

SUMMARY
The immune composition of the tumor microenvironment influences response and resistance to immuno-therapies. While numerous studies have identified somatic correlates of immune infiltration, germline features that associate with immune infiltrates in cancers remain incompletely characterized. We analyze seven million autosomal germline variants in the TCGA cohort and test for association with established immune-related phenotypes that describe the tumor immune microenvironment. We identify one SNP associated with the amount of infiltrating follicular helper T cells; 23 candidate genes, some of which are involved in cytokine-mediated signaling and others containing cancer-risk SNPs; and networks with genes that are part of the DNA repair and transcription elongation pathways. In addition, we find a positive association between polygenic risk for rheumatoid arthritis and amount of infiltrating CD8+ T cells. Overall, we identify multiple germline genetic features associated with tumor-immune phenotypes and develop a framework for probing inherited features that contribute to differences in immune infiltration.
Graphical Abstract
In Brief
The role of inherited variants in influencing the immune composition of the tumor microenvironment is not fully characterized. Shahamatdar et al. identify germline variants, genes, and pathways associated with immune infiltration phenotypes in cancer, which may offer insights into determinants of response to immunotherapy.
INTRODUCTION
Immune checkpoint blockade (ICB) therapies have emerged as impactful treatments for a variety of cancers. The discovery of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1) as important modulators of the adaptive immune system (Tivol et al., 1995; Fife et al., 2009) led to the development of ICB therapies, which target these specific pathways. Antagonism of PD-1 and CTLA-4, negative regulators of T cell activity, stimulates the host immune system to recognize and kill tumor cells. While these therapeutic strategies are effective in a wide variety of cancers, they elicit variable clinical response (Ribas and Wolchok, 2018; Keenan et al., 2019).
Tumor-intrinsic features correlated with ICB clinical activity, such as mutational load and microsatellite instability, have been characterized extensively (Snyder et al., 2014; Gentles et al., 2015; Rizvi et al., 2015; Rooney et al., 2015; Van Allen et al., 2015; Giannakis et al., 2016; Miao and Van Allen, 2016; Charoentong et al., 2017; Miao et al., 2018; Samstein et al., 2019). Numerous lines of evidence indicate that selective response to ICB is also driven by the composition of the tumor microenvironment (TME), particularly the immune infiltration patterns in the TME (Tumeh et al., 2014; Thorsson et al., 2018). A study by Thorsson et al. (2018) analyzed the immunogenomic landscape of over 10,000 tumor samples compiled by The Cancer Genome Atlas (TCGA), reported specific driver mutations correlated with tumor infiltrating leukocyte levels, and demonstrated the prognostic and therapeutic implications associated with the TME composition.
Germline determinants of immune infiltration in solid tumors remain incompletely characterized, although germline features have been found to be associated with immune traits such as anti-tumor response, autoimmune diseases, and baseline white blood cell indices in healthy patients (Orrù et al., 2013; Parkes et al., 2013; Roederer et al., 2015; Astle et al., 2016; Marty et al., 2017; Marty Pyke et al., 2018). Marty et al. (2017) and Marty Pyke et al. (2018) identified germline alleles that affect the anti-tumor immune response and shape the oncogenic mutational landscape of tumors, but the studies focused only on the major histocompatibility complex (MHC). Genome-wide association studies (GWASs) have identified hundreds of germline variants associated with immune-mediated diseases (Parkes et al., 2013). And finally, Astle et al. (2016) found that common autosomal genotypes explain up to 21% of variance in white blood cell indices in a GWAS of 170,000 participants. Despite evidence that germline variants influence the immune system and its response to pathogens and tumors, there is a lack of genome-wide studies that investigate the effects of germline features on shaping the immune composition of the TME.
Recently, Lim et al. (2018) uncovered 103 germline single-nucleotide polymorphisms (SNPs) associated with immune cell abundance in the TME. However, the study overlooked potential confounding effects due to population structure and did not offer insight into how individual variants interact through genes or pathways to affect immune infiltration patterns.
Here, we analyze germline variants and test for association with immune infiltration in solid tumors in a pan-cancer meta-analysis of 30 TCGA cancer cohorts across different genomic scales. We identify SNPs, genes, and networks correlated with immune infiltration patterns, as well as an association between polygenic risk for autoimmune diseases and immune infiltration.
RESULTS
Overview of Association Analyses
In order to characterize how host genetics affect immune infiltration in solid tumors, we analyzed the association between germline variants and 17 phenotypes describing the immune component of the TME across 30 TCGA cohorts (Figure 1A). The genotype data consist of 5,788 samples of European genetic ancestry and 7,070,031 imputed variants. Table S1 describes the 17 molecular phenotypes and sample size per phenotype.
Figure 1. Association Study Approach and GWAS Results.
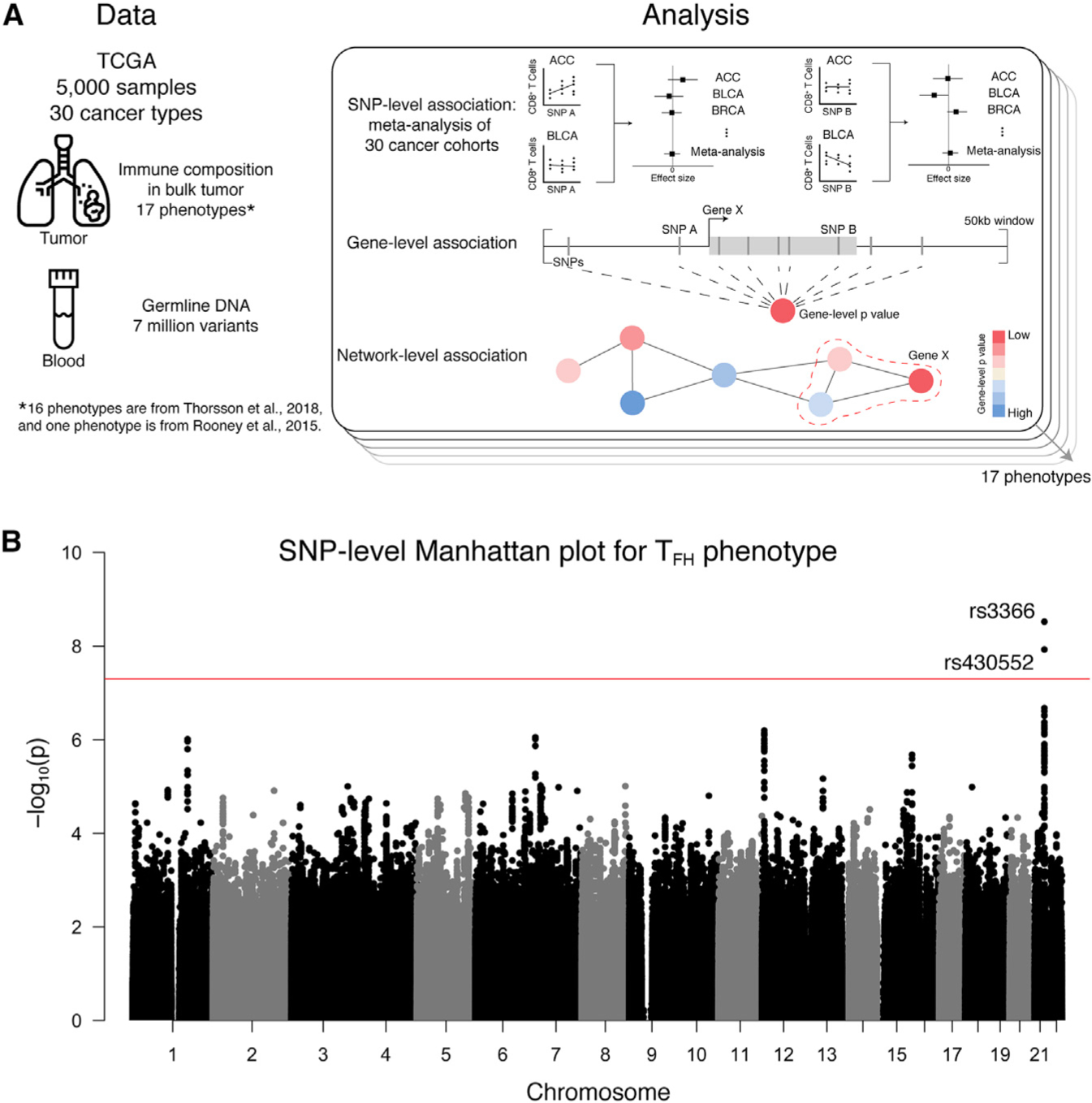
(A) Schematic showing the type and size of dataset for association studies. Association studies are conducted at three genomic scales across all 17 phenotypes. (B) Manhattan plot for GWAS meta-analysis for the TFH cell phenotype. Positions along the chromosomes are on the x axis, and −log10-transformed p values are on the y axis. Every autosome is represented, but some are unlabeled for visualization purposes. The red line indicates genome-wide significance (p < 5 × 10−8). See also Figure S1.
We conducted GWASs of the 17 phenotypes and aggregated SNP-level signals across genes and pathways with gene-level and network-level tests of association. In addition, we asked whether polygenic risks of autoimmune diseases are associated with immune infiltration measures.
SNP-Level Association with Follicular Helper T Cell Phenotype
GWASs conducted on 17 immune infiltration phenotypes reveal two independent associations at genome-wide significance (p <5 3 10−8). rs3366, a variant in the 3′ untranslated region (UTR) of SIK1 (effect size = 0.155, p = 2.99 3 10−9), is associated with the amount of follicular helper T (TFH) cells in bulk tumor (Figure 1B). This SNP currently has no published associations in the GWAS catalog (Buniello et al., 2019). Although the biological role of SIK1 in TFH cells is unknown, there is evidence of differential expression of SIK1 in this cell type (Newman et al., 2015).
rs4819959 is associated with the T helper 17 (Th17) cell signature (effect size = −0.168, p = 2.52 × 10−16). The Th17 cell signature phenotype is defined by the expression of three genes, including IL17RA. The significant SNP is a known expression quantitative trait loci (eQTL) of IL17RA in 31 tissues according to the Genotype-Tissue Expression (GTEx) database (Carithers et al., 2015), meaning the observed association is likely a byproduct of the phenotype definition.
Gene-Level Association Studies Reveal 23 Candidate Genes
We then performed gene-level tests of association with immune infiltration phenotypes using PEGASUS (Nakka et al., 2016). We report gene-level associations at p < 2.8 3 10−6, after Bonferroni correction for 17,563 autosomal genes. These genes are referred to as candidate genes. Because of the small size of the dataset and overlap between genes, we also report suggestive associations at p < 2.9 × 10−5, after Bonferroni correction for 1,703 independent haplotype blocks in the autosomes, consistent with Wojcik et al. (2015) and Gorlova et al. (2018), as defined by Berisa and Pickrell (2016).
We found 24 candidate gene-phenotype relationships, composed of 23 unique genes across 16 phenotypes. There are an additional 54 unique suggestive genes. The results are summarized in Figure 2A; full annotated results can be found in Table S2. We annotated the genes based on (1) gene expression in TCGA bulk tumor and reference immune cell populations (Schmiedel et al., 2018); (2) previously published GWAS hits in the GWAS catalog (Buniello et al., 2019), focusing on traits related to cancer, immunity, or autoimmunity; (3) evidence for promoting oncogenic transformation (Futreal et al., 2004); and (4) correlation between gene expression and tumor purity. The results are summarized in Figure 2A; full results can be found in Table S2.
Figure 2. Summary of Gene-Level Association Results.
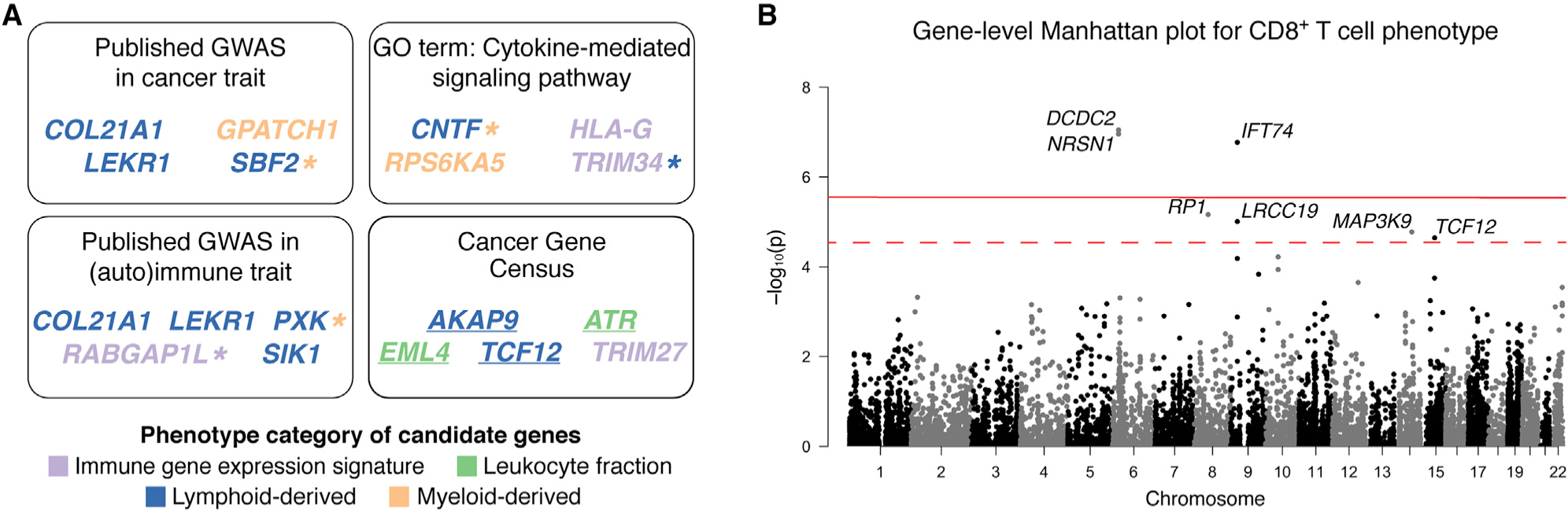
(A) Gene-level association testing identified 23 unique candidate genes. Four candidate genes contained published GWAS SNPs related to cancer traits; five candidate genes contained published GWAS SNPs related to immunity or autoimmune traits. Out of the genes with no previously known associations, the Gene Ontology (GO) term with the most members is shown. Suggestive and candidate genes annotated as casually implicated in cancer by the Cancer Gene Census are also shown. Genes are colored according to the phenotype category for which they are most significant. Genes associated with multiple phenotypes, including suggestive associations, are denoted with a colored asterisk. Genes with only suggestive associations are underlined. See also Table S2. (B) Manhattan plot for gene-level association analysis for the CD8+ T cell phenotype. Each point represents a gene. Positions along the chromosomes are on the x axis, and −log10-transformed p values are on the y axis. The solid red line indicates gene-level significance (p < 2.8 × 10−6), and the dashed red line indicates suggestive significance (p < 2.9 × 10−5).
All 23 candidate genes were expressed in either bulk tumor or reference immune cell populations. In addition, the expression of these genes was either not correlated or only weakly correlated with tumor purity; the correlation coefficients ranged from −0.22 to 0.21. One of the candidate genes, TRIM34, had a negative correlation coefficient that was more than two standard deviations away from the mean correlation coefficients for all genes.
We observed seven genes that contain reported GWAS hits in a related trait according to the GWAS catalog (Buniello et al., 2019). Four of the seven genes (COL21A1, GPATCH1, LEKR1, and SBF2) contain SNPs associated with different cancers, such as small cell lung carcinoma and breast carcinoma (McKay et al., 2017; Wang et al., 2017; Michailidou et al., 2017; Wu et al., 2014; Law et al., 2019). Five of the seven genes (COL21A1, LEKR1, PXK, RABGAP1L, and SIK1) contain SNPs associated with immune or autoimmune traits, such as allergies and systemic lupus erythematosus (Bønnelykke et al., 2013; Ahola-Olli et al., 2017; Alarcón-Riquelme et al., 2016; Kichaev et al., 2019; Ferreira et al., 2017). We refer to genes with no published GWAS hits in traits related to cancer, immunity, or autoimmunity as novel genes. Of the 16 novel candidate genes, the Gene Ontology (GO) term with the most members is the cytokine-mediated signaling pathway. Lastly, four suggestive genes and one candidate gene are annotated by the Cancer Gene Census as casually implicated in cancer (Figure 2A).
We found evidence of genes associated with multiple phenotypes. For example, ZFP91 is associated with the Th17 cell phenotype at gene-level significance and associated with the lymphocytes and macrophages phenotypes at a suggestive level. This gene activates the nuclear factor κB (NF-κB) pathway by stabilizing the NF-κB-inducing kinase, a regulator of the immune system (Jin et al., 2010).
In addition, we identified three candidate genes and four suggestive genes associated with the CD8+ T cell phenotype, an established effector cell in the anti-tumor activity of the immune system (Figure 2B). TCF12 is one of the suggestive genes associated with the CD8+ T cell phenotype. It codes for a transcription factor called HeLa E-box binding protein (HEB), which regulates lineage-specific transcriptional profiles of CD4+CD8+ thymocytes (Emmanuel et al., 2018). The relevance of the other associated genes is not as immediately clear. Two genes, LRRC19 (suggestive association) and IFT74, are related to genes that are involved in the innate immune system (Ng et al., 2011) and recycling of T cell antigen receptors (Finetti et al., 2009), respectively. DCDC2 is aberrantly expressed in prostate tumors (Longoni et al., 2013), and gain-of-function mutations in MAP3K9 (suggestive association) in lung cancer may activate the extracellular signal-regulated kinase (ERK) pathway (Fawdar et al., 2013).
Genes in DNA Repair and Transcription Elongation Pathways Correlated with Leukocyte Fraction
We conducted network propagation analyses (Reyna et al., 2018) to identify gene subnetworks enriched for genes with low gene-level p values whose protein products are topologically connected on a protein-protein interaction network. We found statistically significant subnetworks for the leukocyte fraction phenotype (p < 10−3) with the ReactomeFI 2016 interaction network; two of these subnetworks are highlighted in Figure 3.
Figure 3. Altered Subnetworks in Leukocyte Fraction Phenotype.
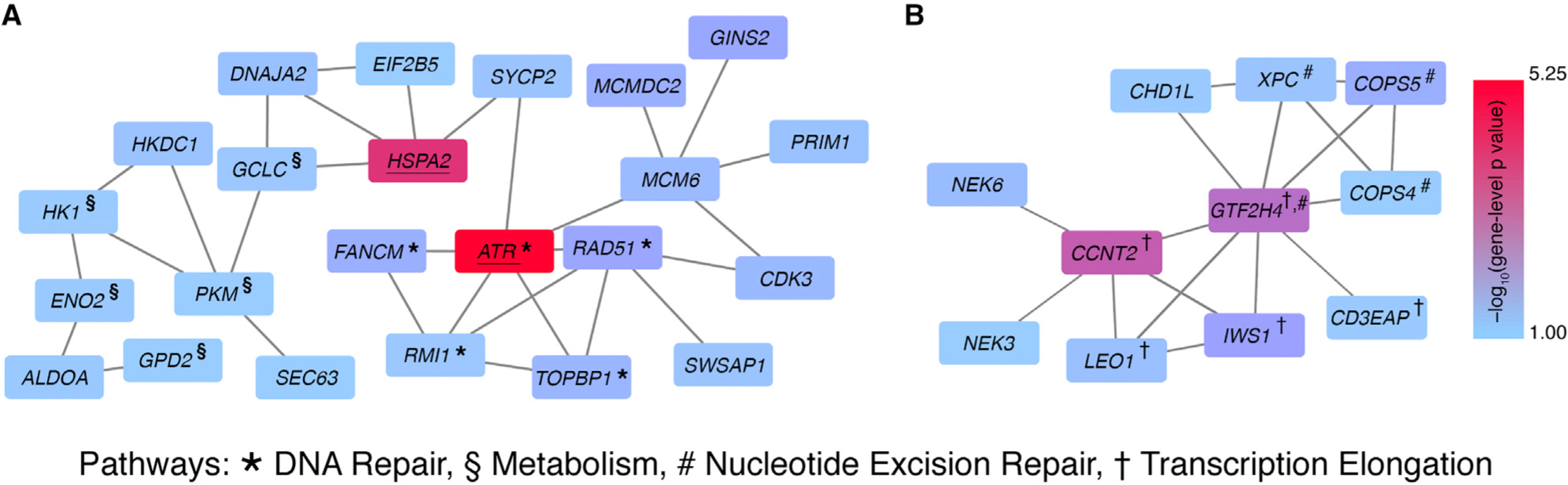
Two statistically significant (p < 0.05) altered subnetworks associated with the leukocyte fraction phenotype in the ReactomeFI 2016 interaction network. Each rectangle represents a gene and is colored according to the gene-level p value. Two genes are connected if their protein products interact in the ReactomeFI 2016 interaction network. Underlined genes are suggestive genes from gene-level analysis.
(A) Two suggestive genes, ATR and HSPA2, are part of a larger subnetwork involved in DNA repair. Genes involved in DNA repair or metabolism are indicated by * and §, respectively.
(B) A subnetwork containing important members of the nucleotide excision repair and transcription elongation pathway, indicated by # and †, respectively.
The second largest connected subnetwork includes two suggestive genes, ATR and HSPA2 (p < 2.81 3 10−5). ATR has been previously implicated in cancer pathogenesis (Futreal et al., 2004). In addition, reported germline ATR variants predispose an individual to cancer (Tanaka et al., 2012). ATR and HSPA2 are connected via SYCP2. Although not significant in our gene-level analysis, somatic mutations in SYCP2 were previously reported to lower regulatory T cell to CD8+ T cell ratios in head and neck cancers (Siemers et al., 2017). Other biologically relevant genes in this subnetwork include FANCM, RAD51, PRIM1, and TOPBP1, which participate in DNA repair pathways.
Components of the subnetwork shown in Figure 3B are involved in the transcription elongation pathway (CCNT2, CD3EAP, GTF2H4, IWS1, and LEO1) and nucleotide excision repair pathway (COPS4, COPS5, GTF2H4, and XPC). None of the genes in this subnetwork had significant gene-level p values, although they are part of a significant subnetwork in the network analysis.
Autoimmune Disease Polygenic Risk Associated with Immune Infiltration Patterns
We investigated if common variants that affect the risk for autoimmune diseases are correlated with immune infiltration (Figure 4A). We calculated polygenic risk scores (PRSs) for five autoimmune disorders: rheumatoid arthritis, inflammatory bowel disease, celiac disease, systemic lupus erythematosus, and multiple sclerosis. These diseases were chosen based on availability of summary statistics in large, well-powered published GWASs (Dubois et al., 2010; Sawcer et al., 2011; Anderson et al., 2011; Okada et al., 2014; Bentham et al., 2015).
Figure 4. PRS Associations with Immune Infiltration.
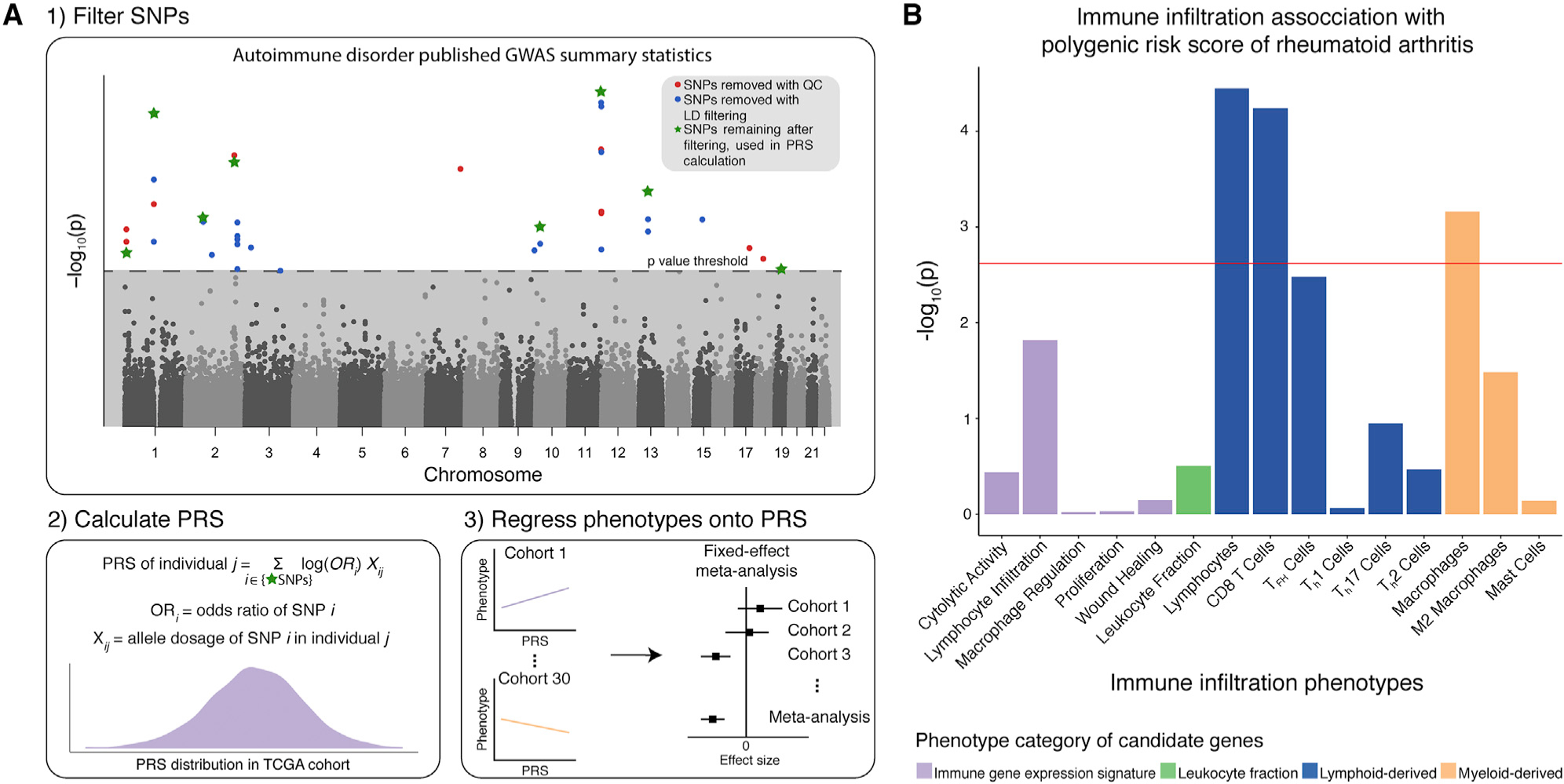
(A) Workflow for calculating polygenic risk scores (PRSs) of autoimmune disorders based on published GWAS summary statistics, followed by regression of the 17 immune infiltration phenotypes onto PRS.
(B) Bar plot showing the strength of association between the phenotypes and PRS for rheumatoid arthritis. The phenotypes are on the x axis, and −log10-transformed p values are on the y axis. Each bar is colored according to the phenotype category. The red line indicates the Bonferroni-corrected significance value (p < 0.0029).
We identified statistically significant associations (p < 0.0029, Bonferroni corrected for 17 immune infiltration phenotypes) between PRS for rheumatoid arthritis and phenotypes: lymphocytes, CD8+ T cells, and macrophages (Figure 4B). The effect sizes are as follows: CD8+ T cells = 0.0088, lymphocytes = 0.0091, and macrophages = −0.0073. It is important to note that the lymphocytes phenotype is defined as the sum of 12 cell types, one of which is amount of CD8+ T cells. To test whether the lymphocyte and CD8+ T cell hits were independent, we subtracted the amount of CD8+ T cells from lymphocytes and repeated the analysis. We no longer observed a significant association between this phenotype and PRS of rheumatoid arthritis (p = 0.0092), demonstrating that the association signal of the lymphocytes phenotype is driven by the CD8+ T cells phenotype.
DISCUSSION
The abundance and composition of immune cell populations in the TME are known to affect response to ICB therapies. Here, we presented a pan-cancer germline analysis of immune infiltration in solid tumors, demonstrating that host genetics are associated with phenotypes describing the immune component of the TME. Through integrative analysis of germline genotype, tumor RNA sequencing (RNA-seq), and tumor DNA methylation data, we identified features at multiple genomic scales (SNP-level, gene-level, and pathway-level) that are correlated with the amount of infiltrating TFH cells and fraction of leukocytes in bulk tumor, among other phenotypes. The 17 immune phenotypes were chosen to capture different facets of the TME, from abundance of particular types of immune cells to gene expression signatures that describe interferon-γ signaling. The association studies described here are sensitive to the precise phenotype definitions.
In our analyses, we found evidence for only one SNP-level association. The sparsity of results from our GWAS analysis is not surprising, as the GWAS framework is underpowered to detect SNP-level associations in complex traits (McClellan and King, 2010; Stranger et al., 2011). The GWAS framework does not account for the genetic heterogeneity often seen in complex traits (McClellan and King, 2010). In addition, we do not have adequate power to detect variants of small effect size because of the small size of our dataset. Gene-level and network-level tests of association overcome these limitations by reducing the multiple hypothesis burden and aggregating SNP-level signals across biologically functional units (Neale and Sham, 2004; Liu et al., 2010; Wu et al., 2010; Nakka et al., 2016; Wang et al., 2010; Reyna et al., 2018).
By combining SNP-level signals and testing for phenotype associations at the gene and pathway levels, we uncovered multiple genes and pathways that are associated with immune infiltration patterns. Out of 23 unique candidate genes, five were previously identified in GWASs on autoimmune disorders or immune-related traits; these results suggest host genomic factors that cause variation or disease in the immune system may also affect immune infiltration of tumors. We found an additional four candidate genes containing SNPs significant in cancer GWASs; these genes may be affecting cancer risk by altering the anti-tumor immune response. There is already evidence for this relationship from GWASs of cancer predisposition, in which cancer-risk SNPs are found to be involved in the immune system (Clifford et al., 2010; Shiels et al., 2012; Peltekova et al., 2014).
We also identified several subnetworks associated with the leukocyte fraction. ATR, a suggestive association from gene-level analysis, and interacting genes were among one of the subnetworks. Germline and somatic mutations in ATR have been reported to play a role in tumorigenesis (Tanaka et al., 2012; Forbes et al., 2017). Somatic ATR mutations have also been shown to modulate the TME in melanomas, recruiting macrophages and blocking T cell recruitment (Chen et al., 2017).
Other significant subnetworks contain genes involved in DNA repair and transcription elongation pathways. Somatic mutations in genes involved in DNA repair can increase the neoantigen load in the TME and affect the response to ICB (Mouw et al., 2017; Knijnenburg et al., 2018). In addition, defective transcription elongation is known to confer resistance to immunotherapy despite increased levels of infiltrating T cells (Modur et al., 2018). We note that these significantly altered subnetworks were found using the ReactomeFI interaction network, and the results using other tested interaction networks were not statistically significant. These results are likely due to differences in network topology, with ReactomeFI being the densest out of the three interaction networks used.
Finally, we showed that the PRS for rheumatoid arthritis is correlated with amount of CD8+ T cells, which may suggest a shared genetic etiology between rheumatoid arthritis and cytotoxic immune response to solid tumors. In the synovial compartment of rheumatic joints, 40% of T cells are CD8+ T cells (McInnes, 2003). Past studies have found associations between rheumatoid arthritis and MHC class I polymorphisms (Raychaudhuri et al., 2012) as well as between amount of CD8+ T cells in synovial fluid and disease activity (Cho et al., 2012), suggesting a potential role for CD8+ T cells in the development and progression of rheumatoid arthritis.
While we applied many quality-control filters to the genotype and phenotype data to remove confounders in our analyses, replication is necessary. However, replication studies are currently not feasible due to a lack of a large, independent, pan-cancer cohort with matched germline and RNA-seq data. The TCGA dataset provided a unique opportunity to conduct integrative association analyses that leverage germline data. The TCGA germline data have been largely underappreciated, besides investigation of predisposition germline variants in cancer (Kim et al., 2013; Palles et al., 2013; Huang et al., 2018). Future studies with larger, integrative datasets are needed to increase statistical power and take advantage of other existing tools to conduct multi-trait GWAS analyses and heritability estimates.
We note that the studied phenotypes were calculated based on sections of tumor tissue at one point in time and therefore do not capture the whole extent of the heterogeneity of the TME. In addition, 16 out of 17 phenotypes were based on bulk RNA-seq data, and 6 of those 16 were derived using a deconvolution method CIBERSORT (Newman et al., 2015). CIBERSORT has several limitations, including reliance on the fidelity of a reference expression panel for deconvolution (Newman et al., 2015). More generally, bulk RNA deconvolution methods have limits to interpretation, as they cannot be used to tease apart the source of gene expression (i.e., if candidate gene is expressed by tumor cell or immune cells). Ideally, future studies will integrate germline and somatic variation with orthogonal measures of immune infiltration patterns (such as single-cell RNA-seq profiling) at different time points, but such study design does not currently exist to validate the reported results.
Follow-up studies incorporating other immune cell populations known to affect response to immunotherapy (such as fraction of infiltrating neutrophils or CD4+ T cells) and joint analysis of germline variants, somatic mutations, and environmental factors will further our understanding of predictors of response to ICB therapies. Ultimately, experimental investigations are also needed to determine the biological mechanisms driving the reported associations.
In conclusion, we report germline variation in SNPs, genes, and pathways associated with immune infiltration patterns. These results highlight the important yet previously overlooked role that inherited variants play in influencing the immune composition of the TME, a crucial step toward understanding predictors of response to ICB therapies.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Sohini Ramachandran (sramachandran@brown.edu). This study did not generate new unique reagents.
METHOD DETAILS
Subject Details
The Cancer Genome Atlas (TCGA) dataset consists of tumor and matched normal samples from over 11,000 patients. The Genomic Data Commons (GDC) legacy archive contains germline data for 11,440 samples from 10,776 unique participants. Samples with the following TCGA project IDs: DLBC, LAML, LCML, MISC, and THYM were excluded as they represent unidentified cancer or cancers derived from immune cells. Samples indicated as problematic by either GDC-issued or TCGA-issued annotations were removed. The reasons for exclusion ranged from mismatched genotypes in tumor and normal samples to incorrect barcodes on aliquots.
Raw Germline Variant Data
Germline variants were derived from the Affymetrix SNP6.0 microarray. Raw CEL files for the TCGA cohort were downloaded from FireCloud (https://software.broadinstitute.org/firecloud/) and the GDC legacy archive (https://portal.gdc.cancer.gov/legacy-archive). Probesets with non-unique mapping in the genome or not mapping to the location provided by Affymetrix (NetAffx Annotation Release 35) were removed.
Germline Variant Calling
Genotype calls from the CEL files were made using Birdseed (Korn et al., 2008) in batches; samples from the same TCGA batch were included in the same run. Because Birdseed recommends more than 50 samples in each run, batches with less than 50 samples were combined with samples from temporally adjacent batches. Genotype calls with Birdseed confidence scores more than 0.1 were removed.
Samples with autosomal SNP missingness > 2% or unexpected sex chromosome genotypes (males with missing Y chromosome calls or females with Y chromosome calls) were removed. Participants with more than two replicate samples were removed. Participants with replicate samples with > 1% discordance among genotype calls were removed. Among these samples, SNPs with missingness > 5%, sex effect (Fisher’s exact p < 10−20) or batch effect (each batch versus all others, Fisher’s exact p < 10−12) were removed. Several participants had two replicate samples remaining after the filtering process. SNPs with > 2% replicate discordance were removed. For each participant, the sample with the higher genotype missingness was removed, and discordant genotypes were excluded.
We imputed genotypes with the Michigan Imputation Server (Das et al., 2016), using data from the Haplotype Reference Consortium (McCarthy et al., 2016) as the reference panel. Loci with imputation quality R2 < 0.8 were excluded.
To prepare the genotype data for association studies, the following additional quality control steps were taken using plink (Chang et al., 2015):
SNPs with minor allele frequency < 1% were removed.
SNPs not in Hardy Weinberg equilibrium (p < 10−6) were removed.
Related individuals (IBD ) were removed.
Samples with missing GDC demographic data (sex and birth year) were removed.
The final genotype data consists of 7,070,031 variants and 5788 samples.
Genetic Ancestry Calculation
Strict ancestry filtering was applied to samples using two techniques: (1) project TCGA samples onto a ten-dimensional principal component (PC)-space derived from principal component analysis (PCA) of all individuals in the 1000 Genomes Project (Auton et al., 2015), and retain only TCGA samples whose five nearest 1000 Genomes neighbors were labeled as “European” and whose mean distance to those neighbors was < 0.1. (2) Run supervised Admixture (Alexander et al., 2009) with K = 3 — using the Utah Residents with Northern and Western European Ancestry (CEU), Yoruba in Ibadan, Nigeria (YRI), and Han Chinese in Beijing, China (CHB) + Japanese in Tokyo, Japan (JPT) populations as reference data — and keep TCGA samples with greater than 90% membership in the CEU cluster.
Phenotype Data
CIBERSORT-derived fraction of 22 types of immune cells (Newman et al., 2015), immune gene expression signatures (Beck et al., 2009; Bindea et al., 2013; Calabrò et al., 2009; Chang et al., 2004; Teschendorff et al., 2010; Wolf et al., 2014), and leukocyte fraction from methylation analysis were downloaded from Thorsson et al. (2018). Cytolytic activity immune signature was added from Rooney et al. (2015). Twenty phenotypes with more than 10% zero values were excluded, with 17 phenotypes remaining. Within each cancer cohort, a rank-based inverse normal transformation was applied to each phenotype. The transformed value of phenotype j for the ith subject in cohort k is:
where rijk is the rank of the ith case in non null observations of phenotype j in cohort k, Njk is the number of non null observations of phenotype j in cohort k, and Ф−1 is the probit function.
SNP-Level and Gene-Level Association Studies
Genome-wide association studies (GWASs) were conducted for 17 phenotypes within each cancer- specific cohort using plink (Chang et al., 2015). The first ten genetic PCs, age, and sex were included in the regression analysis as covariates. We then used METAL (Willer et al., 2010) with a sample size weighting scheme to perform a pan-cancer meta-analysis for each phenotype. SNPs with a calculated p value in all cohort-specific GWASs and a meta-analysis p value less than 5 × 10−8 were reported as significant SNPs. When multiple SNPs in the same haplotype block (r2 > 0.1) were significant, the SNP with the lowest p value is reported. The effect sizes of significant SNPs were calculated using an inverse-variance weighting scheme.
The meta-analysis SNP-level summary statistics were then used as input to the gene-level association test method PEGASUS (Nakka et al., 2016). Gene-level p values are reported for genes with at least one SNP in the gene boundary ± 50kb window (17,563 autosomal genes). Genes with p values less than 2.8 × 10−6 (Bonferroni corrected for 17,563 autosomal genes) were reported as significant. Genes with p values less than 2.9 × 10−5 (Bonferroni corrected for number of independent haplotype blocks in the autosomes, 1703 (Berisa and Pickrell, 2016)) were reported as suggestive.
Candidate Gene Annotation
The candidate genes from the gene-level association studies were annotated using the following methodology:
Mean gene expression (TPM) in each TCGA cohort: RNA-seq data was downloaded for each TCGA cohort from http://firebrowse.org. The patients were subsampled to those included in this study. For each patient, the primary tumor sample was used in these calculations, when available. Otherwise, metastatic tumor samples were used. The TPM values were derived from multiplying the columns labeled “scaled estimates” from files labeled “illuminahiseq rnaseqv2-RSEM genes” by 106.
Mean gene expression (TPM) in immune cells: The mean expression values were downloaded from the DICE database (Schmiedel et al., 2018) for all cell types (https://dice-database.org/download/mean_tpm_merged.csv).
GWAS catalog annotation: Reported associations were downloaded from the GWAS Catalog (http://www.ebi.ac.uk/gwas/) on December 28, 2018. The GWAS traits were recorded from the “MAPPED TRAIT” column, and categorized into immune, autoimmune, or cancer related traits.
Cancer Gene Census annotation: Genes in the Cancer Gene Census were downloaded from https://cancer.sanger.ac.uk/census. In this database, genes are designated as tier 1 or tier 2 depending on the available literature evidence.
Correlation between gene expression and tumor purity: The gene expression (TPM) was calculated for every gene and every sample. See (1) for source of gene expression data. The tumor purity data for each sample was calculated using ABSOLUTE and downloaded from https://api.gdc.cancer.gov/data/4f277128-f793-4354-a13d-30cc7fe9f6b5. The Pearson correlation coefficient per gene was calculated between gene expression and tumor purity across samples.
Network Propagation Analysis
We performed network propagation analysis with Hierarchical HotNet (Reyna et al., 2018) on the −log10-transformed p values from gene-level association testing to identify significantly altered subnetworks. For our analysis, we used the following interaction net-works, which were the most recent versions available as of February 23, 2018.
HINT+HI (Das and Yu, 2012; Rolland et al., 2014): HINT binary + HINT co-complex + HuRI HI
iRefIndex 15.0 (Razick et al., 2008)
ReactomeFI 2016 (Fabregat et al., 2018)
For the ReactomeFI network, we considered the set of interactions with a confidence score of 0.75 (out of 1) or larger. For each network, we restricted our attention to the largest connected subgraph of the network.
To reduce the influence of genes for which we have low confidence of association with a phenotype, we assigned p values of 1 to genes with p values of p > 0.1 and ran Hierarchical HotNet (103 permutations) on these thresholded gene scores. This provides sparser, more interpretable, and higher confidence networks. Similar p value thresholds were applied in similar network analyses (Nakka et al., 2016).
Polygenic Risk Score Analysis
We downloaded the summary statistics from GWASs of five autoimmune traits: celiac disease (Dubois et al., 2010); multiple sclerosis (Sawcer et al., 2011); ulcerative colitis (Anderson et al., 2011); rheumatoid arthritis (Okada et al., 2014); and systemic lupus erythematosus (Bentham et al., 2015). Records with missing odds ratio, p values, and risk alleles were excluded from analysis. For each autoimmune disease, we extracted SNPs at various p value thresholds (p = 1, 10−1, 10−2, 10−3, 10−4, 10−5, 10−6, 10−7, 5 × 10−8) that overlapped with our genotype data, excluding ambiguous and mismatched variants. At each threshold, the SNPs were filtered via linkage disequilibrium (LD) clumping, with a 250kb window and an r2 threshold of 0.1 (Table S3). PRSice (Euesden et al., 2015) was used to calculate the polygenic risk score (PRS) for each autoimmune trait for each sample by summing over the log odds ratio of the selected SNPs, weighted by allele dosage of risk alleles.
The PRS for each disease was regressed against each of the 17 immune infiltration phenotypes within each cancer cohort, using the first 10 PCs, birth year, and sex as covariates. The reported results are from a sample size based meta-analysis of all cancer cohorts. Effect sizes of significant associations (Bonferroni corrected for number of immune infiltration phenotypes tested) were calculated using an inverse-variance weighted analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of all analyses are reported in the Results, figure legends, and Method Details.
DATA AND CODE AVAILABILITY
The raw germline data is available from FireCloud (https://software.broadinstitute.org/firecloud/) and GDC legacy archive (https://portal.gdc.cancer.gov/legacy-archive). The phenotype data is available from the original published sources Rooney et al. (2015) and Thorsson et al. (2018). The software used for the analyses are referenced in the Method Details subsections and Key Resources Table.
KEY RESOURCES TABLE
Supplementary Material
Highlights.
Tumor immune infiltration impacts response to immunotherapy
GWAS identifies inherited genetic variants associated with immune infiltration
Aggregating variants into genes and networks increases power to find associations
Germline associations may offer insight into predictors of response to immunotherapy
ACKNOWLEDGMENTS
We thank the patients who contributed to the TCGA study. The results published here are based upon data generated by the TCGA Research Network. The study was supported by the following funding sources: NSF CAREER DBI-1452622 (S.R.); NIH R01 GM118652 (S.R.); NIH U01 CA217875 (M.A.R.); Brown University Sidney Frank Fellowship (S.S.); PCF-V Foundation Challenge Award (E.M.V.A.); NIH R01 CA227388 (E.M.V.A.); and NIH U01 CA233100 (E.M.V.A.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.039.
DECLARATION OF INTERESTS
E.M.V.A. serves in an advisory/consulting role for the following corporations: Tango Therapeutics, Genome Medical, Invitae, Illumina, Ervaxx, and Janssen. He receives research support from Novartis and Bristol-Myers Squibb. He owns equity in Tango Therapeutics, Genome Medical, Syapse, Microsoft, and Ervaxx. He has received travel reimbursement from Roche/Genentech. He holds institutional patents filed on ERCC2 mutations and chemotherapy response, chromatin mutations and immunotherapy response, and methods for clinical interpretation.
REFERENCES
- Ahola-Olli AV, Würtz P, Havulinna AS, Aalto K, Pitkänen N, Lehtimäki T, Kähönen M, Lyytikäinen LP, Raitoharju E, Seppälä I, et al. (2017). Genome-wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am. J. Hum. Genet 100, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón-Riquelme ME, Ziegler JT, Molineros J, Howard TD, Moreno-Estrada A, Sánchez-Rodríguez E, Ainsworth HC, Ortiz-Tello P, Comeau ME, Rasmussen A, et al. (2016). Genome-wide association study in an Amerindian ancestry population reveals novel systemic lupus erythematosus risk loci and the role of European admixture. Arthritis Rheumatol. 68, 932–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, and Lange K (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet 43, 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, et al. (2016). The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167, 1415–1429.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR; 1000 Genomes Project Consortium (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, Varma S, Marinelli RJ, van de Rijn M, and West RB (2009). The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin. Cancer Res 15, 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, Martín J, Fairfax BP, Knight JC, Chen L, et al. (2015). Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet 47, 1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berisa T, and Pickrell JK (2016). Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatio-temporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Bønnelykke K, Matheson MC, Pers TH, Granell R, Strachan DP, Alves AC, Linneberg A, Curtin JA, Warrington NM, Standl M, et al. ; AAGC (2013). Meta-analysis of genome-wide association studies identifies ten loci influencing allergic sensitization. Nat. Genet 45, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malan-gone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. (2019). The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47 (D1), D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrò A, Beissbarth T, Kuner R, Stojanov M, Benner A, Asslaber M, Ploner F, Zatloukal K, Samonigg H, Poustka A, and Sültmann H (2009). Effects of infiltrating lymphocytes and estrogen receptor on gene expression and prognosis in breast cancer. Breast Cancer Res. Treat 116, 69–77. [DOI] [PubMed] [Google Scholar]
- Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, et al. ; GTEx Consortium (2015). A novel approach to high-quality postmortem tissue procurement: The GTEx Project. Biopreserv. Biobank 13, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, and Brown PO (2004). Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2, E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, and Trajanoski Z (2017). Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262. [DOI] [PubMed] [Google Scholar]
- Chen CF, Ruiz-Vega R, Vasudeva P, Espitia F, Krasieva TB, de Feraudy S, Tromberg BJ, Huang S, Garner CP, Wu J, et al. (2017). ATR mutations promote the growth of melanoma tumors by modulating the immune microenvironment. Cell Rep. 18, 2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho BA, Sim JH, Park JA, Kim HW, Yoo WH, Lee SH, Lee DS, Kang JS, Hwang YI, Lee WJ, et al. (2012). Characterization of effector memory CD8+ T cells in the synovial fluid of rheumatoid arthritis. J. Clin. Immunol 32, 709–720. [DOI] [PubMed] [Google Scholar]
- Clifford RJ, Zhang J, Meerzaman DM, Lyu MS, Hu Y, Cultraro CM, Finney RP, Kelley JM, Efroni S, Greenblum SI, et al. (2010). Genetic variations at loci involved in the immune response are risk factors for hepatocellular carcinoma. Hepatology 52, 2034–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, and Yu H (2012). HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol 6, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. (2016). Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, Heap GA, Adány R, Aromaa A, et al. (2010). Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet 42, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanuel AO, Arnovitz S, Haghi L, Mathur PS, Mondal S, Quandt J, Okoreeh MK, Maienschein-Cline M, Khazaie K, Dose M, and Gounari F (2018). TCF-1 and HEB cooperate to establish the epigenetic and transcription profiles of CD4+CD8+ thymocytes. Nat. Immunol 19, 1366–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euesden J, Lewis CM, and O’Reilly PF (2015). PRSice: polygenic risk score software. Bioinformatics 31, 1466–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome pathway knowledgebase. Nucleic Acids Res. 46 (D1), D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawdar S, Trotter EW, Li Y, Stephenson NL, Hanke F, Marusiak AA, Edwards ZC, Ientile S, Waszkowycz B, Miller CJ, and Brognard J (2013). Targeted genetic dependency screen facilitates identification of actionable mutations in FGFR4, MAP3K9, and PAK5 in lung cancer. Proc. Natl. Acad. Sci. USA 110, 12426–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, Vonk JM, Baurecht H, Marenholz I, Tian C, Hoffman JD, Helmer Q, Tillander A, Ullemar V, van Dongen J, et al. ; 23andMe Research Team; AAGC collaborators; BIOS consortium; LifeLines Cohort Study (2017). Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet 49, 1752–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, and Bluestone JA (2009). Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol 10, 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, and Baldari CT (2009). Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat. Cell Biol 11, 1332–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45 (D1), D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, and Stratton MR (2004). A census of human cancer genes. Nat. Rev. Cancer 4, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. (2015). The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med 21, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. (2016). Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 15, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlova OY, Li Y, Gorlov I, Ying J, Chen WV, Assassi S, Reveille JD, Arnett FC, Zhou X, Bossini-Castillo L, et al. (2018). Gene-level association analysis of systemic sclerosis: A comparison of African-Americans and White populations. PLoS ONE 13, e0189498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, Paczkowska M, Reynolds S, Wyczalkowski MA, Oak N, et al. ; Cancer Genome Atlas Research Network (2018). Pathogenic germline variants in 10,389 adult cancers. Cell 173, 355–370.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Jin HR, Jung HS, Lee SJ, Lee J-H, and Lee JJ (2010). An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-kappaB-inducing kinase via Lys63-linked ubiquitination. J. Biol. Chem 285, 30539–30547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan TE, Burke KP, and Van Allen EM (2019). Genomic correlates of response to immune checkpoint blockade. Nat. Med 25, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, Schoech A, Pasaniuc B, and Price AL (2019). Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet 104, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Minna JD, and White MA (2013). GWAS meets TCGA to illuminate mechanisms of cancer predisposition. Cell 152, 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. ; Cancer Genome Atlas Research Network (2018). Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 23, 239–254.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. (2008). Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet 40, 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law PJ, Timofeeva M, Fernandez-Rozadilla C, Broderick P, Studd J, Fernandez-Tajes J, Farrington S, Svinti V, Palles C, Orlando G, et al. ; PRACTICAL consortium (2019). Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun 10, 2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Chen-Harris H, Mayba O, Lianoglou S, Wuster A, Bhangale T, Khan Z, Mariathasan S, Daemen A, Reeder J, et al. (2018). Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc. Natl. Acad. Sci. USA 115, E11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, Hayward NK, Montgomery GW, Visscher PM, Martin NG, and Macgregor S; AMFS Investigators (2010). A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet 87, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longoni N, Kunderfranco P, Pellini S, Albino D, Mello-Grand M, Pinton S, D’Ambrosio G, Sarti M, Sessa F, Chiorino G, et al. (2013). Aberrant expression of the neuronal-specific protein DCDC2 promotes malignant phenotypes and is associated with prostate cancer progression. Oncogene 32, 2315–2324, 2324.e1–2324.e4. [DOI] [PubMed] [Google Scholar]
- Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, de Prisco N, Ideker T, Hildebrand WH, Font-Burgada J, and Carter H (2017). MHC-I genotype restricts the oncogenic mutational landscape. Cell 171, 1272–1283.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty Pyke R, Thompson WK, Salem RM, Font-Burgada J, Zanetti M, and Carter H (2018). Evolutionary pressure against MHC class II binding cancer mutations. Cell 175, 416–428.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. ; Haplotype Reference Consortium (2016). A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, and King MC (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. [DOI] [PubMed] [Google Scholar]
- McInnes IB (2003). Leukotrienes, mast cells, and T cells. Arthritis Res. Ther 5, 288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, Caporaso NE, Johansson M, Xiao X, Li Y, et al. ; SpiroMeta Consortium (2017). Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet 49, 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D, and Van Allen EM (2016). Genomic determinants of cancer immunotherapy. Curr. Opin. Immunol 41, 32–38. [DOI] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bossé D, Wankowicz SM, Cullen D, et al. (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K, Lindström S, Dennis J, Beesley J, Hui S, Kar S, Lema-çon A, Soucy P, Glubb D, Rostamianfar A, et al. ; NBCS Collaborators; ABCTB Investigators; ConFab/AOCS Investigators (2017). Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modur V, Singh N, Mohanty V, Chung E, Muhammad B, Choi K, Chen X, Chetal K, Ratner N, Salomonis N, et al. (2018). Defective transcription elongation in a subset of cancers confers immunotherapy resistance. Nat. Commun 9, 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw KW, Goldberg MS, Konstantinopoulos PA, and D’Andrea AD (2017). DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 7, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakka P, Raphael BJ, and Ramachandran S (2016). Gene and network analysis of common variants reveals novel associations in multiple complex diseases. Genetics 204, 783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, and Sham PC (2004). The future of association studies: gene-based analysis and replication. Am. J. Hum. Genet 75, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AC, Eisenberg JM, Heath RJ, Huett A, Robinson CM, Nau GJ, and Xavier RJ (2011). Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4631–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. ; RACI consortium; GARNET consortium (2014). Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrù V, Steri M, Sole G, Sidore C, Virdis F, Dei M, Lai S, Zoledziewska M, Busonero F, Mulas A, et al. (2013). Genetic variants regulating immune cell levels in health and disease. Cell 155, 242–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, et al. ; CORGI Consortium; WGS500 Consortium (2013). Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet 45, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes M, Cortes A, van Heel DA, and Brown MA (2013). Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet 14, 661–673. [DOI] [PubMed] [Google Scholar]
- Peltekova VD, Lemire M, Qazi AM, Zaidi SH, Trinh QM, Bielecki R, Rogers M, Hodgson L, Wang M, D’Souza DJ, et al. (2014). Identification of genes expressed by immune cells of the colon that are regulated by colorectal cancer-associated variants. Int. J. Cancer 134, 2330–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, et al. (2012). Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet 44, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razick S, Magklaras G, and Donaldson IM (2008). iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics 9, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyna MA, Leiserson MDM, and Raphael BJ (2018). Hierarchical HotNet: identifying hierarchies of altered subnetworks. Bioinformatics 34, i972–i980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roederer M, Quaye L, Mangino M, Beddall MH, Mahnke Y, Chattopadhyay P, Tosi I, Napolitano L, Terranova Barberio M, Menni C, et al. (2015). The genetic architecture of the human immune system: a bioresource for autoimmunity and disease pathogenesis. Cell 161, 387–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. (2014). A proteome-scale map of the human interactome network. Cell 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet 51, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, et al. ; International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2 (2011). Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, Ha B, Altay G, Greenbaum JA, McVicker G, et al. (2018). Impact of genetic polymorphisms on human immune cell gene expression. Cell 175, 1701–1715.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels MS, Engels EA, Shi J, Landi MT, Albanes D, Chatterjee N, Chanock SJ, Caporaso NE, and Chaturvedi AK (2012). Genetic variation in innate immunity and inflammation pathways associated with lung cancer risk. Cancer 118, 5630–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemers NO, Holloway JL, Chang H, Chasalow SD, Ross-MacDonald PB, Voliva CF, and Szustakowski JD (2017). Genome-wide association analysis identifies genetic correlates of immune infiltrates in solid tumors. PLoS ONE 12, e0179726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Stahl EA, and Raj T (2011). Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 187, 367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Weinel S, Nagy N, O’Driscoll M, Lai-Cheong JE, Kulp-Shorten CL, Knable A, Carpenter G, Fisher SA, Hiragun M, et al. (2012). Germline mutation in ATR in autosomal- dominant oropharyngeal cancer syndrome. Am. J. Hum. Genet 90, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Gomez S, Arenas A, El-Ashry D, Schmidt M, Gehr-mann M, and Caldas C (2010). Improved prognostic classification of breast cancer defined by antagonistic activation patterns of immune response pathway modules. BMC Cancer 10, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. ; Cancer Genome Atlas Research Network (2018). The immune landscape of cancer. Immunity 48, 812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, and Sharpe AH (1995). Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547. [DOI] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet 11, 843–854. [DOI] [PubMed] [Google Scholar]
- Wang H, Schmit SL, Haiman CA, Keku TO, Kato I, Palmer JR, van den Berg D, Wilkens LR, Burnett T, Conti DV, et al. ; Hispanic Colorectal Cancer Study (2017). Novel colon cancer susceptibility variants identified from a genome-wide association study in African Americans. Int. J. Cancer 140, 2728–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y, and Abecasis GR (2010). METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik GL, Kao WH, and Duggal P (2015). Relative performance of gene-and pathway-level methods as secondary analyses for genome-wide association studies. BMC Genet. 16, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DM, Lenburg ME, Yau C, Boudreau A, and van ‘t Veer LJ (2014). Gene co-expression modules as clinically relevant hallmarks of breast cancer diversity. PLoS ONE 9, e88309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Kraft P, Epstein MP, Taylor DM, Chanock SJ, Hunter DJ, and Lin X (2010). Powerful SNP-set analysis for case-control genome-wide association studies. Am. J. Hum. Genet 86, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Kraft P, Stolzenberg-Solomon R, Steplowski E, Brotzman M, Xu M, Mudgal P, Amundadottir L, Arslan AA, Bueno-de-Mesquita HB, et al. (2014). Genome-wide association study of survival in patients with pancreatic adenocarcinoma. Gut 63, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw germline data is available from FireCloud (https://software.broadinstitute.org/firecloud/) and GDC legacy archive (https://portal.gdc.cancer.gov/legacy-archive). The phenotype data is available from the original published sources Rooney et al. (2015) and Thorsson et al. (2018). The software used for the analyses are referenced in the Method Details subsections and Key Resources Table.
KEY RESOURCES TABLE