch J, Goodman KM, Hansen L, Shapiro L, Honig B, et al. (2017). Discovery of an O-mannosylation pathway selectively serving cadherins and protocadherins. Proc. Natl. Acad. Sci. USA 114, 11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Kostadinov D, Chen WV, Maniatis T, and Sanes JR (2012). Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung LC, Urbančič V, Baudet ML, Dwivedy A, Bayley TG, Lee AC, Harris WA, and Holt CE (2013). Coupling of NF-protocadherin signaling to axon guidance by cue-induced translation. Nat. Neurosci 16, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light SEW, and Jontes JD (2017). δ-protocadherins: organizers of neural circuit assembly. Semin. Cell Dev. Biol 69, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wang C, and Redies C (2012). Expression of delta-protocadherins in the spinal cord of the chicken embryo. J. Comp. Neurol 520, 1509–1531. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng W, and Takeichi M (2009). Adherens junction: molecular architecture and regulation. Cold Spring Harb. Perspect. Biol 1, a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modak D, and Sotomayor M (2019). Identification of an adhesive interface for the non-clustered delta1 protocadherin-1 involved in respiratory diseases. Commun. Biol 2, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molumby MJ, Anderson RM, Newbold DJ, Koblesky NK, Garrett AM, Schreiner D, Radley JJ, and Weiner JA (2017). γ-Protocadherins interact with neuroligin-1 and negatively regulate dendritic spine morphogenesis. Cell Rep. 18, 2702–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molumby MJ, Keeler AB, and Weiner JA (2016). Homophilic protocadherin cell–cell interactions promote dendrite complexity. Cell Rep. 15, 1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, and Yagi T (2007). Protocadherin family: diversity, structure, and function. Curr. Opin. Cell Biol 19, 584–592. [DOI] [PubMed] [Google Scholar]
- Mountoufaris G, Canzio D, Nwakeze CL, Chen WV, and Maniatis T (2018). Writing, reading, and translating the clustered protocadherin cell surface recognition code for neural circuit assembly. Annu. Rev. Cell Dev. Biol 34, 471–493. [DOI] [PubMed] [Google Scholar]
- Mountoufaris G, Chen WV, Hirabayashi Y, O’Keeffe S, Chevee M, Nwakeze CL, Polleux F, and Maniatis T (2017). Multicluster Pcdh diversity is required for mouse olfactory neural circuit assembly. Science 356, 411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoludis JM, Lau S-Y, Schärfe CP, Marks DS, Weihofen WA, and Gaudet R (2015). Structure and sequence analyses of clustered protocadherins reveal antiparallel interactions that mediate homophilic specificity. Structure 23, 2087–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoludis JM, Vogt BE, Green AG, Schärfe CP, Marks DS, and Gaudet R (2016). Antiparallel protocadherin homodimers use distinct affinity- and specificity-mediating regions in cadherin repeats 1–4. eLife 5, e18449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble AJ, and Stagg SM (2015). Automated batch fiducial-less tilt-series alignment in Appion using Protomo. J. Struct. Biol 192, 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter J, and Merritt EA (2006). Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr 62, 439–450. [DOI] [PubMed] [Google Scholar]
- Patel SD, Ciatto C, Chen CP, Bahna F, Rajebhosale M, Arkus N, Schieren I, Jessell TM, Honig B, Price SR, and Shapiro L (2006). Type II cadherin ectodomain structures: implications for classical cadherin specificity. Cell 124, 1255–1268. [DOI] [PubMed] [Google Scholar]
- Pederick DT, Richards KL, Piltz SG, Kumar R, Mincheva-Tasheva S, Mandelstam SA, Dale RC, Scheffer IE, Gecz J, Petrou S, et al. (2018). Abnormal cell sorting underlies the unique X-linked inheritance of PCDH19 epilepsy. Neuron 97, 59–66 e55. [DOI] [PubMed] [Google Scholar]
- Redies C, Vanhalst K, and Roy Fv. (2005). delta-Protocadherins: unique structures and functions. Cell. Mol. Life Sci 62, 2840–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich RL, and Myszka DG (2007). Survey of the year 2006 commercial optical biosensor literature. J. Mol. Recognit 20, 300–366. [DOI] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy P, and Bandyopadhyay A (2014). Spatio-temporally restricted expression of cell adhesion molecules during chicken embryonic development. PLoS ONE 9, e96837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein R, Goodman KM, Maniatis T, Shapiro L, and Honig B (2017). Structural origins of clustered protocadherin-mediated neuronal barcoding. Semin. Cell Dev. Biol 69, 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein R, Thu CA, Goodman KM, Wolcott HN, Bahna F, Mannepalli S, Ahlsen G, Chevee M, Halim A, Clausen H, et al. (2015). Molecular logic of neuronal self-recognition through protocadherin domain interactions. Cell 163, 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Suloway C, Shi J, Cheng A, Pulokas J, Carragher B, Potter CS, Zheng SQ, Agard DA, and Jensen GJ (2009). Fully automated, sequential tilt-series acquisition with Leginon. J. Struct. Biol 167, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai K, Kubota M, Shiono K, Tokutsu H, and Suzuki ST (2010). Adhesion properties and retinofugal expression of chicken protocadherin-19. Brain Res. 1344, 13–24. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, and Adams PD (2008). Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr 64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu CA, Chen WV, Rubinstein R, Chevee M, Wolcott HN, Felsovalyi KO, Tapia JC, Shapiro L, Honig B, and Maniatis T (2014). Single-cell identity generated by combinatorial homophilic interactions between a, b, and g protocadherins. Cell 158, 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Kominami K, Waseda M, Komura H, Miyoshi J, Takeichi M, and Takai Y (2011). Nectins establish a checkerboard-like cellular pattern in the auditory epithelium. Science 333, 1144–1147. [DOI] [PubMed] [Google Scholar]
- Uemura M, Nakao S, Suzuki ST, Takeichi M, and Hirano S (2007). OL-Protocadherin is essential for growth of striatal axons and thalamocortical projections. Nat. Neurosci 10, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Vanhalst K, Kools P, Staes K, van Roy F, and Redies C (2005). delta-Protocadherins: a gene family expressed differentially in the mouse brain. Cell. Mol. Life Sci 62, 1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendome J, Felsovalyi K, Song H, Yang Z, Jin X, Brasch J, Harrison OJ, Ahlsen G, Bahna F, Kaczynska A, et al. (2014). Structural and energetic determinants of adhesive binding specificity in type I cadherins. Proc. Natl. Acad. Sci. USA 111, E4175–E4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk T, Cohen O, and Geiger B (1987). Formation of heterotypic adherenstype junctions between L-CAM-containing liver cells and A-CAM–containing lens cells. Cell 50, 987–994. [DOI] [PubMed] [Google Scholar]
- Whelan S, and Goldman N (2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol 18, 691–699. [DOI] [PubMed] [Google Scholar]
- Winkler H, and Taylor KA (2006). Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy 106, 240–254. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Over-view of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Xiao Q, Cosmanescu F, Sergeeva AP, Yoo J, Lin Y, Katsamba PS, Ahlsen G, Kaufman J, Linaval NT, et al. (2018). Interactions between the Ig-superfamily proteins DIP-a and Dpr6/10 regulate assembly of neural circuits. Neuron 100, 1369–1384, e1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Tanaka H, Sugiura H, Okamura K, Sakaguchi T, Tran U, Takemiya T, Mizoguchi A, Yagita Y, Sakurai T, et al. (2007). Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2beta and p38 MAP kinases. Neuron 56, 456–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Lv J, Yang Z, Guo L, Zhang L, Li M, Han W, Chen X, Zhuang H, and Lu F (2014). Protocadherin 9 inhibits epithelial-mesenchymal transition and cell migration through activating GSK-3b in hepatocellular carcinoma. Biochem. Biophys. Res. Commun 452, 567–574. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, and Grueber WB (2013). The molecular basis of self-avoidance. Annu. Rev. Neurosci 36, 547–568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the crystal structure coordinates and structure factors of human pcdh1 EC1–4, pcdh10 EC1–4 monomer, pcdh10 EC1–4 dimer, pcdh17 EC1–4, pcdh18 EC1–4, pcdh19 EC1–4, pcdh8 EC5–6, pcdh10 EC1–6, and Xenopus pcdh 8.1 EC1–6 reported in this paper are PDB: 6VFP, 6VFQ, 6VFW, 6VFT, 6VFR, 6VFU, 6VFV, 6VG4, and 6VG1, respectively. The accession codes for tomograms 1–3 of pcdh1-coated liposomes and tomograms 1–3 of pcdh10-coated liposomes reported in this paper are EMDB: 21188, 21189, 21190, 21191, 21192, 21193.